
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoparticles and DNA – a powerful and growing functional combination in bionanotechnology
Anirban
Samanta
ab and
Igor L.
Medintz
*a
aCenter for Bio/Molecular Science and Engineering, Code 6900, U.S. Naval Research Laboratory, Washington, DC 20375, USA. E-mail: Igor.medintz@nrl.navy.mil
bCollege of Science, George Mason University, Fairfax, Virginia 22030, USA
First published on 15th April 2016
Abstract
Functionally integrating DNA and other nucleic acids with nanoparticles in all their different physicochemical forms has produced a rich variety of composite nanomaterials which, in many cases, display unique or augmented properties due to the synergistic activity of both components. These capabilities, in turn, are attracting greater attention from various research communities in search of new nanoscale tools for diverse applications that include (bio)sensing, labeling, targeted imaging, cellular delivery, diagnostics, therapeutics, theranostics, bioelectronics, and biocomputing to name just a few amongst many others. Here, we review this vibrant and growing research area from the perspective of the materials themselves and their unique capabilities. Inorganic nanocrystals such as quantum dots or those made from gold or other (noble) metals along with metal oxides and carbon allotropes are desired as participants in these hybrid materials since they can provide distinctive optical, physical, magnetic, and electrochemical properties. Beyond this, synthetic polymer-based and proteinaceous or viral nanoparticulate materials are also useful in the same role since they can provide a predefined and biocompatible cargo-carrying and targeting capability. The DNA component typically provides sequence-based addressability for probes along with, more recently, unique architectural properties that directly originate from the burgeoning structural DNA field. Additionally, DNA aptamers can also provide specific recognition capabilities against many diverse non-nucleic acid targets across a range of size scales from ions to full protein and cells. In addition to appending DNA to inorganic or polymeric nanoparticles, purely DNA-based nanoparticles have recently surfaced as an excellent assembly platform and have started finding application in areas like sensing, imaging and immunotherapy. We focus on selected and representative nanoparticle–DNA materials and highlight their myriad applications using examples from the literature. Overall, it is clear that this unique functional combination of nanomaterials has far more to offer than what we have seen to date and as new capabilities for each of these materials are developed, so, too, will new applications emerge.
 Anirban Samanta | Anirban Samanta studied chemistry and obtained his BSc from the University of Calcutta, India in 2007. He received an MSc from the Indian Institute of Technology, Kanpur in 2009. He completed his PhD in 2014 at Arizona State University under the mentorship of Professors Hao Yan and Yan Liu. In the same year he joined the US Naval Research Laboratory in Washington DC as a postdoctoral fellow where he is currently working on DNA directed assembly of photonic materials to build efficient artificial light harvesting systems. |
 Igor L. Medintz | Igor L. Medintz received a Ph.D. in molecular biology from the City University of New York in 1999. Following a National Cancer Institute fellowship at the University of California Berkeley, he joined the Center for Bio/Molecular Science and Engineering of the U.S. Naval Research Laboratory as a National Research Council Fellow in 2002, became a research biologist in 2004 and is currently the Navy's Senior Scientist for Biosensors and Biomaterials. His research focuses on interfacing nanomaterials with biological molecules, understanding how nanoparticles engage in energy transfer, and how biological molecules function at a nanoparticle interface. He has coauthored more than 200 peer-reviewed articles along with receiving 17 patents. |
1. Introduction
The pace of development across all facets of technology continues to accelerate at a nearly unabated rate. The greatest expectations for transformational changes are associated with many of the newer technologies as these changes are far more fundamental in nature and not so incremental or application driven. In terms of transformative technologies, nanotechnology in all its manifestations has perhaps the most to offer as it seeks to create new materials with new properties and functionalities from the bottom up. The first generation of nanotechnologies resulted in the development of a myriad of new “nanomaterials” (NMs) displaying unique optical, magnetic, electronic, mechanical and chemical properties. The physics behind these properties are best described by quantum mechanics in most cases and arise primarily from nanoscale size and quantum-confined effects that are not available to the same materials in bulk form. These include noble metal nanoparticles (NPs) with localized surface plasmon resonances, semiconductor quantum dots (QDs) with size-tunable photoluminescence (PL), and carbon allotropes such as single-wall carbon nanotubes (SWCNTs) and graphene which can manifest ballistic electron transport properties to name just a few prominent examples from a growing list that is far too numerous to detail.1–4 Many NMs, and especially NPs, are also characterized by useful physicochemical properties such as a small size coupled to high surface-to-volume (S/V) ratios and amenability to facile surface modification chemistry such that they can be coupled to other (bio)materials. These NMs are already finding numerous non-biological applications for energy harvesting in solar cells, improved data storage capacity, optical displays, and for chemical catalysis and purification purposes.5–9In parallel to these developments, and in contrast to the above mentioned primarily “hard” NMs, there has also been a tremendous focus on “softer” NMs including those that are biologically-based such as viral capsids along with those derived from biopolymers such as nucleic acids.10–13 Of all of these biomaterials, deoxyribonucleic acid (DNA) in particular appears to have the most to offer for nanotechnology. Indeed, DNA and other nucleic-acid based technologies have already made significant contributions to the diagnostic and genomic revolutions of the last decades.14–20 Here, they were exploited mainly for their sequence recognition and complementarity as primers to access information in the form of genomic sequencing, for polymerase chain reaction (PCR), probes for hybridization arrays, and as sensors within molecular beacon (MB) configurations.21–27 Other DNA utility in the form of aptamers for biorecognition was also developed. These are partially self-complementary single stranded- (ss) DNA which can fold into a unique 3D shape that enables it to recognize non-nucleic acid targets with a high degree of specificity including small and macro-molecules along with cell surface markers in a manner similar to antibodies.28–31 Equally importantly, techniques such as SELEX (systematic evolution of ligands by exponential enrichment) were also developed that allowed aptamers to be both selected for and undergo affinity improvement from large combinatorial libraries of oligonucleotides.32–35 More recently, the advent of structural DNA technology has brought interest in this material to the forefront once again. Along with being the carrier of genetic information, the underlying Watson–Crick base-pairing complementarity, which relies on relatively weak individual hydrogen bonds, also allows for in silico designed de novo DNA structures to be assembled (see also the next section). Moreover, this technology has now matured to the point that almost any 1-, 2-, or 3-dimensional DNA-based nanoscale architecture can be assembled and research is now focused on creating active constructs that are capable of dynamically reconfiguring themselves for sensing, drug delivery, molecular logic, molecular electronics and the like.36–39
One of the ongoing goals of nanotechnology is to combine significantly different NMs into new “value-added” hybrids that are capable of far more than each component material acting independently. Nowhere is this more apparent or epitomized than in bionanotechnology. Using theranostics as a prime example, the overarching goal is to develop multifunctional active composite materials capable of targeted delivery to cells/tissues/organs, combined with sensing of some analyte or endogenous property or process such as enzyme activity, enabling non-invasive contrast or imaging along with localized drug delivery.40–46 The biological molecule (e.g. peptide, aptamer, antibody) would provide the recognition, biocompatibility, active sensing and even drug activity while the NM would host and display the biological on its surface and also provide contrast and the capacity to be activated by radio or near-infrared (NIR) light, for example, to be both visualized and release a drug (e.g. small interfering or siRNA) on demand. Other manifestations would include active NMs for bioremediation, stealth sensors, nanofactories, biocomputers, and the like. These are, in essence, the futuristic and quasi-independent or autonomous nanoscale machines that are so often the focus of science fiction musings.
Our focus here is to provide an overview of the progress towards these goals based on combining the unique properties of NPs with primarily DNA and some other related nucleic acids. The breadth of applications based on the combination of a particular type of NP with a DNA functionality is extraordinary and growing daily. These can range, for example, from utilizing porous NPs decorated with cell-specific DNA aptamers to deliver drugs to an optimized light-harvesting array based on energy transfer (ET).47–51 See Table 1 for a representative but certainly not comprehensive overview of NMs and the potential applications they are being developed for in the context of synergistic activity with DNA. New and unique physicochemical benefits are also being identified from utilizing this combination of materials. For example, NP display of DNA can present very high avidity for enhanced binding and recognition while also offering protection to covalently-attached or adsorbed DNA from nuclease degradation for reasons that are still not fully understood but seem to include display density and NP size.52–55 This has direct beneficial implications for gene delivery and gene therapy and is directly spurring commercial interest and directed research. Beyond classical “hard” NPs, DNA can also be combined with “soft” nanoparticulate materials including viral capsids, other proteinaceous macromolecules, polymers and even DNA-based NPs themselves. Rather than assembling an exhaustive list of every combination and example, we focus instead on highlighting some of the many examples of hybrid NP–DNA materials and their prospective applications to provide the reader with an appreciation of the richness and especially the potential of this field. The review is divided up by the NP materials themselves, with each section providing a brief overview of some relevant NP properties of interest and then a description of demonstrated and potential applications based on incorporation of DNA. Our discussion is richly illustrated with both examples and images from the literature and, due to the overwhelming nature and size of this research field, we can only provide a cursory overview of some materials; we thus apologize in advance for any and all omissions. Given this, we also do not directly discuss many associated topics which are equally important and challenging in their own right including NP synthesis, NP–DNA bioconjugation chemistry, and NP–DNA conjugate analysis/purification unless it is directly relevant to our focus as in the case of QDs and bioconjugation. For the rest of these, the interested reader is referred to some recent reviews and perspectives as a starting point.56–59
| Nanoparticle | Constituents | DNA | Application | Remarks | Ref. |
|---|---|---|---|---|---|
| AuNP | Au | FAM labeled T-rich DNA | Hg2+ detection | ssDNA electrostatically adsorbed on citrate capped AuNPs | 136 |
| AuNR | Au | Leukemia T cell targeting SH-DNA | Photothermal cancer therapy | SH-DNA semi-covalently attached to AuNR surface | 180 |
| Au/Ag hybrid | Au/Ag | Cytosine rich ssDNA | Sulfide detection | Au/Ag attached to cytosine hairpin loop | 195 |
| AgNP | Ag | SH-DNA | DNA sensor | Thiolated DNA adsorbed on AgNP | 245 |
| AgNC | Few Ag atoms | G-rich cocaine binding aptamer | Cocaine detection | Ag bound to G-rich aptamer region | 259 |
| MNP | Fe3O4 | Thrombin binding SH-DNA aptamer | MRI thrombin detection | SH-DNA covalently attached to MNPs | 280 |
| PtNP | Pt | SH-DNA | DNA sensor | SH-DNA non-covalently attached | 297 |
| PdNP | Pd | Thiol and amine functionalized oligos | Electrode fabrication | DNA acted as template for the particle synthesis on ITO surface | 309 |
| QD | CdSe/ZnS | Dye-labeled photonic wire | Light harvesting | Peptide-DNA assembled to QD | 351 |
| SWCNT | Carbon | Ce6 conjugated thrombin binding aptamer | Controlled singlet oxygen generation | ssDNA wrapped onto SWCNT via π stacking interaction | 397 |
| GO | Carbon | Short dsDNA with random sequence | Hydrogel preparation | ssDNA π stacked onto GO surface | 437 |
| Micelle | DNA + PPO | ssDNA covalently attached to PPO | Chemotherapeutic delivery | DNA-b-PPO in micellar structure | 468 |
| Polyacrylamide-NP | Polyacrylamide | Dye-quencher labeled ATP aptamer | Intercellular sensor | Embedded inside the matrix of the NP | 483 |
| Viral NP | Bacteriophage MS2 capsid | Jurkat leukemia T cell specific DNA aptamer | Targeted delivery of photodynamic agents | Covalently linked to unnatural amino acid (paF) on NP exterior surface | 496 |
| Ferritin NPs | hFTN-H/eGFP or DsRed) | Amine modified PDGF specific aptamer | PDGF-BB biosensor | SMCC coupling reduced gFFNPs | 507 |
| UCNP | Yb3+ or Tm3+ doped NaYF4 | Amine-DNA with targeting sequence | DNA sensor | Covalently chemistry | 520 |
| Chalcogenide-NP | CuS | Amine modified targeting DNA | DNA sensor | EDC-based chemistry | 525 |
| Alkaline earth metal NP | Ca(H2PO4)2, CaHPO4 Ca3(PO4)2 | eGFP encoding plasmid DNA | Cell transfection | Adsorbed on the CaP NPs | 531 |
| DNANP | DNA | >100 short oligomers, | Nanobarcoding/multiplexing | Self-assembled structure | 558 |
2. DNA as a nanomaterial
Since DNA is one of the two common elements of the composite materials highlighted here, we begin with a brief overview of some of its properties that are relevant to this discussion. DNA, the carrier of our genetic information, is typically seen in the form of a double helix where two long chains are wound around a common axis in a helical fashion to yield double stranded DNA (dsDNA). Each unit of the double helix is basically a polymer of small repeating units called nucleotides. A single nucleotide has three components, a sugar molecule, a phosphodiester group and a nitrogenous base or nucleobase. For DNA there are four types of nucleobases, Adenine (A), Guanine (G), Cytosine (C) and Thymine (T) and it is the sequence of these four bases that encodes the information (a polymer length of N nucleotides can generate 4N distinct sequences). T and C are pyrimidines while A and G are purines consisting of a pyrimidine fused to an imidazole ring. Bases on the opposite strand of a double helix are hydrogen bonded via the ring nitrogens, carbonyl and the exocyclic amine groups inside the double helix while the sugar and phosphate groups face the water on the outer side. The most prominent hydrogen (H) bonding patterns are those where A binds with T by 2 H bonds and G binds with C via 3 H bonds. Besides this canonical Watson–Crick base pairing, a pair itself can engage in additional hydrogen bonding with another nucleotide, for example, a G–C pair can further interact with another C. These relatively less common hydrogen bondings between more than two nucleotides are called Hoogsteen pairing.60 One of the most notable example of Hoogsteen pairing is a G-quadruplex where four guanine bases get hydrogen bonded in a circular fashion to form a tetrad and multiple tetrads can stack over each other to form the quadruplex (Fig. 1). The G-quadruplex is found mainly in G-rich sequences, especially at telomeres, and it is believed that they have a significant role in genomic stability and replication. Although it is commonly surmised that H bonding is the force that holds the two complementary strands together, the reality is that the dominant contribution comes from the stacking interaction between the adjacent base pairs. Supporting this, Yakovchuk et al. showed that considering all the entropic and enthalpic contribution, G–C pairing has barely any net stabilizing energetic contribution while A–T pairing can sometimes by destabilizing.61 Besides a stronger H bond between a G–C pair, as compared to A–T, the stacking interaction between G–C/G–C is much stronger than that of A–T/A–T (−14.59 kcal mol−1vs. −6.57 kcal mol−1).62 This also explains the sequence dependent strengths and the DNA melting temperature or Tm which is the temperature at which the transition from helical dsDNA to randomly coiled ssDNA occurs within 50% of a given dsDNA duplex. It is worth mentioning that ionic strength also has a profound effect on Tm; increasing ionic strength up to 0.3 M can, in turn, increase Tm; beyond this level it may remain unchanged or even decrease.63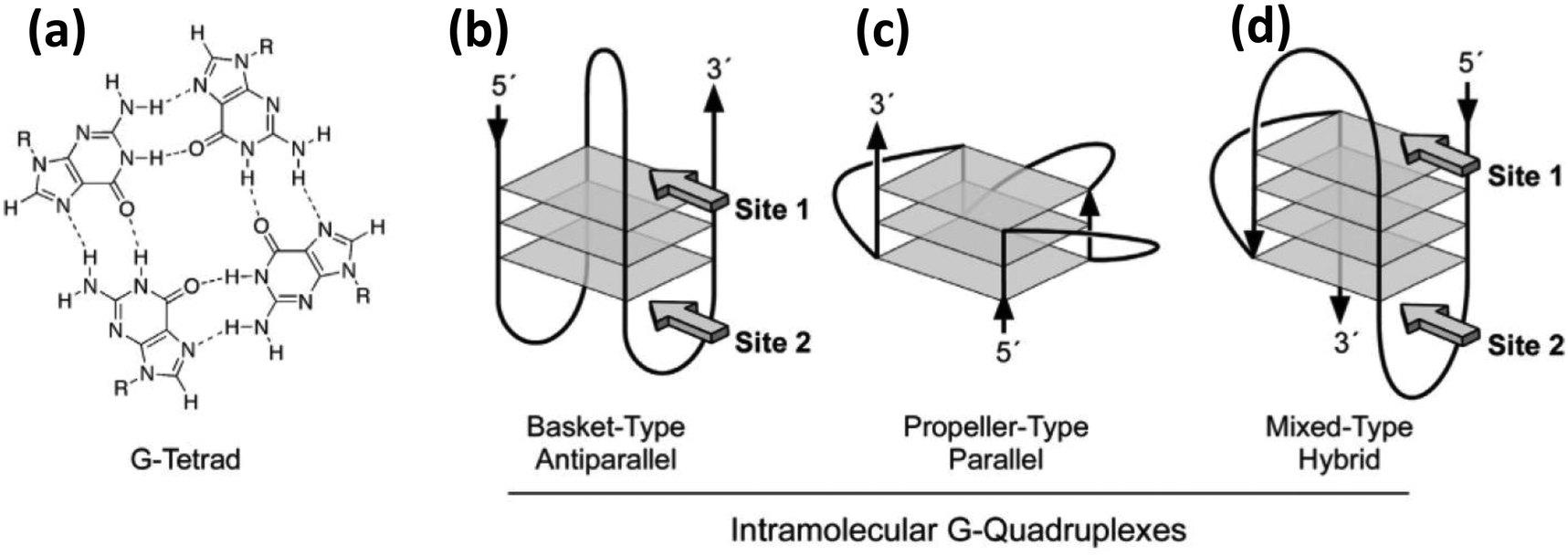 | ||
| Fig. 1 (a) Molecular structure and hydrogen bonding pattern of a G-tetrad. (b) (c) & (d) Show the structure of three different kinds of intramolecular G-quadruplexes. Reprinted with permission from ref. 64. Copyright 2005 American Chemical Society. | ||
Double helical DNA adopts different three-dimensional forms and amongst these the right-handed B form has a diameter of 20 Å, a helical pitch of 34 Å and 10.5 bases per helical turn. This was the structure originally proposed by Watson and Crick and is commonly found in most physiologically relevant conditions.65 While the A-form is right handed, slightly wider and found in water deprived condition, the Z form of DNA is drastically different primarily due to its left handed conformation. Z-DNA appears to be more slender than the B form, and, interestingly, can also be found in physiological environments in short stretches having a sequence of alternate pyrimidine and purine bases.60 Besides natural DNA, the great advancement in synthetic organic chemistry over the last three decades has provided numerous other opportunities to build DNA analogues that differ mostly by structural changes to the backbone and nucleobases. A fairly large library of artificial DNA bases have been synthesized and incorporated into natural backbones in pursuit of either encoding additional information or imparting different properties or roles to the resulting material. This vast and highly active area of research has been carefully reviewed elsewhere.66–68 Like nucleobases, nucleic acid analogues with alternate backbones have also been generated, and interestingly some of them have even been commercialized.69,70 Backbone mimics are produced either by using sugar motifs other than natural ribose or by using molecules other than sugar or even those lacking the phosphate group. Examples of the first kind include pyranosyl-ribonucleic acid or p-RNA,71 threose nucleic acid or TNA,72 conformationally constrained bicyclo and triyclo sugar consisting DNAs,73,74 and, most importantly, locked nucleic acid or LNA.75,76 LNA contains a conformationally restricted ribose ring where the 2′-O and 4′-C are connected via a methylene bridge (Fig. 2c). The heteroduplex it forms with complimentary RNA or DNA tends towards greater affinity and specificity while still assuming an A DNA like conformation. Either alone or in mixed duplexes, LNA are also quite stable against nuclease degradation which makes them an enticing alternative to DNA alone for biosensing, antisense therapy and in microarray techniques.77 Another popular modified DNA is phosphorothioate (ps) DNA, where one of the oxygen atoms on the phosphate group is replaced by a sulfur (Fig. 2b). Examples of the second phosphate-lacking material kind include glycerol nucleic acids or GNA, acyclic threoninol nucleic acid or TNA,78 and peptide nucleic acids or PNA.79,80 PNA lacks any sugar moiety as well as the phosphate group, which is one reason why PNA should realistically speaking not be called a true acid. The PNA backbone is a polymer of modified glycine units and thus it is achiral as well as uncharged (Fig. 2d). In addition, PNA is stable over a wide range of pH and highly resistant towards enzymatic degradation.81 It is also important to mention that the structural DNA revolution, which originated from understanding and then exploiting non-canonical DNA structures such as Holliday junctions and crossovers, has now provided the ability to assemble virtually any 1-, 2-, or 3-dimensional DNA-based structure based on use of DNA origami and similar technologies. Some representative structures are shown in Fig. 2e and f.82 This technology has even progressed to the point of these structures becoming dynamically active and reconfigurable. An excellent primer and overview of some of the possibilities and potential in this area is provided in ref. 82. As will be shown repeatedly below, combining such DNA structures with NPs often leads to new “value-added” materials that demonstrate potent synergistic activity.
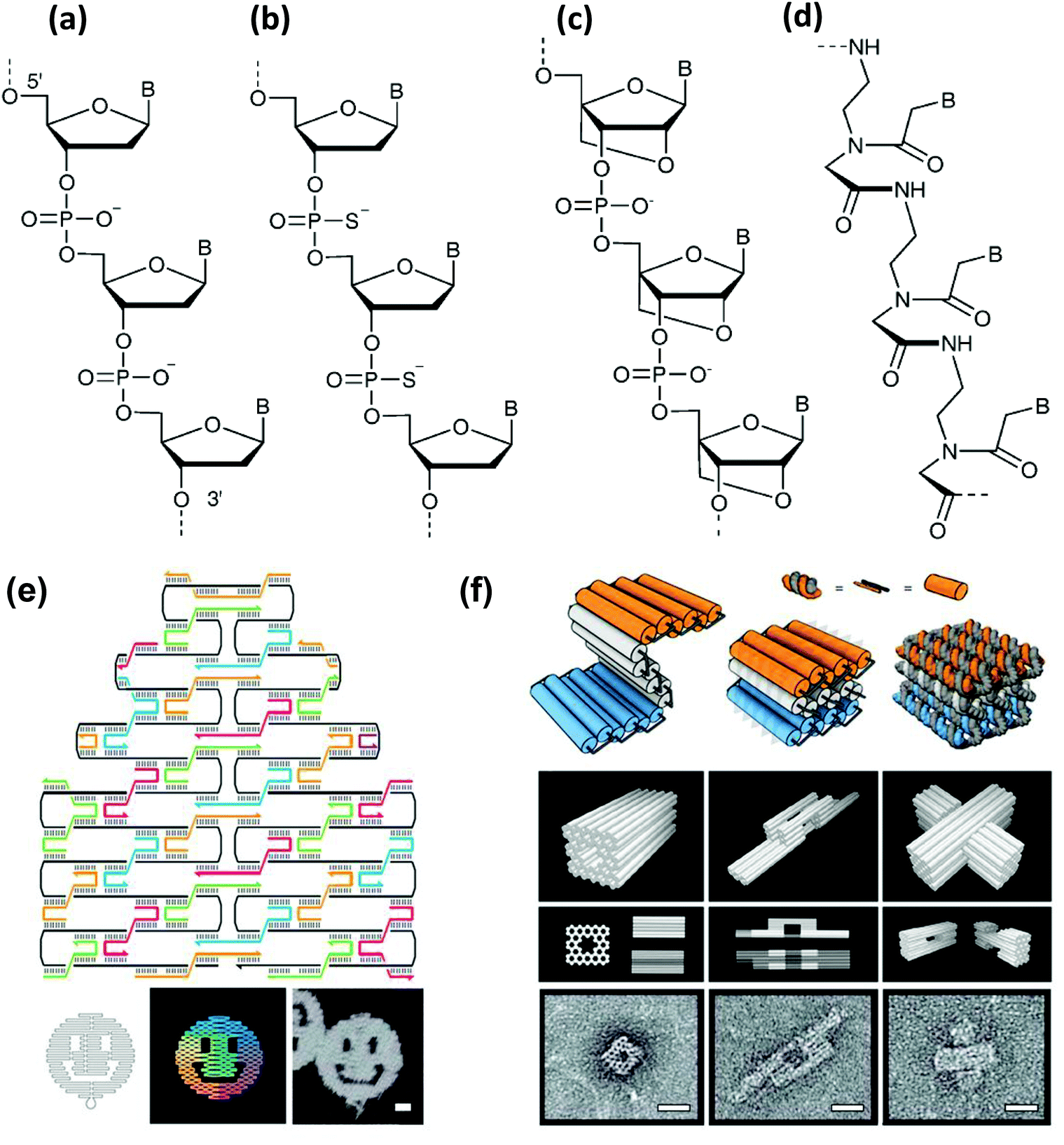 | ||
| Fig. 2 Molecular representation of the backbones of natural and various artificial nucleic materials including: (a) natural phosphodiester, (b) phosphorothioate, (c) locked nucleic acid (LNA) and (d) peptide nucleic acid (PNA). B designates the nitrogenous base position. (a–d) Reproduced from ref. 83 with permission of The Royal Society of Chemistry. (e) Example of a DNA origami. Illustration of the rasterlike pattern of the scaffold strand folded by staple strands, which are collectively used to generate arbitrary 2D patterns – in this case a smiling face. Scale bar, 20 nm. (f) Extension of the origami principle to 3D by using staple strands which promote the formation of pleated sheets of duplexes, which ultimately pack into a honeycomb lattice. Scale bars, 20 nm. (e,f) from ref. 82. Reproduced with permission from AAAS. | ||
3. Gold nanoparticles
Among DNA functionalized NMs, gold nanoparticles (AuNPs) are perhaps the most mature or well-developed and have been undergoing continuous exploration over several decades for various applications including sensing, imaging, catalysis, therapeutics, diagnostics, and drug delivery, to name but a paltry few.84–87 One of the principle physicochemical properties behind its widespread use in nanoparticulate form arises from its surface plasmon resonance (SPR).88–91 This is the collective oscillation of conduction band electrons upon interaction with a wavelength of light that is much larger than the dimension of the NP. The oscillation frequency, which lies in the visible range for gold, strongly depends on the size and shape of the particle and the dielectric constant of its environment. For spherical AuNPs, the resonance frequency does not alter significantly with the change in size, as it does when shape changes and becomes, for example, anisotropic. Cumulatively, this results in the manifestation of several useful properties such as an enhanced localized electric field around the NP surface. Ordinary molecules or fluorophores on the surface of the particles or even in fairly close proximity will generate stronger Raman signals as well as a quenching or, conversely, enhancement of fluorescence when interacting with the unique fields and plasmons.92–96 The interaction of a fluorophore with an AuNP is complex and no single model yet can adequately clarify the accurate picture. The fluorescence quenching efficiency of AuNPs is stronger and works over relatively larger distances than predicted by traditional Förster resonance energy transfer (FRET), which is based on weak electromagnetic interaction between two dipoles. Several theories have emerged to interpret experimental findings including the Gerstein-Nitzan model97 and the CPS-Kuhn model amongst others;98,99 the latter was extended further to obtain the final form of a nanometal surface energy transfer or NSET approach.100 The Gerstein-Nitzan model, which works better for large AuNPs that have a significant scattering contribution, treats the AuNP as a single dipole of a finite size interacting with an oscillator in its excited state. The CPS-Kuhn model describes the system as the interaction between a dipole with a bulk metal or thin film having an array of dipoles. While FRET has an inverse r6 efficiency dependence over donor acceptor separation distance (r), NSET is predicted to have an inverse r4 efficiency dependence between donor fluorophores and AuNP acceptors allowing this interaction to potentially extend over significantly longer separations than FRET. Recently, Strouse has made an attempt at explaining the fluorescence quenching of two dyes in the proximity of AuNPs of various sizes by incorporating size dependent absorption and a dielectric constant to the existing NSET model.100 If the quite complex underlying interactions of NSET processes are ever fully reduced to a predictive framework that accounts for all the relevant parameters and is applicable to the wide varieties of AuNPs that arise with different surface chemistries, sizes and shapes, this could serve as a long-range molecular optical ruler for studying biomolecular events that occur over distances that are now beyond the reach of classical FRET. Indeed, the Strouse group recently reported on telomerase quadruplex folding and the global conformation of a folded RNA ribozyme employing AuNPs within the mathematical formulation of NSET.101,102Countless researchers have contributed to the development of AuNPs in the context of DNA functionalization over the last 50 or more years culminating in seminal demonstrations from both the Mirkin group at Northwestern University and the Alivisatos group at U.C. Berkeley.103,104 The key enabling technology driving attachment of DNA to the AuNPs continues to be the unusually strong binding affinity of alkyl thiols towards gold surfaces.105–108 Today, thiol-modified DNA strands are routinely synthesized and used for loading DNA onto AuNPs of various shapes and sizes. Initially, the electrostatic repulsion originating from the negatively charged groups used in citrate-based AuNP synthesis along with that of the DNA phosphate groups made dense packing of DNA onto AuNPs a challenge. To solve this, the salt concentration of the media was slowly increased to counterbalance the charge.109 In turn, higher DNA loading onto the NP surface ensured colloidal stability by preventing aggregation at high salt concentration, and this is essential for promoting hybridization reactions. Besides colloidal stability, the dense monolayer of highly oriented AuNP-bound nucleic acids exhibit some useful properties like the ability to transfect cells without ancillary transfection agents and cooperative binding to complementary nucleic acid, narrow melting transition with a Tm higher than the particle-free duplex which can lead to better selectivity and sensitivity in various detection and assay schemes.82 The Mirkin group coined the term spherical nucleic acid (SNA) to describe their 3-D topology, although later the terminology has been extended to include cores other than spherical AuNPs, such as Ag, Fe3O4, silica, CdSe QDs or anisotropic AuNPs.82,110 Quite a large number of DNA can be loaded on AuNPs by choosing appropriate DNA sequences (generally thymine rich), appropriate salt aging, proper sonication during the loading process and placement of a spacer, such as polyethylene glycol (PEG) between the thiol moiety and the nucleobases.109 There are far more nuances and ‘rules of thumb’ to the preparation of DNA-functionalized AuNPs for many different applications and the interested reader is referred to ref. 111–114 along with the Mirkin group's extensive work in this area.
3.1. Nucleic acid detection
Two DNA-functionalized AuNPs can be linked if the DNA displayed on each of their surfaces has complementarity to the other or to another third “bridging” DNA and this relatively simple concept has been the principle behind numerous DNA sensors.115,116 Since each NP usually carries multiple copies of a DNA sequence, simple mixing alone can lead to uncontrolled aggregation, which would be reflected vividly in their surface plasmon absorption band and can be quantitatively detected by colorimetric methods. Mirkin and Letsinger first introduced this concept as a highly selective and rapid method of DNA detection in 1997.117 Two different copies of DNA, with specific sequences that are partly complementary to the target DNA, were immobilized onto two different AuNPs. The target DNA worked as a crosslinker leading to NP aggregation, which was accompanied with a color change from red to purple. Looking into the sharp melting transition of the aggregates, the technique can differentiate imperfect DNA targets down to the resolution of a single nucleotide mismatch.118 The reason behind the color change in these aggregates is the intense plasmonic coupling between adjacent particles when the interparticle distance is smaller than the diameter of the NPs. Due to the high extinction coefficient of the materials, the change in color is perceivable to the naked eye without any instrumentation. This makes it a better method than previous detection approaches such as those utilizing PCR or other fluorophore-based assays for situations where supporting instrumentation is not accessible or cost is a concern.118 However, the detection limit of this assay can be quite high, at nearly 10 nM. To circumvent this potential limitation, larger AuNPs have been used.119 One of the main issues arising here is that larger AuNPs are intrinsically difficult to stabilize in buffer solution. Attempting to address this, Bai et al. demonstrated that small AuNPs can still be used which will produce micro-aggregates but the signal can be amplified by subsequent electrodeposition of gold onto them as shown in Fig. 3. The particles now grow and come into contact causing a stronger plasmonic coupling which again results in a detectable blue shift.120 Enhancing the signal by catalytic aggregation, essentially employing a hybridization chain reaction where the target triggers the aggregation but is regenerated after each hybridization step, also increases the sensitivity many fold compared to direct aggregation.121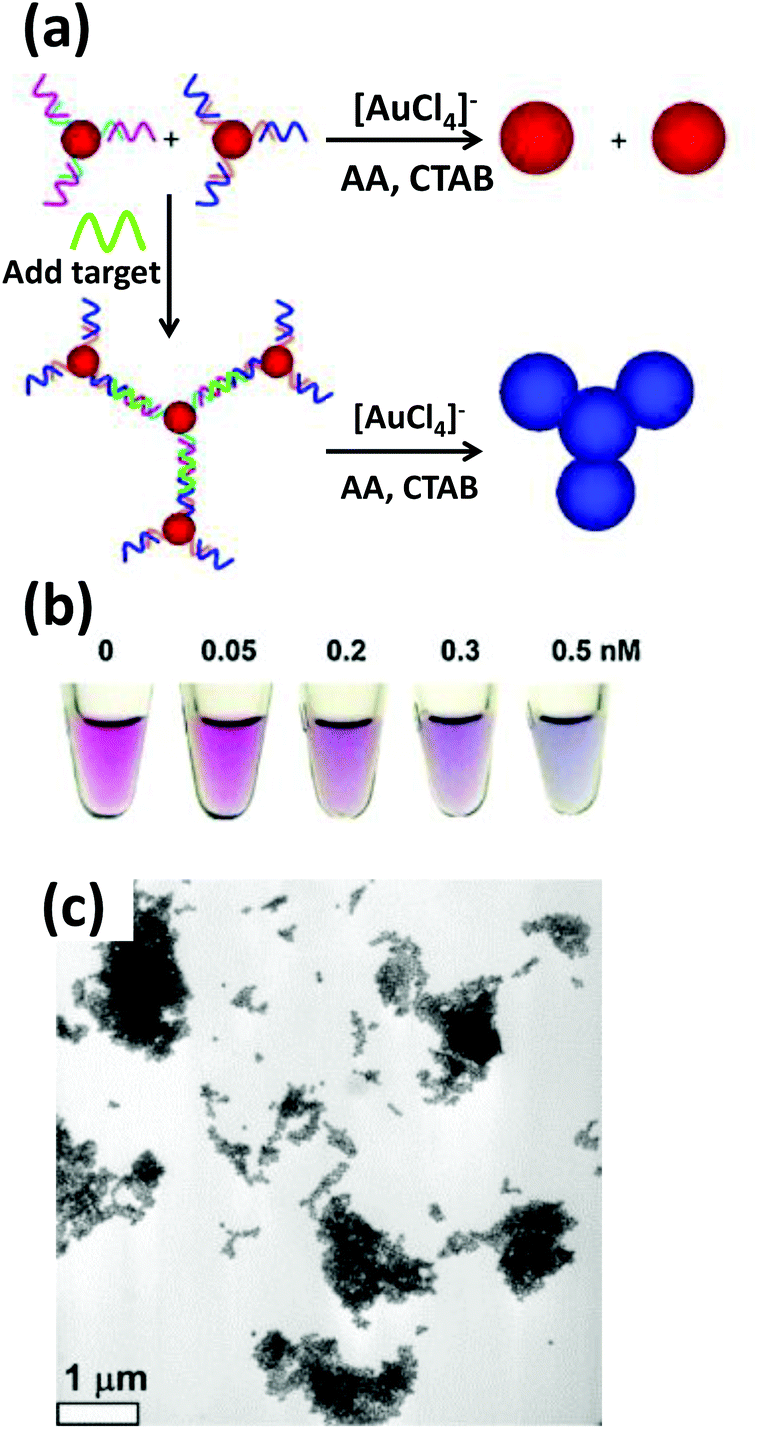 | ||
| Fig. 3 (a) Schematic illustration of a DNA detection assay based on the color change, from purple to blue, due to target DNA driven aggregation of AuNPs followed by their seeded growth. (b) Photograph of the assay solution taken after reduction reaction displaying a stronger blue shift with increasing target DNA concentration. (c) TEM micrograph shows aggregated particles after chemical enlargement. Reprinted with permission from ref. 120. Copyright 2010 Elsevier. | ||
ssDNA and dsDNA adsorb differently onto AuNP surfaces and this too can help with sensing. Due to electrostatic repulsion with the preexisting citrate group on citrate stabilized-AuNPs (the most commonly synthesized and utilized AuNP material), dsDNA with its prominently exposed negatively charged phosphate groups adsorbs less than the ssDNA, which is flexible enough to uncoil its bases and avoid repulsion. More DNA on the surface also confers better stability against aggregation at high salt concentration. Based on this idea of selective adsorption, Li et al. demonstrated a straightforward hybridization assay for detecting untagged oligonucleotides with a detection limit as low as 4.3 nM. The assay was reasonably fast, inexpensive, did not require sophisticated apparatus and was sensitive down to a single base mismatch.122
Besides in vitro DNA sensors, densely coated DNA functionalized AuNPs have also been employed as an intracellular probe for detecting and quantifying over- or under-expressed mRNA. The Mirkin group also developed these materials calling them nano-flares, where 13 nm AuNPs were functionalized with a thiolated DNA comprising an 18-base recognition domain to a specific mRNA sequence.123 The AuNP-appended DNA was partially hybridized to a short DNA sequence modified with Cy5 dye. In the absence of the target mRNA, the dye remained largely quenched by NSET due to the proximity of the Au surface. In the presence of the target, the dye-modified short oligonucleotide was released via strand displacement resulting in restoration of fluorescence. Upon incubation with human breast cancer cells expressing a high level of a specific target mRNA, a strong fluorescence signal was detected while in the control sample displaying a non-complimentary sequence or incubating with negative control mouse endothelial cells, negligible fluorescence was observed. Based on a similar concept, the same group reported a multiplexed nanoflare capable of intracellularly detecting two mRNA at once.124
3.2. Colorimetric detection of miscellaneous analytes
