
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silver nanoparticles confined in carbon nanotubes: on the understanding of the confinement effect and promotional catalysis for the selective hydrogenation of dimethyl oxalate†
Jianwei
Zheng
,
Xinping
Duan
*,
Haiqiang
Lin
,
Zhengqiang
Gu
,
Huihuang
Fang
,
Jianhui
Li
and
Youzhu
Yuan
*
State Key Laboratory of Physical Chemistry of Solid Surfaces, National Engineering Laboratory for Green Chemical Productions of Alcohols-Ethers-Esters, and iChEM, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: xpduan@xmu.edu.cn; yzyuan@xmu.edu.cn; Fax: +86-592-2183047
First published on 15th February 2016
Abstract
A confined Ag nanomaterial in the channels of herringbone multi-walled carbon nanotubes (Ag-in/hCNT) was effectively prepared. The space restriction induces morphological changes of Ag nanoparticles into rough nanowires with an estimated aspect ratio of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 (nm/nm). Dihydrogen activation is enhanced through the vacancy-enriched wire-like Ag nanocatalyst, as well as the confinement effect. The grain boundaries of Ag and rolled-up graphene layers of CNTs are speculated to play vital roles in the diffusion of activated hydrogen species. The Ag-in/hCNT catalyst exhibits an activity that is three times higher than that of Ag nanoparticles located on the CNT exterior walls in DMO hydrogenation. This finding may insinuate that interplanar spaces provide available access to the external surface of CNTs. Designed experiments further confirm the importance of herringbone CNTs with higher reaction rate than parallel CNTs, and confined Ag produces considerably more activated hydrogen species, thereby benefiting the reduction of surface copper nanoparticles or DMO molecules during hydrogenation. This paper presents a study of the effective utilization of hydrogen over herringbone CNT confined Ag and an understanding of the confinement and promotional catalytic effects.
:8 (nm/nm). Dihydrogen activation is enhanced through the vacancy-enriched wire-like Ag nanocatalyst, as well as the confinement effect. The grain boundaries of Ag and rolled-up graphene layers of CNTs are speculated to play vital roles in the diffusion of activated hydrogen species. The Ag-in/hCNT catalyst exhibits an activity that is three times higher than that of Ag nanoparticles located on the CNT exterior walls in DMO hydrogenation. This finding may insinuate that interplanar spaces provide available access to the external surface of CNTs. Designed experiments further confirm the importance of herringbone CNTs with higher reaction rate than parallel CNTs, and confined Ag produces considerably more activated hydrogen species, thereby benefiting the reduction of surface copper nanoparticles or DMO molecules during hydrogenation. This paper presents a study of the effective utilization of hydrogen over herringbone CNT confined Ag and an understanding of the confinement and promotional catalytic effects.
1. Introduction
For heterogeneous supported Ag nanomaterials, the importance of the physiochemical features of Ag nanoparticles (NPs) and their interaction with carriers, such as silica, alumina, and carbon materials, have been intensively recognized.1–3 Among these carriers, carbon nanotubes (CNTs) are distinguished and have triggered extensive research interest for catalytic applications because of their advantageous thermal and electrical properties, high surface area, functionalized surfaces, and unique tubular channels.4,5 Unlike other carbon materials, such as graphite and graphene, CNTs can be envisioned as rolled-up graphene layers forming a one-dimensional tubular structure with abundant sp3 electronic orbits.6 The morphology and electronic structure of CNTs enable them to serve as a specific template for preparing metal CNT hybrids as catalysts.3–6 The combination of metal and CNTs is particularly useful in integrating the properties of the two components, as abundant stereoelectronic orbits allow a strong electronic effect, affecting the absorption behavior of reactants and supported metals.6In addition, a special long-range and one-dimensional channel bestows CNTs with particular properties and widens their applications.6,7 One of the most prominent examples is the utilization of the interior surface of CNTs to encapsulate metals for heterogeneous catalysis. The encapsulated metals were found to exhibit excellent catalytic performance in ethanol synthesis, CO oxidation, and Fischer–Tropsch synthesis.8–10 In terms of the confinement effect, the interaction of reactants and products with CNT walls, optimized redox properties of metal catalysts within the CNTs, and chemical reactions over metallic active sites confined inside the CNT channels can be expected to yield different physiochemical behaviors. Reaction issues involving activity, selectivity, and stability can be improved through reconstruction of the encapsulated metallic CNT composites.6 For instance, Pd NPs were effectively introduced inside multi-walled CNTs with 5–10 nm inner diameter channels for benzene hydrogenation.11 The catalytic activity of confined Ag was considerably higher than that over catalysts supported by activated carbon (AC) and Y zeolite, even though multi-walled CNTs possess markedly smaller specific surface areas than AC and Y zeolite. Pd NPs deposited inside multi-walled CNTs were also utilized as catalysts for the selective hydrogenation of cinnamaldehyde, and the hydrogenation selectivities towards the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds were significantly different from those over the conventional catalyst with similar conversion.12 Depending on the orientation angles of rotation of the graphitic layers relative to the tube axis, multi-walled CNTs can be distinguished as parallel-type and herringbone-type. Despite the widespread use of multi-walled CNTs, the importance of the rolled-up layer structure and the effect and mechanism of confinement on the catalytic performance are barely discussed.
C and CO bonds were significantly different from those over the conventional catalyst with similar conversion.12 Depending on the orientation angles of rotation of the graphitic layers relative to the tube axis, multi-walled CNTs can be distinguished as parallel-type and herringbone-type. Despite the widespread use of multi-walled CNTs, the importance of the rolled-up layer structure and the effect and mechanism of confinement on the catalytic performance are barely discussed.
More recently, a significant integrated technology that involves the coupling of syngas-derived methanol with CO to form dimethyl oxalate and subsequent hydrogenation to yield methyl glycolate (MG) and ethylene glycol (EG) has garnered considerable attention worldwide.13–15 Although the second step of EG production has been successfully scaled up into industrial applications, MG production has been neglected. MG is an important fine chemical with more added value in comparison with EG. It is widely used in the synthesis of numerous pharmaceutical products, fine chemicals, and perfumes. These MG-derived products are biodegradable and environmentally friendly. Therefore, MG presents an enormous potential market. However, the main difficulty of MG synthesis through the DMO intermediate approach lies in the design of effective catalysts with high reactivity and selectivity. Supported copper catalysts are preferentially used for DMO hydrogenation to EG and ethanol.13–16 However, the mono-hydrogenated (unilateral hydrogenated) product, MG, is usually inferior over copper catalysts because of the high thermodynamic constant of the subsequent hydrogenation of MG to EG.17 Silica oxide-supported Ag catalysts were recently reported to possess high MG yield for the chemoselective hydrogenation of DMO to MG.1,17,18 The interaction of Ag with multi-walled CNTs and the implication of confinement in hydrogenation have been scarcely investigated.