Based on the same colorimetric detection principles as above, other cancer cells have been detected as well.125 The DNA used here was a thiol-functionalized aptamer, with specificity to a cancer cell. AuNPs carrying multiple copies of the aptamer bind to the cancer cell surface resulting in an aggregation that leads to a color change from red to purple. Selectivity and sensitivity of this method has also been confirmed with transmission electron microscope (TEM) and this revealed that the AuNPs indeed are attached specifically to the cancer cell surface while leaving the normal cells unperturbed.125There are many other examples of aptamer-functionalized AuNPs being used for various purposes.126–129 One prominent use is the detection of thrombin, an important protease associated with blood coagulation and the principle is the same as stated before – aggregation of AuNPs and its colorimetric detection.126 The assay protocol is usually quite simple, a thiol-modified thrombin binding aptamer is conjugated to AuNPs and the sensor assembly exposed to a solution containing the protein. Thrombin has two binding sites for the aptamer that work as a cross linker inducing aggregation and resulting in a color change detectable to the naked eye. The signal can be further amplified with catalytic enlargement of the AuNPs. The same principle was applied for sensing thrombin on a glass surface modified with thrombin binding aptamer, as outlined in Fig. 4.126 A similar strategy was also followed to detect lysozyme, another enzyme linked to diseases like leukemia and tuberculosis, using lysozyme specific aptamer-modified AuNPs.130 The methodology is again simple and works reliably even within saliva and urine samples. The powerful capability of aptamer-modified AuNPs also helps with detecting small molecules. For example, Liu and coworkers reported on a general method of sensing adenosine or cocaine.131 Here, the aptamer is not covalently linked to the AuNPs, but acts as a crosslinker by hybridizing partly with oligonucleotides displayed from two different AuNPs (Fig. 5). The aptamer changes its conformation in the presence of target releasing the connected AuNPs resulting in a change of color from blue back to red. However, this simply-designed method requires a relatively high concentration of the analytes to release the particles which are aggregated by multiple crosslinks.131 In an original application of this detection scheme that further relied on using dark field microscopy (DFM) and DNA-functionalized AuNPs as contrast agent, Li and coworkers developed a novel combinatorial technique for imaging latent fingerprints and detecting cocaine on them.132 Here AuNPs functionalized with the cocaine specific aptamer aggregate in the presence of cocaine and this is visible as red dots under DFM (Fig. 6). Interestingly, we note that the robustness of these aptamers for recognizing thrombin, lysozyme, cocaine and ATP (adenosine triphosphate) make them versatile recognition elements for prototyping purposes in the development of many other types of sensor as highlighted below.132
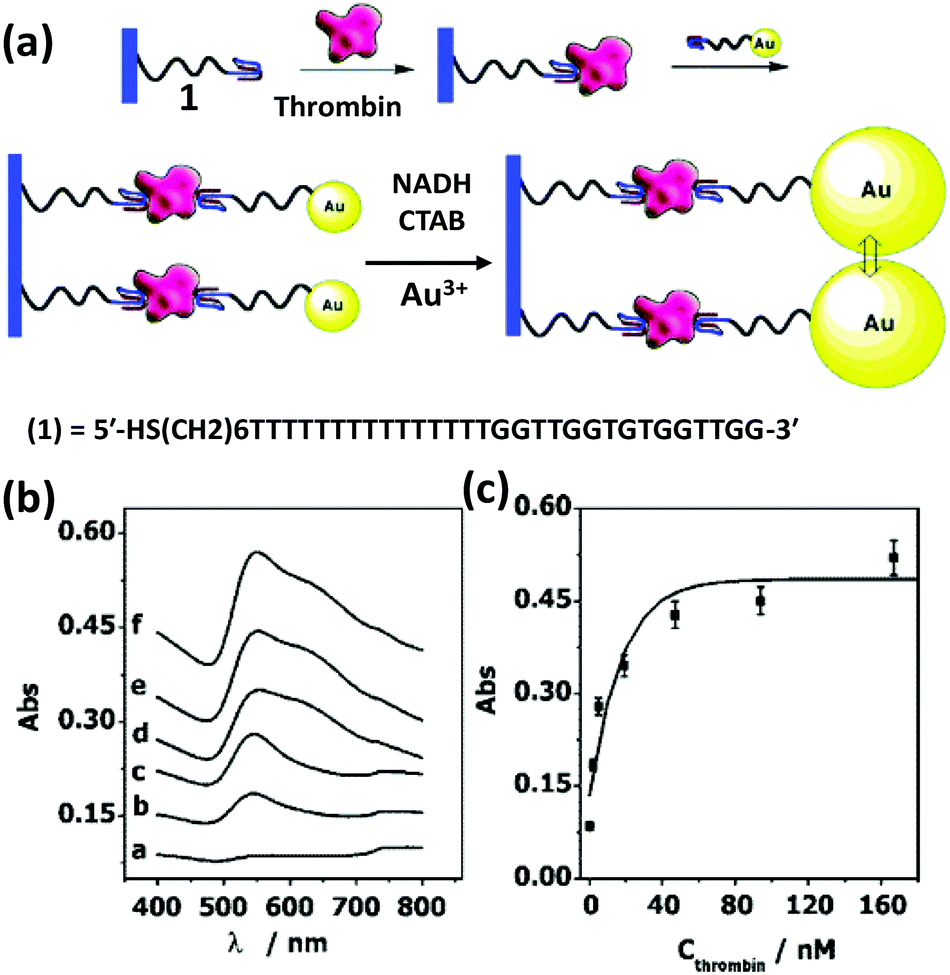 | ||
| Fig. 4 (a) An amplified optical method for detecting thrombin utilizing a thrombin binding aptamer linked to AuNPs. Thrombin acts as a linker here in a sandwich fashion to capture the AuNPs on an aptamer bound glass surface. The plasmonic signal is amplified by enlarging the AuNPs via a seeded growth process. (b,c) Plots show absorbance increases consistently with increasing thrombin concentration: a-0, b-2, c-5, d-19, e-94, and f-167 nM. Reprinted with permission from ref. 126. Copyright 2004 American Chemical Society. | ||
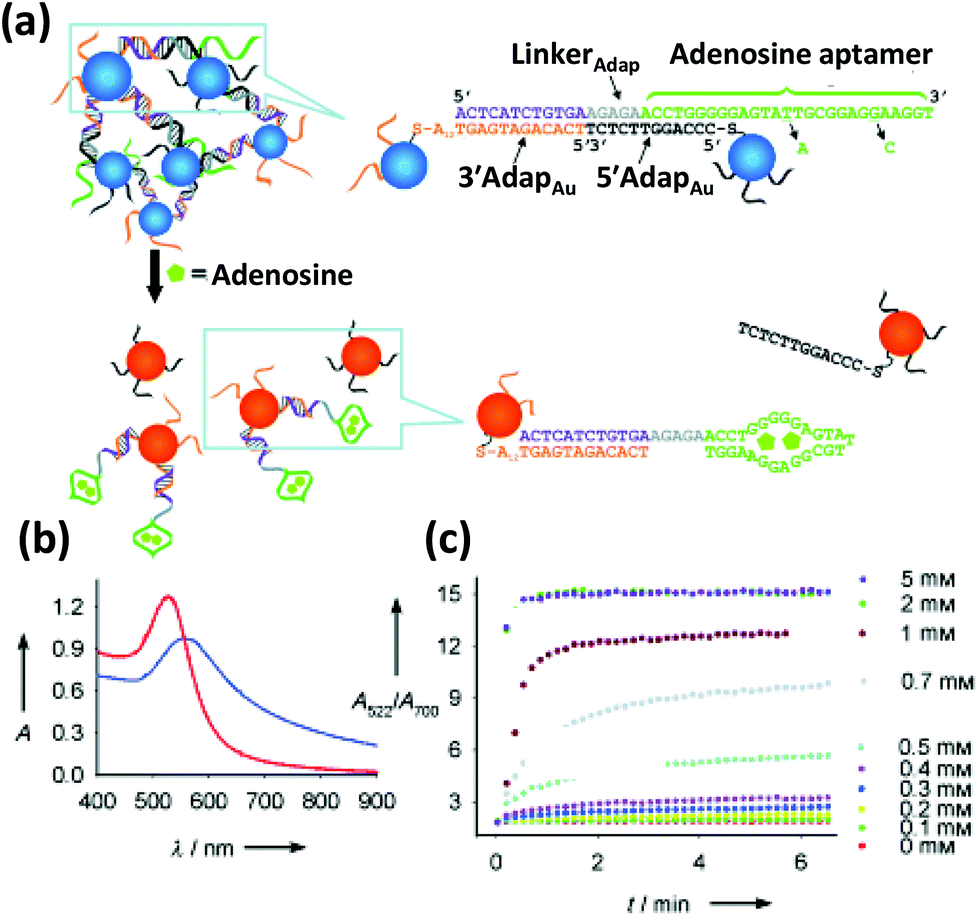 | ||
| Fig. 5 (a) Schematic depicting the colorimetric detection of adenosine. The adenosine binding aptamer, previously bound to a complementary DNA resulting in an aggregation of AuNPs, preferentially binds to its substrate, adenosine, releasing the AuNPs and concomitantly blue shifting its absorption maxima (b). (c) The kinetics of the color change, measured as a ratio of Abs at 522 nm to Abs at 700 nm displays a quick disaggregation at a range of adenosine concentration from 0.1 mM to 5 mM. Reproduced with permission from ref. 131. Copyright 2005 Wiley-VCH. | ||
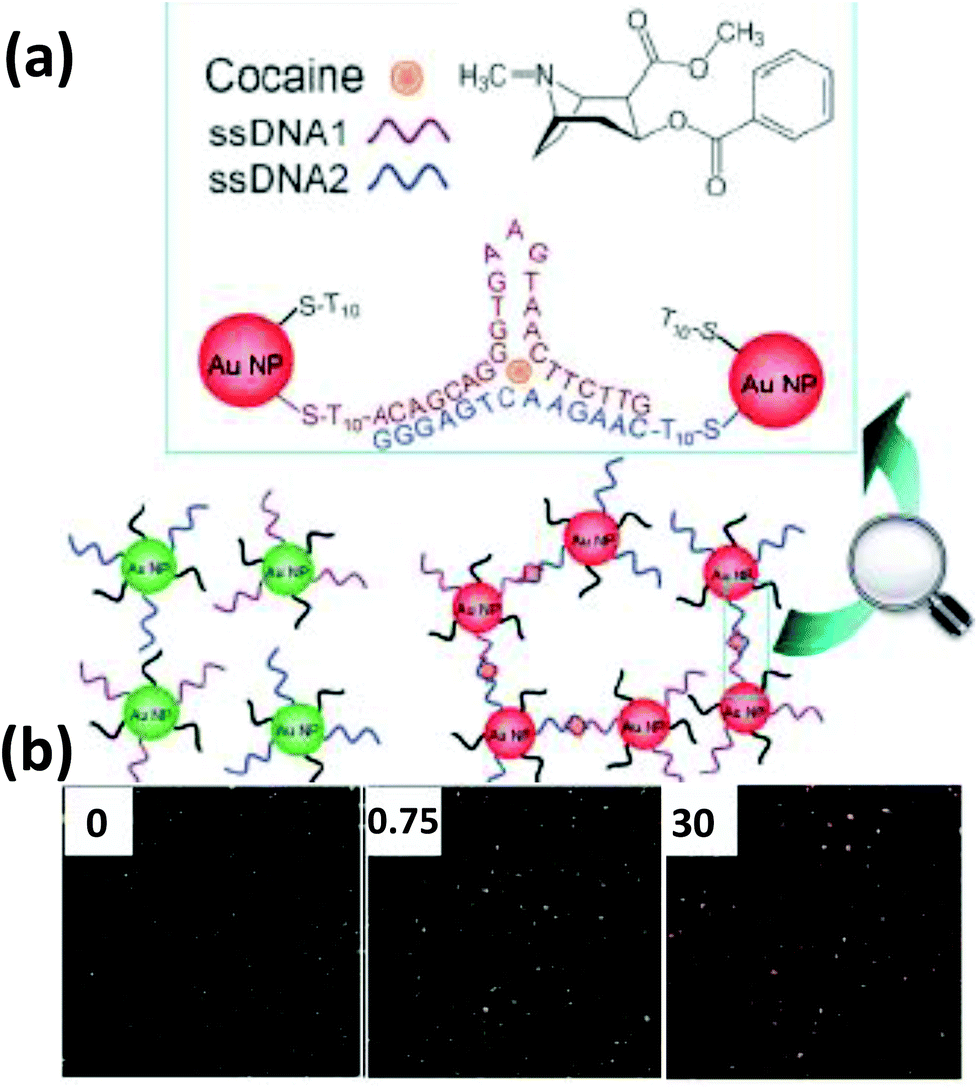 | ||
| Fig. 6 (a) Schematic illustration of a nanoplasmonic strategy for cocaine detection, employing a cocaine binding DNA aptamer attached to AuNPs. Cocaine binds to the aptamer in the specific style shown in the illustration which results in an aggregation visible under DFM. (b) The DFM images show green, orange and red dots representing the greater extents of aggregation with increasing cocaine concentrations (in μg in 10 μl). Reproduced with permission from ref. 132. Copyright 2013 Wiley-VCH. | ||
Not being just limited to detection, molecular-DNA interactions have also been examined with the help of DNA-modified AuNPs. Mirkin's group studied a group of molecules that bind to dsDNA they called the ‘binders’ which include antitumor agents like ellipticine, amsacrine and daunorubicin.133 The driving principle here is that AuNPs carrying complementary DNA sequences aggregate when mixed together in the presence of salt. This process is reversible as dsDNA melts at high temperature and become ssDNA that releases the particles which is accompanied with a color change from purple back to red. But this melting process gets perturbed in the presence of the “binder” molecules; the Tm's change depending on the binding affinity. Molecules with stronger binding affinity prevent the unwinding process and thus increase the Tm which was used as the primary characterization method for how strongly or weakly these molecules interacted with DNA.133
Another significant contribution in this sensing area is the detection of toxic metal ions such as Hg2+ and Pb2+ using DNA-conjugated AuNPs.134–137 This sensing relies on the strong binding affinity of Hg2+ towards thymine nucleobases. Poly-T-adsorbed AuNPs are stable at moderately high salt concentration but in the presence of Hg2+ ions they tend to aggregate. Hg2+ ions form a chelate with thymine nucleotides which changes the overall conformation of the ssDNA from a random coil to a folded structure. This transition reduces the zeta (ζ) potential or net charge on the AuNP surface and, in turn, the electrostatic repulsion. The selectivity of this technique was confirmed in the presence of other metal ions.135 Wang et al. described a slightly different method for the detection of the same ion.136 Here, a fluorophore-modified thymine rich ssDNA was adsorbed onto a 13 nm AuNP surface. In the presence of Hg2+ ions and a complementary oligonucleotide, it formed a dsDNA which released it from the particle surface and induced two changes. The fluorescence of the dye that was initially quenched on the AuNP surface was restored and in the absence of protective DNA on the surface, the AuNPs aggregated resulting in a typical change in color.136 In another sensing configuration, Liu et al. demonstrated a novel strategy for detecting Pb2+ involving DNA-functionalized AuNPs, DNAzymes and their substrate.137 The substrate strand was designed in such a way that at both ends it had complementary sequences to the DNA displayed on the AuNPs surface. Mixing them together caused aggregation again leading to a change in color from red to blue. In the presence of Pb2+ ions, DNAzyme catalytically cleaves the substrate strands unlocking the AuNPs which is also reflected in a reverse color change.137
As mentioned, although AuNPs can effectively quench the fluorescence of proximal fluorophores, the nature of this ET process seems to be different from traditional FRET and appears to work at much larger donor–acceptor separation lengths.95,96,138,139 Based on this quenching capability, several sensing schemes have been developed in the past few years. Huang et al. fabricated an AuNP-based biosensor for platelet derived growth factor (PDGF), with a detection limit of about 3.2 nM.140 PDGF-specific aptamer-modified AuNPs were mixed with the fluorophore N,N-dimethyl-2,7-diazapyrenium dication (DMDAP) which has affinity towards DNA. When the dye was intercalated between the nucleobases, its fluorescence was significantly quenched due to the proximity of the AuNPs. When PDGF bound to its specific aptamer, the intercalated dye was released and its fluorescence was restored.140
3.3. Gene and drug delivery
AuNPs are also an attractive nanoparticulate candidate for drug and gene delivery and, indeed, colloidal gold has a long history of application in medicine.141–146 Advantages for these purposes include minimal toxicity, high cellular uptake despite very high negative ζ potential, facile and well-established synthetic methods capable of producing nearly monodisperse particles, simple thiol-based chemistry along with other chemical (bio)functionalization protocols, and, most importantly, their remarkable stability in serum and intracellular environments against nuclease degradation.52 These unique properties most likely stem from the display of a dense and highly oriented DNA layer as the same citrate capped AuNPs or ones passivated with bovine serum albumin (BSA) show significantly lower cellular uptake.147,148 SNAs with other than an AuNP core or even coreless hollow SNAs, prepared by crosslinking the nucleic acid followed by dissolution of a structural AuNPs, display similarly high transfection properties.149In addition, significant progress has already been made on the area of gene delivery via non-covalent conjugation to AuNPs. Materials functionalized with cationic groups adsorb DNA electrostatically and, as mentioned, can even protect them from enzymatic digestion. For example, Sandhu and coworkers reported transfection of a β-galactoside encoding plasmid with mixed monolayer-protected 2 nm Au clusters into human embryonic kidney (HEK) cells.150 The Au clusters were modified with octane thiol along with a cationic ammonium thiol that helped to bind to the negatively charged DNA. This amphiphilicity also conferred better cellular internalization of the particles. Thomas et al. approached the next stage by delivering plasmid DNA electrostatically adsorbed on AuNPs modified with branched polyethylenimine to COS-1 monkey kidney cells.151 Han et al. demonstrated coupling of an externally initiated actuation for control of subsequent gene delivery. Here, DNA delivery utilized AuNPs modified with a photo-labile cationic group.152 A quaternary ammonium salt group responsible for the positive charge was linked to the particle via a photocleavable o-nitrobenzyl ester group. UV light (>350 nm) cleaved the o-nitrobenzyl ester linkage, releasing the positively charged group and consequently releasing the DNA (Fig. 7a).152
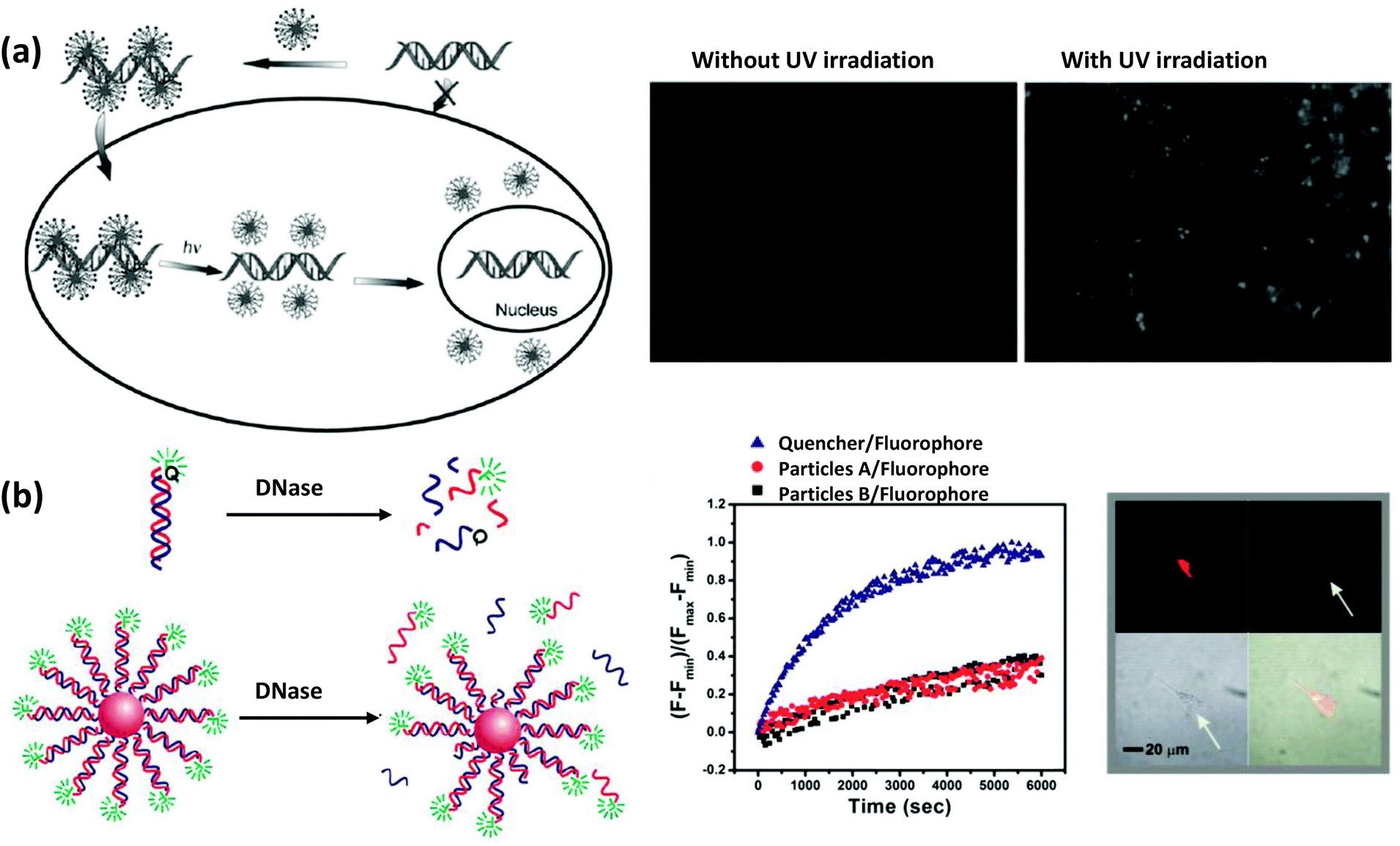 | ||
| Fig. 7 (a) Schematic demonstrating a DNA delivery system to live cells and their release upon UV irradiation employing AuNPs functionalized with a photolabile cationic molecule that electrostatically adsorbs negatively charged DNA. The fluorescence microscopy images show a significant release of the FAM-labeled DNA from AuNPs, which were mostly quenched in proximity to the AuNPs before the UV irradiation.116 Reproduced with permission from ref. 152. Copyright 2006 Wiley-VCH. (b) Schematic depicting the higher resistance power of DNA against DNase degradation when attached to AuNPs. Exposing the bare duplexes, modified with a fluorophore and quencher, to DNase shows a much faster rate of fluorescence recovery compared to the duplexes attached to AuNPs. After 48 h of incubation of AuNPs, conjugated to Cy3- and Cy5.5-modified dsDNA (Cy3 close to the surface and Cy5.5 about 9 nm away from the surface), with C166-eGFP cells, a strong fluorescence signal from Cy5.5 was observed under confocal fluorescence microscope upon excitation at 633 nm (upper left) while negligible fluorescence was observed from Cy3 upon excitation at 546 nm (upper right), indicating their stability in the intercellular environment. The lower left and lower right quadrants display the transmission and composite overlay images. From ref. 153. Reproduced with permission from AAAS. | ||
The Mirkin group reported a novel strategy for intercellular gene regulation using modified SNAs.153 They used a psDNA with complementarity to the mRNA sequence that codes for enhanced green fluorescent protein (eGFP). The strategy behind this antisense therapy approach is that the oligonucleotides will bind by complementarity to mRNA which will then down-regulate the expression of eGFP. Free oligonucleotides are, however, normally susceptible to enzymatic degradation inside the cell which can make this approach quite challenging. It is hypothesized that such enzymatic degradation of free DNA may represent an innate cellular response or protective mechanism arising through evolution to protect against foreign DNA invasion from viruses and the like.154,155 They observed, however, that densely-packed oligonucleotides on the AuNP surface have much better stability against DNase degradation and hybridize more efficiently with complementary mRNA (Fig. 7b). This could be due to two factors, first, the ps backbone itself provides higher nuclease resistance compared to natural phosphodiester backbones.156,157 Secondly, it is believed that high local sodium ion concentration around SNAs is responsible for deactivating proximal enzymes such as nucleases conferring enhanced stability of the nucleic acids as well as preventing unnecessary immune response. Upon internalization of those particles in C166 mouse endothelial cells, they observed less fluorescence confirming a lower degree of eGFP expression. More recently, they improved the system with target specificity by incorporating antibody-labeled DNA that bound only to cells that are overexpressing the particular targeted receptors.158
Besides non-covalent gene delivery, oligonucleotide-functionalized AuNPs have also been covalently linked to anti-cancer drugs and used to deliver them as cargo.159 Lippard's group covalently linked a Pt(IV) complex displaying a carboxylic acid group to AuNP modified with amine functionalized DNA. Upon internalization of the particles, the cytosol reduced Pt(IV) to Pt(II) which released it from the complex and after translocating into the nucleus it formed 1,2-d(GpG) intrastrand crosslinks resulting in cytotoxicity. Interestingly it was observed that such Pt–DNA–AuNPs are far more effective than traditionally used cisplatin in killing cancer cells.159
3.4. Catalytic activity
Another interesting property of AuNPs that is not widely appreciated in the bioapplications community is that they can mimic certain types of enzymatic activity as evidenced by their glucose oxidase- (GOx) type activity.160–162 This has been used for sensing glucose within the context of classic GOx-horseradish peroxidase- (HRP) cascaded reactions and catalysis has been found to directly depend on the amount of available surface area and DNA functionalization; the former almost always plays an important role on the catalytic behavior of the particles in general.162 In this case, denser packing of ssDNA on the particle surface leaves less surface area intact which results in a significantly poorer catalytic activity than that of dsDNA. For utility in a sensing assay, GOx-mediated oxidation of glucose can produce H2O2 which can be utilized for the seed-mediated growth of AuNPs in a reducing HAuCl4 solution (Fig. 8). Utilizing the correspondingly dsDNA coated particles showed an enhanced growth rate as detected by DFM.163 This approach has potential for use in assay formats where enzyme lability can be an issue or where refrigeration is not available. It will also be interesting to see whether the catalytic activity of these NPs can be enhanced in a manner similar to that seen for enzymes attached to NPs.164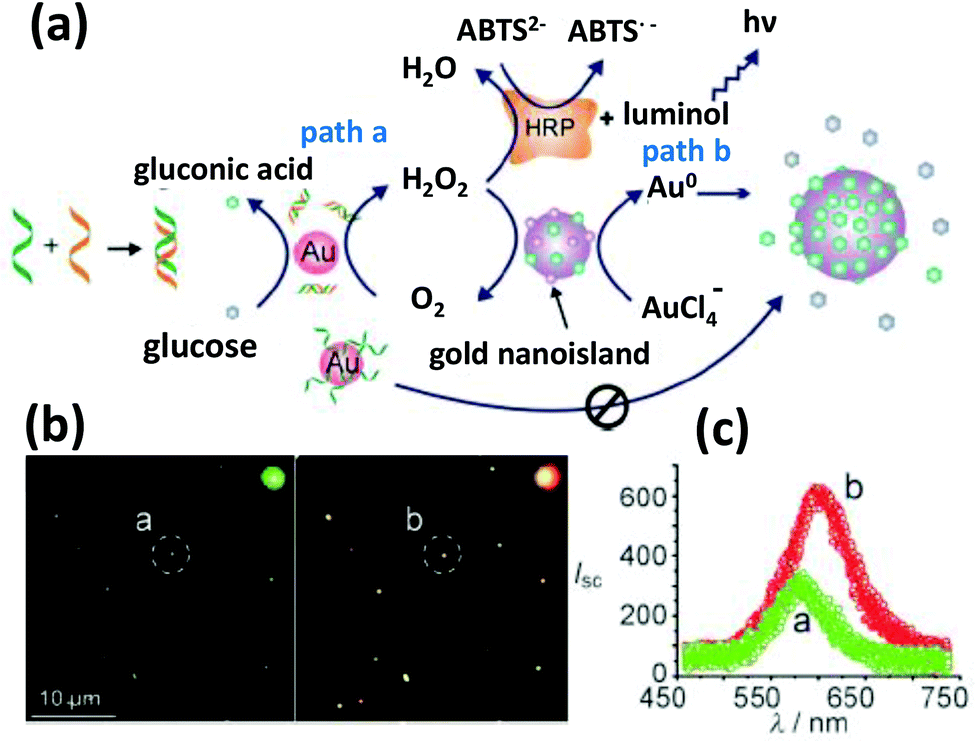 | ||
| Fig. 8 (a) A DNA hybridization detection strategy exploiting the GOx like catalytic activity of AuNPs. The catalytic efficiency is regulated by DNA hybridization, which can be read out via a HRP cascaded luminescence reaction or size enhancement of the AuNPs. (b) The dark field microscopy images show considerably red shifted and intense scattering from larger AuNPs. (c) The light scattering spectra of individual nanoparticles circled in (b). Reproduced with permission from ref. 163. Copyright 2011, Wiley-VCH. | ||
3.5. Electrochemical properties
Because of their outstanding conductivity, AuNPs have also been applied for the detection of various analytes by electrochemical means. Methods of electrochemically detecting DNA with AuNPs have been reviewed in detail before.165,166 In one of the first iterations, Mirkin's group reported a three-component sandwich assay for detecting DNA.167 They prepared a microelectrode array with 20 μm separation that had probe oligomers immobilized in the gaps (Fig. 9). Both the probe DNA and the DNA on the AuNP surface are partly complementary to the target. In the presence of target DNA, AuNPs are captured in the gap. Upon seeded growth of Ag via oxidation of Ag salts, individual electrodes get connected by a string of particles resulting in a sharp drop of circuit resistance.167 Willner's group demonstrated that relatively low concentration DNA detection could be achieved and the sensitivity of the colorimetric aggregation strategy can be significantly improved by associating a redox-active substance with the NPs and then interrogating the resulting aggregates with differential pulse voltammetry.168 The redox-active dye Methylene blue intercalates between the nucleobases of dsDNA and in the presence of target DNA, aggregated particles can act as a conductive matrix with the embedded dye to pass the electrochemical response. Besides just DNA detection, DNA-functionalized AuNPs have also been used for electrochemically studying the interaction mechanisms of several DNA binding drugs such as mythamycin, netropsin, and nogalamycin.169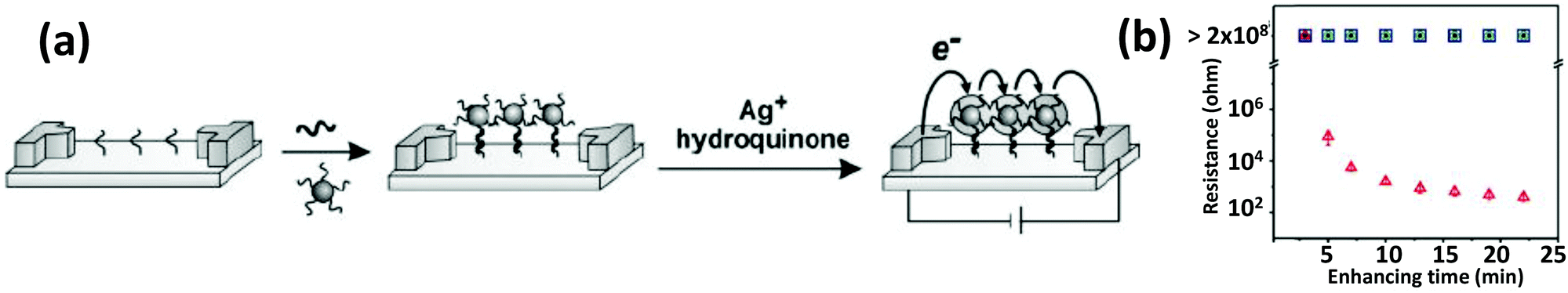 | ||
| Fig. 9 (a) Schematic describing an array-based DNA detection method that relies on the change of conductivity upon binding to AuNP-conjugated DNA. (b) An exponentially sharp decrease in resistance was observed with respect to silver enhancing time. From ref. 167. Reprinted with permission from the AAAS. | ||
3.6. Anisotropic gold nanoparticles
The continued progress on AuNP synthetic methods has now provided access to a library of various anisotropically-shaped AuNPs.170–173 Of all of these different architectures, Au nanorods (AuNRs) have seen the most successful conjugation to DNA along with subsequent employment within various applications.174,175 Because of their anisotropic shape, AuNRs have two distinct plasmon absorption bands, one in the visible range similar to spherical particles which reflects the transverse oscillation of the electrons, while the other is in the NIR region and represents the longitudinal oscillation of electrons (Fig. 10).91,176 When irradiated with NIR light at a wavelength close to their longitudinal plasmonic absorption band, AuNRs are capable of converting the optical energy into heat resulting in a localized heating effect that is far more intense than that of spherical AuNPs. Since NIR light has a better tissue penetration capability, AuNRs have been undergoing concerted prototyping for photothermal therapy (PTT) to kill cancer cells.177,178 In one pertinent demonstration, Huang and coworkers reported on an aptamer-modified AuNR for targeted PTT of cancer cells.179 The custom synthesized nanorods utilized were not purely made of Au but of Au–Ag and were prepared by the seeded growth of Au and Ag in solution containing AuNRs as seed. They observed that the aptamer-functionalized AuNRs bound very efficiently to cancer cells, in fact far better than the free aptamers as the conjugated aptamers are capable of simultaneous multivalent interactions. Upon irradiation with 800 nm laser light, an average 93% death of NR-labeled cells was observed while, in contrast, 87% of control non-NR labeled cells remained intact.141 Wang and coworkers fabricated similar aptamer-functionalized AuNRs but utilized them for multimodal therapy (Fig. 11a).180 The AuNRs here were functionalized with a thiolated-aptamer that specifically targets leukemic T cells. Later the aptamer was hybridized to a short oligomer carrying the photosensitizer molecule Chlorin e6 (Ce6). Upon binding to the cancer cells, the aptamer no longer stayed hybridized to the Ce6-modified short oligomers, but instead formed a MB-type structure that bound to the targeted receptor proteins located on the cancer cell surface. The photosensitizer is non-toxic to cells before light irradiation but when exposed to NIR laser it produced reactive oxygen species (ROS), including singlet oxygen, which are lethal to cells at appropriate dosages. In addition, NIR laser irradiation produced a local thermal effect from the AuNRs. As expected, the combined effect of PTT and photosensitizer therapy (PST) had a more pronounced effect in killing diseased cells.180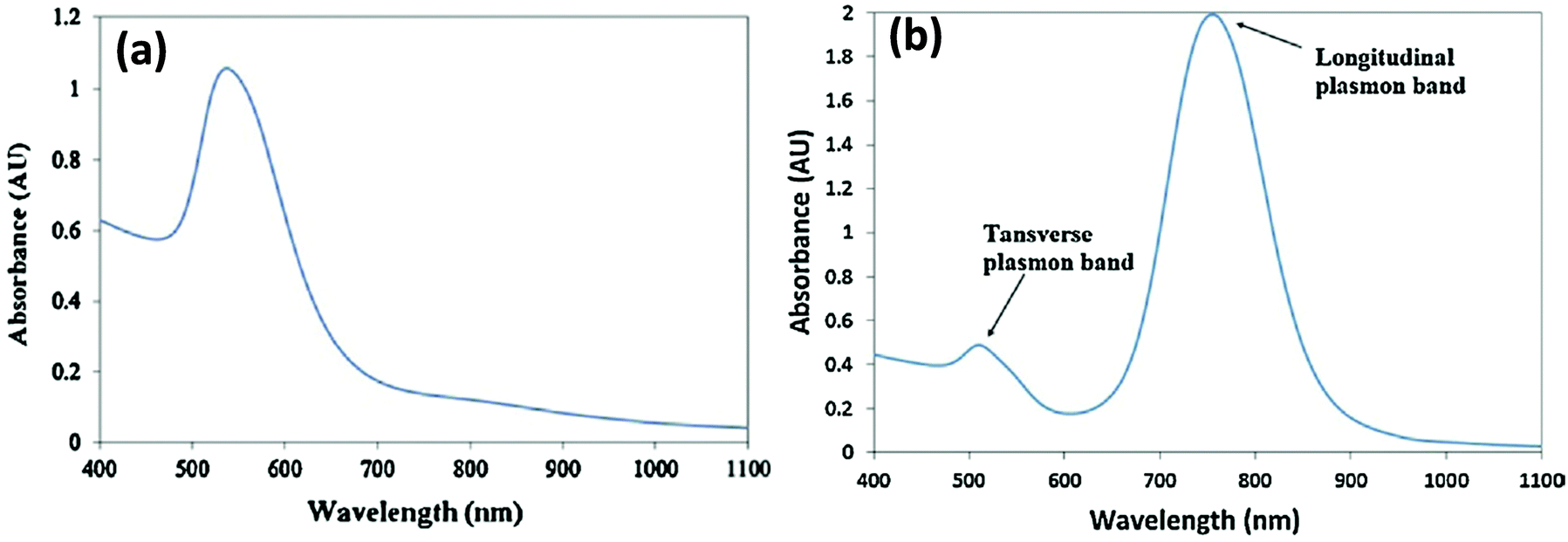 | ||
| Fig. 10 (a) Typical plasmon absorption spectrum of spherical AuNPs (b) Plasmon absorption spectrum of AuNR. Longitudinal and transverse bands correspond to electron oscillation along the long and short axis respectively. Reprinted with permission from ref. 181. Copyright 2011 Elsevier. | ||
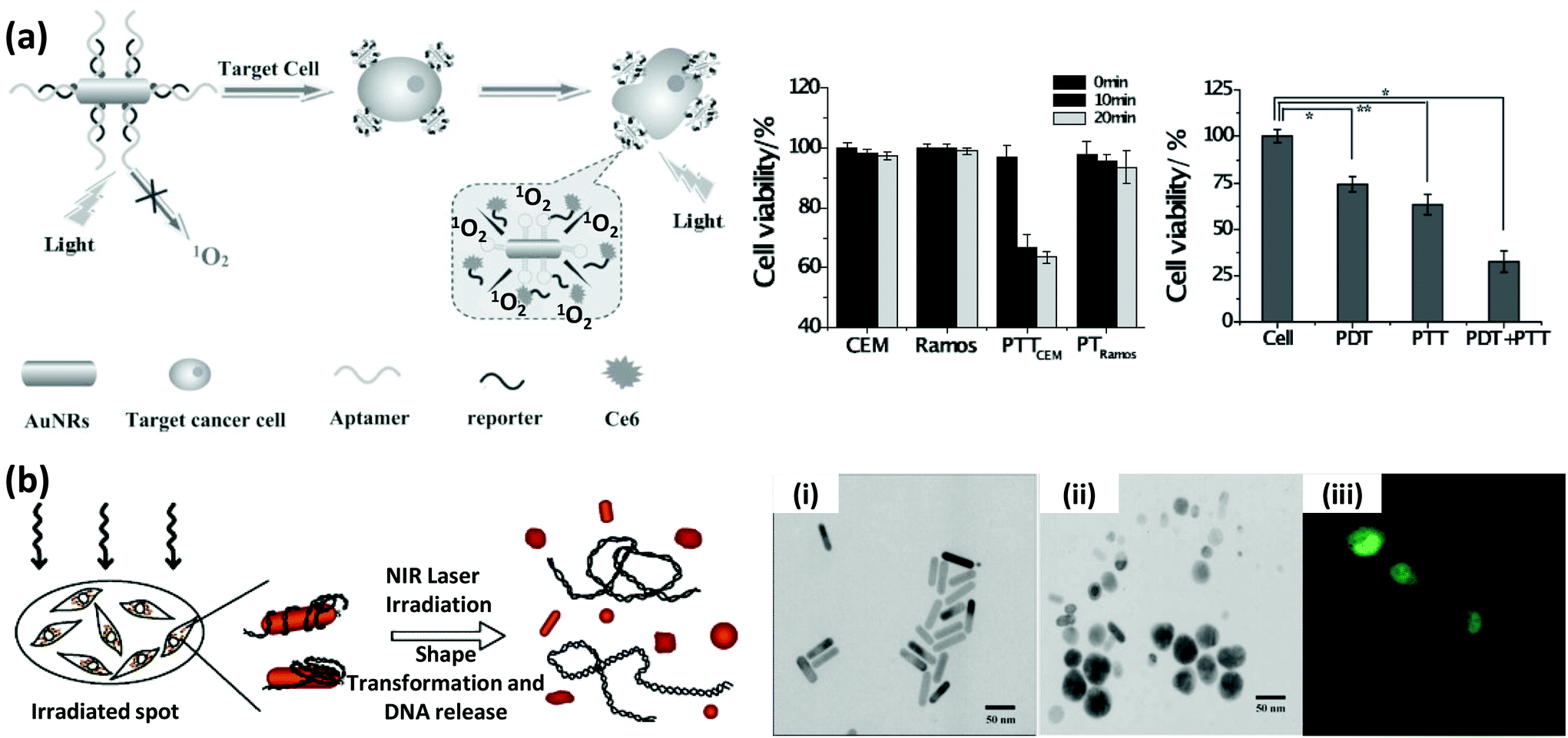 | ||
| Fig. 11 (a) Schematic showing the targeted multimodal therapeutic approach using AuNRs conjugated to a leukemic T cell-binding aptamer. The complementary strand to the aptamer was ligated to a photosensitizer molecule, Ce6. The combined effect of PTT from AuNRs and photodynamic therapy from Ce6 displayed a stronger capability, compared to the individual effects, in killing the target cells (CEM), leaving the non-target cells (Ramos) largely intact. Reproduced with permission from ref. 180. Copyright 2013 Wiley-VCH. (b) eGFP expressing gene sequence attached to AuNRs were delivered, and after NIR laser irradiation, released inside HeLa cells. The NIR irradiation induced a shape transformation, as evidenced by the TEM images before (i) and after (ii) the treatment, which triggered the release of the adsorbed gene. (iii) The confocal microscope image shows the expressed GFP inside the cells. Reprinted with permission from ref. 183. Copyright 2006 American Chemical Society. | ||
The heating effect that arises when AuNRs are irradiated with NIR laser can also induce a unique shape transition in the nanoparticulates from rod to sphere.182 This transition can, in turn, affect the binding of biomolecules onto the surface. Exploiting this property, Chen and coworkers reported a way of remote controlling gene expression.183 For this, eGFP encoding DNA was attached to the AuNRs and exposed to cells and upon irradiation with femto-second 800 nm laser pulses, they measured a release of nearly 70% of control DNA accompanied with the typical shape transformation (Fig. 11b). eGFP expression was observed within 1–2 days after laser irradiation. However, the poor transfection rate of the rod material (<20%) remains a challenge for this technique to be applied within therapeutic approaches.183 Since the SPR band changes with length, diameter and the aspect ratio of the rod, they can also be selectively melted by NIR laser of different wavelengths. Wijaya and coworkers employed this concept to selectively release different DNAs from a mixture of two AuNRs.184 Rods with an absorption band at ∼800 nm were conjugated to DNA modified with fluorescein dye while the ones with absorption at ∼1100 nm were labeled with a rhodamine dye. In a mixture of the two rods, 800 nm laser exposure melted only one type of rod releasing 70% of fluorescein and <10% of the rhodamine. The other laser produced a similar result targeting the other NR-oligo in the converse configuration.184
Besides delivery of DNA, AuNR assemblies have been used for the detection of DNA with surprisingly low limits of detection (LODs) suggesting this could be very useful in medical diagnostics and especially in forensics analysis.185 The sensing mechanism here lies in the chiroplasmonic behavior of self-assembled AuNRs. Two types of thiolated-primers were conjugated to AuNRs having sequences partly complementary to the target DNA. In the presence of target DNA, its PCR replication led to the assembly of the AuNRs. The side-by-side assembly produced a strongly defined circular dichroic (CD) signal in the 500–800 nm region while end-to-end assemblies or simple DNA-modified rods fail to generate the same; this was attributed to a slight twist between the rods in the former case. Since the bi-signate CD signal was linearly dependent on the concentration of the target DNA, this provided for a very sensitive LOD of a few attomoles.185
3.7. Gold nanoclusters
Most gold NMs are notable as fluorescence quenchers but they can also demonstrate size-dependent PL across the spectrum when the size of the crystalline particles is significantly reduced to the 1–2 nm range (Fig. 12).186–189 With typically less than 100 atoms, these NPs, or more familiarly nanoclusters (NCs), develop discrete energy levels that are responsible for the fluorescence.190,191 This relatively new material has drawn significant research interest after it was found that DNA can act as a good stabilizer of these tiny clusters suggesting that they have strong potential to be become a new type of probe and be incorporated into biosensors.186 There are many examples of DNA-templated synthesis of highly fluorescent silver nanoclusters (AgNCs) (vide infra) although having the same type of templating with Au is quite limited. Unlike AgNCs, the fluorescence emission of AuNCs depends more on the reducing agent than on the actual sequence of DNA used. For example, NaBH4 fails to produce any fluorescent NCs, but citrate, being a mild reducing agent, produces blue-emitting NCs while using dimethylamine borane (DMAB) as a reducing agent generates red emitting NCs.192,193 The quantum yield (QY) of the resulting NCs seems to also depend on the DNA sequence with cytosine and guanine rich DNA performing better than those enriched with adenine and thymine.194 DNA-functionalized Au/Ag hybrid NCs have been used for sulfide ion detection since the sulfide ions bind strongly to Au/Ag which triggers a conformational change of the DNA from hairpin to random coil resulting in a quenching of the NC's fluorescence.195 Although there have been several reports involving AuNCs as a sensor or imaging agent,196,197 DNA-functionalized AuNCs have not found broad application here yet. However, it is widely speculated that once the chemistry and photophysics are optimized, AuNCs may help address the photobleaching and oxidation issues commonly found when utilizing AgNCs.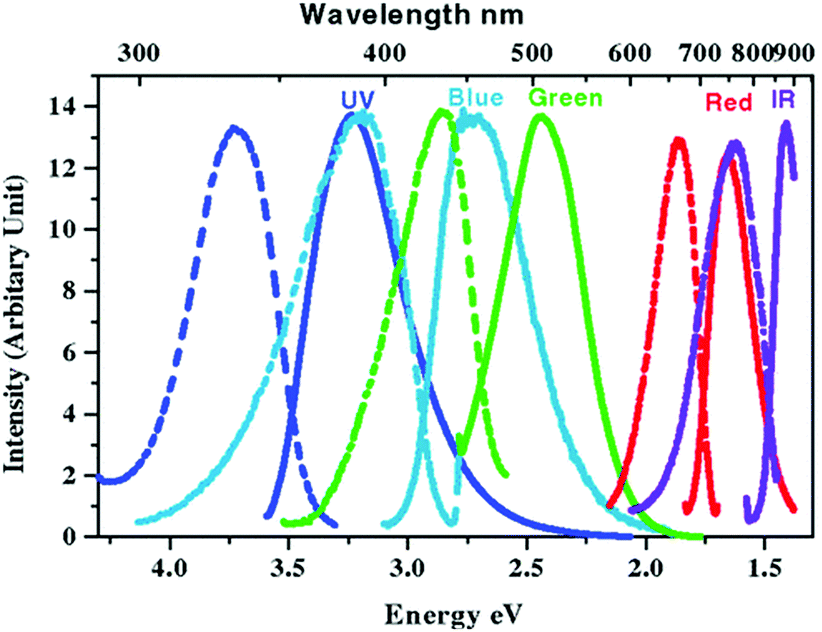 | ||
| Fig. 12 Steady state excitation (dashed) and emission (solid) spectra of different water soluble AuNCs synthesized via slow reduction of HAuCl4 in the presence of poly(amidoamine) dendrimers. Emission maxima shifts to higher wavelengths as the clusters grow larger. Reprinted with permission from ref. 198, Copyright (2004) by the American Physical Society. | ||
3.8. DNA-directed assembly of gold nanoparticles
Another notable and rapidly developing area of research involving DNA-functionalized AuNPs is that of utilizing DNA to direct self-assembly into well-defined architectures. The research so far in this area can be categorized into two primary foci; first is building crystalline lattices by hybridizing NPs bearing complementary sequences; the other one is the spatial organization of the NPs on nanostructures composed entirely of DNA.199–204 DNA-based assembly is attractive as it can bring NPs closer together to within some predefined interacting distance and can also control relative orientation or stoichiometry which can lead to new properties and hence can generate new functional materials.205–207 As mentioned, when AuNPs are aggregated, a change in color from red to purple is observed due to interaction of their SPR at close distance.208 DNA-driven assembly potentially allows for control over the degree of plasmonic interaction by modulating inter-NP distances; this could also be useful as a molecular level plasmonic ruler.139,209 This powerful technology may also allow us to bring two particles nearly within contact distance thus generating a nanogap that can produce an intensely-enhanced local electric field. If a fluorophore molecule is placed in that concentrated electric field, its rate of photoexcitation is increased significantly producing a stronger fluorescence signal.210 Raman scattering, which is well known for its poor scattering cross section, can also generate a much stronger signal in such a nanogap.207DNA structural nanotechnology, which is exploited here to organize NPs into discrete structures, was pioneered by Seeman in the mid 80s and this effectively overturned its classical role as a genetic information carrier since the DNA now functions as an excellent building block for nano-construction. As mentioned earlier, a myriad of DNA structures from simple 2D polymeric assemblies to complex 3D discrete and dynamic structures have been fabricated over the last few years.211–220 Later, it was realized these structures could, in turn, also serve as scaffolds for organizing NPs. Generally two strategies are followed to incorporate NPs into DNA nanostructures: (1) hybridizing the NP bearing one or multiple copies of DNA to preformed DNA structures with ‘capture strands’ having complementary sequences protruding from the NP surface; and (2) during the annealing of structures, replacing a selected strand of a DNA structure with one that is conjugated to a NP in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Most of the early research performed on arranging AuNPs in periodic arrays within polymeric DNA was driven mainly by proof of concept studies.221 In spite of relatively good progress on DNA tile-based arrays, it failed to rapidly meet expectations. This was probably due to two related factors: first, is that the materials were more like a continuous sheet without a proper boundary; and second, lack of control over the range of all the inter-NP distances that one requires for electronic and other photonic uses.
:1 ratio. Most of the early research performed on arranging AuNPs in periodic arrays within polymeric DNA was driven mainly by proof of concept studies.221 In spite of relatively good progress on DNA tile-based arrays, it failed to rapidly meet expectations. This was probably due to two related factors: first, is that the materials were more like a continuous sheet without a proper boundary; and second, lack of control over the range of all the inter-NP distances that one requires for electronic and other photonic uses.
DNA origami, a subset of the DNA structural technology family, has emerged as an alternative tool for assembling NPs with nanometer precision and, in many cases, absolute control over stoichiometry. DNA origami are discrete DNA nanostructures where a long ssDNA scaffold strand, generally the M13 bacteriophage genome, is folded by numerous short staple strands into an array of antiparallel helices that, in turn, shape themselves up into simple geometric structures like rectangles or triangles or even into complex 3D structure with impressive curvature (see Fig. 2e).213–217 Like DNA tile-based arrays, DNA origami was initially exploited to assemble several types of NPs of different sizes including AuNPs, AuNRs, silver nanoparticles (AgNPs) and QDs to primarily show the capability of controlling distance, angle and relative orientation with high yield.206,222,223 This turned out to be a powerful technique capable of engineering new materials in a well-controlled manner. What was postulated earlier, that fluorescence signals can be increased by an impressive amount (>100×) by carefully integrating two AuNPs on a DNA origami and placing a fluorophore in the hotspot was also shown (Fig. 13a).210 Likewise, Thacker and Pilo Pias separately demonstrated that Raman signal from a fluorophore or nonfluorescent molecule (4-aminobenzenethiol) can be enhanced significantly by employing a similar strategy of creating and exploiting an enhancement hot spot (Fig. 13b).224,225 The Liedl group reported that origami can also be used to arrange AuNPs in a helical fashion that subsequently produces a strong CD signal.226 They have also shown that by using different sized AuNPs or by chemically depositing silver on the helical structure, the CD signal can be tuned. AuNRs can also generate a CD signal when assembled in a cross sign fashion on two sides of a 2D origami.227 More recently, Kuzyk et al. reported a reconfigurable plasmonic structure by assembling two AuNRs on two cross-linked DNA origami tubes.228 The angle between the tubes can be modulated by addition of external DNA which ultimately affects the plasmonic interaction between the two tubes resulting in a unique change in the CD signal (Fig. 14). These are excellent examples of the usefulness of DNA-based bottom up approaches to assemble NPs that would otherwise be quite difficult and expensive to achieve when utilizing standard techniques such as E-beam lithography. Implementing these types of constructs as smart sensing systems within the context of an in situ probe may also prove useful for therapeutic applications.
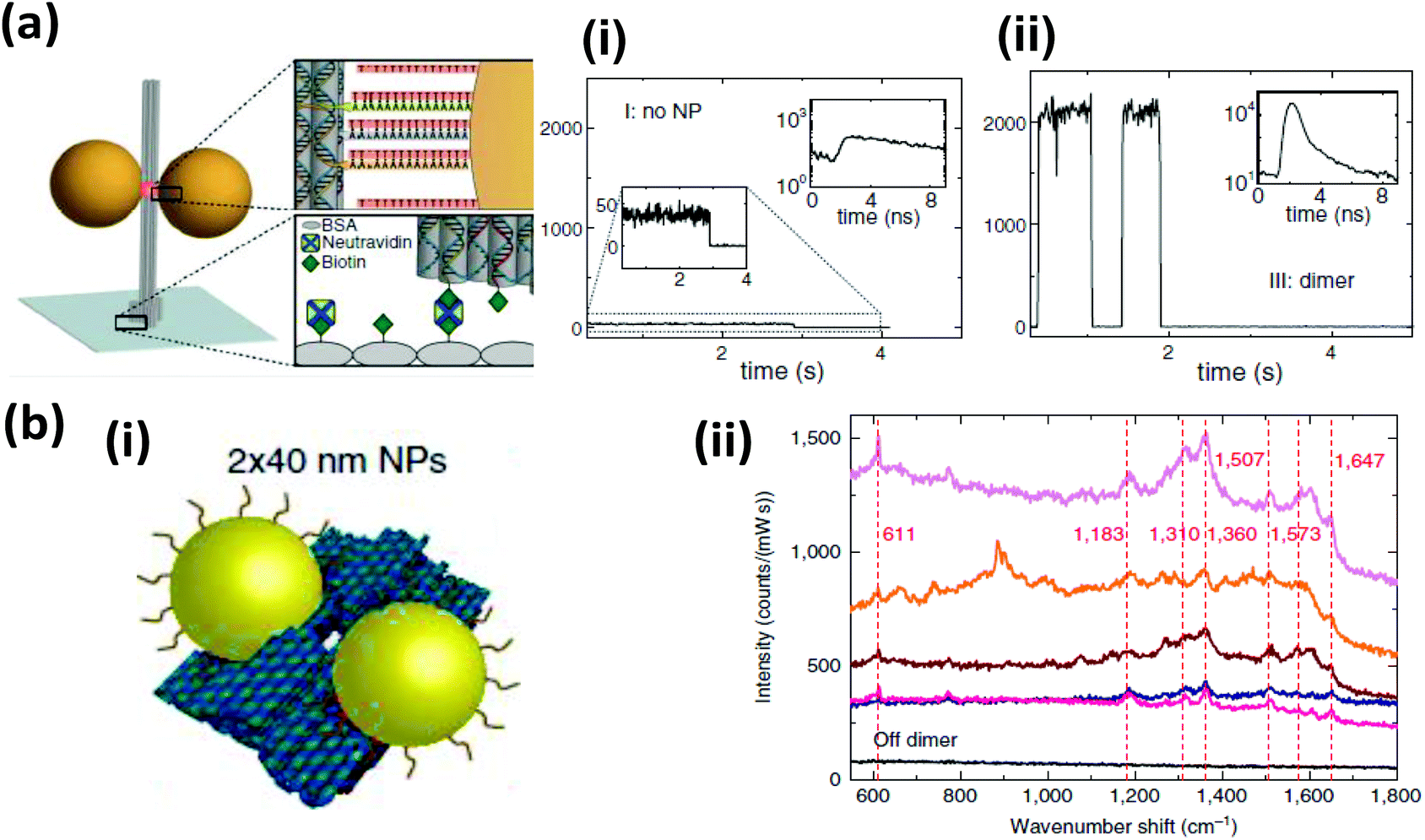 | ||
| Fig. 13 DNA origami directed self-assembled AuNPs displaying unique photonic properties (a) (i) A plasmonic hot spot has been generated by assembling two AuNPs at close proximity on a pillar shaped DNA origami attached to glass surface via biotin–neutravidin interaction. More than a 100 times enhancement in the fluorescence intensity of ATTO647 was observed when the fluorophore was placed between two 100 nm AuNPs separated at a distance of 23 nm. Intensity transient along with fluorescent decay (inset) with (i) no NP and (ii) with a dimer of AuNPs. From ref. 210. Reprinted with permission from AAAS. (b) SERS signal from rhodamine 6G absorbed into DNA origami decorated with two closely spaced 40 nm AuNPs (i). Nearly 5 to 7 times enhancement of Raman signal was obtained with two particles separated at 3.3 ± 1 nm. (ii) Different color corresponds to different dimer structure, stronger signal is associated to shorter distance between the two AuNPs. Reprinted with permission from Macmillan Publishers Ltd: Nature Communications, ref. 224. Copyright 2014. | ||
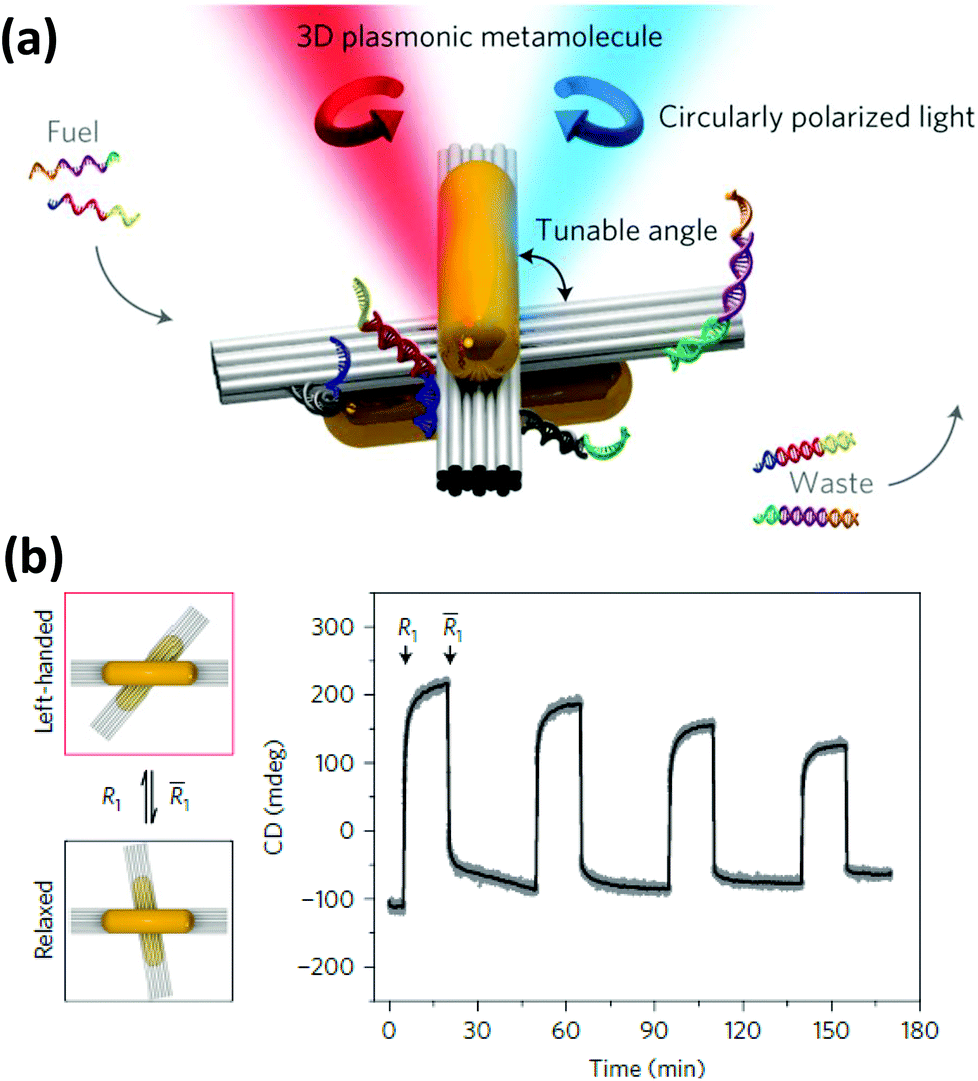 | ||
| Fig. 14 (a) Two AuNRs assembled on a reconfigurable DNA template generated a strong CD signal which was tunable (b) by changing the angle between the two DNA bundles by external DNA input. Reprinted with permission from Macmillan Publishers Ltd: Nature Materials, ref. 228. Copyright 2014. | ||
The other approach to DNA directed NP self-assembly is focused mainly on generating a 3D ordered phase. In 2008, the Mirkin and Gang groups independently showed that DNA-functionalized AuNPs can be an excellent synthon for building novel programmable crystalline materials.229,230 These two groups discovered that not only a well-defined crystal could be generated from the aggregate but the crystal structure can be controlled, either face centered cubic (FCC) or body centered cubic (BCC), simply by changing the DNA sequence.229 In addition, they also demonstrated that lattice parameters can be tailored by altering the length of the DNA anchored to the NPs.231 More recently, they have established some basic “rules-of-thumb” for DNA-mediated crystallization defining the intricate relationship among DNA length, particle size, annealing temperature, etc., that when combined allow us to build NP crystals with tailorable features such as interparticle distance, unit cell lattice parameter, degree of filled space, etc., and these can also ultimately dictate some of the physical properties of the crystal.232–234
Within such structures, energy minimization processes dictate that the maximum possible DNA hybridizes to achieve a conformation that has a maximum number of NPs around each other. In a single component system having self-complementary DNA, the binding affinity to each other is indistinguishable and a close packed structure (FCC) is formed where every particle has 12 neighbors. Whereas in a two component system the binding affinity is not identical since particle A can bind only with particle B, not with itself; here DNA hybridization leads to a non-close packed cubic unit cell (BCC) having eight surrounding neighboring particles. The annealing temperature which could be either below or above the Tm of the hybridizing zone has a very significant role in determining the preferred crystal conformation and its quality.229 In addition to temperature, Gang demonstrated that the flexibility of the interconnecting DNA is critical to the spontaneous crystallization process.230 Mirkin later reported that anisotropic particles like AuNRs or Au prisms can also be crystalized but not in the cubic phase; AuNRs crystalized into a 2D hexagonal lattice while prisms fall in lamellar 1D arrangements.235 More recently, the Gang group reported a generalized method of producing DNA functionalized-NPs where QDs, Fe2O3 NPs, PtNPs along with AuNPs can all serve as programmable atom equivalent that can be used to generate a wide variety of single and multicomponent NP super lattices.236 For an example of where QDs are utilized in a similar type of configuration, see Fig. 27 and its associated text. Overall, this is an especially interesting research area since crystallization by self-assembly is a very delicate process that depends on a fine balance of many noncovalent interactions such as hydrogen bonding, electrostatic, hydrophobic, van der Waals interactions and unpredictable entropic contributions. DNA hybridization can take into account most possible forces that play a role here in a much more simplified way. The only requirement now is a careful control over the annealing temperature gradient to make the energy landscape smoother and more favorable for long-range crystalline order.
4. Silver nanoparticles
4.1. Spherical silver nanoparticles
In comparison to the myriad of reports involving DNA-functionalized AuNPs, examples of AgNPs are far rarer despite the fact that they have higher extinction coefficients than AuNPs of the same size and are better suited for plasmonic applications like enhanced Raman scattering.237–239 The reasons behind this include the lower Ag–S bond energy compared to Au-S, which makes it more difficult to conjugate them to monothiol-modified DNA, strong susceptibility towards oxidation in NP form and a tendency towards irreversible aggregation.240 In an effort to strengthen the attachment chemistry, the number of anchoring points on DNA has been increased by incorporating multithiol moieties. For example, the Mirkin group demonstrated oligonucleotides with a cyclic dithiol functionality could produce AgNPs capable of withstanding saline buffers with salt concentration as high as 1 M.240 Pal et al., experimented with psDNA and following the testing of varying number of ps units reported that 9 ps units per oligo provides the best stability.241 Zhang et al. reported that monothiol-modified DNA can be linked to AgNPs if the pH is reduced to ∼3 which surmounts problems associated with adsorption kinetics at neutral or slightly basic pH.242 Similar to DNA-conjugated AuNPs, AgNPs have found application in DNA detection via sandwich assays and by electrochemical detection.243,244 Thompson described AgNPs decorated with a thiolated-probe DNA as a DNA sensor with a detection sensitivity nearly 50 times higher than similar AuNPs. Higher sensitivity was attributed to the aforementioned AgNP's larger extinction coefficient.245 Similar to an AuNP-based sandwich assay, here, too, the presence of target oligonucleotides induces aggregation by crosslinking NPs and this is accompanied by a change in the ultraviolet-visible (UV-Vis) absorption band and the color of the solution. Employing this same colorimetric detection principle, Xu and coworkers demonstrated that adsorbing label-free DNA onto AgNP surfaces can be used for the detection of small molecules such as coralyne, a therapeutically important drug candidate that binds to poly-A sequences strongly.246 Besides spherical AgNPs, Ag nanocubes have also been conjugated to thiolated DNA.247 Due to their flat surface and sharp edges, Ag nanocubes should also be useful for plasmonic enhancement of fluorescence or Raman signal.4.2. Silver nanoclusters
Unlike AgNPs, DNA-templated AgNCs have drawn significant interest and have created an active area of research focused on both a fundamental scientific understanding of intrinsic properties that give rise to the PL along with practical applications. Although only a few years old, this unprecedented interest is due to their connection with DNA which provides a great degree of versatility. The Dickson group first demonstrated that C-rich DNA sequences can stabilize AgNCs.248 Later it was revealed that different colors of AgNCs can be produced by changing the DNA sequence (Fig. 15).249 Although the mechanism by which the sequence directs the resulting emission color is still poorly understood; it has been shown that DNA not only dictates the emission color, but also affects the NC's photostability and long-term colloidal stability.250–253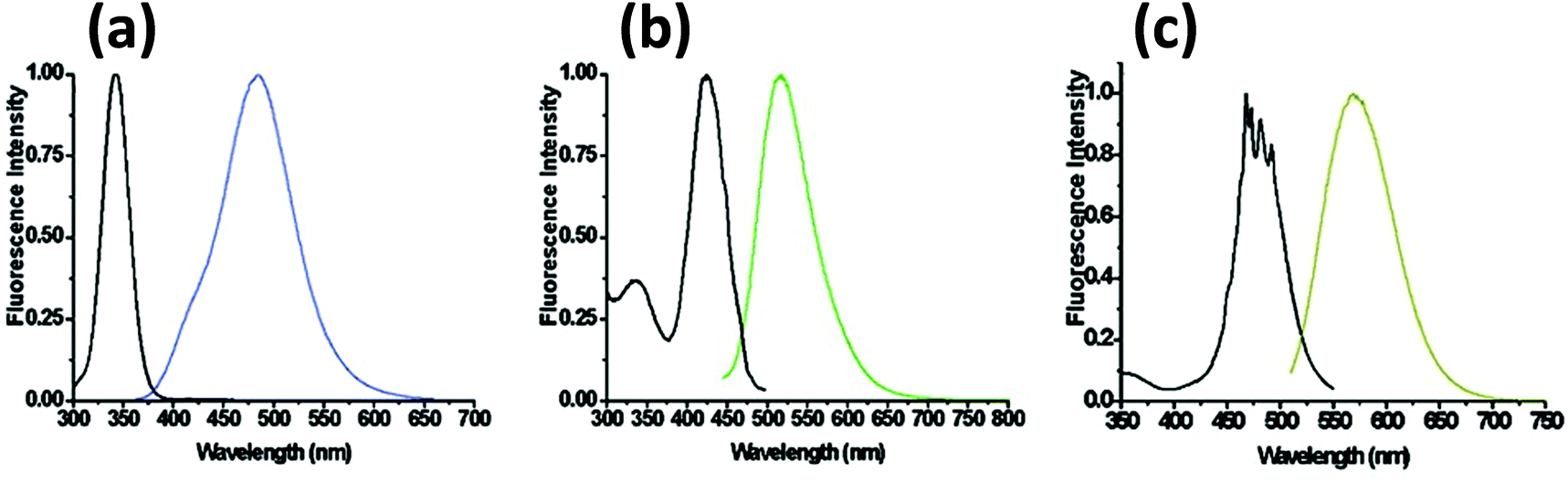 | ||
| Fig. 15 Steady state excitation and emission spectra of DNA templated AgNCs with different DNA sequences. Reprinted with permission from ref. 254. Copyright 2008 American Chemical Society. Sequences utilized include: (a) 5′-CCCTTTAACCCC-3′; (b) 5′-CCCTCTTAACCC-3′; and (c) 5′-CCCTTAATCCCC-3′. | ||
DNA-templated AgNCs have already been implemented in varied ways as sensors for DNA, small molecules or metal ions. The primary mechanism behind this sensing approach lies in the quenching or restoration of the NC's fluorescence in the presence of target analyte. Willner's group integrated DNA-linked AgNCs with graphene oxide (GO) to serve as a sensor for the presence of DNA from infectious pathogens such as HIV, HBV and syphilis.255 It is believed that GO quenches the AgNC PL by electron transfer or FRET mechanisms.256 ssDNA binds to GO with a high affinity via π–π interactions but this is not true for dsDNA. When DNA-appended AgNCs adsorb to GO, its fluorescence is quenched, but in the presence of its target DNA having a complementary sequence, a duplex forms followed by desorption that restores the PL. Combing two different AgNCs emitting visible and NIR light linked to two different recognition probe strands provided for a multiplex sensor. This light-up sensor also works with aptamers in the place of probe strands to detect thrombin and ATP. Here, the aptamer changes its random coil conformation to bind to the specific analyte which triggers desorption from GO and restores PL (Fig. 16). The method can distinguish ATP from other nucleotides such as guanosine 5′ triphosphate (GTP), guanosine 5′ monophosphate (GMP), and uridine 5′ triphosphate (UTP) as the aptamer mostly remained adsorbed onto the GO surface and thus insignificant fluorescence enhancement was observed in the presence of the latter.255
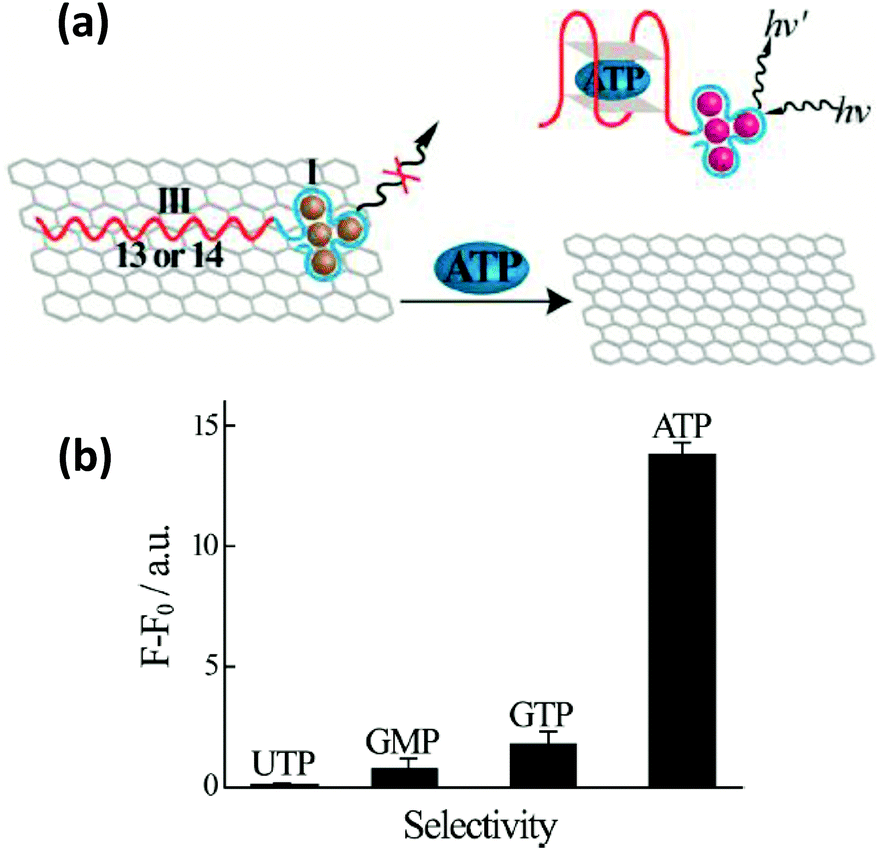 | ||
| Fig. 16 (a) Schematic illustrating an ATP sensor employing a hybrid system consisting of GO and AgNCs bound to an ATP binding DNA aptamer. Desorption of the ATP-aptamer complex from the graphene surface resulted in an increase in the AgNC PL. (b) Selectivity of detecting ATP over other nucleotides, UTP, GMP and GTP, was demonstrated as negligible PL enhancement was observed at 1 mM concentration of the other nucleotides. Reprinted with permission from ref. 255. Copyright 2013 American Chemical Society. | ||
As mentioned, the DNA templating sequence itself is extremely important when preparing fluorescent AgNCs.249,257 From the seminal work of Dickson, it is now known that a C-rich DNA sequence is critical for producing these NCs.248 In addition, some special secondary structure(s) in the sequence appear to play a significant role that can be utilized for the detection of analytes. In one example, Deng et al. exploited specialized motifs for the detection of the toxic metal ion Hg2+.258 They designed two partly-complementary DNA sequences that can potentially produce a C-rich loop when formed into a duplex while leaving several adjacent T–T mismatches. Hg2+ strongly chelates between the two T residues which influences the microenvironment around the loop near the attached AgNC. In the presence of Hg2+, due to the increased stability of the duplex, a much stronger PL was observed providing a detection limit of nearly 10 nM.258 Likewise, Zhou and coworkers incorporated an aptamer for the “turn-on” detection of cocaine as depicted in Fig. 17a.259 The same principle has also been implemented for the detection of single point mutations in DNA, microRNA and PDGF.260
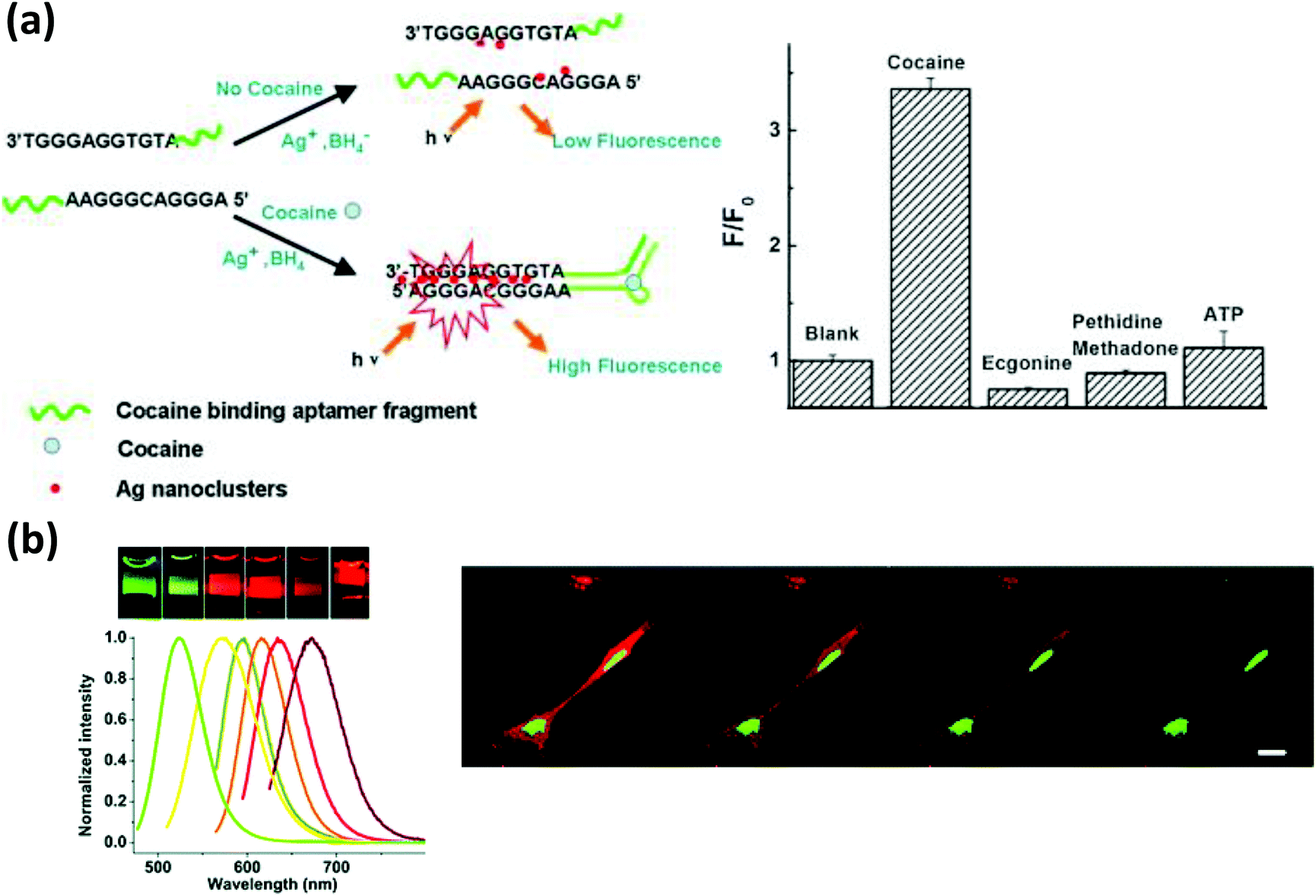 | ||
| Fig. 17 (a) A cocaine detection scheme using DNA-bound AgNCs. Cocaine bound aptamer fragments brought the G rich sequences in close proximity, enhancing the PL intensity of the NCs. Reprinted with permission from ref. 259. Copyright 2011 Elsevier. (b) AgNCs of various colors synthesized in the presence of DNA of varying sequences. The confocal microscope images show better photostability of AgNCs, (green) localized in the nucleus, compared to HCS red (red), localized mainly in the cytoplasm. Reproduced from ref. 261 with permission of The Royal Society of Chemistry. | ||
Because of their small size, favorable optical properties and potential biocompatibility, emissive AgNCs may provide an alternative to the use of QDs for imaging cells and tissues and to study dynamic processes inside cells. As stated earlier, DNA is crucial for the stability of these NCs at high salt concentration but its high charge density, in turn, potentially limits intracellular delivery. Choi and coworkers demonstrated that chemically conjugating a cell penetrating peptide (CPP) to the protective DNA can improve their cytosolic delivery efficiency.261 They conjugated the well-known CPP penetratin to DNA, which had almost no detrimental effects on the spectral properties of the NCs, and then used it to deliver the NCs to NIH 3T3 cells and bovine pulmonary artery derived endothelial cells (Fig. 17b). The peptide-DNA appended NCs entered the cell without causing any significant damage to the cell membrane. The NCs displayed excellent photostability as only 10% fluorescent intensity quenching was observed after 2 hours of continuous exposure while the control HCS red intensity decayed to 60%.261 Yu and coworkers further showed that DNA-encapsulated AgNCs can be easily linked to proteins like avidin and used for labeling cell surfaces.262 They also conjugated DNA to heparin sulfate receptor antibodies which helped them to bind to cells displaying this receptor and which internalized the NCs via the endocytic pathway.
5. Magnetic nanoparticles
Magnetic nanoparticles (MNPs), comprised of mostly magnetite (Fe3O4) and maghemite (γ-Fe3O4) have found application in diverse areas including catalysis, sensing, labeling, magnetic resonance imaging (MRI), and even for electronic data storage.263–268 In an ideal form, they can behave like discrete magnetic domains and high quality materials consistently show high magnetic moments in an applied magnetic field coupled to insignificant residual magnetism after removal of the field. A number of synthetic routes now exist to produce small MNPs especially in the 10–20 nm range which are more suitable for biological applications.269–273 Similar to most other NPs, they can present large S/V ratios and following synthesis they are highly reactive as well as prone to agglomeration which occurs to reduce the surface energy. Hence, an appropriate protective coating is necessary, especially if they are going to be used in a biochemical environment. The coating material could be a polymer like dextran, a PEG surfactant, or even silica which besides protecting them also offers an opportunity to link biomolecules by means of chemical conjugation.56 However, a thick coating can have a degrading effect on their magnetic performance.267,274 A wide variety of biomolecules ranging from small molecules like dopamine to macromolecules like DNA or proteins, have already been ligated to MNPs for many different applications.56 DNA has been attached via chemical conjugation, which can form a covalent bond with the reactive surface functional groups as well as by simple electrostatic adsorption to a positively charged overcoating material, for example polyethyleneimine (PEI). Scherer et al. reported that PEI encapsulated MNPs can be an excellent gene delivery agent to cells and tissues simply by adsorbing the DNA onto them.275 They successfully delivered some DNA vectors to multiple cell lines including lung epithelial cells and blood vessel endothelial cells and observed that the MNPs are quite effective in reducing the dose and time of transfection. DNA has also been ligated to MNPs mostly by making a covalent bond following standard chemistry utilizing 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) or 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (SMCC) coupling or even click chemistry.56 A representative example of the latter was developed by Culter et al. where they produced Fe3O4 NPs with a dense layer of DNA via the copper(I) catalyzed azide–alkyne cycloaddition (CuAAC) click reaction.276 A high DNA surface density offers some interesting new properties in the composite material such as cooperative melting, resistance towards enzymatic degradation of the DNA and very high cellular uptake. Here, the click reaction took place between azide groups on the capping ligand (prepared by reacting succinimidyl 4-azidobutyrate with the amine functionalized particles) and a DNA displaying a terminal alkyne functionality (Fig. 18a). The conjugation was confirmed by rapid formation of aggregates when particles functionalized with complementary DNA were incubated together. The aggregation showed a sharp melting temperature transition which is considered a definitive characteristic feature of NPs with a dense DNA monolayer.276 High cellular uptake of these particles was observed when incubated with HeLa cells without any transfection agent present which was analogous to the results shown for AuNPs having a high DNA surface coverage.277 | ||
| Fig. 18 (a) Superparamagnetic iron oxide nanoparticles (SPIONs) functionalized with a dense layer of DNA by applying ‘click’ chemistry. The particles can be delivered into HeLa cells without any transfection agent as shown in the confocal image (red channel). Inductively-coupled plasma (ICP) spectroscopy analysis shows much higher presence of the densely DNA packed SPIONs in cells compared to simple carboxylic acid functionalized ones. Reprinted with permission from ref. 276. Copyright 2010 American Chemical Society. (b) Schematic showing aptamer-functionalized SPIONs employed for detecting thrombin. The aggregation, caused by thrombin upon binding to multiple aptamer-ligated SPIONs, as reflected in higher hydrodynamic diameter, reduced the T2 relaxation time of adjacent water molecules as evidenced by the T2 weighted MR image. The specificity to thrombin is demonstrated as a negligible alteration in the MR image in the presence of BSA or streptavidin at various concentrations. Reprinted with permission from ref. 280. Copyright 2008 American Chemical Society. (c) Schematic showing an on-demand stimuli responsive delivery system employing porous silica NPs and DNA functionalized SPIONs. The heat generated under the influence of the alternating magnetic field dehybridized the dsDNA leaving the pores open and allowing the trapped dye molecules to diffuse out. The plot describes the reversibility of the system as fluorescein dye was released over multiple cycles. Reprinted with permission from ref. 282. Copyright 2011 American Chemical Society. | ||
It is well known that MNPs can reduce the spin–spin relaxation time of surrounding water protons, and when they form microaggregates the inhomogeneity of the external magnetic field around the clusters is intensified and becomes even more effective in dephasing the spins of nearby water protons leading to an decrease in the T2 relaxation rate.278 Using this mechanism as a sensing basis, Perez and coworkers developed a novel way of detecting DNA along with real-time monitoring of DNA methylation and cleavage.279 They produced 300 to 400 nm micro-aggregates by hybridizing two different MNPs functionalized with complementary DNA that displayed a much reduced T2 relaxation time compared to individual free MNPs in buffer solution. In the presence of an appropriate DNA restriction enzyme, the particles were released from the aggregates which increased the T2 relaxation time to its higher values. They further demonstrated that methylation followed by DNA cleavage can also be monitored via MRI and suggested that the method can be automated for high throughput applications. This magnetic relaxation switching property of MNP aggregates was also employed to detect thrombin with MRI.279 Yigit and coworkers ligated a thiol-modified thrombin-specific aptamer to amine functionalized MNPs via SMCC coupling.280 The aptamer reconfigured into a cruciform conformation to bind thrombin and, since thrombin has more than one binding site, a random crosslinking ended up in an aggregation that affected a larger number of water molecules by reducing their T2 relaxation time. In the presence of thrombin, a clear decrease in the brightness of the T2 weighted MRI was observed (Fig. 18b). This method works in biological fluids like serum and can thus potentially be used for detecting other biological targets with minimal sample clean up and processing requirements.280 Nie et al. employed DNA-functionalized MNPs along with DNA-modified AuNPs for the amplified detection of DNA. This was based on a sandwich hybridization assay technique involving multiple steps including magnetic separation and amplified fluorescence detection.281 Two mismatched DNA were conjugated to the AuNPs at a ratio of 100:1; a portion of the DNA that was present in far lower number was specifically made partly-complementary to the target DNA. DNA on the MNPs was also partly complementary to the target. In the first stage of the assay, the target DNA acted as a linker between the MNPs and AuNPs resulting in a micro-aggregation which was subsequently separated magnetically. The other DNA (called the barcode DNA) which was present on the AuNPs in higher ratios and was previously unhybridized, were now hybridized with a complementary dye-labeled DNA in the second stage and the full construct was again magnetically separated to remove excess dyes. In the final step, denaturation of this entire construct produced free ss-dye-modified DNA, which was detected allowing quantification of the target DNA.281
MNPs can also generate local heating under the influence of an alternating magnetic field since with every pulse the magnetic dipoles try to realign themselves.283 The heating can be strong enough to denture any dsDNA present on the immediate particle surface and hence release drugs or any other molecules intercalated between the base pairs or attached to the complementary DNA. Derfus and coworkers demonstrated that this could be an ideal system for releasing a surface functionality triggered by remote external stimuli.284 They covalently conjugated a 30 base DNA to dextran-stabilized Fe3O4 NPs and subsequently hybridized it to a complementary fluorophore-modified strand. The resulting particles were trapped inside a matrigel plug (an in vitro model of tumor tissue) and they observed that under an electromagnetic pulse (EMP, 400 kHz, 1.25 kW), the dye-modified DNA was largely released from the trapped particles to the surrounding buffer. The MNPs containing a matrigel plug were also implanted inside living mice and under a similar EMP, the same degree of dye release was observed in the surrounding tissue. The penetration capability of the field was quite high approaching greater than 99% in 15 cm thick tissue, which is much higher than light or heat.284 In other utility, Ruiz-Hernandez et al. developed an on-demand, stimuli-responsive therapeutic system with Fe3O4 embedded mesoporous silica NPs and Fe3O4 NPs (Fig. 18c).282 Thiol-modified DNA was ligated via SMCC reaction to the mesoporous silica NPs and the MNPs, both of which displayed surface amine functional groups. When the particles were assembled together by hybridizing the attached complementary DNA, the pore entrances on the silica NPs were largely blocked as assessed from the significantly-reduced nitrogen adsorption profile. To simulate a drug they embedded fluorescein inside the porous silica NPs prior to this self-assembly. When the particles were exposed to an alternating magnetic field, the heat that was generated melted the dsDNA – opening the pore entrance and allowing the trapped dye to diffuse out of the mesoporous silica NPs.282
Besides DNA, Fe3O4 superparamagnetic NPs functionalized with RNA aptamers have been used for targeted negative contrast imaging of prostate cancer cells. Hwang and coworkers developed a multimodal imaging technique with silica-coated cobalt ferrite NPs by attaching fluorescent and radioactive moieties to the particles.285 The silica layer had embedded rhodamine dye present and was chemically ligated to a radioactive 67Ga-citrate chelate complex. They conjugated a DNA aptamer that specifically binds to nucleolin, a protein expressed in significant quantity on the cell surface of some cancer cells. The resulting biofunctionalized particles displayed high specificity in binding nucleolin-expressing C6 cells, as evidenced by the signal obtained from three different sources: MRI, fluorescence, and 67Ga scintigraphic images. The NPs also showed promising result in targeting rat glial tumor C6 cells implanted inside living animal models while the control sample without the right aptamer rapidly cleared out from the body.285 Conversely, Hu et al. employed silica-coated paramagnetic Mn3O4 NPs for T1 weighted MRI via positive contrast.286 As a T1 contrast agent, these NPs performed better than Gd complexes in terms of toxicity and circulation time. The silica-coated MNPs were first functionalized with PEG and then a cancer cell-specific aptamer was covalently ligated to the PEG. The particles showed higher specificity in binding HeLa cells as compared to control normal human hepatic L02 cells while also displaying insignificant toxicity. The efficacy of targeted in vivo imaging was also demonstrated when HeLa cells inoculated inside living mice were detected upon binding these particles with T1-weighted MRI without any noticeable pathogenic change.286
6. Platinum and palladium nanoparticles
Impressive progress has been made over the last decades on the synthesis of platinum nanoparticles (PtNPs) of various shapes and sizes.287–289 The most prominent feature of these NPs is their electrochemical activity which has been successfully used for building low temperature fuel cells and in various other electro-oxidation or reduction reactions.290–292 Catalytic efficiency in these applications depends on many factors including the NP shape, surface ligand and solvent.291,293 There is also a strong interest in improving the catalytic turnover by making binary alloyed nanostructures or integrating them with nanoporous carbon.294–296 Compared to the research attention paid to their electrocatalytic behavior, linking them to biomolecules is relatively scarce. However, it is worth noting that this catalytic property can be employed as a replacement for enzymes meant to provide similar catalytic activity. PtNPs have been functionalized with DNA in the same manner as AuNPs by exchanging labile surface ligands with thiol-modified DNA. The Willner group demonstrated that DNA-modified PtNPs can be used for the amplified electrochemical detection of DNA and thrombin.297 To detect DNA, they immobilized two different oligos that are partly complementary to the target DNA on the PtNPs and the Au electrode, respectively (Fig. 19). In the presence of target DNA, PtNPs are captured onto the Au surface and catalyzed the reduction reaction of H2O2 to H2O which is reflected in the appearance of a noticeable peak using cyclic voltammetry (CV). Detection of thrombin was similar except the protein worked as a connector to the target DNA-PtNP and a thrombin-specific aptamer was immobilized in the place of an ordinary non-specific oligonucleotide. They proposed this method can be generalized for other proteins provided the specific aptamer is attached to the Au electrode surface.297 The same group reported a different DNA readout strategy that generates light due to oxidation of luminol by peroxide using PtNP-catalyzed reduction of H2O2.298 More recently, Kown et al. implemented an identical strategy of DNA detection but with enhanced sensitivity by incorporating an ultramicroelectrode (UME) which is capable of amplifying the current several fold.299 With the use of an UME, they demonstrated a DNA LOD as low as a few zeptomolar which is a relevant target concentration range for some specialized diagnostic and forensic studies. PtNPs are also a potential cancer treatment agent because of their ability to damage DNA. Porcel and coworkers studied the lethal damage that is caused when DNA-bound PtNPs are irradiated with a carbon ion beam.300 They postulated that the damage was caused by the high number of Auger electrons produced by the NPs due to their large surface area. In addition to the self-generated electrons, they also induce radical formation and ionization among surrounding water molecules which can also contribute to DNA damage.300 Interestingly, beyond just a research tool, carbon ion beams actually have clinical use in cancer radiotherapy. Due to relatively low toxicity compared to photons and finite range penetration in tissues, they display better cure rates as a treatment for locally advanced malignant tumors.301 | ||
| Fig. 19 (a) An electrochemical DNA detection scheme using DNA conjugated PtNPs. Target DNA captured the PtNPs onto the electrode substrate. The captured PtNPs acted as an electrocatalyst for the reduction of H2O2 to H2O. (b) Amperometric response corresponding to different target DNA concentration. (c) SEM micrograph shows the captured PtNPs as tiny white spots on the Au electrode surface. Reprinted with permission from ref. 297. Copyright 2006 American Chemical Society. | ||
Palladium nanoparticles (PdNPs) are mostly synthesized in organic media with a hydrophobic capping ligand.302,303 Due to their unique properties, they are currently quite popular for application as catalysts in organic reactions such as hydrogenation, oxidation, carbon–carbon bond formation, etc.304–306 However, there are only a few reports of synthesizing them directly in aqueous media mediated by hydrophilic small molecules or DNA and RNA.307–309 Fang et al. reported a DNA-templated synthesis of PdNPs on an indium tin oxide- (ITO) coated glass surface.309 The resulting NPs exhibited excellent catalytic activity in reducing H2O2 and oxidizing ascorbic acid. Interestingly, it was also observed that the catalytic activity of the NPs is better when dsDNA acted as a template instead of ssDNA. The same type of catalytic activity can be utilized as a way to amplify the sensitivity of biodetection techniques. However, sequence-specific DNA functionalization of PdNPs is more complex because a simple ligand exchange protocol is inadequate in their case. Wang and coworkers covalently conjugated amine-modified DNA to PdNPs synthesized in aqueous media with a hydrophilic 11-mercaptoundecanoic acid capping ligand having an exposed carboxylic acid group.310 The resulting NPs exhibited good stability in saline buffer, were separable by agarose gel electrophoresis, and were further employed for DNA detection in chip-based microarrays (Fig. 20). In the presence of target DNA, complementary DNA-bearing PdNPs were captured, and in the subsequent step they acted as a catalyst for electroless deposition of Co accompanied by a quantifiable change in color.310
 | ||
| Fig. 20 (a) Schematic depicting a DNA detection assay by capturing DNA functionalized PdNPs onto DNA microarrays of fully, partially or non-complementary DNA sequences as capture probes: complementary (first row), one base mismatch (second row), two base mismatch (third row) and a noncomplementary sequence (fourth row). (b) Picture shows different amounts of cobalt staining after electroless deposition of cobalt and (c) the colorimetric analysis shows the capability of this process to distinguish a single base mismatch. Reproduced from ref. 310 with permission of The Royal Society of Chemistry. | ||
7. Quantum dots
Quantum dots (QDs) are luminescent semiconductor nanocrystals composed of a few hundred to a few thousand atoms that display intriguing quantum confinement effects. They are considered a fundamentally new class of fluorophore that offer certain unique and useful properties such as broad absorption coupled to composition and size-dependent, narrow and nearly symmetric PL emission (Fig. 21), high extinction coefficients and QYs along with some of the largest known two photon action cross-sections.311–314 Another remarkable property, and probably the most advantageous one compared to most of the conventional fluorophores, is their strong resistance towards photo-bleaching which can allow imaging for prolonged periods of time. Fluorescence blinking and unresolved questions about toxicity are the two major issues currently associated with QD application in biology. Interestingly, blinking may actually be particularly advantageous for super resolution microscopy. Synthesis of colloidal QDs started maturing in the late 1980s to early 1990s using hydrophobic organic solvents at high temperatures. In contrast to aqueous synthesis, organic synthesis methods are still preferred to produce high-quality, relatively monodisperse materials.315–317 As synthesized or native QDs have already found important application in various electronic devices like light-emitting diodes, transistors and solar cells, however, these organic ligand-capped QDs are insoluble in aqueous solution and inappropriate for direct biological application without additional treatment. Several methods are currently used to make them hydrophilic including ligand exchange, amphiphilic polymer encapsulation or synthesis directly in aqueous media using hydrophilic ligands.318–320 A vast number of synthetic protocols exist in the literature to produce water soluble QDs and these are reviewed in detail elsewhere.321–324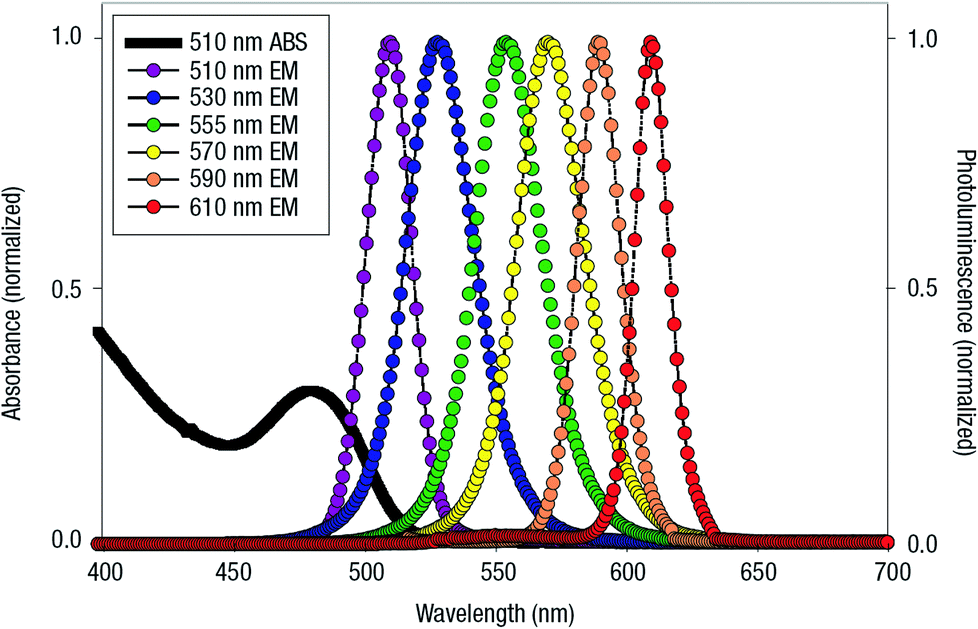 | ||
| Fig. 21 Absorption spectrum of 510 nm emitting QDs and emission spectra of six different QDs with emission maxima at different wavelength. Reprinted with permission from Macmillan Publishers Ltd: Nature Materials, ref. 326. Copyright 2005. | ||
A variety of biomolecules including proteins, small peptides, nucleic acids, and antibodies have already been conjugated to QDs allowing them to be exploited in a diverse range of bioanalytical experimental formats and some of these materials have even been commercialized.325,326 As an alternative to traditional organic fluorophores or fluorescent proteins, bioconjugated QDs have served a significant role in sensing, labeling cellular components and tracking biomolecular events in cells, tissues and even in living organisms despite toxicity concerns; the latter is possibly the most promising and under exploited future application of these NPs.327–337 Here, our discussion will be limited to DNA-functionalized QDs and the reported methods to obtain such conjugates can be categorized primarily into three sections: (i) ligand exchange with thiol- or peptide-modified DNA; (ii) chemical conjugation; and (iii) incorporation of thiolated DNA during QD synthesis in aqueous media. As the chemistry of how DNA is attached to the QDs profoundly affects the subsequent utility and application of the QD–DNA conjugates, we arrange our discussion here by labeling chemistry type and then describe selected examples of how these conjugates were applied.