In this work, confined/unconfined Ag catalysts in different kinds of CNTs were fabricated and applied in DMO hydrogenation to MG for the comparison of dihydrogen activation capacity. Unlike the previously reported confined metal CNT hybrid catalysts,6–12 the filling Ag exists in the morphology of nanowires inside the CNT channel. Unexpectedly, these confined Ag-in/hCNT catalysts were found to exhibit superior activity and selectivity, even though blocking of CNTs may hinder the accessibility of the metals in the inner part of the channels to reactants and significant diffusion resistances for products to exhaust out of the channels. For comparison, Ag catalysts supported on various carriers were prepared to investigate the interaction of Ag NPs and supports and the hydrogenation mechanism. Comprehensive characterization was performed through transmission electron microscopy (TEM), hydrogen temperature-programmed reduction (H2-TPR), X-ray diffraction (XRD), and chemisorption analysis to correlate the geometrical and surface electronic structure with catalytic performance. Finally, a plausible mechanism of hydrogen transfer was proposed.
2. Experimental
2.1 Synthesis of materials
2.2 Physicochemical characterization
2.3 Catalytic performance
The DMO hydrogenation reaction was tested in a stainless steel tubular reactor equipped with a computer-controlled auto-sampling system in a continuous flow mode. Typically, 100 mg of the catalyst precursor (40–60 meshes) was sandwiched with quartz powder (40–60 meshes) and loaded in the center of the reactor. Before the catalytic performance was started, all Ag-based precursors were pre-reduced under a 5% H2–95% N2 atmosphere at 623 K for 4 h with ramping rates of 2 K min−1. The catalyst bed was naturally cooled to 493 K, and pure H2 was fed into the reactor. The system pressure was maintained at 3.0 MPa with the aid of a back-pressure regulator. Subsequently, 0.02 g mL−1 DMO methanol solution was pumped into the catalyst bed with a Series III digital HPLC pump (Scientific Systems, Inc.). The products were analyzed on-line using GC7890-MS5875C. The DMO conversion for the calculation of the reaction rates at different reaction temperatures was around 50%. The apparent activation energy was obtained by the Arrhenius equation.3. Results and discussion
3.1 Comparison of Ag-based materials
Support and size effects are important issues when comparing different materials for heterogeneous catalysis, especially for structure-sensitive reactions. The support carries the nanocrystallite and directly affects the morphology and particle size of the active site. Moreover, the support may also influence substrate sorption because of different properties, such as surface acid–base properties. When the interaction between metal nanocrystallite and the support is weak, the morphology of the nanocrystallite is always spherical with a large size.20 Otherwise, nanocrystallites may be angular and present a small crystallite size. The crystallite size directly accelerates the number of active sites and, consequently, the catalytic ability.21 In general, catalysts with smaller crystallite sizes possess better catalytic activity and reaction rate.Hydrogenation of DMO involves successive hydrogenolysis and hydrogenation of C–O bonds, mainly producing several important chemicals, such as MG, EG, and ethanol (EtOH) at different levels. The reaction scope is shown in Table 1. Previous studies found that the performance of DMO hydrogenation is dependent on the Ag size when the crystallite size is larger than 3.6 nm. Using non-carbon materials as supports, Ag crystallites follow the size effect rule during DMO hydrogenation, which is in accordance with previous studies.17 Ag/H2SiO3 with the smallest crystallite size of 4 nm (Fig. S1†), exhibited the best reaction rate and product yield of 91.3% (56.9% MG and 35.4% EG). However, we unexpectedly found no discernable correlation between the Ag crystallite size and the catalytic activity for Ag-based carbon materials. Ag-in/hCNT with the largest crystallite size of 18 nm presented the best activity and selectivity, which were better than those of the Ag/H2SiO3 catalyst.
|
|
|||||
|---|---|---|---|---|---|
| Carrier | Sizea (nm) | S BET (m2 g−1) | R (mmol h−1) | Yield (%) | |
| MG | EG | ||||
| a Crystallite size was calculated by using the Scherrer equation. b R represents the rate of DMO disappearance. c Activated carbon. d Graphene. e Carbon nanofibers. f Not detected. | |||||
| γ-Al2O3 | 5 | 186 | 46.5 | 37.7 | 39.1 |
| SiO2 | 16 | 312 | 40.7 | 76.5 | 3.7 |
| H2SiO3 | 4 | 201 | 46.9 | 56.9 | 35.4 |
| ACc | 7 | 96 | 7.5 | 14.6 | 0 |
| GAd | ndf | 18 | 5.0 | 9.8 | 0 |
| CNFe | nd | 5 | 1.8 | 3.5 | 0 |
| CNT | 18 | 145 | 50.3 | 98.9 | 0.1 |