7.1. Ligand exchange and direct surface attachment
The first work on DNA attachment to QDs in the form of ligand exchange was reported by Mitchel et al.338 Here, a thiolated DNA was attached to the QD surface by simple mixing and leaving the DNA and mercaptopropionic acid-capped CdSe/ZnS core/shell QDs to react overnight. Attachment was confirmed by incubating together two different QDs functionalized with complementary sequences which resulted in micro-aggregation and a concurrent red shift in the ensemble PL emission.338 However, two major shortcomings related to the stability of the early generation of QD conjugates prepared in this manner were oxidation of the monothiolated ligand to a sulfide in oxygen rich aqueous media and a reverse ligand exchange, which may cause the DNA to decouple from the QD surface.339–342 Polyhistidine- (Hisn) appended DNA may be a better alternative to thiolated DNA for QD attachment since it obviates the need for direct ligand exchange. Hisn sequences have a strong metal driven coordination affinity towards the Zn2+ on ZnS-overcoated QDs which encouraged researchers to recombinantly insert His6 tags into proteins and assembled them onto QDs with an available Zn-rich surface.343–347 Our group has worked extensively on creating His6-modified DNA and have focused on developing this as a robust and reliable chemistry. The nature of the linkage between the peptide and nucleic acid moieties also has a vital role on the overall stability of the constructs. A disulfide linkage was initially used as a chemical bridge between the His-tag and the DNA.348 However, in reductive cytoplasmic environments the disulfide may dissociate into monothiols or be replaced with the myriad other thiols present in the cytosol at higher concentration (e.g. glutathione) and that prompted a switch over to two other His6-oligomer chemical linkers that are devoid of any such problem. Berti et al. synthesized a His6-peptide with a terminal iodoacetyl group and reacted that to thiolated DNA349 while Boeneman et al. placed a hydrazone linker between the two moieties.350 His6-DNA constructs assembled with the hydrazine chemistry were used to study sequential ET from the QD to DNA photonic wires decorated with fluorophore dyes at specific distances.350 Although multi-step ET over a distance of greater than 150 Å was observed, the actual end-to-end transfer efficiency was poor (<1%). More recently Spillmann et al. improved the exciton transfer efficiency to the terminal dye by two orders of magnitude with a careful choice of dyes along with optimizing the separation distances between the dyes and the stoichiometry of the photonic wires onto the QDs (Fig. 22).351 Due to the shorter linkage distance compared to the previously used hydrazone approach, disulfide chemistry was pursued here. Building from Spillmann's findings an even more sophisticated hybrid ET system was recently prototyped.352 Here, QDs were utilized as both a central nanoscaffold and energy harvesting mediator to photophysically connect the DNA photonic wires extending from its surface to the bioluminescent enzyme luciferase (Luc) while still relying on directed self-assembly for the bioconjugation chemistry (Fig. 23). Luc oxidation of Coelenterazine substrate (Coel) provided initial excited state energy which was harvested by the QDs in a bioluminescent RET (BRET) process and then propagated down the DNA photonic wire in a sequential 4-step FRET cascade. The ratio of wires displayed around the QD along with altering substrate concentration provided control over energy flow and its magnitude to the terminal output dye-acceptor. This assembly demonstrated that complex hybrid systems containing biological (enzymes, DNA and peptides), inorganic (QDs), and organic components (dyes, substrate) can all be jointly self-assembled in a predefined manner and function synergistically to generate their own light and then control its directionality.352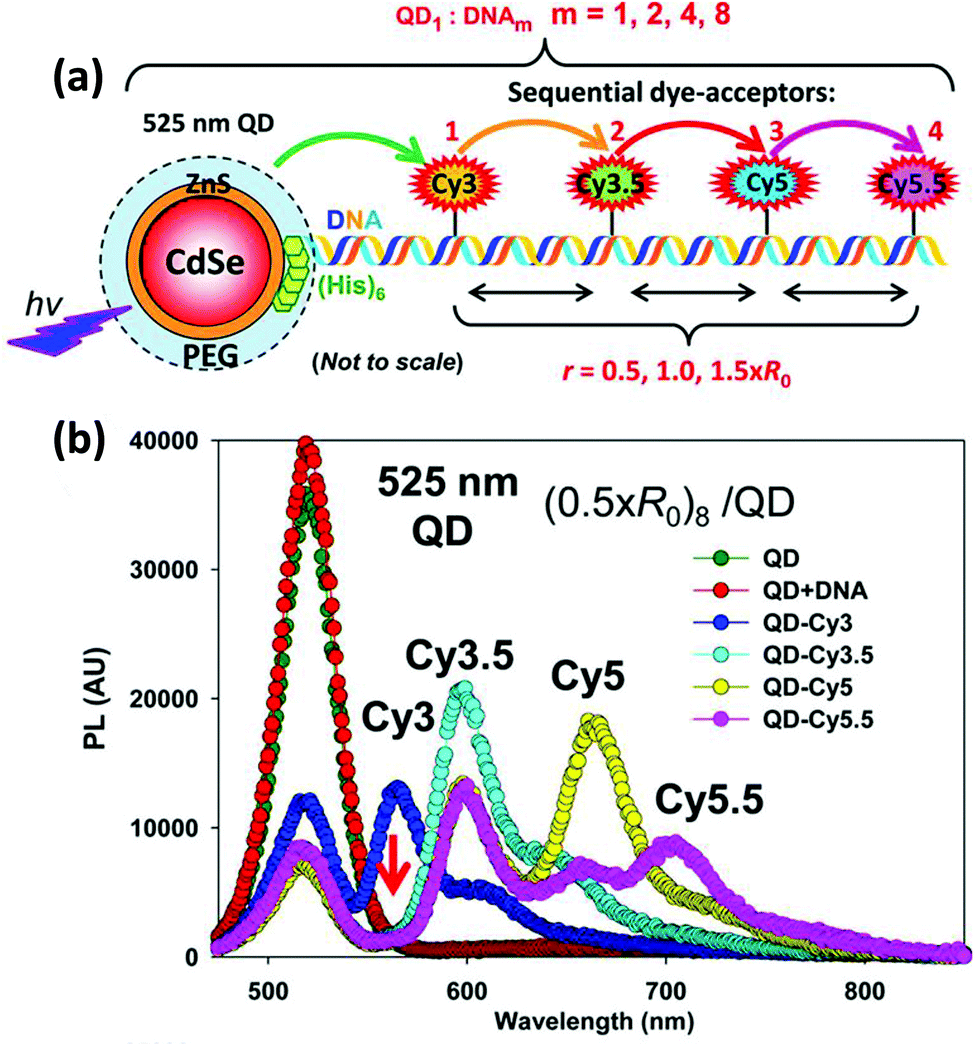 | ||
| Fig. 22 (a) His6-modified DNA-photonic wires on CdSe/ZnS core/shell QDs. Initiated by the excitation of QDs with UV light, a unidirectional energy flow was observed through the spectrally overlapped fluorescent dyes via a sequential multi-step FRET mechanism. (b) The PL spectra shows a representative stepwise FRET progression in the QD–DNA-photonic wire hybrid construct at a QD/DNA ratio of 1:8 as each sequential acceptor dye is added. The red arrow indicates the Cy3 dye acting as a unity FRET relay within the construct. Reprinted with permission from ref. 351. Copyright 2013 American Chemical Society. | ||
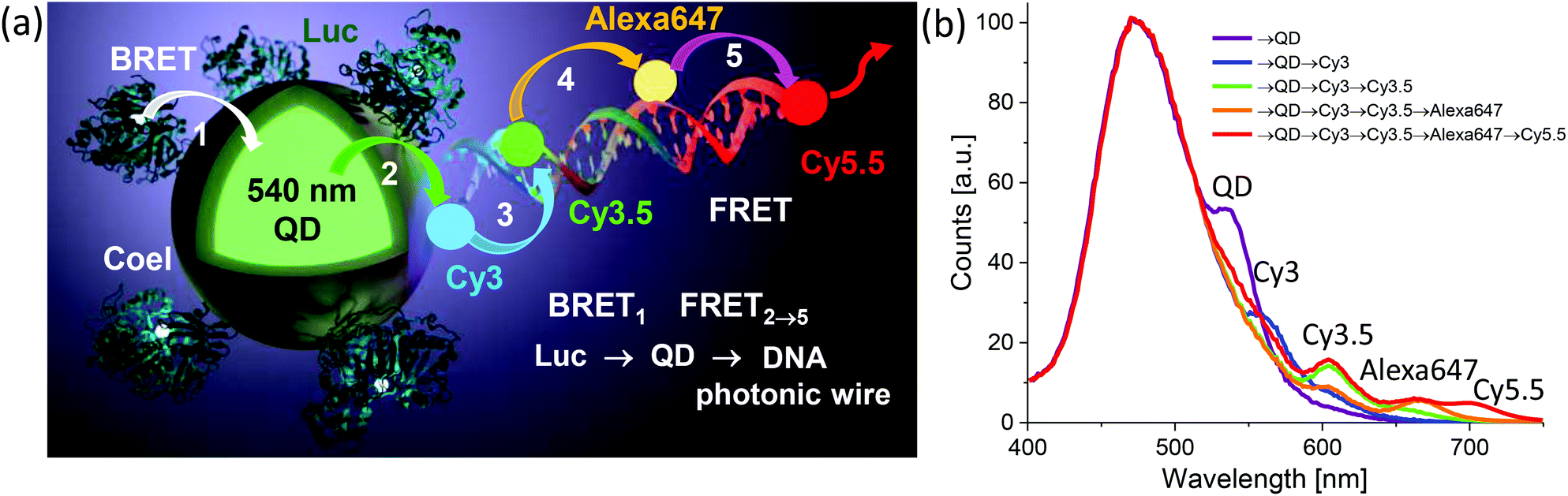 | ||
| Fig. 23 (a) Schematic of the self-assembled enzyme–QD–DNA ET system. Luc appended with terminal His6 ratiometrically coordinates, i.e., with control over display valency or ratio, to the QD. Dye-labeled DNA wires are formed by prehybridization and include a terminal His5-peptido-DNA sequence to facilitate similar QD assembly. Functionally, Coel substrate is enzymatically oxidized by Luc giving rise to excitonic energy that sensitizes the proximal QD by BRET. The QD then redirects this energy and sensitizes the proximal dye on the DNA photonic wire giving rise to a sequential FRET cascade. Assembly number of Luc and photonic wires per QD can be controlled during assembly which, along with Coel concentration, provides control over ET efficiency. (b) The PL spectra show a representative stepwise FRET progression in the QD–DNA-photonic wire hybrid construct at a QD/DNA ratio of 1:8. These consist of the normalized spectra of Luc6-QD–DNA8 constructs as the displayed acceptor dyes are extended along the DNA from Cy3 up to Cy5.5 (→ = BRET/FRET step). Approximate emission maxima for each component indicated by the placement of their name. Reproduced with permission from ref. 352. Copyright 2015 American Chemical Society. | ||
7.2. Chemical ligation
Besides ligand exchange, another popular method of linking DNA to QDs is via a covalent modification, usually by forming a bond directly to a functional group on the preexisting capping ligand. To do so, EDC/NHS (N-hydroxysuccinimide) and sulfo-SMCC coupling chemistry are the most widely practiced strategies.56 In an EDC coupling reaction, for example, an amine/carboxyl-modified DNA is ligated to the converse carboxylic acid/amine-functionalized hydrophilic QDs by amide bond formation. Despite its apparent straightforwardness, drawbacks associated with this approach include a generally poor conjugation yield (<20%). Moreover, a large excess of EDC and sulfo-NHS are needed due to their intrinsic instability in water, which can also be detrimental to the QD PL and this usually requires an extensive number of purification steps.56 In one representative example, Kim and coworkers developed a PDGF sensor by conjugating a PDGF-specific aptamer to carboxylic acid-capped CdSe/ZnS core/shell QDs with an EDC coupling reaction.353 A partially complementary DNA with a black-hole-quencher was then hybridized to the QD-bound aptamer placing the quencher at the nearest possible point from the QD in order to promote maximum possible PL quenching. In the presence of PDGF, the strong binding affinity towards the aptamer triggered its dehybridization from the quencher-bearing strand to reconfigure it into a special secondary structure suitable for the protein pocket. The displacement of the quencher-bearing strand from QD proximity resulted in a restoration of QD PL and served as the sensor output signal.353 He et al. developed a novel strategy of conjugating a long DNA strand, as opposed to the short oligomers that are most often used with QDs, via chemical ligation and subsequent enzymatic extension which they named QD-based PCR as outlined in Fig. 24.354 They first ligated the forward primer specific for a 480 bp sequence fragment of the fatty aldehyde dehydrogenase 1 gene from maize to CdSe/ZnS QDs with an EDC coupling reaction. The PCR product of the primer-conjugated QDs and the cloned plasmid produced QDs that are linked to a single long DNA as evidenced by atomic force microscopy (AFM). It was speculated that an enhanced local steric hindrance stemming from the long strand might have prevented further enzyme attachment during PCR. The product displayed promising results in detecting single copies of the gene in prometaphase maize chromosomes as evidenced in a fluorescence in situ hybridization (FISH) assay.354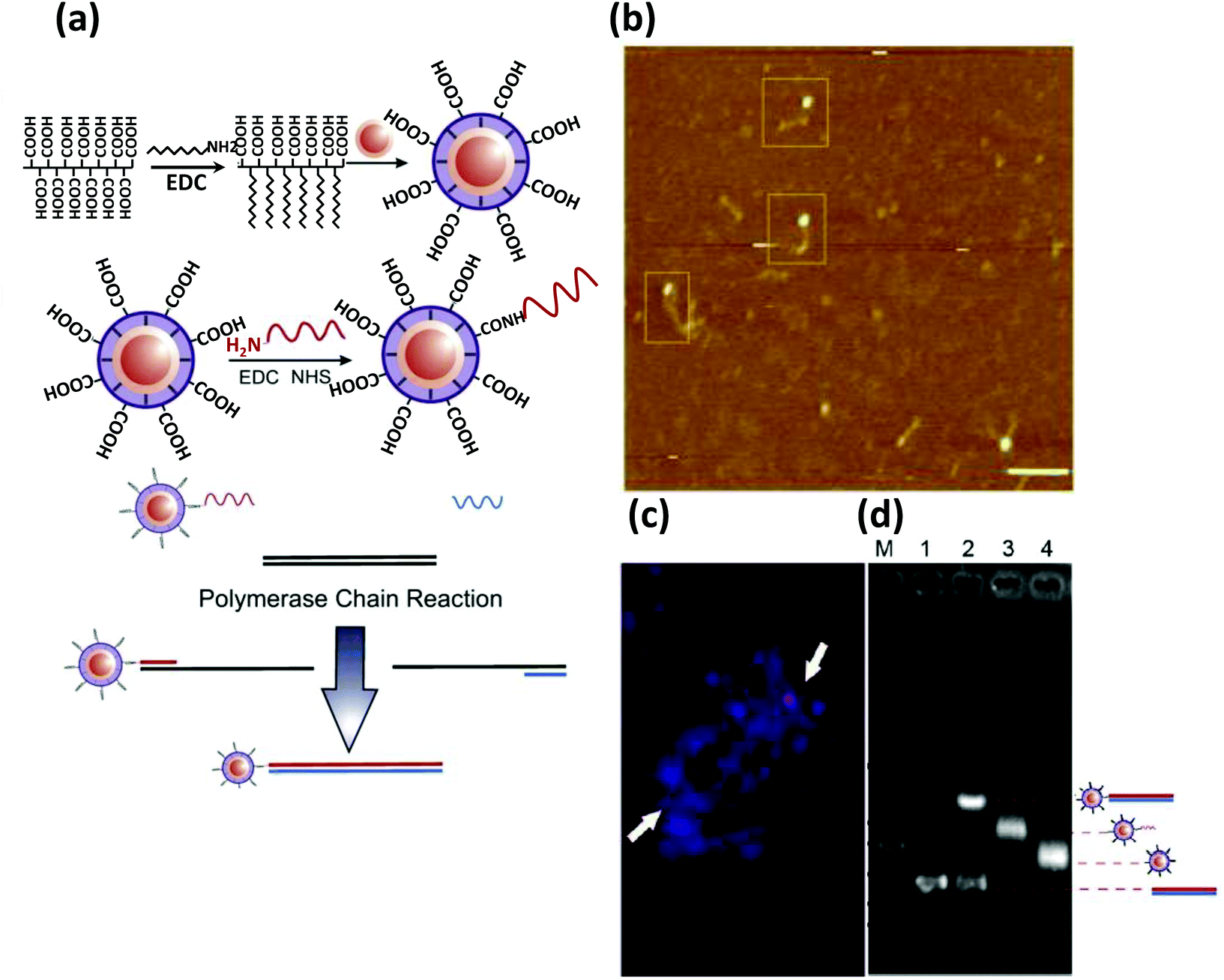 | ||
| Fig. 24 (a) Schematic depicting the strategy for producing QDs conjugated to a single long DNA, by first chemically ligating a short primer sequence to a carboxylic acid group displayed on the QDs followed by its extension via PCR. (b) AFM image shows individual QDs being successfully ligated to single long DNA. (c) FISH signal showing specific binding of the DNA ligated QD to maize metaphase chromosome. (d) Gel electrophoresis shows the retarded mobility of the QDs labeled with the long DNA. Reprinted with permission from ref. 354. Copyright 2011 Elsevier. | ||
Despite being used quite regularly, issues of nonspecific adsorption of DNA to the QD surface remains a matter of concern when using chemical ligation methods especially if the DNA and QD surface have opposite charges.355,356 To circumvent that, Zhou et al. introduced a special custom synthesized capping ligand with a non-fouling ethylene glycol spacer between the surface-binding thiol group and solution facing carboxylic acid group.357 Amine-modified DNA was then ligated via EDC coupling and the resultant QD–DNA conjugates were employed for the detection of label-free DNA via FRET. The QD here acted as the donor while the acceptor fluorophore in this case was ethidium bromide, intercalated between the nucleobases of the dsDNA formed by hybridization with the target.357 Pathak and coworkers addressed the same issue of preventing nonspecific adsorption in a slightly different way by incorporating capping ligands with a hydroxyl group.358 A series of chemical reactions were implemented allowing an amine-modified DNA to be linked to an activated imidazole carbamate functional group. The linked DNA had sequence complementary to the human Y chromosome and when incubated with human sperm cells, a FISH experiment yielded nearly 41% of cells labeled with the QDs as compared to the theoretical total of 50%. A control experiment with a random DNA sequence labeled less than 5% of cells, which validated this approach to reduce nonspecific adsorption.358
7.3. Streptavidin–biotin chemistry
Streptavidin–biotin chemistry is another facile alternative for QD–DNA functionalization and perhaps the most utilized bioconjugation chemistry in current application for this materials combination. Biotinylated DNA can be easily coupled to commercially available streptavidin-coated QDs by simply incubating the two together for a brief period of time. There are several reports in the literature related to their application as FRET-based probes for detecting protein, DNA and other small molecules like ATP.312,359,360 Zhang employed this chemistry to construct a sandwich assay for detecting DNA present in very low concentrations, especially in clinical samples from patients with ovarian serous borderline tumors.361 They used two probe strands that are partially complementary to the target DNA; one of them was modified with a biotin tag while the other one was carrying a Cy5 dye. In the presence of the target, the capture and reporter probe strands were connected via the target strands in a sandwich fashion as shown in Fig. 25. Exposing this hybridized unit to the streptavidin-coated QDs, the biotinylated probe was then readily captured, which brought the Cy5 acceptor into close proximity with the QD allowing for efficient FRET. Owing to the high local probe concentration on the QD scaffold (i.e. high avidity), the sensor was much more responsive than classical MB-based probes with a demonstrated capability of detecting a single copy mutation in clinical samples at the single particle level.361 Levy et al. similarly employed biotinylated-QDs for the detection of thrombin where the sensor also relied on a FRET signal from the QD to a quencher.362 In order to build a turn-on sensor, the quencher was chemically linked to a short oligomer having a complementary sequence to the QD-bound biotinylated DNA. Upon hybridization, the quencher came close to the QD and, because multiple quenchers are assembled around a single QD, this resulted in a dramatic drop in QD PL. The biotinylated DNA used here was a thrombin-specific aptamer, which, in the presence of thrombin, switched to an energetically favorable quadruplex conformation to bind thrombin, concurrently releasing the quencher-carrying strand and restoring QD PL.362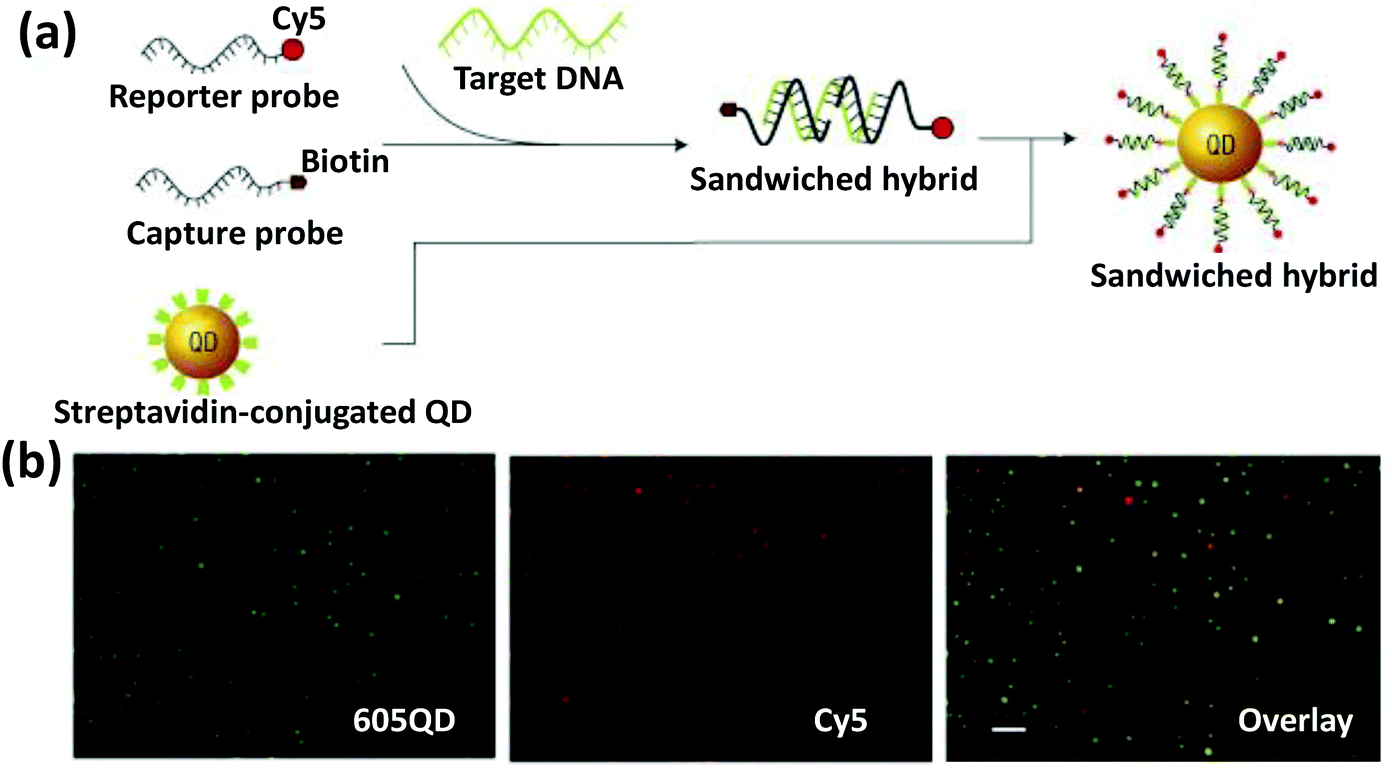 | ||
| Fig. 25 (a) Schematic depicting fabrication of a single-QD-based DNA sensor. FRET sensitized emission from Cy5 indicates the presence of target DNA. (b) The overlaid fluorescence image demonstrates co-localization of QD and Cy5. Reprinted with permission from Macmillan Publishers Ltd: Nature Materials, ref. 362. Copyright 2005. | ||
Aside from designing sensors, QD–DNA building blocks have also been employed to construct novel hybrid NMs governed by DNA hybridization. For example, Fu et al. have shown that complementary DNA-functionalized AuNPs can be hybridized to QDs and discrete hybrid nanostructures generated with different numbers of AuNPs decorated around a QD.363 Bui et al. assembled streptavidin-coated QDs onto a pre-engineered DNA origami tube with biotinylated capture strands protruding out at specific locations.364 A linear array of QDs was constructed with high yield and good control over their inter-particle distance. Nevertheless, in general, the bulkiness of the product resulting from this chemistry remains a matter of concern, especially in cases where precise control over the distance has significant impact on the quantitative outcome. We investigated the effect of a chemoselective ligation strategy versus that of biotin assembly on ET efficiencies from the QD donor to dyes located at specific distances on a dsDNA.365 For this, one set of structures was ligated by DNA–His6 metal affinity coordination and the other via biotin/streptavidin interactions. The data from the His6–DNA hybrids assembled on QDs matched initial predictions showing that the DNA should extend linearly out from the QD. In contrast, the heterogeneity of streptavidin attachment to the QDs in conjunction with its multiple binding sites meant that the DNA was always randomly oriented with respect to the QD.365 Clearly, if a carefully controlled structure with precise spacing is desired, the former approach may be more useful.
7.4. Conjugation during synthesis
The third approach for conjugating DNA to QDs is during their synthesis in polar solvent. There have been some attempts to synthesize hydrophilic QDs in the presence of nucleotides or genomic DNA,366,367 but the discussion here is confined to short oligomers of known sequence. Ma first reported a one-pot synthetic strategy to conjugate DNA during the synthesis of CdTe QDs in water with glutathione as the primary capping ligand.368 The DNA they used had two domains; one called the binding domain having the typical P–O bond of the phosphate group replaced by the phosphorothioate P–S group, and the other called the recognition domain displaying the natural phosphodiester bonds. The DNA appeared to be stable in the synthesis conditions at a pH of nearly 9 and at 100 °C. The QDs were able to detect thrombin, when conjugated to an oligomer having a thrombin-specific aptamer sequence in the recognition domain. Moreover, to demonstrate their applicability in a complex system, cancer cell-specific aptamer-functionalized CdTe particles exhibited high fidelity in binding acute leukemic CCRF-CEM cells over that of Ramos cells derived from Burkitt's lymphoma.368 In 2013, the same group further developed this strategy with the additional capability of controlling the DNA display valency.369 The ps region consisted entirely of guanine bases as it has the highest binding affinity to the QD among the other bases. By altering the length of the binding region, the valency could be varied. For instance, a 20 base long domain ended up having only one DNA bound to the QD surface while a 5 base long domain produced QDs displaying approximately 4 DNA. DNA-directed assembly of these NPs produced unique molecular-like architectures where the individual QD acted as an atom equivalent. Spectroscopically, these structures behaved liked a photonic antennae funneling energy from surrounding green and yellow QDs to the central red QD.369However, CdTe QDs lacking an outer shell do not have a high QY and are prone to degradation by various chemical and photochemical means. Placing a shell of higher bandgap semiconducting material around the QD core particle not only offers higher PL efficiency and long-term stability, but also alters the electronic property of the resulting material. Depending on the bandgap and their relative positioning, the core/shell particles can be either a type I or type II (these have different spatial separation of charge carriers). Apart from enhanced QY in both types, type II QDs are also accompanied with a red shift in their emission spectra with increasing shell thickness while the emission profile remains the same for type I. Wang et al. first introduced the concept of DNA functionalization during shell synthesis by encapsulating oleylamine capped CdSe QDs with ZnS in the presence of thiolated-DNA.370 Deng and colleagues followed the same principle to synthesize a library of DNA-appended type II QDs with emission maxima ranging from UV to the near IR.223 Instead of thiolated-DNA, the DNA they used was modified with a 5′ ps unit, which was believed to be embedded inside the shell. The QDs were stable at high salt concentration and were site specifically organized onto pre-engineered DNA origami with high yield (Fig. 26).223 The same group also conjugated DNA to IR emitting QDs and similarly demonstrated their directed assembly on DNA origami.371 Interestingly, before these developments, the Qbead system – a functional analog where QD encoded DNA-labeled polystyrene beads were meant to be utilized in multicolor hybridization assays, briefly saw some initial commercial testing.372
 | ||
| Fig. 26 Schematic representing DNA functionalization during shell synthesis around a core QD in aqueous solution. The AFM image shows the DNA conjugated QDs are organized via DNA hybridization on triangle shaped DNA nanostructures. Reprinted with permission from ref. 223. Copyright 2012 American Chemical Society. | ||
7.5. Nontraditional methods
Besides the QD-conjugation strategies discussed so far, some nontraditional methods also exist in the literature. One such strategy reported by Kwon had the capability of controlling the stoichiometry of the resulting QD–DNA conjugates.373 It consisted of multiple steps; in the first step, a His6-peptidyl moiety was ligated to polymer-coated QDs via EDC coupling and this was reacted, in the second step, with a Ni2+-chelated nitrilotriacetate- (NTA) modified ssDNA. This is, in essence, the converse of the previous His6-based binding to the ZnS surface of QDs. Due to the nearly stoichiometric yield of metal affinity coordination reactions, the added ratio of DNA to QD was speculated to be the number of DNA bound to each QD.373 In another nonconventional approach, the Mirkin Group used click chemistry to attach a large number of DNA to the QDs and then used them as “programmable atom equivalents” to build higher order crystalline structures.374 A dense layer of DNA on a NP is the prerequisite for DNA-directed crystallization of NPs as it enhances the probability of polyvalent interaction. QDs that are approximately 5 nm or less in diameter fail to fulfill this need due to spatial restrictions. For initial cap exchange, they exploited an azide-functionalized amphiphilic polymer to stabilize the hydrophobic QDs in aqueous solution which also substantially increased the size of the particles and provided additional sites for DNA ligation (Fig. 27). DNA bearing a strained alkyne ring was then used for the click reaction with the azide functionality on the polymer-coated QDs and an estimated 55 DNA was expected to be displayed on the surface of the QDs. DNA-directed assembly of the particles produced a BCC and FCC lattice with a variety of combinations using, for example, different sized QDs, QDs with AuNPs and also with magnetic Fe3O4 NPs.374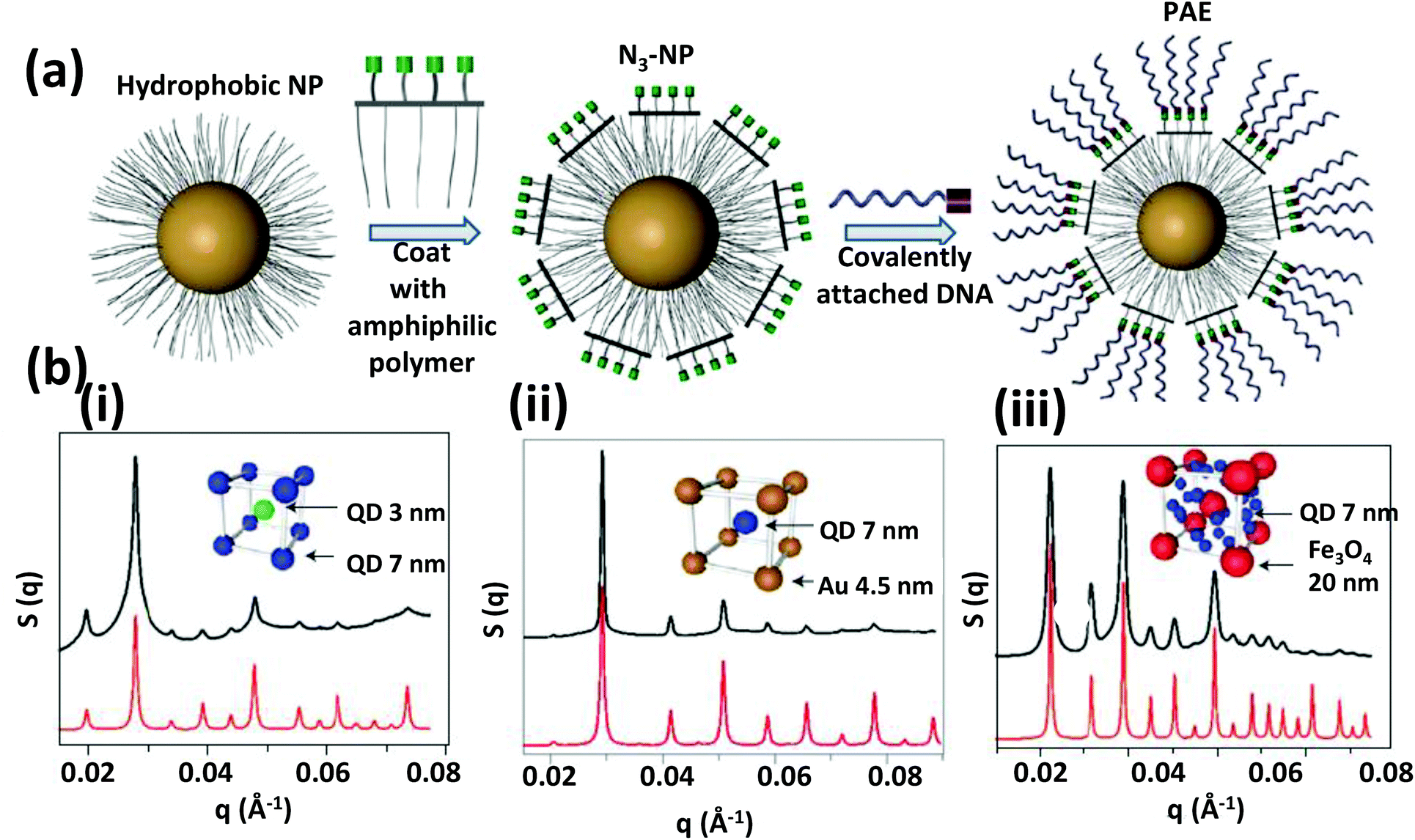 | ||
| Fig. 27 (a) A general scheme for ligating a large number of oligonucleotides to various kinds of hydrophobic NPs. The hydrophobic particles are first made hydrophilic by encapsulating with amphiphilic copolymers. DNA was then covalently linked by applying click chemistry. N3-NP = azide functionalized NP, PAE = programmable atom equivalent. (b) SAXS (small angle X-ray scattering) pattern of binary superlattices produced via sticky-end association of DNA conjugated NPs in different crystalline arrangements. (i) QD (3 nm) – QD (7 nm), CsCl lattice; (ii) QD (7 nm) – Au (4.5 nm), CsCl lattice; (iii) QD (7 nm) – Fe3O4 (20 nm), Cs6C60 lattice. Experimental data are shown in black and predicted scattering pattern in red. Reprinted with permission from Macmillan Publishers Ltd: Nature Materials, ref. 374. Copyright 2013. | ||
8. Carbon nanomaterials
8.1. Carbon nanotubes
Amongst all carbon allotropes developed to date, single walled carbon nanotubes (SWCNTs) are one of the most promising NMs with a strong potential for beneficial impact on material science and engineering.375–377 These typically consist of carbon cylinders with a diameter of ∼0.5 nm to 2 nm, lengths ranging from 10 nm up to a few centimeters, and most importantly a thickness of one atom. In contrast, multiwall carbon nanotubes (MWCNTs) are concentric assemblies of nanotubes with diameters ranging from 2 to 25 nm, and with an interlayer spacing slightly larger than single crystal graphite (0.34 nm and 0.335 nm, respectively). SWCNTs with their seamless nanotubes displaying a honeycomb lattice possess exceptional thermal and electrical conductivity, along with outstanding tensile strength and tenacity (even better than steel!) which makes them highly attractive for engineering applications.378 Additionally, they demonstrate some fundamentally interesting physical properties. For example, they can be either metallic or semiconducting depending on the orientation of the honeycomb lattice, they can have a diameter-dependent band gap and can interact with polarized light in a manner dictated by their chiral handedness.379,380 Being a direct band gap semiconductor, they have been employed both as a detector and light source, although their fluorescence quantum efficiency is usually quite low (<10%) and they have extremely short fluorescence lifetimes of ∼10–100 ps.381–383Solubilization of individual SWCNTs from bundles was an important first step towards understanding their photophysical properties. A variety of chemical compounds have been used to bring them into aqueous solution including surfactants, biomolecules, synthetic polymers, or sometimes simple aromatic polycyclic compounds; these have been reviewed extensively elsewhere.384–387 Here, our interest is in the use of nucleic acids for these purposes. In 2003, Zheng et al. first demonstrated that in the presence of ssDNA, bundles of carbon nanotubes (CNTs) could be brought into aqueous solution as a SWCNT by simple sonication.388 Theoretical modeling suggests that the DNA helically wraps around the CNT surface via π–π interactions between the aromatic bases and the CNT surface.327,389,390 Since then, substantial attention has been paid to such noncovalent interactions to functionalize SWCNTs as they minimally affect the material's π electronic network, unlike covalent conjugation. DNA functionalization has also solved some longstanding problems including separation of semiconducting and metallic CNTs along with separating nanotubes of different chirality. DNA-wrapped semiconducting and metallic nanotubes can be separated on a strong anion exchange column based on the difference of surface charge arising from the DNA phosphate backbone; metallic nanotubes have less surface charge due to the creation of an opposite image charge.388 In addition, separation of CNTs of different chirality was achieved by Tu and coworkers who investigated a large number of short DNA sequences and found nearly 20 with a special affinity towards particular chiral conformations allowing separation in ion exchange media.391 Although the underlying science behind this purification still remains unclear, they observed a pattern of periodic purine-pyrimidine bases on those sequences and speculated they might have formed well-ordered 2D-sheets which subsequently folded into a barrel shape structure around the nanotube that altered the van der Waals and hydrophobic interaction of the nanotubes with the ion exchange resin.391
 | ||
| Fig. 28 (a) (i) Graphic illustration showing conformational change of DNA, wrapped around SWCNTs, from B to Z in the presence of divalent metal ions. (ii) CD spectra show a dramatic change of ellipticity due to the conformational rearrangement in the presence of Hg2+ ion at various concentrations. (iii) Quantification of peak energy of SWCNTs affected by the conformational change of DNA, with respect to various divalent metal ion concentrations. From ref. 393. Reprinted with permission from AAAS. (b) (i) Schematic representing controlled singlet oxygen generation from an aptamer-photosensitizer-SWCNT complex. (ii) Exposing the complex to thrombin significantly increased the fluorescence from singlet oxygen sensor green (SOSG) indicating CNT quenching had largely been reduced. Reprinted with permission from ref. 397. Copyright 2008 American Chemical Society. (c) (i) Schematic showing surface functionalization of SWCNTs with cationic glycopolymers that electrostatically adsorbed negatively charged DNA. (ii) Confocal microscopy image shows an effective uptake of the FITC-DNA labeled nanotubes by HeLa cells after 48 h of incubation. Reprinted with permission from ref. 403. Copyright 2009 American Chemical Society. | ||
Besides quenching fluorescent dyes, SWCNTs are also quite efficient in quenching singlet oxygen generation (SOG), presumably due to a similar mechanism. Photosensitizers that produce singlet oxygen are important therapeutic agents for photodynamic cancer therapy, however, unwanted toxicity due to the high reactivity of singlet oxygen has remained a persistent concern.396 Zhu and coworkers demonstrated that SWCNTs can be useful for controlled generation of singlet oxygen.397 They conjugated the well-known Ce6 photosensitizer to a thrombin-specific aptamer and allowed the conjugate to adsorb on the SWCNTs (Fig. 28b). In the absence of thrombin, the aptamer remained adsorbed onto the SWCNT's surface and due to close proximity to the nanotube surface, 98% quenching of SOG was observed. In the presence of thrombin, the aptamer changed its configuration and subsequently desorbed from the surface to bind its target and this was reflected in a 20-fold increase in SOG. The authors envisioned using a cancer cell-specific aptamer which would preferentially generate singlet oxygen only when bound to cancer cells and thereby reduce nonspecific killing of healthy cells.397
Despite being inherently quite hydrophobic, SWCNTs can actually be transported inside living cells and have even been used as a shuttle for delivering various cargos.398–401 Kam and coworkers observed internalization of Cy3-labelled DNA-wrapped SWCNTs into HeLa cells where they were mostly located in the cytoplasm and presumably in endosomes.402 Interestingly, when the cells were subjected to a NIR laser pulse for a brief period of time, accumulation of Cy3-DNA was observed inside the nucleus as well. They hypothesized that NIR light-driven endosomal rupture released the DNA into the cytoplasm where it eventually diffused across the nuclear membrane into the nucleus. NIR light induces electronic excitation of the SWCNTs and this subsequently cascades down to the surrounding molecules putatively causing significant local heating that triggers the initial endosomal rupture.402
SWCNTs can also protect DNA from enzymatic degradation or intrusion of other nucleic acid-binding proteins which makes them an enticing potential NM for gene delivery. Wu and coworkers studied their protective capability in great detail and postulated several reasons behind such behavior including: physical inaccessibility of the probe to nuclease binding due to formation of small bundles where the probe remains embedded; formation of secondary structure that is unrecognizable to the binding pockets of the proteins; or simply an inhibitory effect from the exposed hydrophobic surface of the SWCNTs.404 Pantarotto et al. showed that positively-charged ammonium-functionalized SWCNTs and MWCNTs can electrostatically adsorb plasmid DNA as observed by TEM.405 The assembled NM, in spite of being positively charged, can easily penetrate into mammalian cells and, as compared to DNA alone as control, this resulted in a nearly 5–10 times higher expression level of an encoded beta-galactosidase marker gene. Ahmed et al. produced cationic SWCNTs by chemically ligating a glycopolymer to the surface which also turned out to yield a good transfection agent.403 They transfected HeLa cells with a green fluorescent protein (GFP)-encoded plasmid by loading it onto SWCNTs and observed high expression of GFP with relatively low concomitant cytotoxicity (Fig. 28c).
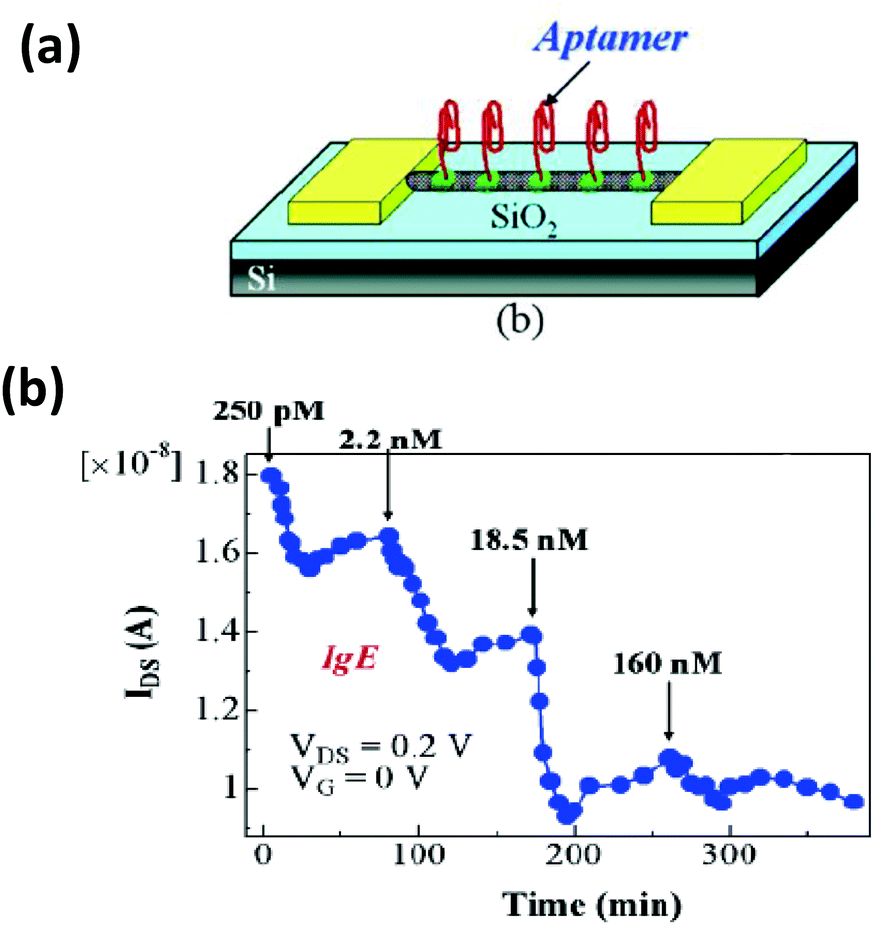 | ||
| Fig. 29 (a) Schematic illustration of aptamer-functionalized CNTs being applied as a protein biosensor based on CNT-FETs. (b) Time dependent source-drain current following addition of different concentration of IgE. Reprinted with permission from ref. 407. Copyright 2007 American Chemical Society. | ||
In contrast to uncontrolled chemical growth issues, DNA-based assembly allows for the controlled assembly of SWCNTs into preengineered patterns. DNA-functionalized SWCNTs have been organized onto DNA nanostructures as well, with the motivation of creating sophisticated nanoscale architecture from individual SWCNTs. This can be primarily accomplished in two ways: (1) by hybridizing complementary strands on to DNA templates and SWCNTs; and (2) via biotin-streptavidin interactions. The former was first reported by Maune and coworkers where they fabricated a cross shape structure of two nucleic acid labeled SWCNTs on a 2D rectangular DNA origami (Fig. 30).408 The SWCNTs used were a mixture of metallic and semiconducting materials, leading to the possibility that in some structures one of the nanotubes is semiconducting and the other one metallic. In the presence of a separating DNA layer, which basically functions as an insulator, the metallic nanotube may induce a FET-type gating on the semiconducting nanotube. Immobilization of these cross-junction structures on electron beam-fabricated electrodes indeed led to observation of FET-like behavior in nearly half of the devices they built.408 In an example of the later assembly type, Eskelinen and coworkers reported that biotinylated-DNA-wrapped SWCNTs can be readily assembled onto pre-engineered DNA origami allowing the protruding streptavidin proteins on the surface to assume some defined pattern.409
 | ||
| Fig. 30 (a) Schematic depiction of DNA-directed assembly of SWCNTs in a cross junction fashion onto a rectangular DNA origami. (b) AFM image of the DNA origami with two SWCNTs placed at right angle on its two surfaces. (c) AFM image shows a SWCNT cross junction structure being placed between electron beam-patterned palladium–gold electrodes. (d) FET like behavior showing the variation of source–drain current with respect to gate voltage. Reprinted with permission from Macmillan Publishers Ltd: Nature Nanotechnology, ref. 408. Copyright 2010. | ||
8.2. Graphene oxide
Graphene is a one-atom thick, 2-D, honeycomb lattice of sp2 hybridized carbon atoms. It is the basic building block of other carbon allotropes such as 0-D fullerenes, 1-D CNTs or 3-D graphite. Graphene oxide (GO) is the oxidized form of graphene having oxygen-linked functional groups such as hydroxyls and epoxys at the basal plane and carboxylic acid groups on the edges. The reduced form of graphene oxide (rGO) resembles graphene except with some imperfections such as missing carbon atoms or residual oxygen which can make it an electrically poor performer compared to native graphene.410 The sp2 electronic network of GO is severely disrupted due to its more abundant pool of sp3 atoms, hence GO does not display the unique electrical and condensed matter effects as graphene does.411 However, GO is highly exciting to chemists as it is an electronically hybrid material having conducting sp2 domains in a non-conducting sp3 matrix which leads to quantum confinement of π electrons. As a consequence it behaves like a semiconductor and also has interesting PL properties. The chemically reactive groups exposed on its plane and edges can interact with small and macro-molecules in a covalent, non-covalent or ionic manner offering potentially versatile routes for biofunctionalization.56,412 GO can also be easily functionalized with biomolecules simply by adsorption because of its ample aromatic surface area.412,413 Lastly, it can be prepared in solution by oxidizing graphite in the presence of strong acids followed by sonication. The intrinsic thickness of GO should be 0.6 nm, however, experimentally it is observed at ∼1 nm which is attributed to the presence of water molecules adsorbed on its oxygen rich surfaces.414–416 The lateral size also varies from a few nanometers to several micrometers.The PL of GO has been exploited for various analytical detection schemes along with cellular imaging applications.417–419 GO can fluoresce in the NIR, visible or ultraviolet region of the spectrum and this can be tuned by controlling the size of the conductive sp2 domain.420 It is believed that a sp2 cluster size of less than 1 nm gives rise to ultraviolet and visible emission while greater than a 2 nm domain with its smaller band gap produces NIR light.421 The electron–hole recombination process is also influenced by several parameters which are still not well understood. For instance, the lateral size of the GO sheet does not influence its emission behavior as significantly as the reduction time does.422 It is also perturbed by the dielectric properties of the environment, as well as the pH of the solution.422 Besides having its own PL properties, it can also act as a fluorescence quencher of QD or dye emission. The mechanism of quenching does not appear to be traditional FRET, rather it is much stronger and possibly involves some other dipole–dipole coupling leading to non-radiative ET, although this too has not been fully elucidated.256,423,424
ssDNA and peptides are readily interfaced onto a graphene surface by π–π stacking interactions and hydrogen bonding.425,426 Interestingly, dsDNA or aptamer complexes do not bind strongly to graphene due to unfavorable thermodynamics. The π stacked nucleobases and hydrogen bonding in dsDNA combine to offer more thermodynamic stability than π stacking of the constituent ssDNA on graphene could provide. Dehybridization appears to be an energetic barrier to π-stacking of the constituent ssDNA strands on the carbon materials and with shielding by the phosphate being only a small contribution. Relying on this phenomenon, GO was employed for DNA detection by Lu and colleagues using a two-step scheme.427 First, the complementary DNA of the target, in their case HIV1 with a FAM fluorophore modification, was adsorbed onto GO resulting in 97% quenching of the FAM fluorescence. In the presence of the target DNA, a duplex forms which subsequently displaces the DNA from the GO surface and fluorescence is restored.427 The method has also been applied for the detection of multiple DNA sequences simultaneously along with microRNA.428 The same principle was also applied for thrombin detection by adsorbing the specific aptamer with a dye modification.429 However, due to extensive loading of the probe strands, specificity could be compromised in this process meaning partially complementary or even a non-complementary strand can displace the adsorbed DNA from the GO surface generating a false signal. Miyahata and coworkers demonstrated that this can be overcome by a toehold-mediated mechanism.430 A toehold is a ssDNA of a few nucleotides dangling from a dsDNA region. Here, the probe strand is partially hybridized to a second DNA strand, called the anchoring strand, which is absorbed onto the GO surface. Target DNA binds to the toehold region and, due to favorable thermodynamics, strand displacement takes place leaving the anchoring strands unhybridized and adsorbed onto GO surface (Fig. 31).430
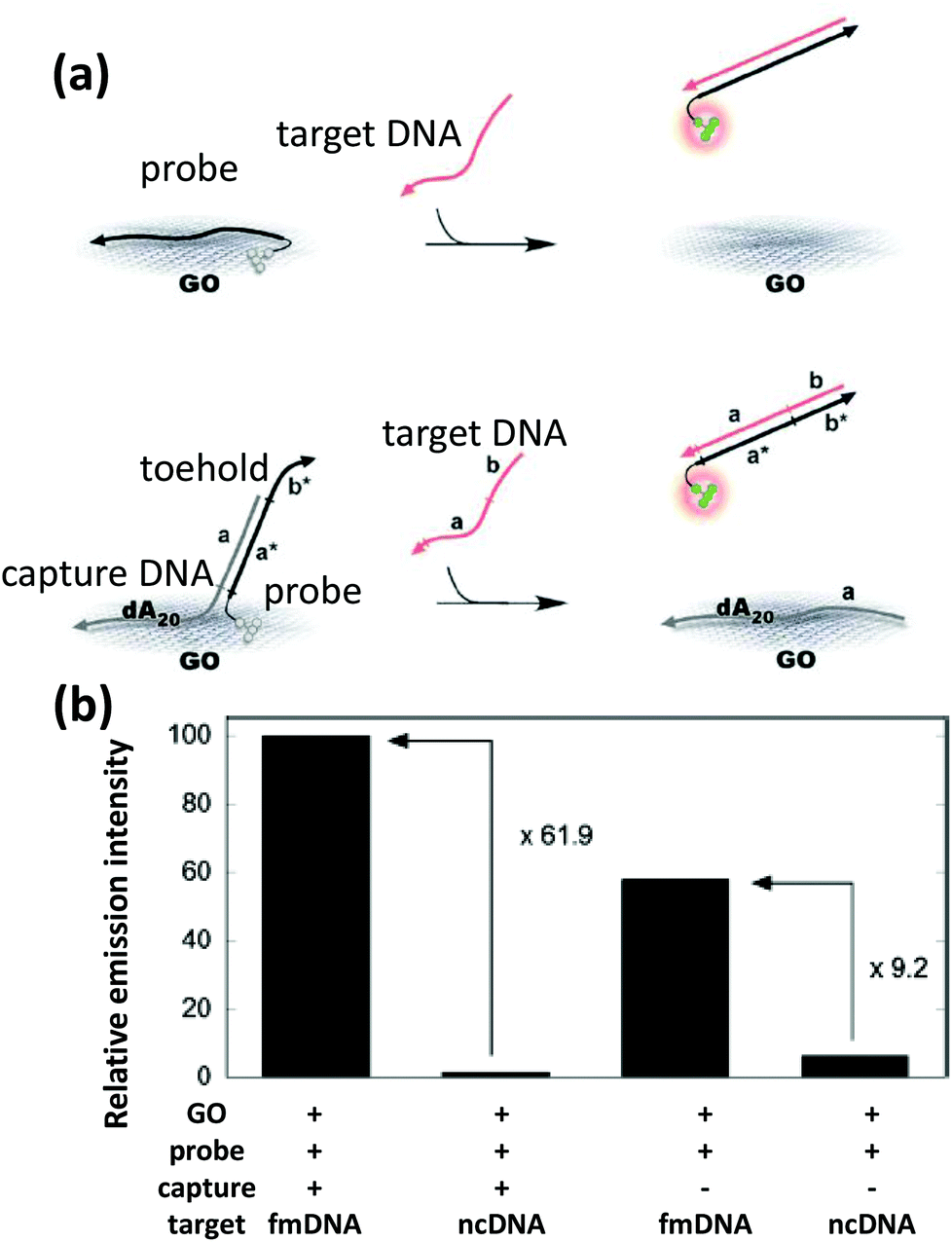 | ||
| Fig. 31 (a) A DNA sensing approach via toehold-mediated strand displacement with better sensitivity than the directly displaced FAM labeled probe DNA from the GO surface. (b) The former method shows nearly 62 times PL enhancement upon target binding while only 9 times enhancement was observed using the latter method (fm – FAM, nc – negative control). Reproduced from ref. 430 with permission of The Royal Society of Chemistry. | ||
It has now also become quite well established that GO can be useful for the detection of small molecules and proteins of special interest with high sensitivity.419 For example, most cocaine-recognizing aptamer sensors suffer from low sensitivity issues stemming from the weak binding affinity of the aptamer. Qiu et al. reported a way to improve the detection limit by integrating graphene and a signal amplification procedure called isothermal circular strand displacement amplification (ICSDA).431 The method requires that the cocaine-specific aptamer be linked to a short piece of DNA, a tool kit for a PCR reaction, SYBR green dye and GO. In the presence of cocaine, the ssDNA transforms into a special conformation to bind cocaine, leaving an unhybridized sequence for the primer to bind. This process triggers a series of reactions which ultimately ends up releasing cocaine from the aptamer complex and forming dsDNA which is detected by the intercalated SYBR green dye fluorescence. In the absence of cocaine, the aptamer-containing oligonucleotide would simply be adsorbed onto GO present in the solution showing no fluorescence from SYBR green.431
Insulin was detected in the same way by restoring fluorescence upon binding and consequent detachment of fluorophore-modified insulin binding aptamer from a GO surface.432 With an added step, sensitivity could be increased by a DNAase digestion that released the insulin to propagate the cycle again. GO has also been used as an intercellular probe. Lu et al. reported that DNA adsorbed onto a graphene surface is capable of withstanding enzymatic degradation.433 They observed that a MB adsorbed on a nanometer-sized GO sheet had virtually no degradation while in the absence of GO, digestion started after 3 min of incubation and was completed within 15 minutes. Transfecting HeLa cells with MB-absorbed nanometer-sized GO allowed detection of mRNA. The delivery of GO was very efficient and fluorescence restoration confirmed the presence of the targeted mRNA.433 Wang et al. utilized a similar approach by immobilizing an ATP binding aptamer and delivered the resulting product into cells where it acted as an intracellular probe for ATP detection (Fig. 32a).434
 | ||
| Fig. 32 (a) (i) Schematic design of an in situ ATP monitoring probe developed by integrating a FAM-labeled ATP aptamer and GO. Differential interference contrast (DIC) (ii & iv) and wide field fluorescence images (iii & v) show the effectiveness of GO as a molecular transporter. In the absence of GO, very few FAM labeled ATP-DNA aptamer gets internalized into cells (ii) while a significant fluorescence signal was obtained from FAM-DNA/GO complex (iv) displaying the ATP distribution inside mouse epithelial cells. Reprinted with permission from ref. 434. Copyright 2010 American Chemical Society. (b) (i) A DNA directed self-assembly method for producing a mechanically strong GO–DNA hydrogel. (ii) SEM image showing the interior microstructure of GO–DNA hydrogel. (iii) Photographs displaying the self-healing capability of the hydrogel simply by heating for a brief period of time. Reprinted with permission from ref. 437. Copyright 2010 American Chemical Society. | ||
With the help of DNA, researchers have produced hybrid NMs associating GO with other NPs like QDs or AuNPs. Dong and coworkers reported on a QD-graphene complex connected by several MBs.435 When the MB-conjugated QDs were mixed with GO, a ssDNA loop region bound to the GO surface. Due to ET from the QD to GO, strong PL quenching was observed. In the presence of target DNA, the loops open up and form dsDNA which releases the QD from the GO surface restoring its PL.435 Besides biochemical assays, such hybrid NMs can have beneficial impact on catalysis or energy storage devices. Apart from chemical nucleation, DNA-directed self-assembly has opened up a new avenue to achieve such nanostructures. Liu and colleagues reported a method of decorating GO with AuNPs on the basal plane.436 This was done in two steps; first thiolated-DNA was adsorbed onto GO and in the second step the resulting DNA-adsorbed GO was incubated with AuNPs. The hybrid nanostructure was separated from free AuNPs by agarose gel electrophoresis and the purified product appeared to be quite stable in saline buffer.436 DNA directed higher order self-assembly of GO into hydrogels was also reported by Xu et al.437 Here, DNA acted as a cross-linker by partially adsorbing onto multiple GO sheets (Fig. 32b). Synthesis of these hydrogels is fairly simple and accomplished by mixing equal volumes of GO and dsDNA at a specified concentration and heating the mixture at 90 °C. At high temperatures, dsDNA unwinds and forms ssDNA which gets adsorbed onto the GO. However, instead of mixing prior to heating, when GO was added directly into a hot solution of ssDNA, a poor quality hydrogel was obtained which was attributed to the fast binding of ssDNA onto individual GO surfaces instead of crosslinking. Due to the cooperative contribution of GO's strong mechanical strength, the resulting hydrogels appeared to have excellent mechanical properties, in spite of the fact that 99% of the gel was actually water. The gel was also a good absorber of a dye and highly stable to harsh chemical conditions like extreme acidic or basic solutions or very high salt concentrations. Another interesting property of the gel was their apparent self-healing capability. When the gel was cut into pieces, simple heating for brief periods of time was sufficient for the blocks to adhere to each other (Fig. 32b-iii).437 These kind of DNA-directed 3D assembled materials could potentially be very useful for tissue engineering or removing organic pollutants.
9. Miscellaneous soft nanoparticles
9.1. Synthetic polymer based nanoparticles
Synthetic polymer-based NPs are of particular relevance to the areas of drug and gene delivery.438–440 Their desirable properties include aqueous solubility or the ability to be solubilized by minor chemical modifications, satisfactory long-term stability in biological environments, capability for controlled release, biodegradability, and more access to the enhanced permeability and retention effect found in many tumors than many other types of traditional hard NPs. This is a fairly large area encompassing a wide variety of hydrophilic, hydrophobic, and hybrid amphiphilic copolymers that are usually produced by precipitation of polymers via emulsion/solvent evaporation, poly-condensation of monomers in aqueous emulsion or by simple self-assembly of copolymers.441–444 To produce hydrophobic polymeric NPs, the most commonly used precursors are poly-lactic acid (PLA),445 poly-lactic-co-glycolic acid (PGLA),446 or polystyrene (PS),447 while for hydrophilic NPs the most favored materials are PEG,448 poly-ethyleneimine (PEI),449 poly-lysine,450 and sometimes poly-methyl methacrylate (PMMA) when appropriately modified.451 Each of them have their respective advantages and disadvantages. For instance, hydrophobic particles are ideal to encapsulate hydrophobic drugs and fluorophores while hydrophilic NPs are more compatible with bodily fluids and can be more easily functionalized with biorecognition or targeting molecules. To facilitate their use, hydrophobic NPs are often attached to a surfactant or some other hydrophilic molecule during the synthesis or via post-synthetic modification to render them colloidally stable or soluble in aqueous solution.452 In terms of biofunctionalization, the majority of the reports involving polymeric NPs describe their conjugation to antibodies,453,454 proteins,455 and peptides456 for in vivo applications; but there are a few reports where DNA has been attached by covalent conjugation or simple electrostatic adsorption. In the latter case, the NPs generally display appropriate amine moieties so as to be positively charged at physiological pH and are therefore suitable for attracting negatively charged DNA. For example, Green reported a series of cationic and hydrolytically degradable poly-β-amino ester-based NPs that adsorbed plasmid DNA and provided for impressive delivery efficacy.457 Interestingly, by slightly varying the end group of the custom synthesized polymers, some structural features and the ζ potential of the resulting self-assembled particles could be tuned and this, in turn, provided for remarkably different transfection efficiencies with model GFP-encoded plasmids in human embryonic stem cells. Huang and colleagues employed the same poly-β-amino ester-derived NPs for the delivery of a suicide gene in the form of a plasmid, encoding a diphtheria toxin protein (DT-A) to ovarian tumor bearing mice.458 Direct administration of the particles to the tumor site significantly reduced the tumor mass, displaying a far more potent effect than the commonly used chemotherapeutic agents cis-platin or paclitaxel (PTX) when used at similar doses. While electrostatically adsorbing DNA to polymeric NPs offers a potential alternative to viral delivery systems, short oligomers have also been covalently ligated to these particles and then applied for targeted delivery of a chemotherapeutic drug. In one example, PTX was encapsulated inside PEG-PGLA NPs during synthesis via an emulsion/solvent evaporation method.459 The PTX-loaded particles were then chemically conjugated to a DNA aptamer that specifically targets nucleolin, a plasma membrane protein overexpressed in many cancer cells. The aptamer-functionalized PTX-loaded particles displayed a substantially higher cytotoxicity in malignant brain glioma cells with an IC50 value 4.5 times lower than that of free PTX.459The other class of synthetic polymer-derived NPs are micelles and closely related liposomes or vesicles. These are formed via the self-assembly of amphiphilic copolymers and are currently considered the most promising “nanocontainers” or nanodelivery vehicle for drug delivery. A wide variety of materials have been exploited to produce micelles of different sizes and shapes and among them some spherical micelles are currently undergoing clinical trials for cancer treatment.460–462 PEG is widely used with aliphatic polyesters to produce diblock copolymers that readily self-assemble into micellar structures at optimal concentration; given this, significant interest has recently emerged on constructing micelles from amphiphilic polymers that contain DNA as their hydrophilic component.463–465 Spherical micelles fabricated from such units will have a hydrophilic corona of DNA around their hydrophobic core in aqueous solution. Additionally, a reactive functional group either on the DNA or another component can allow attachment of other biomolecules. In particular, molecular recognition moieties such as antibodies offer the possibility of targeted delivery. In a pioneering study, Jeong and colleagues chemically-ligated short oligonucleotide sequences to a biodegradable organic polymer.466 The resulting hybrid amphiphilic polymer formed relatively monodisperse micelles in aqueous solution with a hydrophobic core and a corona of ssDNA that was ∼65 nm in diameter. These micelles were readily taken up by mouse fibroblast cells, and as speculated by the authors, possibly by a fluid phase pinocytosis process instead of an absorptive or receptor-mediated endocytic mechanism due to charge-to-charge repulsion between micelles and the cells. A slow release of the oligonucleotide was observed in the cytoplasm, presumably due to the hydrolytic scission of the polymer backbone and, considering some of their presence inside the nucleus, the authors envisioned them as a tool for delivering antisense oligonucleotides to control the expression of a target gene.466 The same group later demonstrated an antisense oligonucleotide delivery system by following a slightly different strategy.467 An antisense c-Raf oligonucleotide linked to a PEG chain was synthesized, which in the presence of the cationic polymer PEI, formed a polyelectrolyte complex micelle that was nearly 70 nm in diameter. This had an oligo-PEI-complex as the inner core surrounded by PEG chains on the surface. The cellular uptake of these micelles was substantially higher than the oligonucleotide alone and displayed a better inhibitory effect against the proliferation of ovarian cancer cells, both in vivo and in vitro.467
Using a different chemical strategy Alemdaroglu et al. prepared a propylene oxide-(PPO) appended oligonucleotide during its solid phase synthesis and in aqueous solution this produced micelles of ∼11 nm in diameter (Fig. 33a).468 The micelles were subsequently employed for targeted delivery of doxorubicin (Dox) to human colon adenocarcinoma cells (Caco) as a conceptual application. To promote cellular targeting, folic acid- (FA) conjugated DNA was hybridized to the coronal ssDNA as these target cells display high levels of folate receptor.469 The resulting particles displayed a high degree of specificity with uptake efficiency being proportional to the number of FA moieties on the micelle rim and on the spatial location of the FA units on the micelles (Fig. 33b). When the micelles displayed 28 targeting units, the uptake efficiency was enhanced nearly 10 times compared to the unmodified micelles. However, when FA was conjugated to the other end of the oligo, and therefore pointing inwards towards the micelle core, the construct largely failed to recognize the target cells. The drug-loaded micelles displayed a high level of cytotoxicity, with a cell viability of ∼24%, in contrast to the micelles lacking drug as control which only showed mild toxicity (75% cell viability).468 The same group employed these PPO–DNA-based micelles for an entirely different purpose, namely to do organic reactions. DNA templated organic reactions have been executed before,470–472 however, the unique aspect of this report was the use of a 3D platform which offered a higher local concentration and both hydrophobic and hydrophilic interfaces.473 Three reactions were performed here: a bimolecular EDC coupling to form an amide bond; a Michael reaction; and a complex trimolecular coupling reaction to produce a fluorescent isoindole from o-phthalaldehyde. The necessary reactive functional groups including a thiol, amine and maleimide were attached to the ssDNA prior to their hybridization to the complementary micellar DNA. The products were characterized via gel electrophoresis which showed that the reaction yield for the first two reactions were above 70%, while the trimolecular coupling yield was 41% which, given the number of participants and rarity of these types of reactions, is quite good. When the necessary functional groups for the third reaction were linked to the 3′ end of the complementary DNA, the reaction took place at the interface of the hydrophobic core and hydrophilic corona, instead of on the rim, and this improved the yield of the last reaction from 41% to 59%. Improved efficiency was attributed to the higher accumulation of hydrophobic o-phthalaldehyde in the hydrophobic core of the micelle increasing its local concentration and facilitating the reaction.473
 | ||
| Fig. 33 (a) A DNA block copolymer based micellar drug delivery system. The red dots at the periphery are FA targeting units and the green dots concentrated around the hydrophobic core are the anticancer drug Dox. (b) Cell viability test shows the FA equipped Dox loaded micelles have the best cell killing capability (column A in the plot). Column B, C and D represent Dox loaded micelles with but not covalently linked FA, Dox loaded micelles and only FA conjugated micelles, respectively. Reproduced with permission from ref. 468. Copyright 2008, Wiley-VCH. | ||
Unlike chemically ligating a biorecognition molecule for targeted delivery such as FA, Kang and coworkers adopted an alternative strategy for targeting by directly incorporating a DNA aptamer as an integral part of their liposome.474 A PEG spacer placed between the hydrophobic domain and the DNA aptamer prevented interparticle fusion as well as improving resistance against enzymatic degradation of DNA in the serum environment. An average of nearly 250 aptamers specific to leukemic CEM-CCRF cancer cells were tethered to each of the liposomes and the resulting multivalent interactions facilitated the translocation of the liposomes across the cell membrane. The aptamer-equipped liposomes loaded with FAM-dextran as a model drug showed high specificity towards binding the target CEM cells compared to non-target NB4 cells (acute promyelocytic leukemia) as evidenced by flow cytometry.474 Structural features of a DNA-block copolymer micelle can also be altered via molecular recognition and this can notably influence their cellular uptake efficiency. Ding and coworkers demonstrated that exposing micelles to a long DNA that has repeated units of complementary sequence to the micellar DNA created a structural transition from a sphere to a rod consisting of two parallel dsDNA held together by the organic polymer.475 The transformation was confirmed by scanning force microscopy where uniform rod-like flat DNA structures with significantly reduced height were observed. The transition was also accompanied with a substantially changed diffusion coefficient as measured via fluorescence correlation spectroscopy. The same group investigated the cellular uptake efficiencies of these rod shaped NPs, and interestingly, found they are internalized nearly 12 times more efficiently than their spherical counterparts.476 The author's suggested the difference could be due to separate uptake mechanisms; the rod-like particles having an exposed hydrophobic polymer could have been internalized by adsorptive endocytosis whereas the spherical particles with the hydrophobic part shielded by the negatively-charged oligos might have been taken up via a fluid phase pinocytosis mechanism.
Polyacrylamide NPs are another popular synthetic polymer based NPs that have demonstrated strong potential as a drug carrier,477 photosensitizer delivery agent,478 and, more importantly, as an in vitro as well as in vivo sensor-displaying platform for various analytes and metabolites.479–482 Nielsen and coworkers employed such NPs to embed an adenine nucleotide responsive aptamer and employed them as an in vivo sensor.483 This aptamer lacks the capability to distinguish between ATP, ADP (adenosine diphosphate) or AMP (adenosine monophosphate) as the phosphate group stays out of the binding pocket, but it can differentiate a GTP (guanosine triphosphate) from an adenine nucleotide. The probe was equipped with a fluorophore and a quencher molecule on the two ends of an oligomer which folds into a hairpin structure bringing the two in close proximity resulting in fluorescence quenching (Fig. 34). The probe was embedded inside 30–35 nm polyacrylamide NPs during their synthesis by an inverse microemulsion process. Prior to incubation of the probe-loaded particles with target yeast cells, they were treated with DNase to remove any aptamer sticking out of the surface. The porous matrix protected the probes trapped inside the particle from DNase degradation while still allowing for diffusion of small molecules like ATP. In the presence of ATP or other adenine nucleotides, the aptamer reconfigured itself to preferentially bind its substrate causing an enhancement in fluorescence.483 The total concentration of adenine nucleotide sensed in the target cells was estimated to be 2.9 mM, which was in the same range as that previously reported based on using yeast cells extract.484
 | ||
| Fig. 34 (a) An aptamer embedded polyacrylamide NP being employed for mapping intracellular distribution of ATP. Cellular ATP binds to the aptamer resulting in a structural rearrangement that separates the fluorophore from the quencher leading to an increase in Texas Red fluorescence as shown in the confocal fluorescence image (b). Reprinted with permission from ref. 483. Copyright 2010 American Chemical Society. | ||
9.2. Viral nanoparticles
Recombinantly and synthetically-engineered viral capsids or virus NPs are an emerging class of NMs that have found application in a variety of areas including biosensing, fluorescence and MR imaging, targeted drug and gene delivery, vaccine generation, light harvesting and even for digital data storage.56,485–490 These proteinaceous self-assembled and hollow nanostructures can be almost perfectly monodisperse in shape and size and amenable for modification due to the display of a diverse range of chemical functionalities (via amino acid side chains) that can later be exploited to attach drugs, probes, contrast agents, biomolecules or to load cargo.56,491 Various kinds of viral NPs with different shapes and sizes can be artificially generated in a relatively easy manner by supramolecular self-assembly of identical protein subunits while remaining free of genomic material. The protein shell, whose primary role is to protect the genomic material tends to be very robust and therefore prevents premature degradation before arriving at the target site during delivery applications. Despite this, viral NPs are still inherently biodegradable and this rules out the possibility of overt bioaccumulation thus lending itself to be an enticing material for cellular delivery purposes.492 Moreover, crystallographic studies provide fairly accurate information about the number of subunits the NPs are comprised of, the spatial positioning of individual amino acid residues and other details such as position of pores through which the interior of the particles are sometimes accessed. For example, the protein shell of the MS2 bacteriophage is composed of 180 identical protein monomers that can be expressed in E. coli and these spontaneously self-assemble into a hollow sphere of 27 nm in diameter with 32 pores on the surface allowing access to the core.493,494 Tong and coworkers demonstrated the potential of these MS2 capsids as targeted drug delivery vesicles by attaching DNA aptamers to the exterior surface.495 To affix the aptamer, an aniline group was introduced on to the outer surface proteins by incorporating the unnatural residue p-aminophenylalanine (paF) using an amber stop codon suppression system. The custom synthesized DNA aptamer was equipped with a phenyldiamine moiety that reacted with the aniline group of paF in a chemoselective fashion leading to attachment of more than 50 aptamers per NP. In addition, a cysteine mutation was engineered onto the interior surface for conjugating AlexaFluor 488 dye as a model cargo. The aptamer targeted protein tyrosine kinase 7, a trans-membrane receptor ubiquitous among different leukemic cells and believed to be responsible for uncontrolled cell proliferation. These double mutant fluorophore and aptamer-decorated viral NPs exhibited high specificity in targeting Jurkat T leukemia cells while random DNA modified particles displayed negligible binding affinity. Moreover, a costaining experiment suggested these particles traffic to the lysosome, which suggests them as cargo carriers for the release of acid-labile prodrugs inside cells.495 In a follow up study, the same group chemically ligated a maleimide-bearing porphyrin molecule to the sulfhydryl groups on the viral NPs interior surface (Fig. 35).496 The goal here was to develop a photodynamic therapeutic NM by exploiting singlet oxygen generated from photoexcited porphyrin. The same Jurkat cell specific aptamer was affixed on the exterior surface of these porphyrin carrying viral NPs for targeted cell killing. 76% of the MS2 exposed Jurkat cells died after irradiation with a 415 nm LED lamp for 30 minutes, while only 3–6% of control cells died under identical illumination conditions.496 The shallow penetration depth of the blue light along with the long irradiation time, however, may remain a challenge for porphyrin mediated photodynamic therapy application in denser tissues.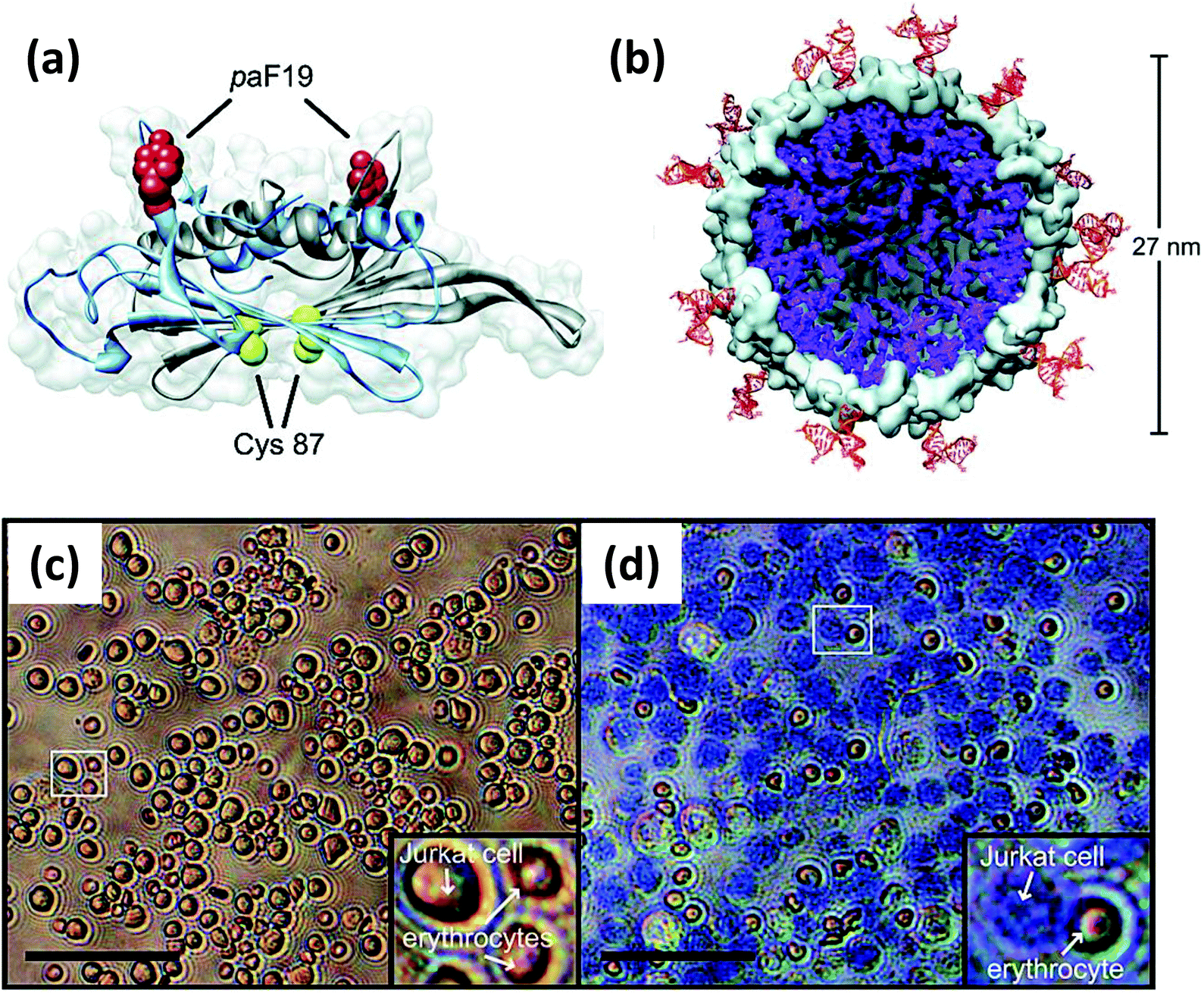 | ||
| Fig. 35 Dual surface modified virus capsids for targeted photodynamic therapy. (a) Sites for exterior and interior modification to the shell protein. The unnatural amino acid p-aminophenyl alanine (paF) residues (red) were exploited to ligate the aptamer on the capsid surface while the cysteine residues (yellow) were used for attaching the porphyrins in the interior surface. (b) Fully modified virus nanoparticles bearing nearly 20 aptamers (red) for targeting Jurkat leukemia T cells. The violet color represents nearly 180 porphyrins installed on the inside surface of the capsids. (c) Targeted killing of Jurkat T cells in the presence of normal erythrocytes. In a live mixture, the Jurkat cells can be distinguished from their size and morphology. (d) Majority of the Jurkat cells are dead (stained by trypan blue) after irradiation at 415 nm, while the erythrocytes are alive and thus unstained. Reprinted with permission from ref. 496. Copyright 2010 American Chemical Society. | ||
Besides chemically ligating the cargo to constituent protein units, an alternative loading possibility was reported by Kwak and colleagues.497 They loaded the molecule of interest to a DNA-amphiphile micelle which, besides acting as the carrier, also served as a template for the surrounding capsid formation. As a model system, they selected cowpea chlorotic mottle virus (CCMV) which can be artificially produced by the self-assembly of 180 identical protein subunits into an icosahedral particle of 18 nm. As a representative example, the authors were successful in trapping a hydrophobic and a hydrophilic fluorophore molecule inside the micelle-encapsulated capsids. A slightly different strategy was followed in these two cases; while hydrophobic pyrene dye was accumulated within the hydrophobic core of the micelles by simple adsorption, the hydrophilic fluorescent dye ROX was introduced by covalently ligating them to an oligo complementary to the micellar DNA.497
9.3. Ferritin nanoparticles
There are numerous other examples of peptide and protein-based NPs in the literature that can be further conjugated to or modified with biomolecules and these have also made important contributions in the areas of targeted cellular delivery and in vitro bioassays.498–500 Ferritin NPs are one such nanoparticulate material. They are a naturally occurring protein that plays a significant role in the homeostasis of ferrous ion levels in many species including humans.501–503 The outer and inner diameters of this cage-shaped protein are 12 and 7 nm, respectively. The hollow interior of apoferritin (ferritin without ferrihydrite, a product of Fe(III) and OH− that fills up the core) has been exploited not only for biological purposes like drug delivery504 or fluorescence and MRI imaging,505 but also for the templated synthesis of inorganic NPs including Fe, Co and Cr oxides.506 Kim and colleagues reported a fluorescent ferritin NP attached to a DNA aptamer which was utilized in a sandwich assay for the detection of platelet derived growth factor B-chain homodimer (PDGF-BB), a cancer biomarker (Fig. 36).507 These novel NPs were produced via bacterial expression of a hybrid gene consisting of human ferritin heavy chain (hFTN-H) and an eGFP or red fluorescent protein (DsRed). The self-assembly of the peptidyl units produced the protein cage along with 24 eGFP (or DsRed) decorated on its surface. A PDGF-BB-specific DNA aptamer was ligated via SMCC coupling to a mutagenically inserted cysteine residue on the eGFP. Interestingly, after the DNA conjugation a slight increase in the fluorescence intensity of the fluorescent protein was noted which was attributed to electrostatic repulsion from the negatively charged DNA which served to keep the proteins away from each other. In a typical sandwich assay, the PDGF-BB protein acted as a linker between the aptamer-modified fluorescent proteins and immobilized-biotinylated aptamer on a streptavidin-coated glass surface. The assay showed an amplified sensitivity nearly three orders of magnitude better compared to those using aptamer-modified eGFP or Cy3 alone.507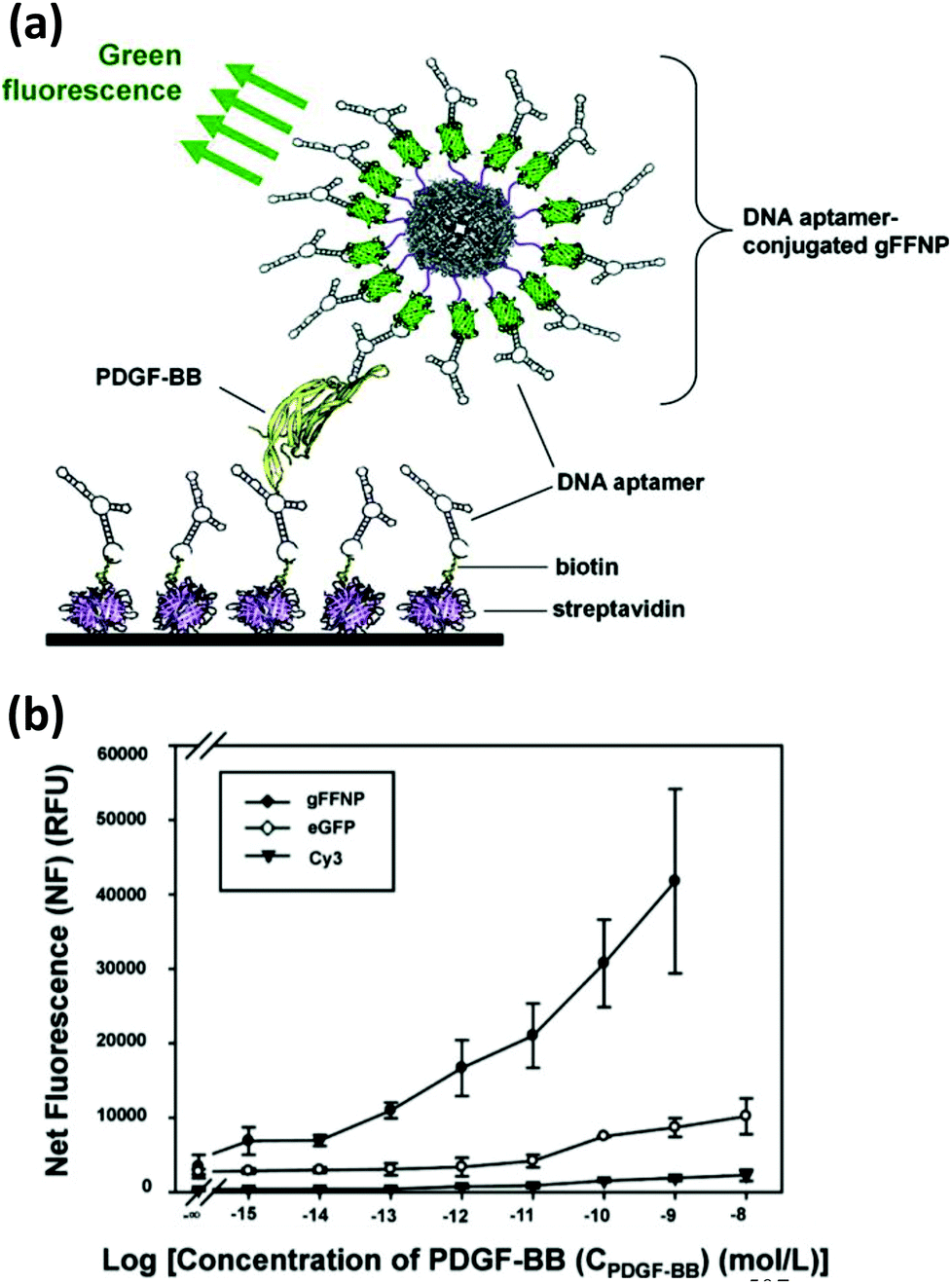 | ||
| Fig. 36 (a) Schematic illustration of a DNA aptamer conjugated fluorescent ferritin NPs being employed as a reporter probe for protein biosensing. The cancer biomarker PDGF-BB captures the aptamer-conjugated ferritin NPs on an appropriately modified glass surface. Green fluorescence retained after washing reflects the amount of PDGF-BB present. (b) The plot shows enhanced sensitivity of the aptamer appended green fluorescent ferritin NPs, compared to the same aptamer ligated to eGFP or Cy3. Reprinted with permission from ref. 507. Copyright 2011 American Chemical Society. | ||
10. Miscellaneous hard nanoparticles
10.1. Lanthanide-based upconversion nanoparticles
Upconversion NPs (UCNPs) are an emerging class of luminophore material that display anti-Stokes emission upon irradiation with infrared light, i.e., blue-shifted emission (higher energy) from a red-shifted excitation (lower energy). They are especially interesting for bioimaging as they do not have certain drawbacks such as the photobleaching seen with traditional organic dyes or the fluorescence intermittency of QDs. Additionally, the infrared light required to excite them has a higher tissue penetration capability with fewer damaging side effects.508–510 These NPs are produced by doping lanthanide ions like Er3+, Tm3+ or Ho3+ as the emitter along with a sensitizer ion, mostly Yb3+, into an inorganic host matrix. They have a ladder like electronic configuration stemming from the 4fn electrons that sequentially absorb photons resulting in a sharp absorption band. Due to sequential photon absorption, they don't require coherent, high power, femtosecond pulsed laser like some other anti-Stokes processes, such as second harmonic generation. In fact, Yb3+, with its large absorption cross section, can be conveniently excited with a low power 980 nm CW laser which can transfer its energy to the activator ions mentioned above. Their emission profile is sharp as well, while the luminescence lifetime is relatively long due to the Laporte and (sometimes) spin-forbidden intra 4fn transition. Unlike QDs, their emission color does not depend on the size of the NPs, rather they are tuned by careful choice of host, dopant and the dopant ratio.511–513 Since most biological molecules poorly absorb NIR light that is used to excite these NPs, the auto-fluorescence contribution to the background signal is dramatically reduced when UCNPs are used, which is another advantage over traditional fluorophore dyes. However, poor QY (∼1–3%) remains a drawback of using this class of luminophore; needless to say, their QY always has to be less than 50% since at least two photons are necessary for the emission of one photon. Various biomolecules have already been coupled to these particles and demonstrated for a variety of in vitro and in vivo assays.514–517In terms of DNA conjugation, Zhang and coworkers constructed a sandwich assay involving Yb3+ and Er3+ doped NaYF4 UCNPs for DNA detection by covalently ligating a short amine-modified DNA, partly complementary to the target strand, to silica-coated UCNPs.518 In the presence of target oligonucleotide, another reporter strand carrying a carboxytetramethylrhodamine (TAMRA) label was captured onto the UCNPs triggering LRET (Luminescence Resonance Energy Transfer) from the UCNP donor to the TAMRA acceptor. Due to the minimal background signal contribution, the process was highly sensitive with a detection limit of 1.3 nM. Enhanced sensitivity and signal was also attainable when using higher laser power due to the power dependency of the UCNP excitation–emission process. Moreover, from the melting transition of the dsDNA as monitored by FRET, the capability of this method to distinguish a single point mutation in the β-globin gene responsible for sickle cell anemia was also demonstrated (Fig. 37).519 Later the same group reported a simplified DNA detection scheme that did not require the dye-labeled reporter strand.520 EDC coupling chemistry was used to attach DNA to water soluble UCNPs and then SYBR green dye was used as the FRET reporter. This intercalating dye is characterized by an enhanced fluorescence QY upon binding to dsDNA. The detection limit in this modified format was 0.1 nM, almost 10 times lower than the initial demonstration.