The abnormal phenomenon implied that, in a reduction and oxidation atmosphere of hydrogen and ester, Ag species is not the only decisive factor that affects catalytic performance. The choice of solid carrier used may significantly influence the catalytic activity. Therefore, the support effect should be considered as an emergent issue for this reaction. A providential support possessing a large surface area, suitable pore structure, and appropriate surface acidity is an ideal carrier for DMO hydrogenation. The use of acidic H2SiO3 and basic Al2O3 as supports for Ag catalysts cannot achieve good selectivity. However, high MG selectivity was preserved for Ag-supported inert carbon materials and silica oxides. The results indirectly demonstrated that carriers may affect the absorption of the substrate and cause differences in product selectivity. When considering the absorption properties of support materials, the size effect will be indistinct. Among various carbon material-supported Ag, CNT-supported Ag with the largest crystallite size showed superior catalytic activity. The specificity of CNTs should be carefully considered. CNTs possess a large external surface with oxidized vacancies and localized double bonds, electron transfer between the metal and CNTs, special ability in hydrogen storage, confined space of the nanochannel, relatively low limitations for mass transfer, and high thermal conductivity.22 Given their intrinsic features, CNTs play a beneficial role in numerous catalytic applications.6–13
3.2 Morphology of Ag-in/hCNT and Ag-out/hCNT and their catalytic performance
The morphologies of Ag located in or out of the CNT hybrid channels were investigated by TEM. Interestingly, in the case of the Ag-in/hCNT catalyst, an estimated 80% of the observed Ag NPs was found in the internal channels of CNTs in long wire shapes, as observed from the TEM snapshots (Fig. 1a). Large Ag crystallites were restricted and had to block the channels, further reducing the exposed surface. In theory, this blocking of Ag crystallites should present harsh internal diffusion limitations and minimal active surface area. Therefore, regardless of the view of mass transfer or point of size effect, the pore-plugging Ag-in/hCNT catalyst should exhibit low catalytic activity, which was contrary to the experimental results.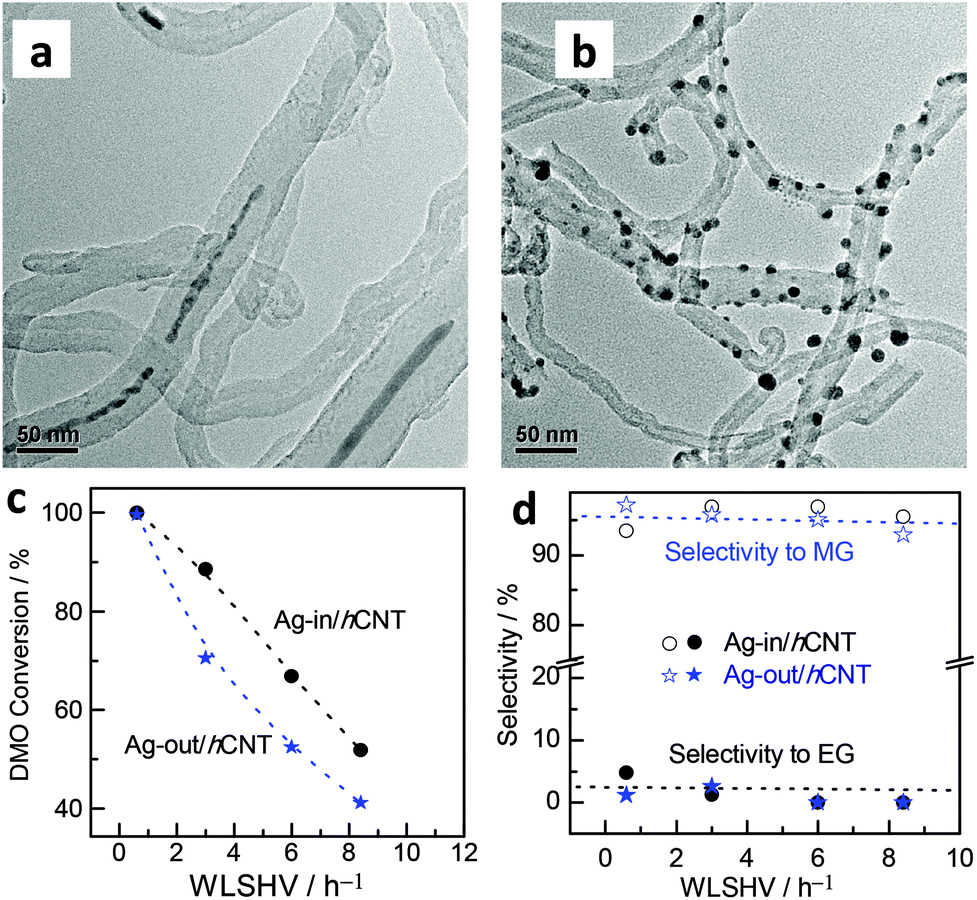 | ||
| Fig. 1 TEM images of (a) Ag-in/hCNT and (b) Ag-out/hCNT; (c) conversion of DMO and (d) MG selectivity using Ag catalysts. | ||
By contrast, Ag crystallites implanted on the external surface of CNTs (Fig. 1b) were prepared by the classical excessive impregnation method. Ag crystallites grew on the external surface without confinement into CNTs. The average particle size was calculated from XRD based on the Scherrer equation. Ag-out/hCNT catalysts possess a smaller crystallite Ag size of 11 nm, whereas the Ag crystallite in the Ag-in/hCNT catalyst is approximately 18 nm. This value cannot conclude the accurate specification of Ag nanowires, but we can calculate the specification by translating the nanowires into a spherical model. Based on 4/3 × πr3 = L × π(d/2)2, where the spherical radius (r) is 9 nm and the CNT channel size (d) is about 8 nm, the average nanowire length (L) is obtained as 60 nm. In addition, the morphology of most Ag crystallites is spherical, indicating that the interaction between outer Ag and CNT is weak. By contrast, confined Ag, which is shaped into wires, may strengthen the interaction of Ag and CNTs.
However, abnormal activity was observed (Fig. 1c). At a weight liquid hourly space velocity (WLHSV) of 0.6 h−1, both catalysts exhibited an ideal performance, with 100% conversion of DMO. Ag crystallites confined inside the channel of CNTs displayed a higher level of conversion and reaction rate compared with Ag-out/hCNT under elevated LHSV. However, activity tests showed that MG was the dominant product (Fig. 1d), regardless of whether Ag crystallites were in or out of CNTs. Therefore, minimal internal diffusion limitations existed to inhibit the process of chemoselective hydrogenation of DMO to MG. Considering that high conversion was obtained over Ag-in/hCNT, the hydrogenation of CO bonds should mainly occur on the outer surface of CNTs so that the reaction could proceed, regardless of the occurrence of pore plugging.
3.3 Ability to activate dihydrogen
Although hydrogen is not prone to forming bonds with Ag as the d-orbital of Ag is filled,23 especially for larger Ag nanocrystallites with few defects and grain boundaries, the metallic Ag could absorb hydrogen in our case, rendering CO hydrogenation reaction possible. The electronic structure of metallic Ag should be subtly modified. H2 chemisorption (Fig. 2a) was conducted to compare the activation ability of Ag dependent on the located position relative to CNTs. Notably, Ag confined inside CNTs possessed almost twice the amount of H2 sorption than that of outside Ag. We further used Arrhenius plots to derive apparent activation energies (Ea) (Fig. 2b). Ea was 349 kJ mol−1 for Ag-out/hCNT, whereas the value for Ag-in/hCNT was approximately 221 kJ mol−1. Ag located inside the channel of CNTs benefits more for H2 activation, which is crucial for hydrogenation reactions. The confined environment can enrich the reactant atmosphere.6 In our case, H2 may be concentrated inside the channels of CNTs, facilitating the hydrogenation reaction toward hydroproducts. Both H2 chemisorption and apparent activation energies revealed that confined Ag possessed stronger ability to activate dihydrogen and facilitated hydrogenation.