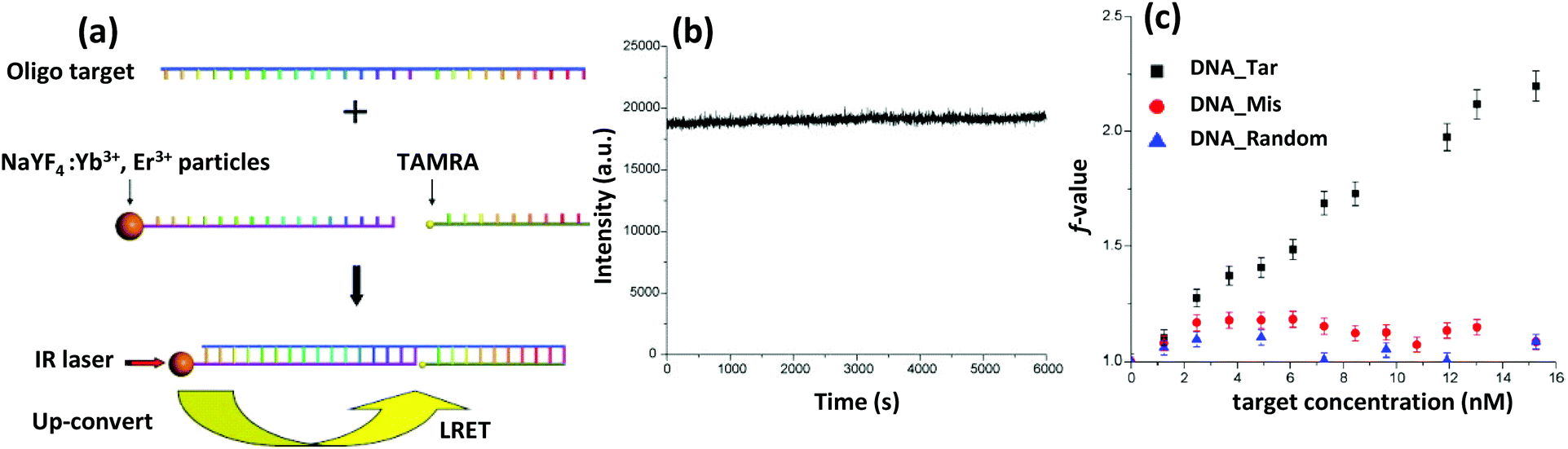 | ||
| Fig. 37 (a) Schematic design of a LRET based DNA sensor using UCNPs (λmax, em 537 nm). (b) Plot demonstrates photostability of the UCNPs, irradiated with 975 nm diode laser light at ∼500 mW for a prolonged period of time. (c) The plot describes the sensitivity of the sensor to single nucleotide mismatch. The f value is defined as f = (I575/I537)/(I575,0/I537,0) − 1, where I575/I537 is the normalized TAMRA (λmax, em 575 nm) emission at a given target DNA concentration and I575,0/I537,0 is the corresponding value for the blank solution. Reprinted with permission from ref. 519. Copyright 2009 Elsevier. | ||
10.2. Metal chalcogenides
In addition to the previously mentioned QDs, some transition and post-transition metal chalcogenide NPs also display interesting optical, magnetic and electrochemical properties, but due to the potential for severe toxicity, their in vivo application is still strictly limited. However, there are several reports where CuS or PbS NPs have been exploited for in vitro electrochemical sensing, both with and without direct bioconjugation, and also alone or in association with other NMs.521–524 For example, Ding and coworkers covalently ligated amine-functionalized DNA to mercaptoacetic acid-stabilized CuS nanoparticles via EDC chemistry and employed the resulting particles as a DNA sensor by integrating them within a flow-injection chemiluminescence system.525 In the presence of the target DNA, the CuS NPs were captured onto a DNA-anchored glass electrode surface in a sandwich fashion. Following washing, the CuS NPs were dissolved in a mildly acidic solution and the concentration of Cu2+ was monitored by the chemiluminescence generated from a luminol-H2O2-Cu2+ system. Sensitivity of the biosensor was improved using an electrochemical preconcentration process performed with anodic stripping voltammetry, which encompasses depositing the initially dissolved Cu2+ ions from the captured CuS NPs on a Pt electrode surface followed by resuspending them into a smaller volume. Here, the chemiluminescence intensity was directly proportional to the cupric ions, which in return, was again proportional to the amount of target DNA.525 The sensitivity of this method was further improved by modifying the reporter strand to carry AuNPs decorated with multiple CuS NPs connected via a ssDNA as depicted in Fig. 38.526 Since a single 20 nm AuNP could accommodate more than 70 CuS NPs around it, the sensitivity of the method was greatly amplified with a detection limit approaching a few attomolar. In the same way, a AuNP-PbS NP hybrid system was constructed and demonstrated as a sensitive DNA biosensor where the concentration of Pb2+ was quantified by anodic stripping voltammetry with a reported detection limit of a few picomolar.523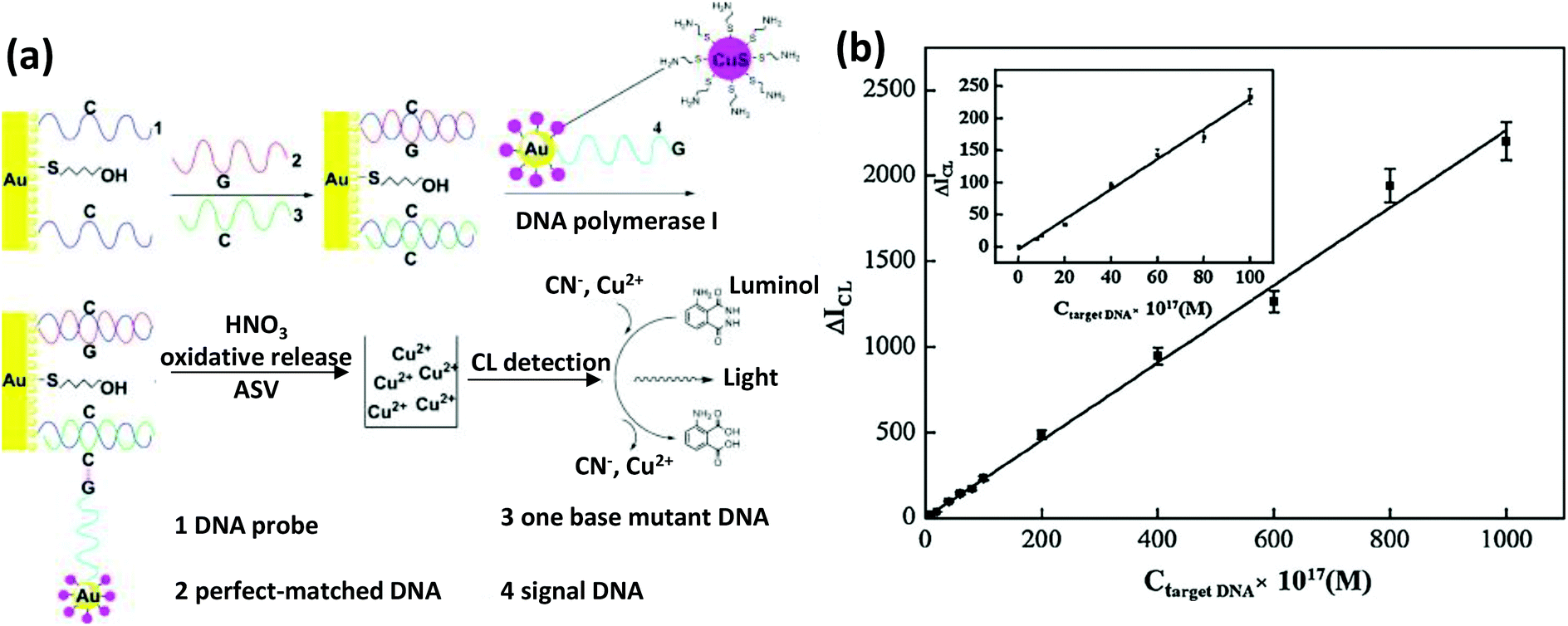 | ||
| Fig. 38 (a) An single nucleotide polymorphism (SNP) detection scheme employing AuNPs and CuS NPs. The CuS-AuNPs hybrid NP probes are coupled to the mutated site in the presence of DNA polymerase. The SNP was quantified by the chemoluminescence intensity from luminol – CN−–Cu2+ ions (the cupric ions were dissolved from the hybrid). (b) Calibration curve for measuring mutated DNA concentration with respect to chemiluminescent (CL) intensity. Reprinted with permission from ref. 526. Copyright 2010 Elsevier. | ||
10.3. Alkaline earth metal nanoparticles
While transition metal chalcogenide NPs are inappropriate for in vivo application, alkaline earth metal-based NPs tend to be quite suitable for biological use due to their significantly reduced toxicity. Calcium phosphate NPs (CaPNPs) are especially interesting among others of this class as they can be prepared and bioconjugated in a facile and inexpensive manner. They have already been exploited as a fluorescence contrast agent, drug delivery vesicle and, most importantly, as a non-viral vector for gene delivery.527–531 Under the generalized term of CaPNPs, they consist of either Ca(H2PO4)2, CaHPO4 or Ca3(PO4)2 and are predominantly prepared by precipitation of inorganic salts containing PO43− and Ca2+-counter ions, such as calcium nitrate (Ca(NO3)2) and diammonium hydrogen phosphate ((NH4)2HPO4). Unlike other NPs that usually require an appended recognizable biomolecule like an aptamer or antibody to introduce target specificity, the pH dependent solubility of CaPNPs itself has instead been directly exploited for targeting type activity. These NPs are stable at physiological pH but dissolve in acidic environments and since such an environment is found around many solid tumors, CaPNPs are useful for targeted delivery of drugs or genes to these cancer cells. However, one major problem associated with CaPNPs is their poor long-term colloidal stability as they grow in size when stored after synthesis. Several reports have described methods to inhibit the continual growth process and these include addition of Mg2+ ions, encapsulation with block copolymers, use of PEG or simply by adsorbing DNA.532–535 Plasmid DNA or simple oligonucleotides get readily adsorbed onto the CaPNPs surface during a standard coprecipitation process which, besides functioning as a solubilizing ligand for colloidal stability, can also enhance the intrinsically poor transfection efficiency of these particles.536–538 The problem of rapid NP growth during coprecipitation was addressed by Kakizawa et al. by introducing a PEG-poly-aspartic acid (PEG-PAA) block copolymer which adsorbed to the NP surface forming a core–shell structure with a hybrid core of CaP and plasmid DNA surrounded by a PEG shell.532 Besides providing colloidal stability, the presence of a PEG corona substantially increased intrinsic resistance towards nuclease degradation. This was shown with in vitro analysis after incubating luciferase-encoding plasmid-carrying NPs with DNase I. A significantly higher expression of luciferase gene in HEK293 and HeLa cells was noted compared to unmodified particles.532 To prevent similar nuclease degradation, Sokolova and colleagues described a novel strategy of creating triple-layered CaP-DNA NPs using a three-step synthesis.531 In the first step, the plasmid DNA-coated NP was initially formed via coprecipitation of (NH4)2HPO4 and Ca(NO3)2 in the presence of the plasmid DNA. In the second step, the resulting NPs were subjected to another coprecipitation round with the same reagents to build a layer of CaP. Finally, the NPs were mixed with nonspecific DNA to provide the required colloidal stability. This yielded final NPs that were 10–20 nm in diameter while displaying an enhanced transfection efficiency (from 3.5% to 10% compared to standard CaP) for an eGFP-encoded plasmid in multiple cell lines.53111. DNA nanoparticles
DNA nanoparticles (DNANPs) are fundamentally different from the other conventional inorganic, polymer or protein-derived NPs discussed previously. DNANPs can be designed and simulated in silico by applying the basic principles of Watson–Crick base pairing and the helical geometry of dsDNA. They are then produced by annealing the computer-suggested ssDNA sequence at a specific stoichiometric ratio in saline buffer and characterized directly via gel-electrophoresis and AFM/TEM. This simple method has produced nanostructures with various well-defined configurations and conformations, some with the capability of reconfiguring themselves based on the influence of external stimuli like DNA, mRNA, proteins, or pH and even some that are enzymatically replicable.211,212,219,539–543 Originally envisioned by Prof. Seeman nearly three decades ago, the primary goal of DNA nanotechnology was to build 3D networks from branched immobile DNA motifs to organize target molecules, such as proteins for crystal studies. Although the ultimate goal of crystalizing a protein in a DNA scaffold is yet to be realized, well-defined DNA-assembled 2D and 3D crystalline frameworks have already been produced with a considerable degree of success.544,545 To date, a fairly large collection of discrete DNANPs with varying degrees of complexity can be found in the literature with some produced to display a specific property or to execute a certain task and some driven by the sheer interest of generating novel nanoscale patterns or geometric shapes with an increasing degree of sophistication.12 This structural library can be categorized into three distinctive subsections: (1) small DNA tiles formed by the self-assembly of a few ssDNA molecules, which if designed wisely with self-complementary unhybridized strands (commonly known as sticky ends), can grow into finite or seemingly infinite periodic 2D arrays or a 3D network via sticky end association; (2) discrete 3D polyhedra, from a simple tetrahedron to something as structurally rich as a buckyball; and (3) scaffolded-DNA origami produced by folding a long ssDNA (most commonly the M13mp18 bacteriophage genome of ∼7000 nucleotides length) with numerous predesigned short oligonucleotides, called staple strands, into a desired, discrete 2D or 3D architectures. All of these are powerful templates for organizing multiple nanoscale entities with nanometer precision which, along with unique addressability, are the primary reasons behind the growing popularity of these exquisite nanostructures. Thanks to both modern organic chemistry and molecular biology, a rich variety of bioconjugation methods have been developed over the years to modify DNA with biorecognition molecules and other NPs.58,546 ssDNA-functionalized NPs or biomolecules can be easily incorporated into DNA nanostructures by complementary base pairing interactions targeted to protruding sticky ends, and the resulting hybrid nanoassemblies have found application in a wide variety of areas including biosensing, cellular delivery and even for preliminary theranostic purposes.547,548 It has also opened up a new paradigm for fundamental studies, such as understanding the interaction mechanisms between discretely placed photonic elements.194,209,21011.1. Biosensing and molecular logic devices
Biocompatible DNA nanostructures are obviously ideal for exploitation as both biosensing platforms and biosensors themselves and the most convenient approach to achieve this is by incorporating a target-specific aptamer as a probe; this is easily accomplished by simply extending a constituent strand with an aptamer sequence. Pei and coworkers reported a DNA self-assembled tetrahedron shaped nanostructure functioning as a central DNANP which could serve as an electrochemical sensor for the detection of target DNA (Fig. 39a).549 The rigid pyramidal DNA nanostructure was synthesized by the self-assembly of four self-complementary ssDNA in less than two minutes with a remarkable yield of nearly 85%. Among the four constituent strands, three were conjugated to thiol moieties while the fourth strand was carrying the extended probe sequence. The thiol groups were conjugated to DNA strands in such a fashion that in the self-assembled structure they occupied the vertex positions and therefore acted as anchors to firmly and rapidly adsorb the tetrahedron onto an Au electrode surface, leaving the probe available and displayed at the top. A standard sandwich assay was then performed where the target strand acted as a linker between the probe and a biotin-modified reporter strand which was bound to avidin-HRP. Presence of the target DNA was transduced into electrochemical signal by the catalyzed electroreduction of H2O2 by HRP in the presence of the redox active cosubstrate TMB (3,3′,5,5′-tetramethylbenzidine). The same principle was applied for detecting the often targeted thrombin by conjugating a thrombin-specific aptamer as the probe to the fourth strand, leaving the rest of the design intact. The assay could potentially be applied in biological fluids as the DNA tetrahedron-coated Au electrode surface displayed minimal nonspecific adsorption to other proteins present in serum. The detection limit for target DNA and thrombin was 1 pM and 100 pM respectively, the latter being significantly lower than typical ssDNA aptamer-based sensors.549 Later, the same group extended the tetrahedron DNA nanostructures for the detection of cocaine by incorporation of a biotinylated cocaine-targeting aptamer as the reporter strand.550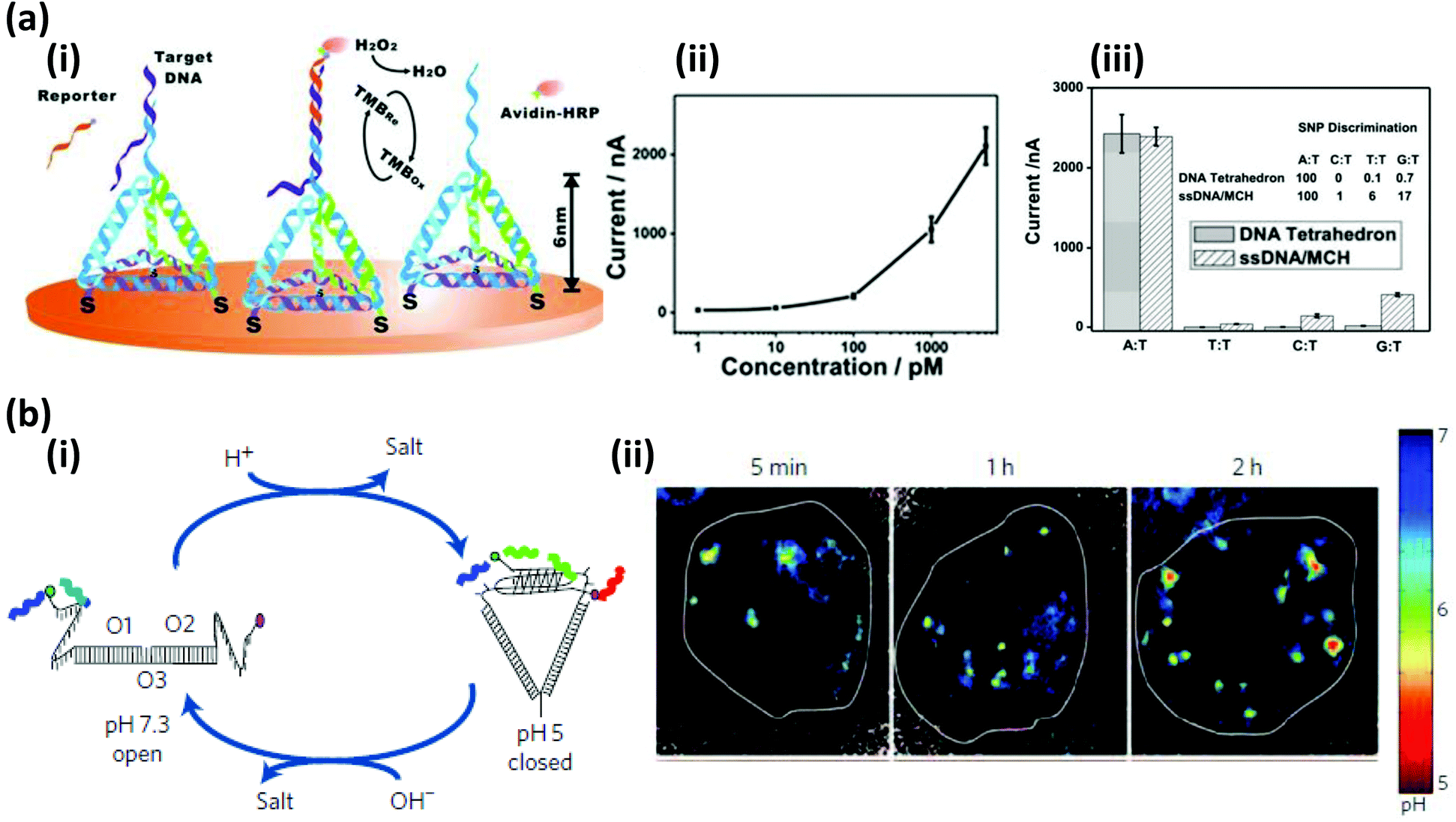 | ||
| Fig. 39 (a) (i) Schematic representation of an electrochemical DNA sensor employing a 3D DNA nanostructure immobilized on a gold electrode surface. (ii) Amperometric measurement displays stronger electrochemical signal with increasing target DNA concentration. (iii) The sensor's capability of discriminating a single base mismatch is demonstrated. The fully complementary strand gives rise to much stronger current than three other single base mismatches (T:T, C:T and G:T). Reproduced with permission from ref. 549. Copyright 2010, Wiley-VCH. (b) (i) A DNA nanodevice utilized for mapping spatial and temporal changes of pH inside living cell. Schematic shows the working mechanism involving an I-switch that, depending on the pH, tunes the extent of FRET between two fluorescent dyes AF488 and AF647. (ii) Time dependent pseudo color confocal images of donor (AF488)/acceptor (AF647) intensity in haemocytes, scaled with respect to pH. Reprinted with permission from Macmillan Publishers Ltd: Nature Nanotechnology, ref. 551. Copyright 2009. | ||
Besides static structures, intricate design capabilities have produced a rich library of DNA nanostructures that are physically reconfigurable by external stimuli and some of these have even been employed as logic-based sensing devices, both in vitro and in vivo. For example, Pei and coworkers synthesized a library of DNA tetrahedrons to construct AND, OR, XOR, and INH logic gates. Based on their programmable configuration, the structures displayed a remarkable response to several targets like protons (H+), mercury ions, ATP and, of course, complementary DNA strands.552 The presence of the target triggered a conformational change from a relaxed to a taut state, which was read out via a change in FRET signal between dyes attached to the DNA. These scaffolded logic gates also displayed promising activity inside a cellular environment mapping the spatial distribution of ATP in the cytoplasm of HeLa cells. External stimuli-driven conformational changes of DNA nanostructures have been further exploited by Modi and coworkers to map the spatial and temporal pH dynamics inside living cells (Fig. 39b).551 The simple DNA nanostructure that they developed was equipped with cytosine rich sticky ends that formed an i-motif complex under acidic condition, bringing a FRET pair closer to each other. The nanodevice was readily internalized in Drosophila haemocytes, presumably by anionic ligand binding receptor pathways, and changes from an initially strong to a weak FRET signal was associated with the rapid acidification rate of the early endosomes followed by a later, far slower rate during their maturation into liposomes.551
Along with sensing, such dynamic DNA nanostructures and especially those displaying a central cavity to safeguard a cargo could be advantageous for targeted delivery. Andersen et al. reported a DNA origami box with a hollow core, and a controllable lid that could be opened and locked by ssDNA inputs via a simple toehold-mediated strand displacement. The hollow core was large enough to contain a ribosome or a polio virus and could easily be loaded with drugs or antibody for use as a delivery vehicle.542 Douglas et al. reported a more advanced hollow barrel shaped DNA origami with the unique capability of sensing a specific cell surface and structurally reconfiguring itself on that basis (Fig. 40).553 This origami was loaded with a fluorescently-labeled antibody against human leukocyte antigen on the inner surface, while the outer surface was decorated with aptamer encoded logic gates that dictated the open and closed states of the barrel. In buffer solution, the origami remained in a locked state where the two identical units of the barrel were connected by DNA aptamer-based locks. Encountering the right combination of proteins expressed in target cell lines, the aptamer preferentially bound to its target protein displacing the partially-hybridized complementary strand and this resulted in a drastic structural reconfiguration which unlocked the barrel and exposed the antibody payloads; this, in turn, led to the origami itself finally attaching to the cell surface.553 More recently, Amir et al. applied this same principle to construct a complex logic gate system employing a batch of similar DNA origami nanostructure as above equipped with carefully manipulated ‘keys’ and ‘locks’.554 Besides an external DNA input, the opening, closing and recognition of the robot-like origami were controlled by the specific stoichiometric presence of other origami referred to colloquially as the positive regulator, negative regulator, effector robot, etc. The gates performed impressively in the haemolymph of a living adult Blaberus dicoidalis (tropical cockroach) which is known to have low systemic or endogenous nuclease activity and was thus compatible with prototyping such DNA nanostructures. Comparing the Reynolds number in the human circulatory system (1 in arteriols, ∼4000 in aorta and vena, 1–5 in insect's haemocoel), the authors were optimistic about the application of these DNA robots in human if the quantity of the robot was scaled up and they were made more nuclease resistant.554
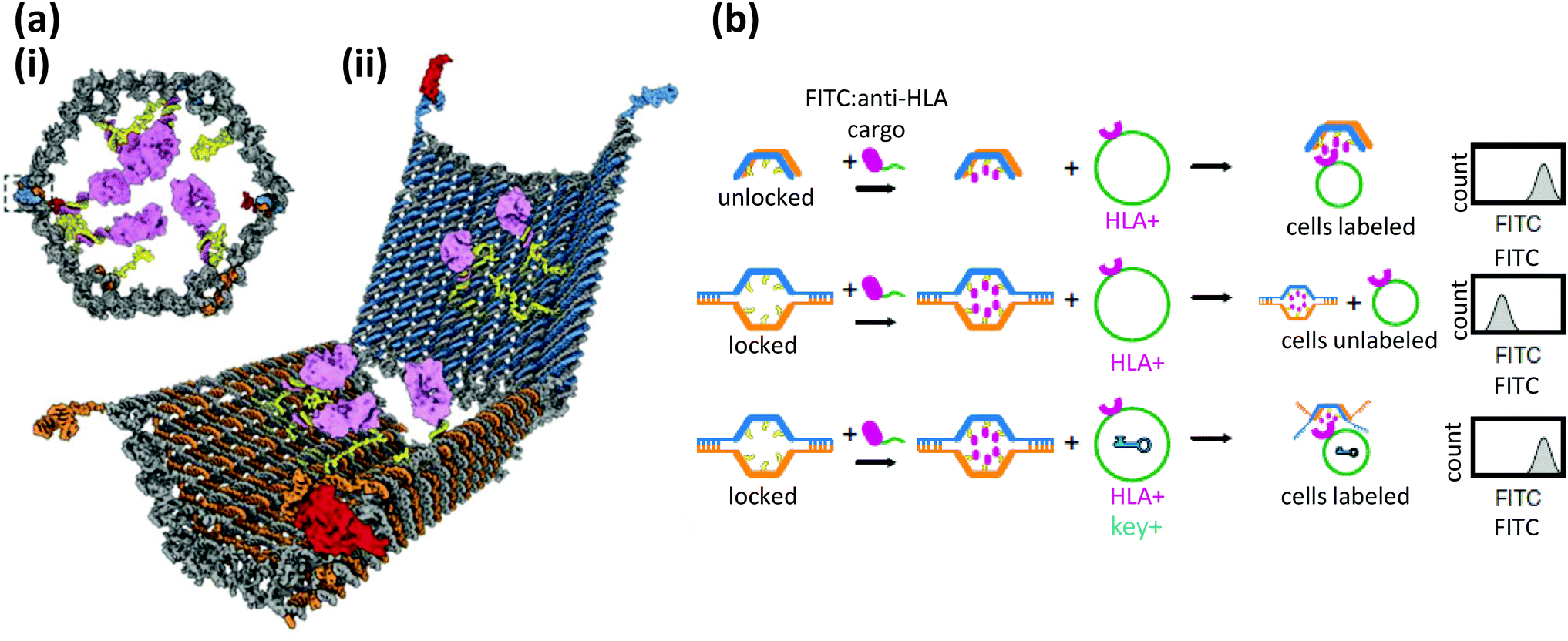 | ||
| Fig. 40 (a) Schematic drawing of an aptamer-gated DNA nanorobot capable of transporting molecular payloads to specific targets. (i) The front orthographic view shows the protein loaded nanorobot in locked configuration. (ii) Perspective view of the DNA nanorobot in unlocked configuration with exposed antibody payloads. (b) Activation scheme of the nanorobot loaded with antibody fragments to human leukocyte antigen HLA-A/B/C. Specific protein keys are necessary for opening the aptamer gated locks which then allow the nanorobots to bind to cells expressing HLA-A/B/C antigens. From ref. 553. Reprinted with permission from AAAS. | ||
11.2. Multiplexed detection
The unique programmability of DNA nanostructures also offers opportunities to build sophisticated multifunctional detection platforms by integrating multiple non-interacting DNA or RNAs aptamers onto a single DNA nanostructure. For example Ke et al. used a DNA origami to construct a nanoscopic DNA chip that was capable of simultaneously detecting multiple label-free RNA and which they envisioned could be an alternative to assays based on DNA microarrays or real-time PCR (RT-PCR).555 They employed a rectangular DNA origami with three different capture strands organized repeatedly in a specific geometrical pattern. Upon binding to the RNA target, the local stiffening was clearly imaged via AFM. The method, besides showing exceptionally high specificity, displayed high sensitivity, with a potential LOD down to a few thousand molecules.555 For multiplex detection, fluorophore-modified DNA nanostructures can have an especially useful impact. The wide commercial availability of fluorescent dye-labeled DNA has made the task of modifying a DNA nanostructure with fluorophores routine and quite straightforward.546,556 Multiple fluorescent dyes can be easily incorporated into DNA nanostructures at a specific stoichiometric ratio or in a certain geometrical pattern, which can serve as a distinct code. For example, Li and coworkers reported a DNA dendrimer-like nanostructure that worked as a functional probe with the capability of simultaneously detecting multiple pathogenic DNA present in a complex clinical sample.557 The Y-shaped dendrimer was fabricated by the self-assembly of three other smaller but similarly-shaped dendrimers with unhybridized sticky ends and varying ratios of a green and red fluorescent dye affixed at the periphery. Unlike DNA microarrays, which work on the principle of positional encoding, the Y-shaped dendrimer acted as a fluorescent intensity encoded nanobarcode. In order to amplify the output of fluorescent signal intensity, the dendrimers were concentrated by capturing on an avidin-functionalized polystyrene microbead using a sandwich assay where the target DNA acted as a linker between the preassembled biotinylated DNA on the microbead and the reporter barcode. Based on the ratio of the dyes on the reporter dendrimer, the microbeads displayed different colors which was vividly differentiated under a fluorescence microscope and later processed by computer. The method was rapid and highly sensitive with a detection limit approaching a few hundred attomoles.557 A geometric encoding approach was also adopted by Lin and colleagues to construct a fluorescent barcode with an enhanced multiplexing capability (Fig. 41).558 In order to unambiguously detect the geometrical pattern of the fluorescent spots, maintaining the robustness of the underlying scaffold is extremely important and this was achieved by using a 800 nm long tubular DNA origami. The excellent addressability of the DNA origami was exploited to organize three different fluorescent dyes at three well-separated and distinguishable locations in multiple numbers. Using permutations and combinations of the three dyes, 27 distinct (33) barcodes were initially composed. Later admixing of two different fluorophores at one zone allowed three extra pseudo colors to be generated, which increased the size of the barcode library from 27 to 216 (63). The color identity and the relative spatial position could be clearly resolved by TIRF microscopy (total internal reflection fluorescence microscopy). By increasing the number of fluorescent zones, the size of the barcode library could also be further increased, however, due to the reduced spacing between the fluorophores upon increasing the zone numbers, super-resolution microscopy was required to distinctively identify the pattern. The DNA-PAINT (point accumulation for imaging in nanoscale topography) super-resolution technique was employed and resolved two fluorescent spots separated by a distance of 42 nm.559 The capability of this barcode to be used as an in situ imaging probe was also demonstrated by tagging surface proteins in yeast cells (Fig. 41c).557 | ||
| Fig. 41 (a) Detailed 3D illustration of a zone of a DNA self-assembled rod like nanostructure decorated with fluorescent dyes at specific locations that can act as fluorescent barcode. (b) A representative superimposed TIRF microscopy image of an equimolar mixture of 27 distinct barcode structures. (c) TIRF images of yeast cells tagged with DNA nanobarcodes (green-red-green). Reprinted with permission from Macmillan Publishers Ltd: Nature Chemistry, ref. 558. Copyright 2012. | ||
11.3. Vaccine display, gene delivery, green regulation and others
Another promising research frontier emerging in recent years involves appending immunogenic molecules to DNA nanostructures to stimulate a strong and long-term immune response.560–562 DNA nanostructures are inherently biocompatible, have low auto-immunogenicity, satisfactory cell permeability and, most importantly, their extraordinary programmability can allow antigen and adjuvant to be brought closer together on one platform which can augment an antibody response. The pioneering report in this area came from Li et al. where a DNA tetrahedron conjugated to a cytosine-phosphate-guanine (CpG) motif (known for their strong immune-stimulatory activity) was delivered to macrophage like RAW274.7 cells without the aid of any transfection agent. A high level of secretion of pro-inflammatory cytokines including tumor necrosis factor, TNF-α, was subsequently observed.560 While the CpG-free tetrahedron displayed negligible stimulation, the CpG-modified tetrahedron showed a ∼9–18 times higher level of TNF-α expression compared to ssDNA-CpG. Similarly, Schüller and colleagues delivered a hollow 30 helix origami tube decorated with 62 CpG units to spleen cells and observed a much higher cytokine production than just CpG-conjugated oligonucleotides when using Lipofectamine as a carrier and delivery system.561 Liu and coworkers approached the next level by fabricating a vaccine complex where CpG units were appended as adjuvant at the vertices of a DNA tetrahedron and streptavidin (STV) used as a model antigen affixed in the inner core (Fig. 42).562 The vaccine complex was injected into a BALB/c mouse model and over a period of seventy days the serum of the immunized mouse developed a significantly higher level of anti-STV IgG antibodies compared to free STV and CpG control complexes. The authors envisioned that such programmable DNA nanostructures could be a promising platform for developing far more complex vaccines by constructing multivalent and multi-specific antigen-adjuvant complexes or by incorporating various other immunogenic epitomes.562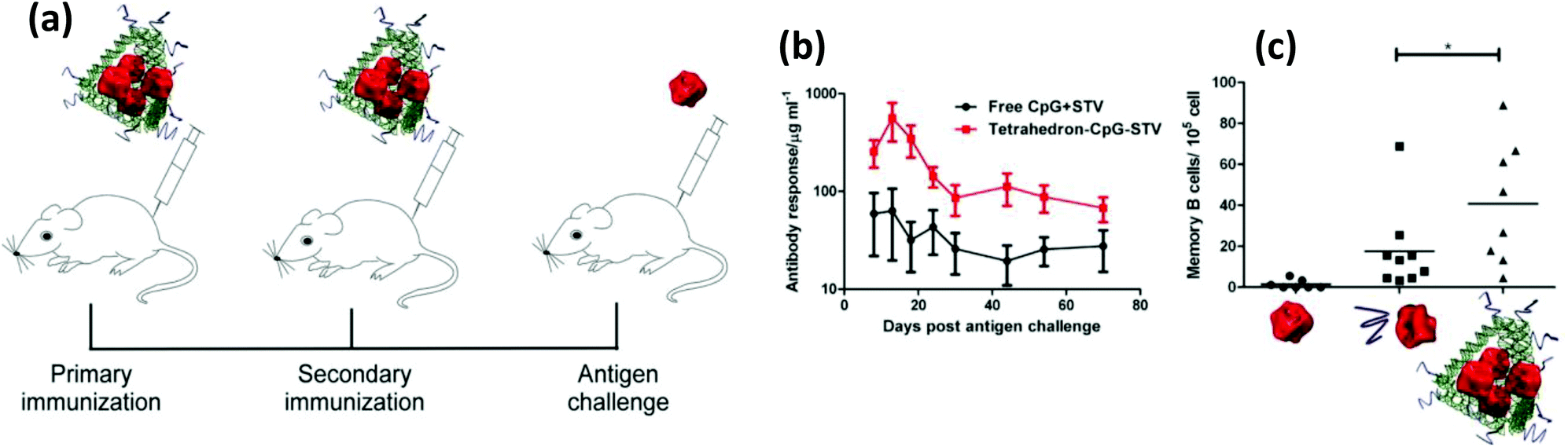 | ||
| Fig. 42 Self-assembled DNA nanostructure being used as a scaffold for constructing a synthetic vaccine complex. (a) The red color streptavidin proteins are captured inside the green tetrahedron shaped DNA nanostructure decorated with CpG units (purple color) at the vertices. (b) Immunization of the BALB/c mice (scheme a) with the DNA scaffolded vaccine complex has stronger antibody response (higher Anti-STV IgG level) compared to free components. (c) Specific memory B cell response was also higher when the DNA-assembled vaccine was used. Reprinted with permission from ref. 562. Copyright 2012 American Chemical Society. | ||
The ability of designer DNA nanostructures to also be a good nucleic acid delivery agent was demonstrated by Lee and coworkers.547 Unlike most NPs, which are generally somewhat heterogeneous in size (except viral capsids), the high degree of homogeneity in size and shape is another unique feature of DNA nanostructures and this can also enhance permeability and retention time and thus overall delivery performance. In addition to a uniform population, its programmable nature offers a capability to append cargo or targeting ligands with unmatched control over stoichiometry and spatial orientation. Lee produced a DNA tetrahedron structure by the self-assembly of six ssDNAs to deliver siRNA meant to silence target genes in tumor cells. Each edge of the 10 nm length DNA tetrahedron had an ss overhang which was exploited to bind to short oligomers-conjugated with anti-luciferase siRNA. In order to achieve the best targeted delivery of the siRNA, they screened various cancer cell-targeting ligands, including FA and nearly 30 different peptides, by conjugating them to the tetrahedron with different stoichiometries or relative orientations. It was observed that the FA-conjugated tetrahedron was best in showing the maximum inhibitory effect of luciferase expression in model HeLa cells. Interestingly, the orientation and location of the targeting ligand, especially when they are closely spaced, had a profound effect on the gene silencing efficacy although the cellular uptake efficiencies were the same. It was also noted that a minimum of three FA tags were necessary for proper targeting and, when they were arranged on one face of the tetrahedron, the gene-silencing efficacy was increased. This suggests that the local density of the FA moieties and how they interact with the FA receptor on cell surfaces might, in turn, have an impact on intracellular trafficking pathways and therefore on the gene silencing.547
Besides just DNA tetrahedra, numerous other wireframe DNA polyhedra nanostructures, even some with reconfigurable sizes, have been reported by several groups.211,540,563–568 These biocompatible and fully addressable nanostructures, sometimes even with accessible pores, can be extremely useful for encapsulating and delivering drugs, proteins or other therapeutic materials to target sites or to build novel nanomaterials. Historically, the Seeman group first reported a cube and truncated octahedron like structures synthesized from DNA via a series of ligation and purification steps.540,563 The Sleiman group later came up with a less cumbersome approach for producing sophisticated DNA polyhedra by first creating polygons, with ssDNA as arms and particular organic molecule as vertices, and later connecting them as top and bottom faces via specific linking strands.566 The ss edges of these cage-like structures were available for integrating biomolecules of interest. Edwardson attached dendrimeric alkyl chains by first chemically conjugating them to a DNA and then hybridizing the hybrid conjugate to the ss edges of a cube like nanostructure.569 Depending on the number and spatial orientation, the engagement behavior of the hydrophobic side chains, whether inter- or intra-molecularly, could be controlled. For example, four amphiphiles on the same face of the cube associated inter-molecularly leading to formation of a cage-dimer while eight of them, one at each vertex, interacted intra-molecularly forming a micellar environment within the cage. Those scaffolded micelles could be exploited for loading small hydrophobic molecules, such as Nile Red in this case, as cargo and releasing them in response to external DNA stimuli.569 To further demonstrate the potential usability of the wireframe cages, they recently employed a 3D DNA prism, synthesized in the same fashion, to integrate discrete number of antisense oligonucleotide to induce gene silencing.570 The prismatic nanostructures with 4 and 6 antisense units displayed significantly higher stability against nuclease degradation in serum and superior firefly luciferase gene knockdown efficiency compared to ss-antisense oligonucleotides alone. The Sleiman group also showed that the polygons can further be connected longitudinally to produce nanotubes with tunable geometry, size and stiffness, which they envisioned can be used for selectively loading and releasing NP cargos.571 Lo and colleagues actually encapsulated AuNPs in such a nanotube with alternating large and small capsules that resulted in a ‘pea-pod’ like structure.572 The tubes could be opened in the presence of external DNA via strand displacement leading to release of the captured AuNPs. Hamblin et al. reported that somewhat analogous DNA nanotubes, prepared in a slightly different way, could be easily transported inside HeLa cells and display significantly higher resistance against enzymatic degradation than the component oligonucleotides alone.573
12. Conclusions and perspective
Looking cumulatively over what has been covered above, although not comprehensive by any means, still provides an appreciation of the breadth of application space where both NPs and DNA have worked together synergistically. It is also readily apparent that this material partnership is steadily growing both more sophisticated and capable with time. By this we mean that newer applications seek to exploit more of the unique capabilities that each material can provide while also utilizing them to accomplish de novo tasks and to create new types of functional materials in concert with other building blocks. For example, using the recent example described by Dwyer et al.,352 in Fig. 23, we note the use of the QD as both: a (i) central nanoplatform or host for displaying multiple different types of biologicals – luciferase and DNA – with control over ratio or valency; and (ii) an energy harvesting acceptor for the luciferase-generated sensitization along with a relay for ET to the photonic wires. Here the DNA provides a chemical linkage to the QDs in the form of an appended peptide while also providing programmable and site-specific placement of the acceptor dye cascade. This combined functionality, where the assembly is capable of generating and then directing excitonic energy in a somewhat controlled manner, is clearly far more sophisticated in comparison to the first reports of DNA-modified QDs being utilized as hybridization probes. Other examples of a similar vein include the ability to generate DNA functionalized-NPs that are then incorporated into single and multicomponent NP super lattices or use of DNA nanostructures for epitope and even adjuvant display in vaccine development.236 The intersection of multiple active materials together such as DNA and catalysts or enzymes also has very strong potential.574–576 We are also seeing the intersection of theory and experiment giving rise to fascinating new materials such as DNA-based switchable chiral plasmonic structures and catenated DNA machines that can control spectroscopic properties such as plasmon coupling.207,224,226,228,577 Composite materials that bring together multiple NP materials to work in concert can also be expected in the near term.A critical point about progress to be appreciated is the continuity and connectivity of research in this field. For example, considering the work in Fig. 23 described above,352 this did not occur de novo, rather, many of the initial concepts that gave rise to this construct originated with the work of the Maye group who, in turn, were inspired by the seminal work of Rao.578–580 The above examples also, in essence, provide a roadmap as to what we can expect in the future from this working materials combination. Clearly, there will be continuous and significant improvements to all their current roles in diagnostics and the like. More importantly though, their applications will grow in terms of both sophistication and complexity while also demonstrating ever new capabilities. Moreover, these will be in concert with other types of NMs and biologicals along with application to in vivo environments such as cells, tissues and model organisms.551 The latter will also bring with it a host of other important issues that need to be addressed that sometimes arise from the constituent materials in the NPs. For example, use of semiconductor QDs in live organisms is always tempered by concerns of toxicity. Fortunately, concerted research continues to address this issue.581–584 In a similar vein, we can expect that many other related issues, such as improving NP–DNA bioconjugation and better targeted cellular and tissue delivery may also be addressed by continuing research avenues.585–587 We can also start to expect what some would consider former science fiction predictions to become reality in the form of nanomachines and nanodevices capable of quasi-autonomous exploits. Along with the NPs themselves, here too the DNA will also provide multiple roles in terms of recognizing other DNA or materials, site-specifically displaying fluorophores or other active molecules even to include far larger enzymes,574–576 joining NPs and other materials into complex 3-D structures and perhaps even encoding and storing important information – the utility DNA was originally selected for by evolution.
Abbreviations
| ADP | Adenosine diphosphate |
| AFM | Atomic force microscopy |
| AgNC | Silver nanocluster |
| AgNP | Silver nanoparticle |
| AMP | Adenosine monophosphate |
| ATP | Adenosine triphosphate |
| AuNP | Gold nanoparticle |
| AuNR | Gold nanorod |
| BCC | Body centered cubic |
| BSA | Bovine serum albumin |
| Caco | Colon adenocarcinoma |
| CaPNP | Calcium phosphate nanoparticle |
| CCMV | Cowpea chlorotic mottle virus |
| CD | Circular dichroism |
| CNT | Carbon nanotube |
| CpG | Cytosine-phosphate-guanine |
| CPP | Cell penetrating peptide |
| CV | Cyclic voltammetry |
| C/V | Surface to volume ratio |
| Ce6 | Chlorin e6 |
| DFM | Dark field microscopy |
| DMAB | Dimethylamine borane |
| DNA | Deoxyribonucleic acid |
| Dox | Doxorubicin |
| ds | Double stranded |
| ET | Energy transfer |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide |
| eGFP | Enhanced green fluorescent protein |
| EMP | Electromagnetic pulse |
| FA | Folic acid |
| FCC | Face centered cubic |
| FISH | Fluorescence in situ hybridization |
| FRET | Förster resonance energy transfer |
| FET | Field effect transistor |
| GFP | Green fluorescent protein |
| GTP | Guanosine triphosphate |
| GO | Graphene oxide |
| GOx | Glucose oxidase |
| HEK | Human embryonic kidney cells |
| hFTN-H | Human ferritin heavy chain |
| HRP | Horseradish peroxidase |
| ICSDA | Isothermal circular strand displacement amplification |
| IgE | Immunoglobin E |
| ITO | Indium tin oxide |
| LED | Light emitting diode |
| LNA | Locked nucleic acid |
| LOD | Limit of detection |
| LRET | Luminescence resonance energy transfer |
| MB | Molecular beacon |
| MNP | Magnetic nanoparticles |
| MRI | Magnetic resonance imaging |
| MWCNT | Multiwall carbon nanotube |
| NC | Nanocluster |
| NHS | N-Hydroxysuccinimide |
| NIR | Near infrared |
| NM | Nanomaterial |
| NP | Nanoparticle |
| PAINT | Point accumulation for imaging in nanoscale topography |
| paF | p-Aminophenylalanine |
| PCR | Polymerase chain reaction |
| PDGF | Platelet derived growth factor |
| PDGF-BB | Platelet derived growth factor B-chain homodimer |
| PdNP | Palladium nanoparticles |
| PEG | Polyethylene glycol |
| PEI | Polyethyleneimine |
| PL | Photoluminescence |
| PNA | Peptide nucleic acid |
| PPO | Propylene oxide |
| PTX | Paclitaxel |
| ps | Phosphorothioate |
| psDNA | Phosphorothioate DNA |
| QD | Quantum dot |
| QY | Quantum yield |
| RET | Resonance energy transfer |
| rGO | Reduced graphene oxide |
| ROS | Reactive oxygen species |
| RT | PCR-real time polymerase chain reaction |
| siRNA | Small interfering RNA |
| SNA | Spherical nucleic acid |
| SOG | Singlet oxygen generation |
| SPR | Surface plasmon resonance |
| SMCC | 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide |
| SWCNT | Single wall carbon nanotube |
| ss | Single stranded |
| TAMRA | Carboxytetramethylrhodamine |
| TEM | Transmission electron microscopy |
| TIRF | Total internal reflection fluorescence |
| TMB | 3,3′,5,5′-Tetramethylbenzidine |
| TNF | Tumor necrosis factor |
| UCNP | Upconversion nanoparticle |
| UME | Ultramicroelectrode |
| UV-Vis | Ultraviolet-visible |
| ζ | Zeta (for zeta potential) |
Acknowledgements
The authors acknowledge the Naval Research Laboratory Nanosciences Institute and DTRA JSTO MIPR # B112582M.References
- M. I. Stockman, Phys. Today, 2011, 64, 39–44 CrossRef.
- M. Nirmal and L. Brus, Acc. Chem. Res., 1999, 32, 407–414 CrossRef CAS.
- M. Pumera, Chem. Soc. Rev., 2010, 39, 4146–4157 RSC.
- T. W. Odom, J. L. Huang, P. Kim and C. M. Lieber, Nature, 1998, 391, 62–64 CrossRef CAS.
- A. J. Nozik, M. C. Beard, J. M. Luther, M. Law, R. J. Ellingson and J. C. Johnson, Chem. Rev., 2010, 110, 6873–6890 CrossRef CAS PubMed.
- S. Singamaneni, V. N. Bliznyuk, C. Binek and E. Y. Tsymbal, J. Mater. Chem., 2011, 21, 16819–16845 RSC.
- A. N. Shipway, E. Katz and I. Willner, ChemPhysChem, 2000, 1, 18–52 CrossRef CAS PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- N. Savage and M. S. Diallo, J. Nanopart. Res., 2005, 7, 331–342 CrossRef CAS.
- J. M. Hu, G. Q. Zhang and S. Y. Liu, Chem. Soc. Rev., 2012, 41, 5933–5949 RSC.
- H. S. Peng and D. T. Chiu, Chem. Soc. Rev., 2015, 44, 4699–4722 RSC.
- F. Zhang, J. Nangreave, Y. Liu and H. Yan, J. Am. Chem. Soc., 2014, 136, 11198–11211 CrossRef CAS PubMed.
- I. Yildiz, S. Shukla and N. F. Steinmetz, Curr. Opin. Biotechnol., 2011, 22, 901–908 CrossRef CAS PubMed.
- S. D. Patil, D. G. Rhodes and D. J. Burgess, APPS J., 2005, 7, E61–E77 CAS.
- O. P. Kallioniemi, A. Kallioniemi, J. Piper, J. Isola, F. M. Waldman, J. W. Gray and D. Pinkel, Genes, Chromosomes Cancer, 1994, 10, 231–243 CrossRef CAS PubMed.
- L. Schena, F. Nigro, A. Ippolito and D. Gallitelli, Eur. J. Plant Pathol., 2004, 110, 893–908 CrossRef CAS.
- M. J. Heller, Annu. Rev. Biomed. Eng., 2002, 4, 129–153 CrossRef CAS PubMed.
- D. R. Call, M. K. Borucki and F. J. Loge, J. Microbiol. Methods, 2003, 53, 235–243 CrossRef CAS PubMed.
- S. Yang and R. E. Rothman, Lancet Infect. Dis., 2004, 4, 337–348 CrossRef CAS PubMed.
- R. T. Ranasinghe and T. Brown, Chem. Commun., 2005, 44, 5487–5502 RSC.
- C. Debouck and P. N. Goodfellow, Nat. Genet., 1999, 21, 48–50 CrossRef CAS PubMed.
- J. W. Liu, Z. H. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed.
- Y. Krishnan and F. C. Simmel, Angew. Chem., Int. Ed., 2011, 50, 3124–3156 CrossRef CAS PubMed.
- D. Bhatia, S. Sharma and Y. Krishnan, Curr. Opin. Biotechnol., 2011, 22, 475–484 CrossRef CAS PubMed.
- L. Berti, J. Xie, I. L. Medintz, A. N. Glazer and R. A. Mathies, Anal. Biochem., 2001, 292, 188–197 CrossRef CAS PubMed.
- I. Medintz, W. W. Wong, L. Berti, L. Shiow, J. Tom, J. Scherer, G. Sensabaugh and R. A. Mathies, Genome Res., 2001, 11, 413–421 CrossRef CAS PubMed.
- I. L. Medintz, B. M. Paegel, R. G. Blazej, C. A. Emrich, L. Berti, J. R. Scherer and R. A. Mathies, Electrophoresis, 2001, 22, 3845–3856 CrossRef CAS PubMed.
- W. Mok and Y. F. Li, Sensors, 2008, 8, 7050–7084 CrossRef CAS.
- W. Z. Zhou, P. J. J. Huang, J. S. Ding and J. Liu, Analyst, 2014, 139, 2627–2640 RSC.
- D. Li, S. P. Song and C. H. Fan, Acc. Chem. Res., 2010, 43, 631–641 CrossRef CAS PubMed.
- A. A. Lubin and K. W. Plaxco, Acc. Chem. Res., 2010, 43, 496–505 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Biomol. Eng., 2007, 24, 381–403 CrossRef CAS PubMed.
- X. H. Fang and W. H. Tan, Acc. Chem. Res., 2010, 43, 48–57 CrossRef CAS PubMed.
- S. C. B. Gopinath, Anal. Bioanal. Chem., 2007, 387, 171–182 CrossRef CAS PubMed.
- M. Ye, J. Hu, M. Y. Peng, J. Liu, J. Liu, H. X. Liu, X. L. Zhao and W. H. Tan, Int. J. Mol. Sci., 2012, 13, 3341–3353 CrossRef CAS PubMed.
- A. V. Pinheiro, D. R. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS PubMed.
- C. Lin, Y. Liu and H. Yan, Biochemistry, 2009, 48, 1663–1674 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- P. K. Lo, K. L. Metera and H. F. Sleiman, Curr. Opin. Chem. Biol., 2010, 14, 597–607 CrossRef CAS PubMed.
- J. Xie, S. Lee and X. Y. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS PubMed.
- M. A. Hahn, A. K. Singh, P. Sharma, S. C. Brown and B. M. Moudgil, Anal. Bioanal. Chem., 2011, 399, 3–27 CrossRef CAS PubMed.
- P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 53–63 CrossRef CAS PubMed.
- A. Dowd, D. Pissuwan and M. B. Cortie, Trends Biotechnol., 2010, 32, 571–577 CrossRef PubMed.
- Y. Cui and J. Irudayaraj, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 387–407 CrossRef CAS PubMed.
- J. B. Delehanty, J. C. Breger, K. B. Gemmill, M. H. Stewart and I. L. Medintz, Ther. Delivery, 2013, 4, 1411–1429 CrossRef CAS PubMed.
- M. Nazarenus, Q. Zhang, M. G. Soliman, P. del Pino, B. Pelaz, S. Carregal-Romero, J. Rejman, B. Rothen-Rutishauser, M. J. D. Clift, R. Zellner, G. U. Nienhaus, J. B. Delehanty, I. L. Medintz and W. J. Parak, Beilstein J. Nanotechnol., 2014, 5, 1477–1490 CrossRef PubMed.
- L. Yang, X. B. Zhang, M. Ye, J. H. Jiang, R. H. Yang, T. Fu, Y. Chen, K. M. Wang, C. Liu and W. H. Tan, Adv. Drug Delivery Rev., 2011, 63, 1361–1370 CrossRef CAS PubMed.
- T. Chen, M. I. Shukoor, Y. Chen, Q. A. Yuan, Z. Zhu, Z. L. Zhao, B. Gulbakan and W. H. Tan, Nanoscale, 2011, 3, 546–556 RSC.
- T. C. Chiu and C. C. Huang, Sensors, 2009, 9, 10356–10388 CrossRef CAS PubMed.
- H. Xing, N. Y. Wong, Y. Xiang and Y. Lu, Curr. Opin. Chem. Biol., 2012, 16, 429–435 CrossRef CAS PubMed.
- S. Buckhout-White, C. M. Spillmann, W. R. Algar, A. Khachatrian, J. S. Melinger, E. R. Goldman, M. G. Ancona and I. L. Medintz, Nat. Commun., 2014, 5, 5615, DOI:10.1038/ncomms6615.
- D. S. Seferos, A. E. Prigodich, D. A. Giljohann, P. C. Patel and C. A. Mirkin, Nano Lett., 2009, 9, 308–311 CrossRef CAS PubMed.
- G. Han, C. T. Martin and V. M. Rotello, Chem. Biol. Drug Des., 2006, 67, 78–82 CAS.
- C. H. Lu, C. L. Zhu, J. Li, J. J. Liu, X. Chen and H. H. Yang, Chem. Commun., 2010, 46, 3116–3118 RSC.
- Y. R. Wu, J. A. Phillips, H. P. Liu, R. H. Yang and W. H. Tan, ACS Nano, 2008, 2, 2023–2028 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- S. Avvakumova, M. Colombo, P. Tortora and D. Prosperi, Trends Biotechnol., 2014, 32, 11–20 CrossRef CAS PubMed.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS PubMed.
- I. Medintz, Nat. Mater., 2006, 5, 842–842 CrossRef CAS PubMed.
- B. Lewin, Genes VII, Oxford University Press, Oxford, U.K., 2000 Search PubMed.
- P. Yakovchuk, E. Protozanova and M. D. Frank-Kamenetskii, Nucleic Acids Res., 2006, 34, 564–574 CrossRef CAS PubMed.
- R. L. Orstein, R. Rein, D. L. Breen and R. D. Macelroy, Biopolymers, 1978, 10, 2341–2360 CrossRef.
- K. Hamaguchi and E. P. Geiduschek, J. Am. Chem. Soc., 1962, 84, 1329–1338 CrossRef CAS.
- E. M. Rezler, J. Seenisamy, S. Bashyam, M.-Y. Kim, E. White, W. D. Wilson and L. H. Hurley, J. Am. Chem. Soc., 2005, 127, 9439–9447 CrossRef CAS PubMed.
- J. D. Watson and F. H. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- A. A. Henry and F. E. Romesberg, Curr. Opin. Chem. Biol., 2003, 7, 727–733 CrossRef CAS PubMed.
- I. Hirao, Curr. Opin. Chem. Biol., 2006, 10, 622–627 CrossRef CAS PubMed.
- E. T. Kool, Acc. Chem. Res., 2002, 35, 936–943 CrossRef CAS PubMed.
- A. D. Mesmaeker, K.-H. Altmann, A. Waldner and S. Wendeborn, Curr. Opin. Struct. Biol., 1995, 5, 343–355 CrossRef PubMed.
- C. Briones and M. Moreno, Anal. Bioanal. Chem., 2012, 402, 3071–3089 CrossRef CAS PubMed.
- M. Bolli, R. Micura and A. Eschenmoser, Chem. Biol., 1997, 4, 309–320 CrossRef CAS PubMed.
- K.-U. Schöning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Science, 2000, 290, 1347–1351 CrossRef.
- D.-R. Ahn, A. Egger, S. Pitsch and C. Lehmann, Chem. – Eur. J., 2002, 8, 5312–5322 CrossRef CAS.
- R. Steffens and C. Leumann, J. Am. Chem. Soc., 1997, 119, 11548–11549 CrossRef CAS.
- B. Vester and J. Wengel, Biochemistry, 2004, 43, 13233–13241 CrossRef CAS PubMed.
- H. Kaur, B. R. Babu and S. Maiti, Chem. Rev., 2007, 107, 4672–4697 CrossRef CAS PubMed.
- J. Kurreck, E. Wyszko, C. Gillen and V. A. Erdmann, Nucleic Acids Res., 2002, 30, 1911–1918 CrossRef CAS PubMed.
- H. Asanuma, T. Toda, K. Murayama, X. Liang and H. Kashida, J. Am. Chem. Soc., 2010, 132, 14702–14703 CrossRef CAS PubMed.
- P. E. Nielsen and G. Haaima, Chem. Soc. Rev., 1997, 26, 73–78 RSC.
- B. Hyrup and P. E. Nielson, Bioorg. Med. Chem., 1996, 4, 5–23 CrossRef CAS PubMed.
- V. V. Demidov, V. N. Potaman, M. D. Frank-Kamenetskii, M. Egholm, O. Buchard, S. H. Sonnichsen and P. E. Nielsen, Biochem. Pharmacol., 1994, 48, 1310–1313 CrossRef CAS PubMed.
- M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 1260901, DOI:10.1126/science.1260901.
- Y. Singh, P. Murat and E. Defrancq, Chem. Soc. Rev., 2010, 39, 2054–2070 RSC.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- E. C. Dreaden, A. M. Alkilany, X. H. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- X. H. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayad, Plasmonics, 2007, 2, 107–118 CrossRef CAS.
- H. W. Liao, C. L. Nehl and J. H. Hafner, Nanomedicine, 2006, 1, 201–208 CrossRef CAS PubMed.
- B. Sepulveda, P. C. Angelome, L. M. Lechuga and L. M. Liz-Marzan, Nano Today, 2009, 4, 244–251 CrossRef CAS.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410–8426 CrossRef CAS.
- M. D. Porter, R. J. Lipert, L. M. Siperko, G. Wang and R. Narayanana, Chem. Soc. Rev., 2008, 37, 1001–1011 RSC.
- L. Guerrini and D. Graham, Chem. Soc. Rev., 2012, 41, 7085–7107 RSC.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171–194 CrossRef CAS PubMed.
- E. Dulkeith, A. C. Morteani, T. Niedereichholz, T. A. Klar, J. Feldmann, S. A. Levi, F. van Veggel, D. N. Reinhoudt, M. Moller and D. I. Gittins, Phys. Rev. Lett., 2002, 89, 203002 CrossRef CAS PubMed.
- E. Dulkeith, M. Ringler, T. A. Klar, J. Feldmann, A. M. Javier and W. J. Parak, Nano Lett., 2005, 5, 585–589 CrossRef CAS PubMed.
- J. I. Gersten and A. Nitzan, J. Chem. Phys., 1981, 75, 1139–1152 CrossRef CAS.
- R. R. Chance, A. Prock and R. Silbey, J. Chem. Phys., 1975, 62, 2245–2253 CrossRef CAS.
- H. Kuhn, J. Chem. Phys., 1970, 53, 101–108 CrossRef CAS.
- C. J. Breshike, R. A. Riskowski and G. F. Strouse, J. Phys. Chem. C, 2013, 117, 23942–23949 CAS.
- R. E. Armstrong, R. A. Riskowski and G. F. Strouse, Photochem. Photobiol., 2015, 91, 732–738 CrossRef CAS PubMed.
- R. A. Riskowski, R. E. Armstrong, N. L. Greenbaum and G. F. Strouse, ACS Nano, 2016, 10, 1926–1938 CrossRef CAS PubMed.
- A. P. Alivisatos, K. P. Johnsson, X. G. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez and P. G. Schultz, Nature, 1996, 382, 609–611 CrossRef CAS PubMed.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- C. D. Bain, J. Evall and G. M. Whitesides, J. Am. Chem. Soc., 1989, 111, 7155–7164 CrossRef CAS.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086 CrossRef CAS.
- H. Gronbeck, A. Curioni and W. Andreoni, J. Am. Chem. Soc., 2000, 122, 3839–3842 CrossRef.
- C. S. Weisbecker, M. V. Merritt and G. M. Whitesides, Langmuir, 1996, 12, 3763–3772 CrossRef CAS.
- S. J. Hurst, A. K. R. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2006, 78, 8313–8318 CrossRef CAS PubMed.
- C. H. J. Choi, L. Hao, S. P. Narayan, E. Auyeung and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7625–7630 CrossRef CAS PubMed.
- J. Deka, R. Mech, L. Ianeselli, H. Amenitsch, F. Cacho-Nerin, P. Parisse and L. Casalis, ACS Appl. Mater. Interfaces, 2015, 7, 7033–7040 CAS.
- W. T. Zhao, L. Lin and I. M. Hsing, Bioconjugate Chem., 2009, 20, 1218–1222 CrossRef CAS PubMed.
- S. M. Xu, H. Yuan, A. Xu, J. Wang and L. J. Wu, Langmuir, 2011, 27, 13629–13634 CrossRef CAS PubMed.
- F. Seela, P. Ding and S. Budow, Bioconjugate Chem., 2011, 22, 794–807 CrossRef CAS PubMed.
- C. S. Thaxton, D. G. Georganopoulou and C. A. Mirkin, Clin. Chim. Acta, 2006, 363, 120–126 CrossRef CAS PubMed.
- H. I. Peng and B. L. Miller, Analyst, 2011, 136, 436–447 RSC.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–1081 CrossRef CAS PubMed.
- J. J. Storhoff, R. Elghanian, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 1959–1964 CrossRef CAS.
- J. Liu and Y. Lu, Chem. Commun., 2007, 46, 4872–4874 RSC.
- X. Bai, C. Y. Shao, X. G. Han, Y. L. Li, Y. F. Guan and Z. X. Deng, Biosens. Bioelectron., 2010, 25, 1984–1988 CrossRef CAS PubMed.
- H. M. Huttanus, E. Graugnard, B. Yurke, W. B. Knowlton, W. Kuang, W. L. Hughes and J. Lee, Biosens. Bioelectron., 2013, 50, 382–386 CrossRef CAS PubMed.
- H. X. Li and L. Rothberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14036–14039 CrossRef CAS PubMed.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CrossRef CAS PubMed.
- A. E. Prigodich, P. S. Randeria, W. E. Briley, N. J. Kim, W. L. Daniel, D. A. Giljohann and C. A. Mirkin, Anal. Chem., 2010, 84, 2062–2066 CrossRef PubMed.
- C. D. Medley, J. E. Smith, Z. Tang, Y. Wu, S. Bamrungsap and W. H. Tan, Anal. Chem., 2008, 80, 1067–1072 CrossRef CAS PubMed.
- V. Pavlov, Y. Xiao, B. Shlyahovsky and I. Willner, J. Am. Chem. Soc., 2004, 126, 11768–11769 CrossRef CAS PubMed.
- J. H. Lee, M. V. Yigit, D. Mazumdar and Y. Lu, Adv. Drug Delivery Rev., 2010, 62, 592–605 CrossRef CAS PubMed.
- L. Yang, X. Zhang, M. Ye, J. Jiang, R. Yang, T. Fu, Y. Chen, K. Wang, C. Liu and W. Tan, Adv. Drug Delivery Rev., 2011, 63, 1361–1370 CrossRef CAS PubMed.
- H. Xing, W. Ngo Yin, Y. Xiang and Y. Lu, Curr. Opin. Chem. Biol., 2012, 16, 429–435 CrossRef CAS PubMed.
- X. Y. Wang, Y. Xu, Y. Chen, L. M. Li, F. Liu and N. Li, Anal. Bioanal. Chem., 2011, 400, 2085–2091 CrossRef CAS PubMed.
- J. W. Liu and Y. Lu, Angew. Chem., Int. Ed., 2006, 45, 90–94 CrossRef CAS PubMed.
- K. Li, W. W. Qin, F. Li, X. C. Zhao, B. W. Jiang, K. Wang, S. H. Deng, C. H. Fan and D. Li, Angew. Chem., Int. Ed., 2013, 52, 11542–11545 CrossRef CAS PubMed.
- M. S. Han, A. K. R. Lytton-Jean, B. K. Oh, J. Heo and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 1807–1810 CrossRef CAS PubMed.
- C. W. Liu, Y. T. Hsieh, C. C. Huang, Z. H. Lin and H. T. Chang, Chem. Commun., 2008, 2242–2244 RSC.
- D. Li, A. Wieckowska and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 3927–3931 CrossRef CAS PubMed.
- H. Wang, Y. X. Wang, J. Y. Jin and R. H. Yang, Anal. Chem., 2008, 80, 9021–9028 CrossRef CAS PubMed.
- J. W. Liu and Y. Lu, J. Am. Chem. Soc., 2003, 125, 6642–6643 CrossRef CAS PubMed.
- C. J. Breshike, R. A. Riskowski and G. F. Strouse, J. Phys. Chem. C, 2013, 117, 23942–23949 CAS.
- T. Pons, I. L. Medintz, K. E. Sapsford, S. Higashiya, A. F. Grimes, D. S. English and H. Mattoussi, Nano Lett., 2007, 7, 3157–3164 CrossRef CAS PubMed.
- C. C. Huang, S. H. Chiu, Y. F. Huang and H. T. Chang, Anal. Chem., 2007, 79, 4798–4804 CrossRef CAS PubMed.
- E. H. Jeong, G. Jung, C. A. Hong and H. Lee, Arch. Pharm. Res., 2014, 37, 53–59 CrossRef CAS PubMed.
- R. Cao-Milan and L. M. Liz-Marzan, Expert Opin. Drug Delivery, 2014, 11, 741–752 CrossRef CAS PubMed.
- L. A. Dykman and N. G. Khlebtsov, Acta Nat., 2011, 3, 34–55 CAS.
- Y. Ding, Z. W. Jiang, K. Saha, C. S. Kim, S. T. Kim, R. F. Landis and V. M. Rotello, Mol. Ther., 2014, 22, 1075–1083 CrossRef CAS PubMed.
- L. Vigderman and E. R. Zubarev, Adv. Drug Delivery Rev., 2011, 65, 663–676 CrossRef PubMed.
- A. J. Mieszawska, W. J. M. Mulder, Z. A. Fayad and D. P. Cormode, Mol. Pharm., 2013, 10, 831–847 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, P. C. Patel, J. E. Millstone, N. L. Rosi and C. A. Mirkin, Nano Lett., 2007, 7, 3818–3821 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- J. I. Cutler, K. Zhang, D. Zhang, E. Auyeung, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2011, 133, 9254–9257 CrossRef CAS PubMed.
- K. K. Sandhu, C. M. McIntosh, J. M. Simard, S. W. Smith and V. M. Rotello, Bioconjugate Chem., 2002, 13, 3–6 CrossRef CAS PubMed.
- M. Thomas and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9138–9143 CrossRef CAS PubMed.
- G. Han, C. C. You, B. J. Kim, R. S. Turingan, N. S. Forbes, C. T. Martin and V. M. Rotello, Angew. Chem., Int. Ed., 2006, 45, 3165–3169 CrossRef CAS PubMed.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS PubMed.
- W. Yang, Q. Rev. Biophys., 2011, 44, 1–93 CrossRef CAS PubMed.
- M. E. Gleave and B. P. Monia, Nat. Rev. Cancer, 2005, 5, 468–479 CrossRef CAS PubMed.
- T. Shimayama, F. Nishikawa, S. Nishikawa and K. Taira, Nucleic Acids Res., 1993, 21, 2605–2611 CrossRef CAS PubMed.
- B. P. Monia, J. F. Johnston, H. Sasmor and L. L. Cummins, J. Biol. Chem., 1996, 271, 14533–14540 CrossRef CAS PubMed.
- K. Zhang, L. L. Hao, S. J. Hurst and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 16488–16491 CrossRef CAS PubMed.
- S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652–14653 CrossRef CAS PubMed.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS PubMed.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1–7 CrossRef CAS.
- W. J. Luo, C. F. Zhu, S. Su, D. Li, Y. He, Q. Huang and C. H. Fan, ACS Nano, 2010, 4, 7451–7458 CrossRef CAS PubMed.
- X. X. Zheng, Q. Liu, C. Jing, Y. Li, D. Li, W. J. Luo, Y. Q. Wen, Y. He, Q. Huang, Y. T. Long and C. H. Fan, Angew. Chem., Int. Ed., 2011, 50, 11994–11998 CrossRef CAS PubMed.
- B. J. Johnson, W. R. Algar, A. P. Malanoski, M. G. Ancona and I. L. Medintz, Nano Today, 2014, 9, 102–131 CrossRef CAS.
- S. J. Guo and E. K. Wang, Anal. Chim. Acta, 2007, 598, 181–192 CrossRef CAS PubMed.
- J. M. Pingarron, P. Yanez-Sedeno and A. Gonzalez-Cortes, Electrochim. Acta, 2008, 53, 5848–5866 CrossRef CAS.
- S. J. Park, T. A. Taton and C. A. Mirkin, Science, 2002, 295, 1503–1506 CrossRef CAS PubMed.
- D. Li, Y. Yan, A. Wieckowska and I. Willner, Chem. Commun., 2007, 34, 3544–3546 RSC.
- C. Z. Li, Y. L. Liu and J. H. T. Luong, Anal. Chem., 2005, 77, 478–485 CrossRef CAS PubMed.
- C. J. Murphy, T. K. San, A. M. Gole, C. J. Orendorff, J. X. Gao, L. Gou, S. E. Hunyadi and T. Li, J. Phys. Chem. B, 2005, 109, 13857–13870 CrossRef CAS PubMed.
- J. Perez-Juste, I. Pastoriza-Santos, L. M. Liz-Marzan and P. Mulvaney, Coord. Chem. Rev., 2005, 249, 1870–1901 CrossRef CAS.
- S. S. Shankar, A. Rai, B. Ankamwar, A. Singh, A. Ahmad and M. Sastry, Nat. Mater., 2004, 3, 482–488 CrossRef CAS PubMed.
- C. J. Johnson, E. Dujardin, S. A. Davis, C. J. Murphy and S. Mann, J. Mater. Chem., 2002, 12, 1765–1770 RSC.
- L. Tong, Q. S. Wei, A. Wei and J. X. Cheng, Photochem. Photobiol., 2009, 85, 21–32 CrossRef CAS PubMed.
- L. Vigderman, B. P. Khanal and E. R. Zubarev, Adv. Mater., 2012, 24, 4811–4841 CrossRef CAS PubMed.
- H. J. Chen, L. Shao, Q. Li and J. F. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- E. B. Dickerson, E. C. Dreaden, X. H. Huang, I. H. El-Sayed, H. H. Chu, S. Pushpanketh, J. F. McDonald and M. A. El-Sayed, Cancer Lett., 2008, 269, 57–66 CrossRef CAS PubMed.
- X. H. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- Y. F. Huang, H. T. Chang and W. H. Tan, Anal. Chem., 2008, 80, 567–572 CrossRef CAS PubMed.
- J. Wang, M. X. You, G. Z. Zhu, M. I. Shukoor, Z. Chen, Z. L. Zhao, M. B. Altman, Q. Yuan, Z. Zhu, Y. Chen, C. Z. Huang and W. H. Tan, Small, 2013, 9, 3678–3684 CrossRef CAS PubMed.
- J. Cao, T. Sun and K. T. V. Grattan, Sens. Actuators, B, 2014, 195, 332–351 CrossRef CAS.
- S. Link, C. Burda, M. B. Mohamed, B. Nikoobakht and M. A. El-Sayed, J. Phys. Chem. A, 1999, 103, 1165–1170 CrossRef CAS.
- C. C. Chen, Y. P. Lin, C. W. Wang, H. C. Tzeng, C. H. Wu, Y. C. Chen, C. P. Chen, L. C. Chen and Y. C. Wu, J. Am. Chem. Soc., 2006, 128, 3709–3715 CrossRef CAS PubMed.
- A. Wijaya, S. B. Schaffer, I. G. Pallares and K. Hamad-Schifferli, ACS Nano, 2009, 3, 80–86 CrossRef CAS PubMed.
- W. Ma, H. Kuang, L. G. Xu, L. Ding, C. L. Xu, L. B. Wang and N. A. Kotov, Nat. Commun., 2013, 4, 2689, DOI:10.1038/ncomms3689.
- L. Y. Chen, C. W. Wang, Z. Q. Yuan and H. T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS PubMed.
- R. C. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- E. Oh, J. B. Delehanty, K. E. Sapsford, K. Susumu, R. Goswami, J. B. Blanco-Canosa, P. E. Dawson, J. Granek, M. Shoff, Q. Zhang, P. L. Goering, A. Huston and I. L. Medintz, ACS Nano, 2011, 5, 6434–6448 CrossRef CAS PubMed.
- E. Oh, F. K. Fatemi, M. Currie, J. B. Delehanty, T. Pons, A. Fragola, S. Leveque-Fort, R. Goswami, K. Susumu, A. L. Huston and I. L. Medintz, Part. Part. Syst. Charact., 2013, 30, 453–466 CrossRef CAS.
- G. Li and R. C. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- H. F. Qian, M. Z. Zhu, Z. K. Wu and R. C. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- T. A. C. Kennedy, J. L. MacLean and J. W. Liu, Chem. Commun., 2012, 48, 6845–6847 RSC.
- G. Y. Liu, Y. Shao, K. Ma, Q. H. Cui, F. Wu and S. J. Xu, Gold Bull., 2012, 45, 69–74 CrossRef CAS.
- G. Y. Liu, Y. Shao, F. Wu, S. J. Xu, J. Peng and L. L. Liu, Nanotechnology, 2013, 24, 015503 CrossRef PubMed.
- W. Y. Chen, G. Y. Lan and H. T. Chang, Anal. Chem., 2011, 83, 9450–9455 CrossRef CAS PubMed.
- V. Venkatesh, A. Shukla, S. Sivakumar and S. Verma, ACS Appl. Mater. Interfaces, 2014, 6, 2185–2191 CAS.
- X. M. Yang, S. S. Zhu, Y. Dou, Y. Zhuo, Y. W. Luo and Y. J. Feng, Talanta, 2014, 122, 36–42 CrossRef CAS PubMed.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef PubMed.
- A. Samanta, S. Banerjee and Y. Liu, Nanoscale, 2015, 7, 2210–2220 RSC.
- X. N. Zhang, R. Wang and G. Xue, Soft Matter, 2015, 11, 1862–1870 RSC.
- Y. Chen and W. L. Cheng, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 587–604 CrossRef CAS PubMed.
- C. Knorowski and A. Travesset, Curr. Opin. Solid State Mater. Sci., 2011, 15, 262–270 CrossRef CAS.
- H. A. Becerril and A. T. Woolley, Chem. Soc. Rev., 2009, 38, 329–337 RSC.
- S. J. Tan, M. J. Campolongo, D. Luo and W. L. Cheng, Nat. Nanotechnol., 2011, 6, 268–276 CrossRef CAS PubMed.
- Z. Chen, X. Lan, Y. C. Chiu, X. X. Lu, W. H. Ni, H. W. Gao and Q. B. Wang, ACS Photonics, 2015, 2, 392–397 CrossRef CAS.
- S. Pal, Z. T. Deng, H. N. Wang, S. L. Zou, Y. Liu and H. Yan, J. Am. Chem. Soc., 2011, 133, 17606–17609 CrossRef CAS PubMed.
- P. Kuhler, E. M. Roller, R. Schreiber, T. Liedl, T. Lohmuller and J. Feldmann, Nano Lett., 2014, 14, 2914–2919 CrossRef PubMed.
- Z. X. Wang and L. N. Ma, Coord. Chem. Rev., 2009, 253, 1607–1618 CrossRef CAS.
- A. Samanta, Y. D. Zhou, S. L. Zou, H. Yan and Y. Liu, Nano Lett., 2014, 14, 5052–5057 CrossRef CAS PubMed.
- G. P. Acuna, F. M. Moller, P. Holzmeister, S. Beater, B. Lalkens and P. Tinnefeld, Science, 2012, 338, 506–510 CrossRef CAS PubMed.
- R. P. Goodman, M. Heilemann, S. Doose, C. M. Erben, A. N. Kapanidis and A. J. Turberfield, Nat. Nanotechnol., 2008, 3, 93–96 CrossRef CAS PubMed.
- Y. He, Y. Tian, A. E. Ribbe and C. Mao, J. Am. Chem. Soc., 2006, 128, 15978–15979 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- F. A. Aldaye and H. F. Sleiman, J. Am. Chem. Soc., 2007, 129, 13376–13377 CrossRef CAS PubMed.
- P. Yin, H. M. T. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CrossRef CAS PubMed.
- K. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. Johnson-Buck, J. Nangreave, S. Taylor, R. Pei, M. N. Stojanovic, N. G. Walter, E. Winfree and H. Yan, Nature, 2010, 465, 206–210 CrossRef CAS PubMed.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS PubMed.
- Y. Ke, L. L. Ong, W. M. Shih and P. Yin, Science, 2012, 338, 1177–1183 CrossRef CAS PubMed.
- F. Zhang, Y. Liu and H. Yan, J. Am. Chem. Soc., 2013, 135, 7458–7461 CrossRef CAS PubMed.
- S. Buckhout-White, J. C. Claussen, J. S. Melinger, Z. Dunningham, M. G. Ancona, E. R. Goldman and I. L. Medintz, RSC Adv., 2014, 4, 48860–48871 RSC.
- C. X. Lin, Y. Liu, S. Rinker and H. Yan, ChemPhysChem, 2006, 7, 1641–1647 CrossRef CAS PubMed.
- B. Q. Ding, Z. T. Deng, H. Yan, S. Cabrini, R. N. Zuckermann and J. Bokor, J. Am. Chem. Soc., 2010, 132, 3248–3249 CrossRef CAS PubMed.
- Z. T. Deng, A. Samanta, J. Nangreave, H. Yan and Y. Liu, J. Am. Chem. Soc., 2012, 134, 17424–17427 CrossRef CAS PubMed.
- V. V. Thacker, L. O. Herrmann, D. O. Sigle, T. Zhang, T. Liedl, J. J. Baumberg and U. F. Keyser, Nat. Commun., 2014, 5, 3448, DOI:10.1038/ncomms4448.
- M. Pilo-Pais, A. Watson, S. Demers, T. H. LaBean and G. Finkelstein, Nano Lett., 2014, 14, 2099–2104 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, Z. Y. Fan, G. Pardatscher, E. M. Roller, A. Hogele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed.
- X. Lan, Z. Chen, G. L. Dai, X. X. Lu, W. H. Ni and Q. B. Wang, J. Am. Chem. Soc., 2013, 135, 11441–11444 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, H. Zhang, A. O. Govorov, T. Liedl and N. Liu, Nat. Mater., 2014, 13, 862–866 CrossRef CAS PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS PubMed.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS PubMed.
- M. M. Maye, M. T. Kumara, D. Nykypanchuk, W. B. Sherman and O. Gang, Nat. Nanotechnol., 2010, 5, 116–120 CrossRef CAS PubMed.
- R. J. Macfarlane, M. R. Jones, A. J. Senesi, K. L. Young, B. Lee, J. S. Wu and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 4589–4592 CrossRef CAS PubMed.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS PubMed.
- R. J. Macfarlane, M. N. O'Brien, S. H. Petrosko and C. A. Mirkin, Angew. Chem., Int. Ed., 2013, 52, 5688–5698 CrossRef CAS PubMed.
- M. R. Jones, R. J. Macfarlane, B. Lee, J. A. Zhang, K. L. Young, A. J. Senesi and C. A. Mirkin, Nat. Mater., 2010, 9, 913–917 CrossRef CAS PubMed.
- Y. G. Zhang, F. Lu, K. G. Yager, D. van der Lelie and O. Gang, Nat. Nanotechnol., 2013, 8, 865–872 CrossRef CAS PubMed.
- K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS PubMed.
- W. E. Doering and S. M. Nie, J. Phys. Chem. B, 2002, 106, 311–317 CrossRef CAS.
- S. L. Zou and G. C. Schatz, Chem. Phys. Lett., 2005, 403, 62–67 CrossRef CAS.
- J. S. Lee, A. K. R. Lytton-Jean, S. J. Hurst and C. A. Mirkin, Nano Lett., 2007, 7, 2112–2115 CrossRef CAS PubMed.
- S. Pal, J. Sharma, H. Yan and Y. Liu, Chem. Commun., 2009, 6059–6061 RSC.
- X. Zhang, M. R. Servos and J. W. Liu, Chem. Commun., 2012, 48, 10114–10116 RSC.
- H. Li, Z. Y. Sun, W. Y. Zhong, N. Hao, D. K. Xu and H. Y. Chen, Anal. Chem., 2010, 82, 5477–5483 CrossRef CAS PubMed.
- Y. Liu and C. Z. Huang, Analyst, 2013, 137, 3434–3436 RSC.
- D. G. Thompson, A. Enright, K. Faulds, W. E. Smith and D. Graham, Anal. Chem., 2008, 80, 2805–2810 CrossRef CAS PubMed.
- X. W. Xu, J. Wang, F. Yang, K. Jiao and X. R. Yang, Small, 2009, 5, 2669–2672 CrossRef CAS PubMed.
- H. G. Park, J. H. Joo, H. G. Kim and J. S. Lee, J. Phys. Chem. C, 2012, 116, 2278–2284 CAS.
- J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207–5212 CrossRef CAS PubMed.
- E. G. Gwinn, P. O'Neill, A. J. Guerrero, D. Bouwmeester and D. K. Fygenson, Adv. Mater., 2008, 20, 279–283 CrossRef CAS.
- J. Sharma, R. C. Rocha, M. L. Phipps, H. C. Yeh, K. A. Balatsky, D. M. Vu, A. P. Shreve, J. H. Werner and J. S. Martinez, Nanoscale, 2012, 4, 4107–4110 RSC.
- K. Morishita, J. L. MacLean, B. W. Liu, H. Jiang and J. W. Liu, Nanoscale, 2013, 5, 2840–2849 RSC.
- G. Y. Lan, W. Y. Chen and H. T. Chang, RSC Adv., 2011, 1, 802–807 RSC.
- G. Y. Lan, W. Y. Chen and H. T. Chang, Analyst, 2011, 136, 3623–3628 RSC.
- C. I. Richard, S. Choi, J.-C. Hsiang, Y. Antoku, T. Vosch, A. Bongiorno, Y.-L. Tzeng and R. M. Dickson, J. Am. Chem. Soc., 2008, 130, 5038–5039 CrossRef PubMed.
- X. Liu, F. Wang, R. Aizen, O. Yehezkeli and I. Willner, J. Am. Chem. Soc., 2013, 135, 11832–11839 CrossRef CAS PubMed.
- S. Ding, A. A. Cargill, S. R. Das, I. L. Medintz and J. C. Claussen, Sensors, 2015, 15, 14766–14787 CrossRef CAS PubMed.
- W. Guo, J. Yuan, Q. Dong and E. Wang, J. Am. Chem. Soc., 2010, 132, 932–934 CrossRef CAS PubMed.
- L. Deng, Z. Zhou, J. Li, T. Li and S. Dong, Chem. Commun., 2011, 47, 11065–11067 RSC.
- Z. Zhou, Y. Du and S. Dong, Biosens. Bioelectron., 2011, 28, 33–37 CrossRef CAS PubMed.
- P. Shah, A. Rorvig-Lund, S. Ben Chaabane, P. W. Thulstrup, H. G. Kjaergaard, E. Fron, J. Hofkens, S. W. Yang and T. Vosch, ACS Nano, 2012, 6, 8803–8814 CrossRef CAS PubMed.
- S. Choi, J. Yu, S. A. Patel, Y.-L. Tzeng and R. M. Dickson, Photochem. Photobiol. Sci., 2011, 10, 109–115 CAS.
- J. Yu, S. Choi, C. I. Richards, Y. Antoku and R. M. Dickson, Photochem. Photobiol., 2008, 84, 1435–1439 CrossRef CAS PubMed.
- C. Fang and M. Zhang, J. Mater. Chem., 2009, 19, 6258–6266 RSC.
- A.-H. Lu, E. L. Salabas and F. Schueth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- R. Abu-Reziq, H. Alper, D. S. Wang and M. L. Post, J. Am. Chem. Soc., 2006, 128, 5279–5282 CrossRef CAS PubMed.
- N. A. Frey, S. Peng, K. Cheng and S. Sun, Chem. Soc. Rev., 2009, 38, 2532–2542 RSC.
- X. Sun, Y. Huang and D. E. Nikles, Int. J. Nanotechnol., 2004, 1, 328–346 CrossRef CAS.
- S. H. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS PubMed.
- S. Si, A. Kotal, T. K. Mandal, S. Giri, H. Nakamura and T. Kohara, Chem. Mater., 2004, 16, 3489–3496 CrossRef CAS.
- X. Sun, C. Zheng, F. Zhang, Y. Yang, G. Wu, A. Yu and N. Guan, J. Phys. Chem. C, 2009, 113, 16002–16008 CAS.
- N. Mizutani, T. Iwasaki, S. Watano, T. Yanagida and T. Kawai, Curr. Appl. Phys., 2010, 10, 801–806 CrossRef.
- C. C. Huang, K. Y. Chuang, C. P. Chou, M. T. Wu, H. S. Sheu, D. B. Shieh, C. Y. Tsai, C. H. Su, H. Y. Lei and C. S. Yeh, J. Mater. Chem., 2011, 21, 7472–7479 RSC.
- W. Wu, Q. He and C. Jiang, Nanoscale Res. Lett., 2008, 3, 397–415 CrossRef CAS PubMed.
- F. Scherer, M. Anton, U. Schillinger, J. Henkel, C. Bergemann, A. Kruger, B. Gansbacher and C. Plank, Gene Ther., 2002, 9, 102–109 CrossRef CAS PubMed.
- J. I. Cutler, D. Zheng, X. Xu, D. A. Giljohann and C. A. Mirkin, Nano Lett., 2010, 10, 1477–1480 CrossRef CAS PubMed.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS PubMed.
- P. Gillis, F. Moiny and R. A. Brooks, Magn. Reson. Med., 2002, 47, 257–263 CrossRef PubMed.
- J. M. Perez, T. O'Loughin, F. J. Simeone, R. Weissleder and L. Josephson, J. Am. Chem. Soc., 2002, 124, 2856–2857 CrossRef CAS PubMed.
- M. V. Yigit, D. Mazumdar and Y. Lu, Bioconjugate Chem., 2008, 19, 412–417 CrossRef CAS PubMed.
- L.-B. Nie, X.-L. Wang, S. Li and H. Chen, Anal. Sci., 2009, 25, 1327–1331 CrossRef CAS PubMed.
- E. Ruiz-Hernandez, A. Baeza and M. Vallet-Regi, ACS Nano, 2011, 5, 1259–1266 CrossRef CAS PubMed.
- J.-P. Fortin, F. Gazeau and C. Wilhelm, Eur. Biophys. J. Biophys. Lett., 2008, 37, 223–228 CrossRef CAS PubMed.
- A. M. Derfus, G. von Maltzahn, T. J. Harris, T. Duza, K. S. Vecchio, E. Ruoslahti and S. N. Bhatia, Adv. Mater., 2007, 19, 3932–3936 CrossRef CAS.
- D. W. Hwang, H. Y. Ko, J. H. Lee, H. Kang, S. H. Ryu, I. C. Song, D. S. Lee and S. Kim, J. Nucl. Med., 2010, 51, 98–105 CrossRef CAS PubMed.
- H. Hu, A. Dai, J. Sun, X. Li, F. Gao, L. Wu, Y. Fang, H. Yang, L. An, H. Wu and S. Yang, Nanoscale, 2013, 5, 10447–10454 RSC.
- C. Wang, H. Daimon, T. Onodera, T. Koda and S. Sun, Angew. Chem., Int. Ed., 2008, 47, 3588–3591 CrossRef CAS PubMed.
- T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 272, 1924–1926 CAS.
- L. Nguyen Viet, C. Nguyen Duc, T. Hayakawa, H. Hirata, G. Lakshminarayana and M. Nogami, Nanotechnology, 2010, 21, 035605 CrossRef PubMed.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS PubMed.
- H. Ye, J. A. Crooks and R. M. Crooks, Langmuir, 2007, 23, 11901–11906 CrossRef CAS PubMed.
- Z. L. Liu, L. M. Gan, L. Hong, W. X. Chen and J. Y. Lee, J. Power Sources, 2005, 139, 73–78 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2004, 108, 5726–5733 CrossRef CAS.
- H. Yang, W. Vogel, C. Lamy and N. Alonso-Vante, J. Phys. Chem. B, 2004, 108, 11024–11034 CrossRef CAS.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- C. Wang, H. Daimon and S. Sun, Nano Lett., 2009, 9, 1493–1496 CrossRef CAS PubMed.
- R. Polsky, R. Gill, L. Kaganovsky and I. Willner, Anal. Chem., 2006, 78, 2268–2271 CrossRef CAS PubMed.
- R. Gill, R. Polsky and I. Willner, Small, 2006, 2, 1037–1041 CrossRef CAS PubMed.
- S. J. Kwon and A. J. Bard, J. Am. Chem. Soc., 2012, 134, 10777–10779 CrossRef CAS PubMed.
- E. Porcel, S. Liehn, H. Remita, N. Usami, K. Kobayashi, Y. Furusawa, C. Le Sech and S. Lacombe, Nanotechnology, 2012, 21, 085103 CrossRef PubMed.
- M. Muramatsu and A. Kitagawa, Rev. Sci. Instrum., 2012, 83, 02B909, DOI:10.1063/1.3671744.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS PubMed.
- S. W. Kim, J. Park, Y. Jang, Y. Chung, S. Hwang, T. Hyeon and Y. W. Kim, Nano Lett., 2003, 3, 1289–1291 CrossRef CAS.
- H. P. Hemantha and V. V. Sureshbabu, Org. Biomol. Chem., 2011, 9, 2597–2601 CAS.
- Z. Hou, N. Theyssen, A. Brinkmann and W. Leitner, Angew. Chem., Int. Ed., 2005, 44, 1346–1349 CrossRef CAS PubMed.
- Y. Li, X. Fan, J. Qi, J. Ji, S. Wang, G. Zhang and F. Zhang, Nano Res., 2010, 3, 429–437 CrossRef CAS.
- S. Sawoo, D. Srimani, P. Dutta, R. Lahiri and A. Sarkar, Tetrahedron, 2009, 65, 4367–4374 CrossRef CAS.
- S. Sharma, B. Kim and D. Lee, Langmuir, 2012, 28, 15958–15965 CrossRef CAS PubMed.
- C. Fang, Y. Fan, J. M. Kong, G. J. Zhang, L. Linn and S. Rafeah, Sens. Actuators, B, 2007, 126, 684–690 CrossRef CAS.
- Z. Wang, H. Li, S. Zhen and N. He, Nanoscale, 2012, 4, 3536–3542 RSC.
- B. K. Andrasfalvy, G. L. Galinanes, D. Huber, M. Barbic, J. J. Macklin, K. Susumu, J. B. Delehanty, A. L. Huston, J. K. Makara and I. L. Medintz, Nat. Methods, 2014, 11, 1237–1241 CrossRef CAS PubMed.
- E. Petryayeva, W. R. Algar and I. L. Medintz, Appl. Spectrosc., 2013, 67, 215–252 CrossRef CAS PubMed.
- W. R. Algar, K. Susumu, J. B. Delehanty and I. L. Medintz, Anal. Chem., 2011, 83, 8826–8837 CrossRef CAS PubMed.
- S. J. Rosenthal, J. C. Chang, O. Kovtun, J. R. McBride and I. D. Tomlinson, Chem. Biol., 2011, 18, 10–24 CrossRef CAS PubMed.
- J. Chang and E. R. Waclawik, RSC Adv., 2014, 4, 23505–23527 RSC.
- V. Lesnyak, N. Gaponik and A. Eychmuller, Chem. Soc. Rev., 2013, 42, 2905–2929 RSC.
- C. M. Evans, L. C. Cass, K. E. Knowles, D. B. Tice, R. P. H. Chang and E. A. Weiss, J. Coord. Chem., 2012, 65, 2391–2414 CrossRef.
- S. B. Brichkin and E. V. Chernykh, High Energy Chem., 2011, 45, 1–12 CrossRef CAS.
- K. Susumu, E. Oh, J. B. Delehanty, J. B. Blanco-Canosa, B. J. Johnson, V. Jain, W. J. Hervey, W. R. Algar, K. Boeneman, P. E. Dawson and I. L. Medintz, J. Am. Chem. Soc., 2011, 133, 9480–9496 CrossRef CAS PubMed.
- K. Susumu, E. Oh, J. B. Delehanty, F. Pinaud, K. B. Gemmill, S. Walper, J. Breger, M. J. Schroeder, M. H. Stewart, V. Jain, C. M. Whitaker, A. L. Huston and I. L. Medintz, Chem. Mater., 2014, 26, 5327–5344 CrossRef CAS.
- K. C. Kang, W. B. Tan and Y. Zhang, J. Biomed. Nanotechnol., 2006, 2, 165–172 CrossRef CAS.