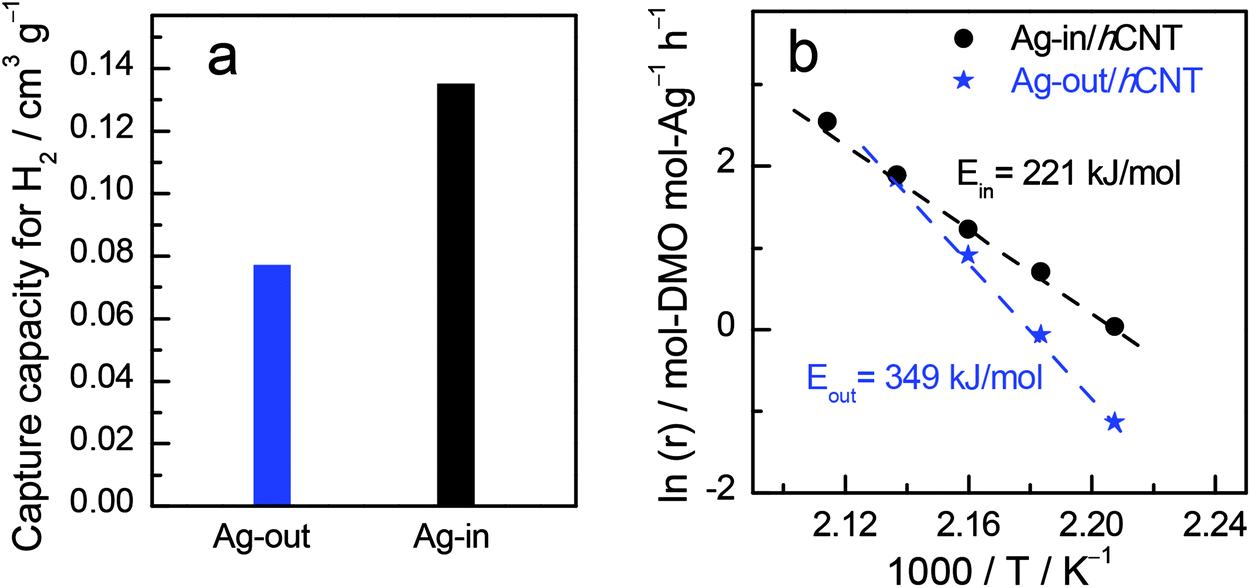 | ||
| Fig. 2 Comparison of (a) H2 chemisorption and (b) apparent activation energies. The symbol r means the reaction rate, moles of DMO converted by per mole of Ag per hour (mol-DMO per mol-Ag per h). | ||
3.4 Probe into the texture structure for H2 activation
As shown in Fig. 1 and 3, confined Ag NPs were oppressed and stretched into wires by the interior wall of CNTs, whereas free Ag atoms integrated into spherical particles on the exterior wall of CNTs. As sketched from high-resolution TEM, both Ag nanowires inside CNTs and Ag particles outside CNTs possessed grain boundaries, in which atom vacancies were enriched. The former exhibited a rougher surface, with more grain boundaries. Balluffi and Seigle showed that sufficient vacancies can be produced at transverse grain boundaries because of the tension acting across the boundary.24 The stress from the interior walls of CNTs and the confined environment cause the stretching of Ag particles, leading to the emergence of atom dislocation and the consequent formation of grain boundaries and vacancies. Grain boundaries are recommended as rapid diffusion paths for hydrogen species, and atomic hydrogen species will emerge and transfer onto vacancies. The activation of H2 is further improved at accessorial vacancies in the grain boundaries. We previously proved that metallic Ag is structurally sensitive to the activity of DMO hydrogenation. Vacancies can be regarded as open volume defects and are able to trap hydrogen species.25 The concentrated hydrogen species is beneficial for DMO hydrogenation. This finding can explain why Ag-in/hCNT possesses stronger H2 chemisorption and reduced activation energy barrier.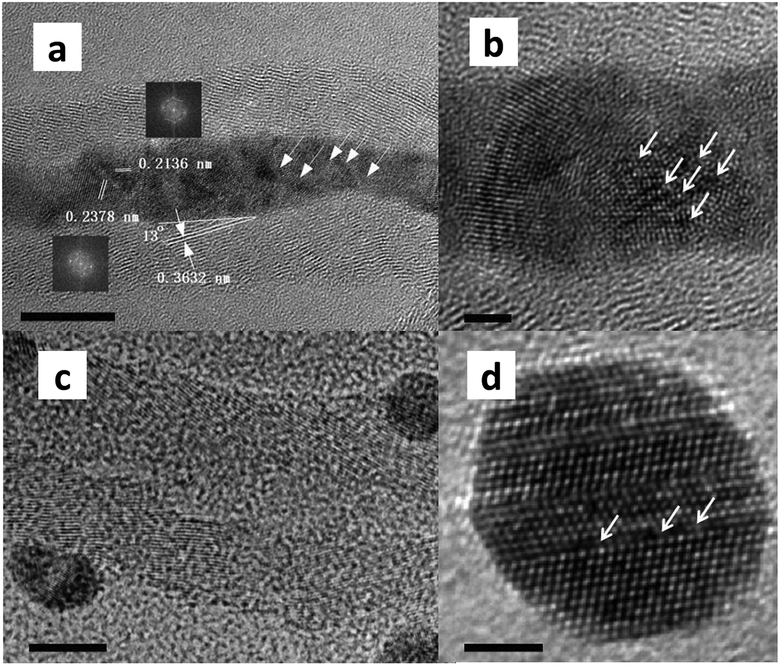 | ||
| Fig. 3 High-resolution TEM images of (a) and (b) Ag-in/hCNT; (c) and (d) Ag-out/hCNT. The bar of (a) and (c) is 10 and 2 mm for (b) and (d). The inserted figures show the FFT changes of Ag crystallites and were used to calculate lattice spacing. The arrows show the vacancies in Ag crystallites. | ||
3.5 Approach of H2 diffusion
However, the mechanism underlying the reaction of activated hydrogen species on the Ag nanowires with DMO molecule remains unclear. Given that no entries are available for the diffusion of a comparatively larger molecule DMO, we can easily speculate that CNTs should be able to connect the activated hydrogen species and the atmospheric DMO. Previous reports revealed that carbon materials play an important role in H2 transfer and spillover.26–29 Both theoretical and experimental studies have demonstrated that activated hydrogen species derived from dihydrogen can flow onto graphitic carbon surfaces proximal to a metallic subnanoparticle and subsequently diffuse to other sites of the materials.29 Yang used carbon materials to bridge the metal and the receptor to enhance hydrogen spillover and depicted that activated hydrogen species can be transferred and stored in the interval layer of graphitic sheets.27 Among the different kinds of carbon materials, multi-walled CNTs possess rolled-up graphitic layers and are classified as parallel-type and herringbone-type on the basis of the rotational axis. As exhibited by high-resolution TEM, the graphitic layers of Ag-out/hCNT and Ag-in/hCNT were tilted by 13° relative to the one-dimensional tunnel axis, demonstrating the herringbone structure of CNTs with 0.36 nm of interlamellar spacing. The terminals of these graphitic layers were in intimate contact with Ag nanowires for Ag-in/hCNT. Thus, the activated hydrogen atoms could migrate from confined Ag nanowires into the interlayer, thereby rendering the activated hydrogen atoms available to react with DMO molecules. To confirm that the internal activated hydrogen atoms on Ag nanowires could approach the external receptor DMO through the interlayer in the herringbone CNTs, we deliberately adopted two designed experiments.As the first strategy, Ag nanowires confined in the pores of parallel CNTs and SBA-15 (Fig. 4) were evaluated for comparison. Notably, confined Ag in parallel CNTs (Ag-in/pCNT) showed an R value of 14.1 mmol h−1 for the DMO hydrogenation. This activity was considerably lower than the activity of Ag located in the herringbone CNTs. The major difference of these two catalysts was the architectural structure of CNTs. Although atomic hydrogen species can diffuse and be stored in interplanar graphitic layers, the species can hardly pass through graphitic layers with only 0.142 nm of carbon–carbon bonds, unless on the domain enriched with defect sites. Chen et al. investigated the hydrogen-migrated behavior using KOH to partially etch graphitic carbon on the surfaces of parallel CNTs.30 They found that hydrogen atoms easily diffuse and migrate from the external surface into the interplanar spacing via defect sites on the surface of activated CNTs, causing hydrogen spillover enhancement. Therefore, the diffusion of activated hydrogen atoms on Ag nanowires in parallel CNTs was tightly strained by the multiple walls of graphitic layers, resulting in poor hydrogenation performance. On the contrary, activated hydrogen atoms on Ag nanowires could directly migrate through the terminal of the epitaxial interplanar space. The enhanced performance insinuated that the herringbone CNTs exhibited a better capacity for hydrogen spillover than parallel CNTs. In addition, we speculated that the stored hydrogen species in the interplanar space of CNTs could be further utilized for the hydrogenation of DMO. Although the comparison of herringbone CNTs and parallel CNTs is barely discussed, herringbone CNTs are recognized as a better support in most cases that have appeared.31 SBA-15 is a well-known inert support with low acidity that possesses 2D channels, but the silica layers are impenetrable. As shown in Fig. 4c, Ag nanowires blocked the channels of SBA-15, which may extremely limit the activation of dihydrogen and the diffusion of activated hydrogen species and DMO. Thus, Ag nanowires in SBA-15 predictably showed only trace activity. This finding also indicated that carbon medium material was necessary for hydrogen diffusion and spillover for the confined Ag nanowires.
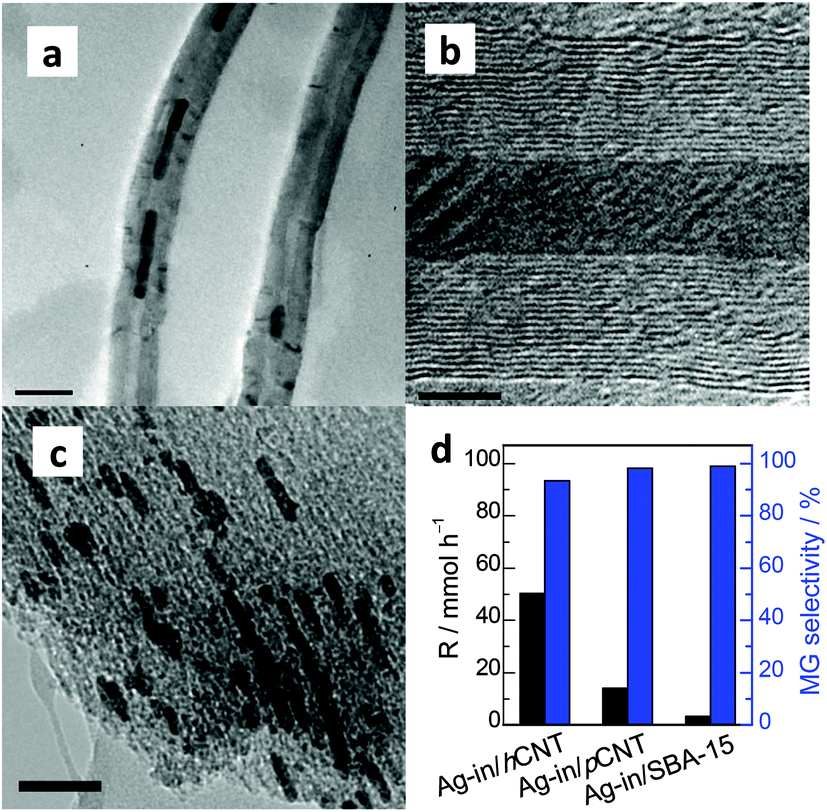 | ||
| Fig. 4 (a) TEM images of parallel CNT confined Ag nanowires. (b) High-resolution TEM of Ag-in/pCNT and (c) Ag-in/SBA-15. (d) Performance of herringbone CNTs, parallel CNTs, and SBA-15 confined Ag catalysts. The bar of (a) and (c) is 50 nm and (b) is 5 nm. | ||
Activated hydrogen species can be transferred and stored in the interplanar space. However, whether stored hydrogen will tarry in the interplanar space or diffuse to the outer surface and be used for hydrogenation remains unsure. For confirmation, we prepared Cu-out-Ag-in/hCNT (Fig. 5b) loaded with copper NPs onto the external surface of Ag-in/hCNT catalysts. The TPR technique (Fig. 5c) was employed to investigate the reductive behavior of outside copper NPs, and a mass spectrometer was used as a detector to ensure accuracy. The result showed that Ag-in/hCNT presented negligible reductive signal, indicating the spontaneous reductive behavior of Ag crystallites supported on CNTs during calcination. Similar spontaneous reductive phenomena have been frequently observed for numerous noble metals supported on CNTs.32,33 Meanwhile, copper NPs supported on CNTs maintained the oxidative state and showed a reductive peak maximum at 538 K. Interestingly, the reductive peak shifted to 514 K when Ag nanowires were introduced into CNTs. Copper NPs supported on Ag-in/hCNT could be easily reduced compared with copper NPs carried by pure CNTs. Hydrogen species detained in the interplanar space of CNTs could diffuse to the outer copper NPs through the terminal and reduce them.6 Moreover, hydrogen species activated by Ag-in/hCNT could be further utilized for the reduction of copper NPs located on the external surface of CNTs, although the Ag nanowires and copper NPs were separated by the multi-walled CNTs.
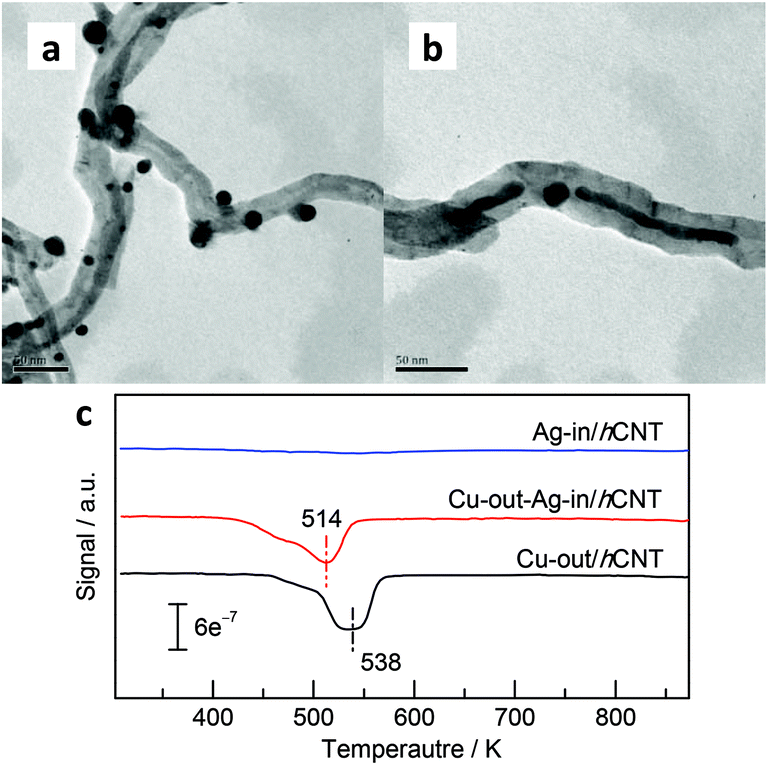 | ||
| Fig. 5 TEM images of (a) Cu-out/hCNT and (b) Cu-out-Ag-in/hCNT. (c) TPR profiles of Ag-in/hCNT, Cu-out/hCNT, and Cu-out-Ag-in/hCNT. | ||
3.6 Plausible scheme of confinement and promotional effects via CNT intermediate confined Ag
The possible confinement and promotional effects are compared in Scheme 1. On the basis of the above analysis, the confinement effect caused compressive deformation for Ag crystallite confined in the internal channel of CNTs. As a result, Ag crystallites were shaped in wires containing grain boundaries with abundant vacancies. Despite the weak interaction of Ag and dihydrogen, activation of dihydrogen could be further enhanced on Ag vacancies, and grain boundaries could provide approaches for hydrogen diffusion. Interactions may be developed between encapsulated nanomaterials and CNT surfaces. The curvature of CNT walls is reported to cause the π electron density of graphene layers to shift from the concave inner surfaces to the convex outer surface, resulting in an electric potential difference.34,35 As a result, the molecules and nanomaterials on the exterior walls of CNTs probably displayed different properties and chemical reactivity from those confined within CNTs. According to the different electronegativities of Ag and carbon (C) (1.93 versus 2.55), interactions of Ag and C may cause electron transfer from metallic Ag to CNTs, resulting in electron-deficient Ag crystallites. The curvature of CNTs may cause the interior walls to become more electrophilic, thereby aggravating the electron-deficient properties, even for the herringbone CNTs, which had interior walls that leaned to the internal Ag nanowires. Experiments and DFT calculations have proven that electron-deficient metallic sites are beneficial for the activation of dihydrogen.36,37 With the combination of geometric and electronic effects, the confined Ag should present stronger capacity to activate dihydrogen. This finding has been confirmed by H2 chemisorption characterization and apparent activation energy tests.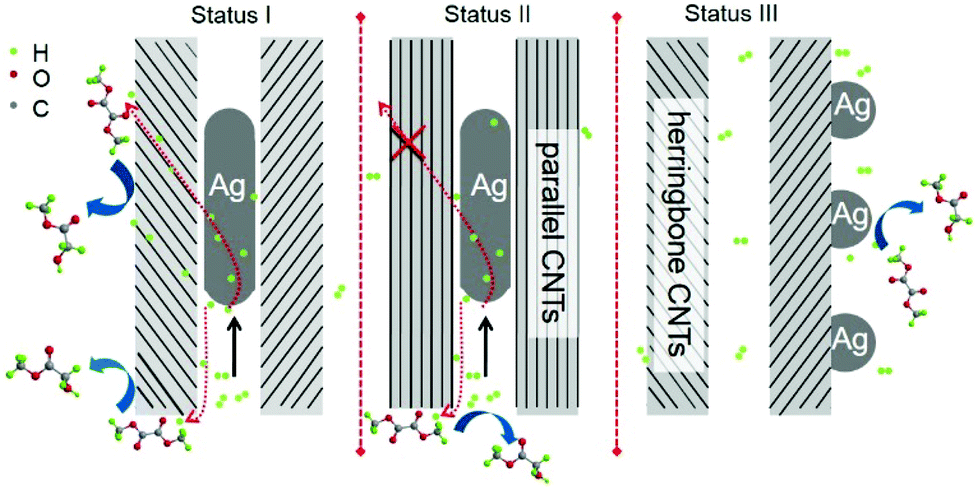 | ||
| Scheme 1 Plausible scheme for the confinement and promotional effects of Ag/C hybrids. | ||
It was reported that the electrons of the encapsulated CoNi clusters can penetrate through three covered parallel graphene layers, making hydrogen species adsorption available on covered graphene shells.38 Calculations showed that the electronic potential difference decreased from −0.5 eV to 0 eV when the graphene cover increased from one to three layers. Thus, the effect of enclosed metals in graphene shells consisting of more than three layers will decline. In the present work, the electronic penetration effect of Ag-in/pCNT with approximately 20 layers of graphite was probably suppressed and negligible. The activation of dihydrogen may be influenced by the shape of nanomaterials and the activated hydrogen species may facilitate the hydrogenation reaction through hydrogen spillover. In contrast to parallel CNTs, the herringbone CNTs obtained showed not only the confinement effect but also the relatively exoteric access of activated hydrogen species. Design experiments proved that activated hydrogen species on Ag nanowires in the channels of herringbone CNTs could be transferred and utilized by the outer surface species. By contrast, activated hydrogen species in the channel of parallel CNTs could hardly transfer to the outer space, except back to the original inlet. Predictably, the direct contact of interplanar spacing terminals to Ag species offered an approach for hydrogen diffusion. Based on the previous literature,34,35 the CNT surface is beneficial for the π–π interaction between CO bonds of DMO and the basic π sites on CNT surfaces, enabling the activated hydrogen species to diffuse from the internal Ag to hydrogenate the carbonyl group of DMO. Life test (Fig. 6) was conducted and it was observed that the activity of Ag-in/hCNT maintained at a high MG yield >97% for more than 200 h, revealing high catalytic stability. This indicated that the performance aided by the confinement effect and promotional effect could last and the Ag-in/hCNT catalyst showed a potential industrial application for MG synthesis from DMO hydrogenation.
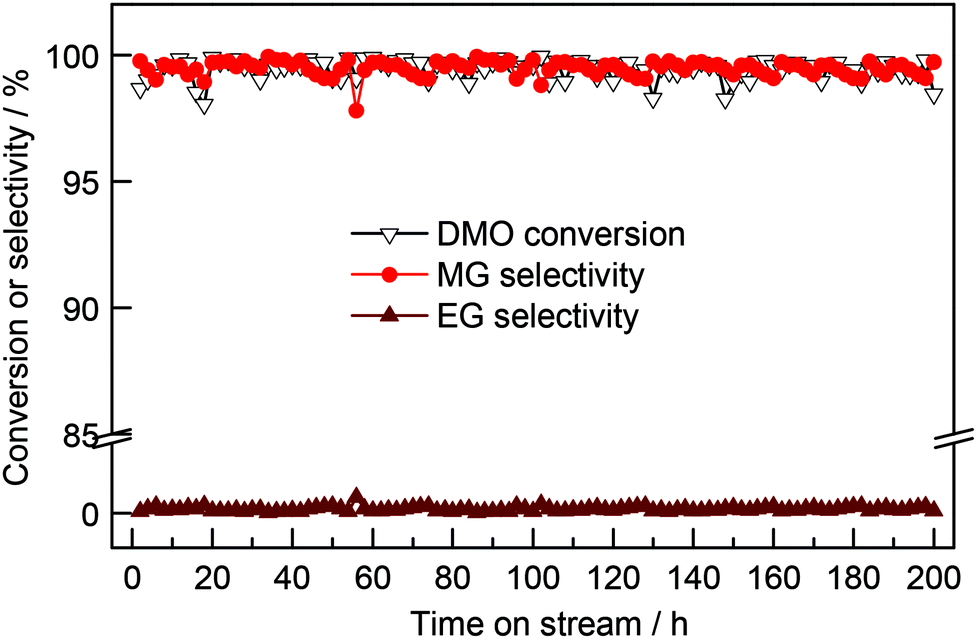 | ||
| Fig. 6 Catalytic performance of Ag-in/hCNT as a function of reaction time. Reaction conditions: T = 493 K, P(H2) = 3.0 MPa, H2/DMO = 80, WLHSV = 0.6 h−1. | ||
4. Conclusions
In conclusion, herringbone CNT confined Ag NPs with nanowire-like morphology, Ag-in/hCNTs, were first prepared and found to exhibit superior catalytic performance for DMO hydrogenation to MG. The Ag-in/hCNT catalyst exhibited approximately three times higher activity than Ag NPs located on the CNT exterior walls (Ag-out/hCNT) for DMO hydrogenation. Moreover, Ag confined in the channel showed excellent durability over 200 h with high MG yield greater than 97%. High-resolution TEM showed that confined Ag crystallites were shaped into wires, whereas free Ag particles were spherical, indicating the stronger interaction between confined Ag nanocrystals and CNTs. The confinement effect was found to induce a geometric effect, which made the surface of Ag rough and enriched with grain boundaries and vacancies. These grain boundaries and vacancies may be beneficial for the activation and diffusion of hydrogen species, helping in increasing the chemisorption of hydrogen and decreasing the apparent activated energy. The activated hydrogen could diffuse to the interplanar spaces of herringbone CNTs, as verified by the promoted reduction of copper NPs on the external surface of herringbone CNTs. Subsequently, hydrogen diffusion from confined Ag to herringbone CNTs could implement the hydrogenation of DMO. This work provides protocols to realize confined Ag within CNTs and understand the active hydrogen spillover (migration), benefiting fundamental research on hydrogenation and industrial applications for MG synthesis.Acknowledgements
We acknowledge the financial support from the Natural Science Foundation of China (91545115, 21473145, 21403178, and 21303141), the Postgraduate Basic Innovative Research Program of Xiamen University (201412G001), and the Program for Innovative Research Team in Chinese Universities (no. IRT_14R31).Notes and references
- A. Y. Yin, X. Y. Guo, W. L. Dai and K. N. Fan, Chem. Commun., 2010, 46, 4348 RSC.
- H. H. Liu, G. K. Chuah and S. Jaenicke, Phys. Chem. Chem. Phys., 2015, 17, 15012 RSC.
- C. Gao, W. W. Li, Y. Z. Jin and H. Kong, Nanotechnology, 2006, 17, 2882 CrossRef CAS.
- P. Luksirikul, K. Tedsree, M. G. Moloney, M. L. Green and S. C. Tsang, Angew. Chem., Int. Ed., 2012, 51, 6998 CrossRef CAS PubMed.
- P. Zhang, C. L. Shao, Z. Y. Zhang, M. Y. Zhang, J. B. Mu, Z. C. Guo and Y. C. Liu, Nanoscale, 2011, 3, 3357 RSC.
- X. L. Pan and X. H. Bao, Acc. Chem. Res., 2011, 44, 553 CrossRef CAS PubMed.
- S. J. Guo, X. L. Pan, H. L. Gao, Z. Q. Yang, J. J. Zhao and X. H. Bao, Chem. – Eur. J., 2010, 16, 5379 CrossRef CAS PubMed.
- X. L. Pan, Z. L. Fan, W. Chen, Y. J. Ding, H. Y. Luo and X. H. Bao, Nat. Mater., 2007, 6, 507 CrossRef CAS PubMed.
- E. Castillejos, R. Chico, R. Bacsa, S. Coco, P. Espinet, M. Pérez-Cadenas, A. Guerrero-Ruiz, I. Rodríguez-Ramos and P. Serp, Eur. J. Inorg. Chem., 2010, 5096 CrossRef CAS.
- W. Chen, Z. L. Fan, X. L. Pan and X. H. Bao, J. Am. Chem. Soc., 2008, 130, 9414 CrossRef CAS PubMed.
- A. M. Zhang, J. L. Dong, Q. H. Xu, H. K. Rhee and X. L. Li, Catal. Today, 2004, 93–95, 347 CrossRef CAS.
- J.-P. Tessonnier, L. Pesant, G. Ehret, M. J. Ledoux and C. Pham-Huu, Appl. Catal., A, 2005, 288, 203 CrossRef CAS.
- L. Zhang, L. P. Han, G. F. Zhao, R. J. Chai, Q. F. Zhang, Y. Liu and Y. Lu, Chem. Commun., 2015, 51, 10547 RSC.
- J. W. Zheng, J. F. Zhou, H. Q. Lin, X. P. Duan, C. T. Williams and Y. Z. Yuan, J. Phys. Chem. C, 2015, 119, 13758 CAS.
- C. Wen, Y. Y. Cui, W. L. Dai, S. Xie and K. N. Fan, Chem. Commun., 2013, 49, 5195 RSC.
- H. R. Yue, Y. J. Zhao, S. Zhao, B. Wang, X. B. Ma and J. L. Gong, Nat. Commun., 2013, 4, 2339 Search PubMed.
- J. W. Zheng, H. Q. Lin, X. L. Zheng, X. P. Duan and Y. Z. Yuan, Catal. Commun., 2013, 40, 129 CrossRef CAS.
- A. Y. Yin, C. Wen, W. L. Dai and K. N. Fan, Appl. Catal., B, 2011, 108–109, 90 CrossRef CAS.
- B. Xue, P. Chen, Q. Hong, J. Y. Lin and K. L. Tan, J. Mater. Chem., 2001, 11, 2378 RSC.
- J. Y. Liu, ChemCatChem, 2011, 3, 934 CrossRef CAS.
- J. Guerra and M. A. Herrero, Nanoscale, 2010, 2, 1390 RSC.
- R. Kumar, E. Gravel, A. Hagege, H. Li, D. V. Jawale, D. Verma, I. N. Namboothiri and E. Doris, Nanoscale, 2013, 5, 6491 RSC.
- G. Vilé, D. Baudouin, I. N. Remediakis, C. Copéret, N. López and J. Pérez-Ramírez, ChemCatChem, 2013, 5, 3750 CrossRef.
- R. W. Balluffi and L. L. Seigle, Acta Metall., 1957, 5, 449 CrossRef CAS.
- A. Pundt and R. Kirchheim, Annu. Rev. Mater. Res., 2006, 36, 555 CrossRef CAS.
- Y. W. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 8136 CrossRef CAS PubMed.
- A. J. Lachawiec, J. G. S. Qi and R. T. Yang, Langmuir, 2005, 21, 11418 CrossRef CAS PubMed.
- L. F. Wang and R. T. Yang, Catal. Rev., 2010, 52, 411 CAS.
- L. Chen, A. C. Cooper, G. P. Pez and H. S. Cheng, J. Phys. Chem. C, 2007, 111, 18995 CAS.
- C. H. Chen and C. C. Huang, Microporous Mesoporous Mater., 2008, 109, 549 CrossRef CAS.
- X. L. Liang, X. Dong, G. D. Lin and H. B. Zhang, Appl. Catal., B, 2009, 88, 315 CrossRef CAS.
- B. M. Quinn, C. Dekker and S. G. Lemay, J. Am. Chem. Soc., 2005, 127, 6146 CrossRef CAS PubMed.
- P. Singh, G. Lamanna, C. Menard-Moyon, F. M. Toma, E. Magnano, F. Bondino, M. Prato, S. Verma and A. Bianco, Angew. Chem., Int. Ed., 2011, 50, 9893 CrossRef CAS PubMed.
- Z. Guo, Y. T. Chen, L. S. Li, X. M. Wang, G. L. Haller and Y. Yang, J. Catal., 2010, 276, 314 CrossRef CAS.
- Y. B. Yan, J. W. Miao, Z. H. Yang, F. X. Xiao, H. B. Yang, B. Liu and Y. Yang, Chem. Soc. Rev., 2015, 44, 3295 RSC.
- M. Bron, D. Teschner, A. Knop-Gericke, F. C. Jentoft, J. Krohnert, J. Hohmeyer, C. Volckmar, B. Steinhauer, R. Schlogl and P. Claus, Phys. Chem. Chem. Phys., 2007, 9, 3559 RSC.
- Y. Xu, J. Greeley and M. Mavrikakis, J. Am. Chem. Soc., 2005, 127, 12823 CrossRef CAS PubMed.
- J. Deng, P. J. Ren, D. H. Deng and X. H. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the XRD patterns of Ag on different carriers. See DOI: 10.1039/c5nr08651e |
| This journal is © The Royal Society of Chemistry 2016 |