- N. Tomczak, R. R. Liu and J. G. Vancso, Nanoscale, 2013, 5, 12018–12032 RSC.
- W. W. Yu, E. Chang, R. Drezek and V. L. Colvin, Biochem. Biophys. Res. Commun., 2006, 348, 781–786 CrossRef CAS PubMed.
- A. F. E. Hezinger, J. Tessmar and A. Gopferich, Eur. J. Pharm. Biopharm., 2008, 68, 138–152 CrossRef CAS PubMed.
- R. Gill, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 7602–7625 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- H. J. Gao and Y. Kong, Annu. Rev. Mater. Res., 2004, 34, 123–150 CrossRef CAS.
- M. E. Akerman, W. C. W. Chan, P. Laakkonen, S. N. Bhatia and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12617–12621 CrossRef CAS PubMed.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Adv. Mater., 2004, 16, 961–966 CrossRef CAS.
- X. Y. Wu, H. J. Liu, J. Q. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. F. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- A. P. Alivisatos, W. W. Gu and C. Larabell, in Annu. Rev. Biomed. Eng, 2005, vol. 7, pp. 55–76 Search PubMed.
- B. R. Liu, Y. W. Huang, H. J. Chiang and H. J. Lee, J. Nanosci. Nanotechnol., 2010, 10, 7897–7905 CrossRef CAS PubMed.
- Y. P. Ho and K. W. Leong, Nanoscale, 2010, 2, 60–68 RSC.
- A. M. Smith, H. W. Duan, A. M. Mohs and S. M. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef CAS PubMed.
- K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC.
- S. Silvi and A. Credi, Chem. Soc. Rev., 2015, 44, 4275–4289 RSC.
- G. P. Mitchell, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1999, 121, 8122–8123 CrossRef CAS.
- W. R. Algar and U. J. Krull, ChemPhysChem, 2007, 8, 561–568 CrossRef CAS PubMed.
- V. V. Breus, C. D. Heyes, K. Tron and G. U. Nienhaus, ACS Nano, 2009, 3, 2573–2580 CrossRef CAS PubMed.
- J. Aldana, N. Lavelle, Y. J. Wang and X. G. Peng, J. Am. Chem. Soc., 2005, 127, 2496–2504 CrossRef CAS PubMed.
- J. Aldana, Y. A. Wang and X. G. Peng, J. Am. Chem. Soc., 2001, 123, 8844–8850 CrossRef CAS PubMed.
- A. R. Clapp, I. L. Medintz, J. M. Mauro, B. R. Fisher, M. G. Bawendi and H. Mattoussi, J. Am. Chem. Soc., 2004, 126, 301–310 CrossRef CAS PubMed.
- A. R. Clapp, I. L. Medintz, H. T. Uyeda, B. R. Fisher, E. R. Goldman, M. G. Bawendi and H. Mattoussi, J. Am. Chem. Soc., 2005, 127, 18212–18221 CrossRef CAS PubMed.
- K. Boeneman, J. B. Delehanty, K. Susumu, M. H. Stewart and I. L. Medintz, J. Am. Chem. Soc., 2010, 132, 5975–5977 CrossRef CAS PubMed.
- H. Lu, O. Schops, U. Woggon and C. M. Niemeyer, J. Am. Chem. Soc., 2008, 130, 4815–4827 CrossRef CAS PubMed.
- J. B. Blanco-Canosa, M. Wu, K. Susumu, E. Petryayeva, T. L. Jennings, P. E. Dawson, W. R. Algar and I. L. Medintz, Coord. Chem. Rev., 2014, 263, 101–137 CrossRef.
- I. L. Medintz, L. Berti, T. Pons, A. F. Grimes, D. S. English, A. Alessandrini, P. Facci and H. Mattoussi, Nano Lett., 2007, 7, 1741–1748 CrossRef CAS PubMed.
- L. Berti, P. S. D'Agostino, K. Boeneman and I. L. Medintz, Nano Res., 2009, 2, 121–129 CrossRef CAS.
- K. Boeneman, D. E. Prasuhn, J. B. Blanco-Canosa, P. E. Dawson, J. S. Melinger, M. Ancona, M. H. Stewart, K. Susumu, A. Huston and I. L. Medintz, J. Am. Chem. Soc., 2010, 132, 18177–18190 CrossRef CAS PubMed.
- C. M. Spillmann, M. G. Ancona, S. Buckhout-White, W. R. Algar, M. H. Stewart, K. Susumu, A. L. Huston, E. R. Goldman and I. L. Medintz, ACS Nano, 2013, 7, 7101–7118 CrossRef CAS PubMed.
- C. L. Dwyer, S. A. Diaz, S. A. Walper, A. Samanta, K. Susumu, E. Oh, S. Buckhout-White and I. L. Medintz, Chem. Mater., 2015, 28, 8205–8215 Search PubMed.
- G. I. Kim, K. W. Kim, M. K. Oh and Y. M. Sung, Nanotechnology, 2009, 20, 175503 CrossRef PubMed.
- S. B. He, B. H. Huang, J. J. Tan, Q. Y. Luo, Y. Lin, J. Li, Y. Hu, L. Zhang, S. H. Yan, Q. Zhang, D. W. Pang and L. J. Li, Biomaterials, 2011, 32, 5471–5477 CrossRef CAS PubMed.
- W. R. Algar and U. J. Krull, Langmuir, 2006, 22, 11346–11352 CrossRef CAS PubMed.
- W. Zhang, Y. Yao and Y. S. Chen, J. Phys. Chem. C, 2011, 115, 599–606 CAS.
- D. J. Zhou, L. M. Ying, X. Hong, E. A. Hall, C. Abell and D. Klenerman, Langmuir, 2008, 24, 1659–1664 CrossRef CAS PubMed.
- S. Pathak, S. K. Choi, N. Arnheim and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4103–4104 CrossRef CAS PubMed.
- N. Hildebrandt, K. D. Wegner and W. R. Algar, Coord. Chem. Rev., 2014, 273, 125–138 CrossRef.
- X. Qiu and N. Hilderbrandt, ACS Nano, 2015, 9, 8449–8457 CrossRef CAS PubMed.
- C. Y. Zhang, H. C. Yeh, M. T. Kuroki and T. H. Wang, Nat. Mater., 2005, 4, 826–831 CrossRef CAS PubMed.
- M. Levy, S. F. Cater and A. D. Ellington, ChemBioChem, 2005, 6, 2163–2166 CrossRef CAS PubMed.
- A. H. Fu, C. M. Micheel, J. Cha, H. Chang, H. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2004, 126, 10832–10833 CrossRef CAS PubMed.
- H. Bui, C. Onodera, C. Kidwell, Y. Tan, E. Graugnard, W. Kuang, J. Lee, W. B. Knowlton, B. Yurke and W. L. Hughes, Nano Lett., 2010, 10, 3367–3372 CrossRef CAS PubMed.
- K. Boeneman, J. R. Deschamps, S. Buckhout-White, D. E. Prasuhn, J. B. Blanco-Canosa, P. E. Dawson, M. H. Stewart, K. Susumu, E. R. Goldman, M. Ancona and I. L. Medintz, ACS Nano, 2010, 4, 7253–7266 CrossRef CAS PubMed.
- S. Hinds, B. J. Taft, L. Levina, V. Sukhovatkin, C. J. Dooley, M. D. Roy, D. D. MacNeil, E. H. Sargent and S. O. Kelley, J. Am. Chem. Soc., 2006, 128, 64–65 CrossRef CAS PubMed.
- L. Levina, W. Sukhovatkin, S. Musikhin, S. Cauchi, R. Nisman, D. P. Bazett-Jones and E. H. Sargent, Adv. Mater., 2005, 17, 1854–1857 CrossRef CAS.
- N. Ma, E. H. Sargent and S. O. Kelley, Nat. Nanotechnol., 2009, 4, 121–125 CrossRef CAS PubMed.
- G. Tikhomirov, S. Hoogland, P. E. Lee, A. Fischer, E. H. Sargent and S. O. Kelley, Nat. Nanotechnol., 2011, 6, 485–490 CrossRef CAS PubMed.
- Q. B. Wang, Y. Liu, Y. G. Ke and H. Yan, Angew. Chem., Int. Ed., 2008, 47, 316–319 CrossRef CAS PubMed.
- A. Samanta, Z. T. Deng and Y. Liu, Nanoscale, 2014, 6, 4486–4490 RSC.
- P. S. Eastman, W. M. Ruan, M. Doctolero, R. Nuttall, G. De Feo, J. S. Park, J. S. F. Chu, P. Cooke, J. W. Gray, S. Li and F. Q. F. Chen, Nano Lett., 2006, 6, 1059–1064 CrossRef CAS PubMed.
- H. Kwon, S. Hong, H. Kim, Y. Choi, J. Kim and R. Song, Chem. Commun., 2010, 46, 8959–8961 RSC.
- C. Zhang, R. J. Macfarlane, K. L. Young, C. H. J. Choi, L. L. Hao, E. Auyeung, G. L. Liu, X. Z. Zhou and C. A. Mirkin, Nat. Mater., 2013, 12, 741–746 CrossRef CAS PubMed.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
- E. T. Thostenson, Z. F. Ren and T. W. Chou, Compos. Sci. Technol., 2001, 61, 1899–1912 CrossRef CAS.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105–1136 CrossRef CAS PubMed.
- C. C. Chang, I. K. Hsu, M. Aykol, W. H. Hung, C. C. Chen and S. B. Cronin, ACS Nano, 2010, 4, 5095–5100 CrossRef CAS PubMed.
- S. Nanot, E. H. Haroz, J. H. Kim, R. H. Hauge and J. Kono, Adv. Mater., 2012, 24, 4977–4994 CrossRef CAS PubMed.
- E. L. Ivchenko and B. Spivak, Phys. Rev. B: Condens. Matter, 2002, 66, 155404 CrossRef.
- F. Wang, G. Dukovic, L. E. Brus and T. F. Heinz, Phys. Rev. Lett., 2004, 92, 177401–177404 CrossRef PubMed.
- J. Crochet, M. Clemens and T. Hertel, J. Am. Chem. Soc., 2007, 129, 8058–8059 CrossRef CAS PubMed.
- S. Reich, M. Dworzak, A. Hoffmann, C. Thomsen and M. S. Strano, Phys. Rev. B: Condens. Matter, 2005, 71, 033402 CrossRef.
- O. V. Kharissova, B. I. Kharisov and E. G. D. Ortiz, RSC Adv., 2013, 3, 24812–24852 RSC.
- T. Premkumar, R. Mezzenga and K. E. Geckeler, Small, 2012, 8, 1299–1313 CrossRef CAS PubMed.
- B. I. Kharisov, O. V. Kharissova, H. L. Gutierrez and U. O. Mendez, Ind. Eng. Chem. Res., 2009, 48, 572–590 CrossRef CAS.
- S. Z. Zu, X. X. Sun, Y. Q. Liu and B. H. Han, Chem. – Asian J., 2009, 4, 1562–1572 CrossRef CAS PubMed.
- M. Zheng, A. Jagota, E. D. Semke, B. A. Diner, R. S. McLean, S. R. Lustig, R. E. Richardson and N. G. Tassi, Nat. Mater., 2003, 2, 338–342 CrossRef CAS PubMed.
- S. Manohar, T. Tang and A. Jagota, J. Phys. Chem. C, 2007, 111, 17835–17845 CAS.
- R. R. Johnson, A. T. C. Johnson and M. L. Klein, Nano Lett., 2008, 8, 69–75 CrossRef CAS PubMed.
- X. Tu, S. Manohar, A. Jagota and M. Zheng, Nature, 2009, 460, 250–253 CrossRef CAS PubMed.
- E. S. Jeng, A. E. Moll, A. C. Roy, J. B. Gastala and M. S. Strano, Nano Lett., 2006, 6, 371–375 CrossRef CAS PubMed.
- D. A. Heller, E. S. Jeng, T. K. Yeung, B. M. Martinez, A. E. Moll, J. B. Gastala and M. S. Strano, Science, 2006, 311, 508–511 CrossRef CAS PubMed.
- R. Yang, J. Jin, Y. Chen, N. Shao, H. Kang, Z. Xiao, Z. Tang, Y. Wu, Z. Zhu and W. Tan, J. Am. Chem. Soc., 2008, 130, 8351–8358 CrossRef CAS PubMed.
- R. Yang, Z. Tang, J. Yan, H. Kang, Y. Kim, Z. Zhu and W. Tan, Anal. Chem., 2008, 80, 7408–7413 CrossRef CAS PubMed.
- W. M. Sharman, C. M. Allen and J. E. van Lier, Drug Discovery Today, 1999, 4, 507–517 CrossRef CAS PubMed.
- Z. Zhu, Z. Tang, J. A. Phillips, R. Yang, H. Wang and W. Tan, J. Am. Chem. Soc., 2008, 130, 10856–10857 CrossRef CAS PubMed.
- S. Dhar, Z. Liu, J. Thomale, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 11467–11476 CrossRef CAS PubMed.
- N. W. S. Kam, T. C. Jessop, P. A. Wender and H. J. Dai, J. Am. Chem. Soc., 2004, 126, 6850–6851 CrossRef CAS PubMed.
- D. Cai, J. M. Mataraza, Z. H. Qin, Z. P. Huang, J. Y. Huang, T. C. Chiles, D. Carnahan, K. Kempa and Z. F. Ren, Nat. Methods, 2005, 2, 449–454 CrossRef CAS PubMed.
- R. Singhal, Z. Orynbayeva, R. V. K. Sundaram, J. J. Niu, S. Bhattacharyya, E. A. Vitol, M. G. Schrlau, E. S. Papazoglou, G. Friedman and Y. Gogotsi, Nat. Nanotechnol., 2011, 6, 57–64 CrossRef CAS PubMed.
- N. W. S. Kam, M. O'Connell, J. A. Wisdom and H. J. Dai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11600–11605 CrossRef CAS PubMed.
- M. Ahmed, X. Jiang, Z. Deng and R. Narain, Bioconjugate Chem., 2009, 20, 2017–2022 CrossRef CAS PubMed.
- Y. Wu, J. A. Phillips, H. Liu, R. Yang and W. Tan, ACS Nano, 2008, 2, 2023–2028 CrossRef CAS PubMed.
- D. Pantarotto, R. Singh, D. McCarthy, M. Erhardt, J. P. Briand, M. Prato, K. Kostarelos and A. Bianco, Angew. Chem., Int. Ed., 2004, 43, 5242–5246 CrossRef CAS PubMed.
- S. Park, M. Vosguerichian and Z. Bao, Nanoscale, 2013, 5, 1727–1752 RSC.
- K. Maehashi, T. Katsura, K. Kerman, Y. Takamura, K. Matsumoto and E. Tamiya, Anal. Chem., 2007, 79, 782–787 CrossRef CAS PubMed.
- H. T. Maune, S.-P. Han, R. D. Barish, M. Bockrath, W. A. Goddard III, P. W. K. Rothemund and E. Winfree, Nat. Nanotechnol., 2010, 5, 61–66 CrossRef CAS PubMed.
- A.-P. Eskelinen, A. Kuzyk, T. K. Kaltiaisenaho, M. Y. Timmermans, A. G. Nasibulin, E. I. Kauppinen and P. Torma, Small, 2011, 7, 746–750 CrossRef CAS PubMed.
- C. Gomez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Nano Lett., 2007, 7, 3499–3503 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- Y. Wang, Z. Li, J. Wang, J. Li and Y. Lin, Trends Biotechnol., 2011, 29, 205–212 CrossRef CAS PubMed.
- J. S. Park, N.-I. Goo and D.-E. Kim, Langmuir, 2014, 30, 12587–12595 CrossRef CAS PubMed.
- O. Akhavan, Carbon, 2010, 48, 509–519 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2009, 25, 5957–5968 CrossRef CAS PubMed.
- A. Buchsteiner, A. Lerf and J. Pieper, J. Phys. Chem. B, 2006, 110, 22328–22338 CrossRef CAS PubMed.
- L. Gao, C. Lian, Y. Zhou, L. Yan, Q. Li, C. Zhang, L. Chen and K. Chen, Biosens. Bioelectron., 2014, 60, 22–29 CrossRef CAS PubMed.
- J.-L. Li, B. Tang, B. Yuan, L. Sun and X.-G. Wang, Biomaterials, 2013, 34, 9519–9534 CrossRef CAS PubMed.
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- G. Eda, Y.-Y. Lin, C. Mattevi, H. Yamaguchi, H.-A. Chen, I. S. Chen, C.-W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- Z. Luo, P. M. Vora, E. J. Mele, A. T. C. Johnson and J. M. Kikkawa, Appl. Phys. Lett., 2009, 94, 111909 CrossRef.
- E. Morales-Narvaez, B. Perez-Lopez, L. Baptista Pires and A. Merkoci, Carbon, 2012, 50, 2987–2993 CrossRef CAS.
- J. C. Claussen, W. R. Algar, N. Hildebrandt, K. Susumu, M. G. Ancona and I. L. Medintz, Nanoscale, 2013, 5, 12156–12170 RSC.
- M. Wu, R. Kempaiah, P.-J. J. Huang, V. Maheshwari and J. Liu, Langmuir, 2011, 27, 2731–2738 CrossRef CAS PubMed.
- J. Katoch, S. N. Kim, Z. Kuang, B. L. Farmer, R. R. Nalk, S. A. Tatulian and M. Ishigami, Nano Lett., 2012, 12, 2342–2346 CrossRef CAS PubMed.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS PubMed.
- Y. Tu, W. Li, P. Wu, H. Zhang and C. Cai, Anal. Chem., 2013, 85, 2536–2542 CrossRef CAS PubMed.
- H. Chang, L. Tang, Y. Wang, J. Jiang and J. Li, Anal. Chem., 2013, 82, 2341–2346 CrossRef PubMed.
- T. Miyahata, Y. Kitamura, A. Futamura, H. Matsuura, K. Hatakeyama, M. Koinuma, Y. Matsumoto and T. Ihara, Chem. Commun., 2013, 49, 10139–10141 RSC.
- L. Qiu, H. Zhou, W. Zhu, L. Qiu, J. Jiang, G. Shen and R. Yu, New J. Chem., 2013, 37, 3998–4003 RSC.
- Y. Pu, Z. Zhu, D. Han, H. Liu, J. Liu, J. Liao, K. Zhang and W. Tan, Analyst, 2011, 136, 4138–4140 RSC.
- C.-H. Lu, C.-L. Zhu, J. Li, J.-J. Liu, X. Chen and H.-H. Yang, Chem. Commun., 2010, 46, 3116–3118 RSC.
- Y. Wang, Z. Li, D. Hu, C.-T. Lin, J. Li and Y. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS PubMed.
- H. Dong, W. Gao, F. Yan, H. Ji and H. Ju, Anal. Chem., 2010, 82, 5511–5517 CrossRef CAS PubMed.
- J. Liu, Y. Li, Y. Li, J. Li and Z. Deng, J. Mater. Chem., 2010, 20, 900–906 RSC.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed.
- Z. Wang, G. Niu and X. Y. Chen, Pharm. Res., 2014, 31, 1358–1376 CrossRef CAS PubMed.
- M. Yokoyama, J. Drug Targeting, 2014, 22, 576–583 CrossRef CAS PubMed.
- Y. A. Zhong, F. H. Meng, C. Deng and Z. Y. Zhong, Biomacromolecules, 2014, 15, 1955–1969 CrossRef CAS PubMed.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS.
- Z. H. Cao and U. Ziener, Nanoscale, 2013, 5, 10093–10107 RSC.
- K. M. Ho, W. Y. Li, C. H. Wong and P. Li, Colloid Polym. Sci., 2010, 288, 1503–1523 CAS.
- T. Akagi, M. Baba and M. Akashi, Polymer, 2007, 48, 6729–6747 CrossRef CAS.
- F. Rancan, D. Papakostas, S. Hadam, S. Hackbarth, T. Delair, C. Primard, B. Verrier, W. Sterry, U. Blume-Peytavi and A. Vogt, Pharm. Res., 2009, 26, 2027–2036 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymer, 2011, 3, 1377–1397 CAS.
- O. Lunov, T. Syrovets, C. Loos, J. Beil, M. Delecher, K. Tron, G. U. Nienhaus, A. Musyanovych, V. Mailander, K. Landfester and T. Simmet, ACS Nano, 2011, 5, 1657–1669 CrossRef CAS PubMed.
- J. Cheng, B. A. Teply, I. Sherifi, J. Sung, G. Luther, F. X. Gu, E. Levy-Nissenbaum, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Biomaterials, 2007, 28, 869–876 CrossRef CAS PubMed.
- M. Bivas-Benita, S. Romeijn, H. E. Junginger and G. Borchard, Eur. J. Pharm. Biopharm., 2004, 58, 1–6 CrossRef CAS PubMed.
- S. H. Kim, J. H. Jeong, K. W. Chun and T. G. Park, Langmuir, 2005, 21, 8852–8857 CrossRef CAS PubMed.
- L. Yuan, Y. Wang, M. Pan, G. L. Rempel and Q. Pan, Eur. Polym. J., 2013, 49, 41–48 CrossRef CAS.
- C. Vauthier and K. Bouchemal, Pharm. Res., 2009, 26, 1025–1058 CrossRef CAS PubMed.
- M.-Y. Bai and S.-Z. Liu, Colloids Surf., B, 2014, 117, 346–353 CrossRef CAS PubMed.
- R. Vivek, R. Thangam, V. NipunBabu, C. Rejeeth, S. Sivasubramanian, P. Gunasekaran, K. Muthuchelian and S. Kannan, ACS Appl. Mater. Interfaces, 2014, 6, 6469–6480 CAS.
- W. Bao, R. Liu, Y. Wang, F. Wang, G. Xia, H. Zhang, X. Li, H. Yin and B. Chen, Int. J. Nanomed., 2015, 10, 557–566 CAS.
- C. Zhang, X. Zheng, X. Wan, X. Shao, Q. Liu, Z. Zhang and Q. Zhang, J. Controlled Release, 2014, 192, 317–324 CrossRef CAS PubMed.
- J. J. Green, B. Y. Zhou, M. M. Mitalipova, C. Beard, R. Langer, R. Jaenisch and D. G. Anderson, Nano Lett., 2008, 8, 3126–3130 CrossRef CAS PubMed.
- Y.-H. Huang, G. T. Zugates, W. Peng, D. Holtz, C. Dunton, J. J. Green, N. Hossain, M. R. Chernick, R. F. Padera Jr., R. Langer, D. G. Anderson and J. A. Sawicki, Cancer Res., 2009, 69, 6184–6191 CrossRef CAS PubMed.
- J. Guo, X. Gao, L. Su, H. Xia, G. Gu, Z. Pang, X. Jiang, L. Yao, J. Chen and H. Chen, Biomaterials, 2011, 32, 8010–8020 CrossRef CAS PubMed.
- A. J. ten Tije, J. Verweij, W. J. Loos and A. Sparreboom, Clin. Pharmacokinet., 2003, 42, 665–685 CrossRef CAS PubMed.
- Y. Bae and K. Kataoka, Adv. Drug Delivery Rev., 2009, 61, 768–784 CrossRef CAS PubMed.
- J. C. Kraft, J. P. Freeling, Z. Wang and R. J. Y. Ho, J. Pharm. Sci., 2014, 103, 29–52 CrossRef CAS PubMed.
- J. H. Jeong, S. W. Kim and T. G. Park, Bioconjugate Chem., 2003, 14, 473–479 CrossRef CAS PubMed.
- S. Katayose and K. Kataoka, Bioconjugate Chem., 1997, 8, 702–707 CrossRef CAS PubMed.
- X. Jiang, W. Qu, D. Pan, Y. Ren, J.-M. Williford, H. Cui, E. Luijten and H.-Q. Mao, Adv. Mater., 2013, 25, 227–232 CrossRef CAS PubMed.
- J. H. Jeong and T. G. Park, Bioconjugate Chem., 2001, 12, 917–923 CrossRef CAS PubMed.
- J. H. Jeong, S. W. Kim and T. G. Park, J. Controlled Release, 2003, 93, 183–191 CrossRef CAS PubMed.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Adv. Mater., 2008, 20, 899–902 CrossRef CAS.
- G. Toffoli, C. Cernigoi, A. Russo, A. Gallo, M. Bagnoli and M. Boiocchi, Int. J. Cancer, 1997, 74, 193–198 CrossRef CAS PubMed.
- Z. J. Gartner, M. W. Kanan and D. R. Liu, Angew. Chem., Int. Ed., 2002, 41, 1796–1800 CrossRef CAS.
- M. W. Kanan, M. M. Rozenman, K. Sakurai, T. M. Snyder and D. R. Liu, Nature, 2004, 431, 545–549 CrossRef CAS PubMed.
- D. Summerer and A. Marx, Angew. Chem., Int. Ed., 2002, 41, 89–90 CrossRef CAS.
- F. E. Alemdaroglu, K. Ding, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2006, 45, 4206–4210 CrossRef CAS PubMed.
- H. Kang, M. B. O'Donoghue, H. Liu and W. Tan, Chem. Commun., 2010, 46, 249–251 RSC.
- K. Ding, F. E. Alemdaroglu, M. Boersch, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2007, 46, 1172–1175 CrossRef CAS PubMed.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Macromol. Rapid Commun., 2008, 29, 326–329 CrossRef CAS.
- M. Qin, H. J. Hah, G. Kim, G. Nie, Y.-E. K. Lee and R. Kopelman, Photochem. Photobiol. Sci., 2011, 10, 832–841 CAS.
- M. Kuruppuarachchi, H. Savoie, A. Lowry, C. Alonso and R. W. Boyle, Mol. Pharm., 2011, 8, 920–931 CrossRef CAS PubMed.
- H. Sun, A. M. Scharff-Poulsen, H. Gu, I. Jakobsen, J. M. Kossmann, W. B. Frommer and K. Almdal, ACS Nano, 2008, 2, 19–24 CrossRef CAS PubMed.
- A. K. Poulsen, A. M. Scharff-Poulsen and L. F. Olsen, Anal. Biochem., 2007, 366, 29–36 CrossRef CAS PubMed.
- H. Sun, R. V. Benjaminsen, K. Almdal and T. L. Andresen, Bioconjugate Chem., 2012, 23, 2247–2255 CrossRef CAS PubMed.
- H. A. Clark, M. Hoyer, M. A. Philbert and R. Kopelman, Anal. Chem., 1999, 71, 4831–4836 CrossRef CAS PubMed.
- L. J. Nielsen, L. F. Olsen and V. C. Ozalp, ACS Nano, 2010, 4, 4361–4370 CrossRef CAS PubMed.
- P. Richard, B. Teusink, M. B. Hemker, K. VanDam and H. V. Westerhoff, Yeast, 1996, 12, 731–740 CrossRef CAS PubMed.
- J. D. Lewis, G. Destito, A. Zijlstra, M. J. Gonzalez, J. P. Quigley, M. Manchester and H. Stuhlmann, Nat. Med., 2006, 12, 354–360 CrossRef CAS PubMed.
- L. Liepold, S. Anderson, D. Willits, L. Oltrogge, J. A. Frank, T. Douglas and M. Young, Magn. Reson. Med., 2007, 58, 871–879 CrossRef CAS PubMed.
- Y. Ren, S. M. Wong and L. Y. Lim, Bioconjugate Chem., 2007, 18, 836–843 CrossRef CAS PubMed.
- D. J. Manayani, D. Thomas, K. A. Dryden, V. Reddy, M. E. Siladi, J. M. Marlett, G. J. A. Rainey, M. E. Pique, H. M. Scobie, M. Yeager, J. A. T. Young, M. Manchester and A. Schneemann, PLoS Pathog., 2007, 3, 1422–1431 CAS.
- R. A. Miller, A. D. Presley and M. B. Francis, J. Am. Chem. Soc., 2007, 129, 3104–3109 CrossRef CAS PubMed.
- R. J. Tseng, C. L. Tsai, L. P. Ma and J. Y. Ouyang, Nat. Nanotechnol., 2006, 1, 72–77 CrossRef CAS PubMed.
- C. M. Soto and B. R. Ratna, Curr. Opin. Biotechnol., 2010, 21, 426–438 CrossRef CAS PubMed.
- N. F. Steinmetz, Nanomedicine, 2010, 6, 634–641 CAS.
- K. Valegard, L. Liljas, K. Fridborg and T. Unge, Nature, 1990, 345, 36 CrossRef CAS PubMed.
- E. W. Kovacs, J. M. Hooker, D. W. Romanini, P. G. Holder, K. E. Berry and M. B. Francis, Bioconjugate Chem., 2007, 18, 1140–1147 CrossRef CAS PubMed.
- G. J. Tong, S. C. Hsiao, Z. M. Carrico and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 11174–11178 CrossRef CAS PubMed.
- N. Stephanopoulos, G. J. Tong, S. C. Hsiao and M. B. Francis, ACS Nano, 2010, 4, 6014–6020 CrossRef CAS PubMed.
- M. Kwak, I. J. Minten, D. M. Anaya, A. J. Musser, M. Brasch, R. J. M. Nolte, K. Mullen, J. Cornelissen and A. Herrmann, J. Am. Chem. Soc., 2010, 132, 7834–7835 CrossRef CAS PubMed.
- K. Numata, M. R. Reagan, R. H. Goldstein, M. Rosenblatt and D. L. Kaplan, Bioconjugate Chem., 2011, 22, 1605–1610 CrossRef CAS PubMed.
- Y. Q. Wu, J. A. MacKay, J. R. McDaniel, A. Chilkoti and R. L. Clark, Biomacromolecules, 2009, 10, 19–24 CrossRef CAS PubMed.
- A. MaHam, Z. W. Tang, H. Wu, J. Wang and Y. H. Lin, Small, 2009, 5, 1706–1721 CrossRef CAS PubMed.
- S. C. Andrews, A. K. Robinson and F. Rodriguez-Quinones, FEMS Microbiol. Rev., 2003, 27, 215–237 CrossRef CAS PubMed.
- P. Arosio and S. Levi, Free Radicals Biol. Med., 2002, 33, 457–463 CrossRef CAS PubMed.
- N. C. Andrews and P. J. Schmidt, in Annu. Rev. Physiol, 2007, vol. 69, pp. 69–85 Search PubMed.
- M. M. Liang, K. L. Fan, M. Zhou, D. M. Duan, J. Y. Zheng, D. L. Yang, J. Feng and X. Y. Yan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14900–14905 CrossRef CAS PubMed.
- K. Li, Z. P. Zhang, M. Luo, X. Yu, Y. Han, H. P. Wei, Z. Q. Cui and X. E. Zhang, Nanoscale, 2012, 4, 188–193 RSC.
- M. Uchida, S. Kang, C. Reichhardt, K. Harlen and T. Douglas, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 834–845 CrossRef CAS PubMed.
- S.-E. Kim, K.-Y. Ahn, J.-S. Park, K. R. Kim, K. E. Lee, S.-S. Han and J. Lee, Anal. Chem., 2011, 83, 5834–5843 CrossRef CAS PubMed.
- S. Wu, G. Han, D. J. Milliron, S. Aloni, V. Altoe, D. V. Talapin, B. E. Cohen and P. J. Schuck, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10917–10921 CrossRef CAS PubMed.
- Q. Zhan, J. Qian, H. Liang, G. Somesfalean, D. Wang, S. He, Z. Zhang and S. Andersson-Engels, ACS Nano, 2011, 5, 3744–3757 CrossRef CAS PubMed.
- G. Chen, J. Shen, T. Y. Ohulchanskyy, N. J. Patel, A. Kutikov, Z. Li, J. Song, R. K. Pandey, H. Agren, P. N. Prasad and G. Han, ACS Nano, 2012, 6, 8280–8287 CrossRef CAS PubMed.
- S. Heer, O. Lehmann, M. Haase and H. U. Gudel, Angew. Chem., Int. Ed., 2003, 42, 3179–3182 CrossRef CAS PubMed.
- S. Heer, K. Kompe, H. U. Gudel and M. Haase, Adv. Mater., 2004, 16, 2102–2105 CrossRef CAS.
- F. Wang and X. G. Liu, J. Am. Chem. Soc., 2008, 130, 5642–5643 CrossRef CAS PubMed.
- H. Hu, L. Q. Xiong, J. Zhou, F. Y. Li, T. Y. Cao and C. H. Huang, Chem. – Eur. J., 2009, 15, 3577–3584 CrossRef CAS PubMed.
- L. Q. Xiong, Z. G. Chen, Q. W. Tian, T. Y. Cao, C. J. Xu and F. Y. Li, Anal. Chem., 2009, 81, 8687–8694 CrossRef CAS PubMed.
- M. Wang, W. Hou, C. C. Mi, W. X. Wang, Z. R. Xu, H. H. Teng, C. B. Mao and S. K. Xu, Anal. Chem., 2009, 81, 8783–8789 CrossRef CAS PubMed.
- T. Rantanen, M. L. Jarvenpaa, J. Vuojola, R. Arppe, K. Kuningas and T. Soukka, Analyst, 2009, 134, 1713–1716 RSC.
- P. Zhang, S. Rogelj, K. Nguyen and D. Wheeler, J. Am. Chem. Soc., 2006, 128, 12410–12411 CrossRef CAS PubMed.
- M. Kumar, Y. Guo and P. Zhang, Biosens. Bioelectron., 2009, 24, 1522–1526 CrossRef CAS PubMed.
- M. Kumar and P. Zhang, Langmuir, 2009, 25, 6024–6027 CrossRef CAS PubMed.
- G. D. Liu, T. M. H. Lee and J. S. Wang, J. Am. Chem. Soc., 2005, 127, 38–39 CrossRef CAS PubMed.
- Y. J. Yang, J. F. Zi and W. K. Li, Electrochim. Acta, 2014, 115, 126–130 CrossRef CAS.
- W. Sun, J. H. Zhong, P. Qin and K. Jiao, Anal. Biochem., 2008, 377, 115–119 CrossRef CAS PubMed.
- J. A. Hansen, R. Mukhopadhyay, J. O. Hansen and K. V. Gothelf, J. Am. Chem. Soc., 2006, 128, 3860–3861 CrossRef CAS PubMed.
- C. Ding, H. Zhong and S. Zhang, Biosens. Bioelectron., 2008, 23, 1314–1318 CrossRef CAS PubMed.
- C. Ding, Z. Wang, H. Zhong and S. Zhang, Biosens. Bioelectron., 2010, 25, 1082–1087 CrossRef CAS PubMed.
- T. T. Morgan, H. S. Muddana, E. I. Altinoglu, S. M. Rouse, A. Tabakovic, T. Tabouillot, T. J. Russin, S. S. Shanmugavelandy, P. J. Butler, P. C. Eklund, J. K. Yun, M. Kester and J. H. Adair, Nano Lett., 2008, 8, 4108–4115 CrossRef CAS PubMed.
- H. T. Schmidt, B. L. Gray, P. A. Wingert and A. E. Ostafin, Chem. Mater., 2004, 16, 4942–4947 CrossRef CAS.
- E. I. Altinoglu, T. J. Russin, J. M. Kaiser, B. M. Barth, P. C. Eklund, M. Kester and J. H. Adair, ACS Nano, 2008, 2, 2075–2084 CrossRef CAS PubMed.
- I. Roy, S. Mitra, A. Maitra and S. Mozumdar, Int. J. Pharm., 2003, 250, 25–33 CrossRef CAS PubMed.
- V. V. Sokolova, I. Radtke, R. Heumann and M. Epple, Biomaterials, 2006, 27, 3147–3153 CrossRef CAS PubMed.
- Y. Kakizawa and K. Kataoka, Langmuir, 2002, 18, 4539–4543 CrossRef CAS.
- T. Welzel, I. Radtke, W. Meyer-Zaika, R. Heumann and M. Epple, J. Mater. Chem., 2004, 14, 2213–2217 RSC.
- E. H. Chowdhury, M. Kunou, M. Nagaoka, A. K. Kundu, T. Hoshiba and T. Akaike, Gene, 2004, 341, 77–82 CrossRef CAS PubMed.
- F. Pittella, M. Zhang, Y. Lee, H. J. Kim, T. Tockary, K. Osada, T. Ishii, K. Miyata, N. Nishiyama and K. Kataoka, Biomaterials, 2011, 32, 3106–3114 CrossRef CAS PubMed.
- H. Tolou, Anal. Biochem., 1993, 215, 156–158 CrossRef CAS PubMed.
- C. Chen and H. Okayama, Mol. Cell. Biol., 1987, 7, 2745–2752 CrossRef CAS PubMed.
- F. L. Graham and A. J. Vandereb, Virology, 1973, 52, 456–467 CrossRef CAS PubMed.
- H. Yan, S. H. Park, G. Finkelstein, J. H. Reif and T. H. LaBean, Science, 2003, 301, 1882–1884 CrossRef CAS PubMed.
- J. H. Chen and N. C. Seeman, Nature, 1991, 350, 631–633 CrossRef CAS PubMed.
- Y. He, T. Ye, M. Su, C. Zhang, A. E. Ribbe, W. Jiang and C. D. Mao, Nature, 2008, 452, 198–202 CrossRef CAS PubMed.
- E. S. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, W. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. P. Oliveira, J. S. Pedersen, V. Birkedal, F. Besenbacher, K. V. Gothelf and J. Kjems, Nature, 2009, 459, 73–76 CrossRef CAS PubMed.
- C. X. Lin, X. Wang, Y. Liu, N. C. Seeman and H. Yan, J. Am. Chem. Soc., 2007, 129, 14475–14481 CrossRef CAS PubMed.
- Y. He, Y. Chen, H. P. Liu, A. E. Ribbe and C. D. Mao, J. Am. Chem. Soc., 2005, 127, 12202–12203 CrossRef CAS PubMed.
- J. P. Zheng, J. J. Birktoft, Y. Chen, T. Wang, R. J. Sha, P. E. Constantinou, S. L. Ginell, C. D. Mao and N. C. Seeman, Nature, 2009, 461, 74–77 CrossRef CAS PubMed.
- S. Buckhout-White, M. Ancona, E. Oh, J. R. Deschamps, M. H. Stewart, J. B. Blanco-Canosa, P. E. Dawson, E. R. Goldman and I. L. Medintz, ACS Nano, 2012, 6, 1026–1043 CrossRef CAS PubMed.
- H. Lee, A. K. R. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS PubMed.
- G. P. Acuna, M. Bucher, I. H. Stein, C. Steinhauer, A. Kuzyk, P. Holzmeister, R. Schreiber, A. Moroz, F. D. Stefani, T. Liedl, F. C. Simmel and P. Tinnefeld, ACS Nano, 2012, 6, 3189–3195 CrossRef CAS PubMed.
- H. Pei, N. Lu, Y. L. Wen, S. P. Song, Y. Liu, H. Yan and C. H. Fan, Adv. Mater., 2010, 22, 4754–4758 CrossRef CAS PubMed.
- Y. L. Wen, H. Pei, Y. Wan, Y. Su, Q. Huang, S. P. Song and C. H. Fan, Anal. Chem., 2011, 83, 7418–7423 CrossRef CAS PubMed.
- S. Modi, M. G. Swetha, D. Goswami, G. D. Gupta, S. Mayor and Y. Krishnan, Nat. Nanotechnol., 2009, 4, 325–330 CrossRef CAS PubMed.
- H. Pei, L. Liang, G. B. Yao, J. Li, Q. Huang and C. H. Fan, Angew. Chem., Int. Ed., 2012, 51, 9020–9024 CrossRef CAS PubMed.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed.
- Y. Amir, E. Ben-Ishay, D. Levner, S. Ittah, A. Abu-Horowitz and I. Bachelet, Nat. Nanotechnol., 2014, 9, 353–357 CrossRef CAS PubMed.
- Y. G. Ke, S. Lindsay, Y. Chang, Y. Liu and H. Yan, Science, 2008, 319, 180–183 CrossRef CAS PubMed.
- L. Berti, I. L. Medintz, J. Tom and R. A. Mathies, Bioconjugate Chem., 2001, 12, 493–500 CrossRef CAS PubMed.
- Y. G. Li, Y. T. H. Cu and D. Luo, Nat. Biotechnol., 2005, 23, 885–889 CrossRef CAS PubMed.
- C. Lin, R. Jungmann, A. M. Leifer, C. Li, D. Levner, G. M. Church, W. M. Shih and P. Yin, Nat. Chem., 2012, 4, 832–839 CrossRef CAS PubMed.
- R. Jungmann, M. S. Avendano, J. B. Woehrstein, M. J. Dai, W. M. Shih and P. Yin, Nat. Methods, 2014, 11, 313–318 CrossRef CAS PubMed.
- J. Li, H. Pei, B. Zhu, L. Liang, M. Wei, Y. He, N. Chen, D. Li, Q. Huang and C. H. Fan, ACS Nano, 2011, 5, 8783–8789 CrossRef CAS PubMed.
- V. J. Schuller, S. Heidegger, N. Sandholzer, P. C. Nickels, N. A. Suhartha, S. Endres, C. Bourquin and T. Liedl, ACS Nano, 2011, 5, 9696–9702 CrossRef PubMed.
- X. W. Liu, Y. Xu, T. Yu, C. Clifford, Y. Liu, H. Yan and Y. Chang, Nano Lett., 2012, 12, 4254–4259 CrossRef CAS PubMed.
- Y. Zhang and N. C. Seeman, J. Am. Chem. Soc., 1994, 116, 1661–1669 CrossRef CAS.
- R. P. Goodman, I. A. T. Schaap, C. F. Tardin, C. M. Erben, R. M. Berry, C. F. Schmidt and A. J. Turberfield, Science, 2005, 310, 1661–1665 CrossRef CAS PubMed.
- W. M. Shih, J. D. Quispe and G. F. Joyce, Nature, 2004, 427, 618–621 CrossRef CAS PubMed.
- F. A. Aldaye and H. F. Sleiman, J. Am. Chem. Soc., 2007, 129, 13376–13377 CrossRef CAS PubMed.
- Y. He, T. Ye, M. Su, C. Zhang, A. E. Ribbe, W. Jiang and C. Mao, Nature, 2008, 452, 198–201 CrossRef CAS PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- T. G. W. Edwardson, K. M. M. Carneiro, C. K. McLaughlin, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2013, 5, 868–875 CrossRef CAS PubMed.
- J. F. Fakhoury, C. K. Mclaughlin, T. W. Edwardson, J. W. Conway and H. F. Sleiman, Biomacromolecules, 2014, 15, 276–282 CrossRef CAS PubMed.
- F. A. Aldaye, P. K. Lo, P. Karam, C. K. McLaughlin, G. Cosa and H. F. Sleiman, Nat. Nanotechnol., 2009, 4, 349–352 CrossRef CAS PubMed.
- P. K. Lo, P. Karam, F. A. Aldaye, C. K. McLaughlin, G. D. Hamblin, G. Cosa and H. F. Sleiman, Nat. Chem., 2010, 2, 319–328 CrossRef CAS PubMed.
- G. D. Hamblin, K. M. M. Carneiro, J. F. Fakhoury, K. E. Bujold and H. F. Sleiman, J. Am. Chem. Soc., 2012, 134, 2888–2891 CrossRef CAS PubMed.
- J. L. Fu, M. H. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516–5519 CrossRef CAS PubMed.
- J. L. Fu, Y. R. Yang, A. Johnson-Buck, M. H. Liu, Y. Liu, N. G. Walter, N. W. Woodbury and H. Yan, Nat. Nanotechnol., 2014, 9, 531–536 CrossRef CAS PubMed.
- M. H. Liu, J. L. Fu, C. Hejesen, Y. H. Yang, N. W. Woodbury, K. Gothelf, Y. Liu and H. Yan, Nat. Commun., 2013, 4, 2127, DOI:10.1038/ncomms3127.
- J. Elbaz, A. Cecconello, Z. Fan, A. O. Govorov and I. Willner, Nat. Commun., 2012, 4, 2000, DOI:10.1038/ncomms3000.
- R. Alam, D. M. Fontaine, B. R. Branchini and M. Maye, Nano Lett., 2012, 12, 3251–3256 CrossRef CAS PubMed.
- R. Alam, J. Zylstra, D. M. Fontaine, B. R. Branchini and M. M. Maye, Nanoscale, 2013, 5, 5303–5306 RSC.
- M. K. So, C. J. Xu, A. M. Loening, S. S. Gambhir and J. H. Rao, Nat. Biotechnol., 2006, 24, 339–343 CrossRef CAS PubMed.
- C. E. Bradburne, J. B. Delehanty, K. Boeneman Gemmill, B. C. Mei, H. Mattoussi, K. Susumu, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2013, 24, 1570–1158 CrossRef CAS PubMed.
- E. Oh, R. Liu, A. Nel, K. B. Gemill, M. Bilal, Y. Cohen and I. L. Medintz, Nat. Nanotechnol., 2016 DOI:10.1038/NNANO.2015.338 , In Press.
- K. M. Tsoi, Q. Dai, B. A. Alman and W. C. W. Chan, Acc. Chem. Res., 2013, 46, 662–671 CrossRef CAS PubMed.
- K.-T. Yong, W.-C. Law, R. Hu, L. Ye, L. Liu, M. T. Swiharte and P. N. Prasad, Chem. Soc. Rev., 2013, 42, 1236–1250 RSC.
- K. Boneman Gemmill, J. B. Delehanty, J. Blanco-Canosa, K. Susumu, M. H. Stewart, E. Oh, A. L. Huston, G. Dawson, S. Ingale, R. Walters, M. Domowicz, J. R. Deschamps, W. R. Algar, S. C. DiMaggio, J. Manono, C. M. Spillmann, D. Thompson, T. L. Jennings, P. E. Dawson and I. L. Medintz, ACS Nano, 2013, 7, 3778–3796 CrossRef PubMed.
- J. Breger, J. B. Delehanty and I. L. Medintz, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 131–151 CrossRef CAS PubMed.
- L. D. Field, J. B. Delehanty, Y. Chen and I. L. Medintz, Acc. Chem. Res., 2015, 48, 1380–1390 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |