
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic polymers for DNA origami coating – examining their binding efficiency and tuning the enzymatic reaction rates†
Jenny K.
Kiviaho‡
a,
Veikko
Linko‡
a,
Ari
Ora
a,
Tony
Tiainen
b,
Erika
Järvihaavisto
a,
Joona
Mikkilä
a,
Heikki
Tenhu
 b,
Nonappa
c and
Mauri A.
Kostiainen
*a
b,
Nonappa
c and
Mauri A.
Kostiainen
*a
aBiohybrid Materials Group, Department of Biotechnology and Chemical Technology, Aalto University, P.O. Box 16100, 00076 Aalto, Finland. E-mail: mauri.kostiainen@aalto.fi
bLaboratory of Polymer Chemistry, Department of Chemistry, University of Helsinki, P.O. Box 55, 00014 Helsinki, Finland
cMolecular Materials Group, Department of Applied Physics, Aalto University, P.O. Box 15100, 00076 Aalto, Finland
First published on 12th May 2016
Abstract
DNA origamis are fully tailored, programmable, biocompatible and readily functionalizable nanostructures that provide an excellent foundation for the development of sophisticated drug-delivery systems. However, the DNA origami objects suffer from certain drawbacks such as low cell-transfection rates and low stability. A great deal of studies on polymer-based transfection agents, mainly focusing on polyplex formation and toxicity, exists. In this study, the electrostatic binding between a brick-like DNA origami and cationic block-copolymers was explored. The effect of the polymer structure on the binding was investigated and the toxicity of the polymer–origami complexes evaluated. The study shows that all of the analyzed polymers had a suitable binding efficiency irrespective of the block structure. It was also observed that the toxicity of polymer–origami complexes was insignificant at the biologically relevant concentration levels. Besides brick-like DNA origamis, tubular origami carriers equipped with enzymes were also coated with the polymers. By adjusting the amount of cationic polymers that cover the DNA structures, we showed that it is possible to control the enzyme kinetics of the complexes. This work gives a starting point for further development of biocompatible and effective polycation-based block copolymers that can be used in coating different DNA origami nanostructures for various bioapplications.
Introduction
Structural DNA nanotechnology has made considerable progress especially during the past decade.1 New techniques to design and construct precise DNA nano-objects have emerged since the pioneering work of Nadrian Seeman.2,3 Currently, the most versatile and common method to produce DNA nano-objects is the DNA origami technique, which is based on the thermal annealing of long single-stranded DNA with complementary short oligonucleotides to form stable and well-defined two and three-dimensional nano-objects.4–7As the DNA origami technique enables precise design and production8 of nanometer-scale shapes, it is employed in various applications such as protein and DNA analysis,9–11 microscopy,12 plasmonics,13 molecular engineering14,15 and nanoscale patterning.16–18 Furthermore, DNA origami is an extremely promising platform for creating sophisticated drug-delivery vehicles and molecular devices for bionanotechnology.19–21 DNA origamis, and DNA nano-objects in general, are found to be biocompatible, non-toxic and only mildly immunogenic, and moreover, they are able to enter cells with and without the help of transfection agents.21–27 The origamis are reported to tolerate various cell lysates and nucleases, which suggests that they could remain intact under physiological conditions to some extent.8,28 Additionally, DNA origamis exhibit enhanced permeability and retention (EPR) effect, which results in passive tumor-targeting and an accumulation of nano-objects into the tumor region.24 This makes DNA origami an attractive candidate for anti-cancer applications. It has also been suggested that DNA origami drug carriers could serve as a means to circumvent the drug resistance of certain cancer types.23
Although cells internalize bare DNA origamis, the cell uptake is significantly increased when transfection agents are applied. The uptake of origamis is improved considerably when e.g. the commercial lipid-based transfection agent, Lipofectamine®, is employed.29 Similarly, by coating the origami with virus capsid proteins, the cell uptake can be enhanced.30 In addition, the pharmacokinetic bioavailability can be increased by encapsulating origamis with lipid membranes.31 This implies that DNA origamis require an auxiliary substance in order to efficiently enter the cells, and moreover, coating/encapsulation could be the key factor in enhancing their stability in biologically relevant environments. Numerous studies on traditional non-viral carriers, including cationic polymers and dendrimers, show that synthetic organic carriers can prolong the in vivo half-life and enhance the cell transfection of drugs or genetic material.32,33 Thus, it is justified to hypothesize that these materials could also improve the cell transfection of DNA origami nano-objects.
Cationic polymers are a particularly interesting class of transfection agents, since they can be readily synthesized and a variety of functionalities can be incorporated into the polymer chains. Even stimuli-responsive behavior is achievable through copolymer structures.34–36 Cationic polymers are associated with relatively low immunogenicity and reasonable transfection efficiency, but unfortunately they exhibit some cytotoxicity. For example poly(l-lysine) (PLL), poly(2-dimethylaminoethyl methacrylate) (PDMAEMA) and branched and linear polyethylenimine (PEI) are widely studied polycations. Additionally, PLL demonstrates low transfection efficiency and no buffering capacity.37,38 PEI, in contrast, is an effective transfection agent, but associated with high cytotoxicity and haemolytic activity.39–41 PDMAEMA and PDMAEMA-based polyplexes, on the other hand are found less toxic than PEI and the corresponding polyplexes, while the transfection efficiency is comparable to that of branched PEI.42,43 In addition, PDMAEMA-based polymers do not induce red blood cell aggregation nor demonstrate haemolytic activity in whole blood.41
Despite the lower toxicity and non-haemolytic nature, PDMAEMA still faces certain limitations regarding biocompatibility. Furthermore, bare PDMAEMA–DNA complexes may possess poor colloidal stability. These limitations could be overcome by incorporating additional blocks to the polymer structure. Suitable blocks could decrease the toxicity of the system and even increase the colloidal stability and blood circulation times.38 A good candidate for reducing the cytotoxicity is a hydrophilic polymer widely employed in biomedical applications, poly(ethylene glycol) (PEG). PEG is a good example of a polymer that provides protection for nanoparticles and proteins against the reticulo-endothelial system, thus prolonging the circulation time. It also prevents aggregation and improves particle stability.44
In this work, 2-(dimethylamino)ethyl methacrylate was polymerized employing poly(ethylene glycol)-based macroinitiators to explore the electrostatic binding between a brick-like DNA origami and the block copolymers (Fig. 1).
 | ||
| Fig. 1 Scaffold strand folds into desired shape with the help of staple strands. Each cylinder on the origami represents a DNA double helix. DNA origami possesses a negative surface charge, which enables the electrostatic binding of the cationic polymers. | ||
The PEG-moiety was incorporated into the polymers to expectedly improve the biocompatibility of PDMAEMA and to provide additional protection and colloidal stability to the proposed DNA origami–polymer particles. Two different block copolymer structures, AB-type diblock and ABA-type triblock structures, and PDMAEMA homopolymer (HP) were studied. The aim of the work is to understand the effect of polymer structure on the binding and to investigate if the utilization of the polymers in biomedical applications is feasible. Furthermore, to test the conceivable applications of the polymer–DNA complexes, tubular DNA origamis were loaded with enzymes and coated with varying amounts of polymers in order to control the enzymatic reaction rates.
Results and discussion
Polymer properties
To study the electrostatic binding of cationic PDMAEMA-based polymers on the anionic DNA origami surface, a set of polymers were synthesized. Polymers with different structures were prepared, including PDMAEMA homopolymer (HP), PDMAEMA-PEG (AB-type) diblock copolymer and PDMAEMA-PEG-PDMAEMA (ABA-type) triblock copolymer (Fig. 2).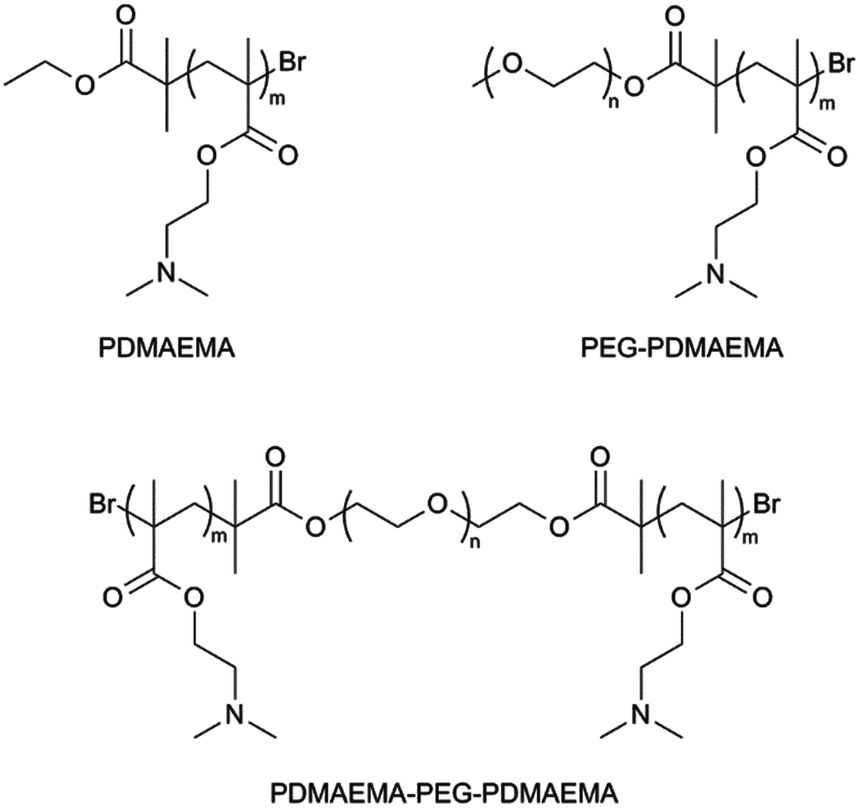 | ||
| Fig. 2 Three polymers were prepared utilizing the Atom Transfer Radical Polymerization (ATRP) technique. Homopolymer PDMAEMA was prepared using a commercial ATRP initiator, ethyl α-bromoisobutyrate. The copolymers were synthesized using the PEG-macroinitiators. Two separate copolymer structures were synthesized, PDMAEMA-PEG (AB-type) and PDMAEMA-PEG-PDMAEMA (ABA-type). | ||
All the polymers were synthesized by employing the Atom Transfer Radical Polymerization (ATRP) technique. ATRP enables fast reactions under mild conditions and allows production of polymers with narrow molecular weight distributions and set molecular weights. Low polydispersity indices (PDIs) and the ability to produce polymers with desired molecular weights is especially important in biomedical applications where it is crucial to generate well-defined polymer bioconjugates.45–47 PEG was introduced to the polymers by using PEG monomethyl ether (mPEG, 5000 g mol−1) and difunctional PEG (4000 g mol−1) macroinitiators in DMAEMA polymerization.
Both the macroinitiator and polymer syntheses were successful. The structural integrity of macroinitiators was confirmed with 1H NMR and IR spectroscopy, size exclusion chromatography (SEC) and matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometry (specifications on characterization can be found in the ESI†). 1H NMR and IR confirmed the presence of the initiator groups on mPEG and PEG chains after esterification. SEC affirmed that esterification reaction did not result in uncontrolled chain breaking or other side reactions as the polydispersity index (PDI) of both PEGs remained narrow. Also the masses obtained from MALDI-ToF spectroscopy corresponded to the theoretical mass values of the macroinitiators.
1H NMR, IR, MALDI-ToF and SEC data also confirmed the successful block copolymer syntheses. Polymers with desired molecular weights and relatively low PDIs were obtained (Table 1). The total molecular weights of the polymers vary, but the size of the PDMAEMA block in polymers is roughly the same, ∼6000 g mol−1. By fixing the length of the PDMAEMA block, the amine to phosphate ratio between the polymer block and origami remains the same, while the effect of PEG blocks and different polymer structures on binding can be studied.
| Polymer | DP (PDMAEMA) | DP (PEG) | M n (g mol−1) |
M
w/Mn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
|---|---|---|---|---|
| a Calculated from 1H NMR. b Determined with MALDI-ToF. c Measured with SEC. DP = degree of polymerization. | ||||
| PDMAEMA (HP) | 35b | — | 5700b | 1.31 |
| PEG macroinitiator | — | 115b | 5200b | 1.04 |
| PEG-PDMAEMA (AB) | 38a | 120a | 11300a |
1.12 |
| PEG difunctional macroinitiator | — | 95b | 4600b | 1.03 |
| PDMAEMA-PEG-PDMAEMA (ABA) | ∼39/blocka | 95 | 18800c |
1.29 |
Polymer binding to DNA origami
A DNA origami nanostructure, dubbed 60-helix bundle (60HB), was prepared and characterized as reported previously.48 Agarose gel electrophoresis and transmission electron microscopy (TEM) were used to verify the quality of the folding (Fig. 3). Fig. 3A shows how the mobility of a pure scaffold strand (left lane) in gel differs from that of completely folded origami (right lane). The additional band on the right lane corresponds to the excess staple strands present in the folding mixture. The agarose gel in Fig. 3A was run prior to spin filtering which was used to remove the excess staple strands. | ||
| Fig. 3 (A) Agarose gel was run to verify the quality of the folding. Left lane: scaffold strand M13mp18 as a reference. Right lane: DNA origami and excess staple strands. (B) TEM image of the 60HB structures. | ||
TEM micrographs were taken to confirm the proper assembly of the origamis. Fig. 3B shows the structural details of the DNA origami and highlights the precise folding.
The binding of the polymers on the origami surface was studied with gel electrophoresis, ethidium bromide assay and TEM. TEM micrographs reveal that by adding the polymer, the structural details of the origami surface disappear, and the objects become rounded (smooth edges) (Fig. 4A). The TEM, however, did not reveal any significant differences between the structures of separate origami–polymer complexes.
 | ||
| Fig. 4 (A) TEM micrographs showing (left to right) native origami (60HB), PDMAEMA-PEG-PDMAEMA-coated origami (ABA) with npolymer/norigami = 200, PDMAEMA-coated origami (HP) with npolymer/norigami = 600 and PEG-PDMAEMA-coated origami (AB) with npolymer/norigami = 600. (B) Native 60HB origami (leftmost lane) was run in an agarose gel along with 60HB–polymer complexes. The amine to phosphate ratio is denoted as (nN+/nP−). | ||
The gel electrophoresis, in contrast, displays slight differences in the complex formation between separate polymers. The agarose gel clearly shows that polymers bind on the origami surface and alter the charge, size and shape thus changing the mobility of the origamis in the gel (Fig. 4B).
The molar amounts of the polymer required to immobilize the 60HB origami in the gel varies between the ABA, AB and homopolymer. The ABA-type copolymer changes the 60HB origami mobility with lower polymer concentration than the AB and homopolymer (HP). A molar ratio of npolymer/norigami = 400 for ABA was enough to immobilize the complexes, whereas the ratio of npolymer/norigami = 1200 was needed for the other two polymers to reach a similar effect. In order to take the cationic block length and number into account, the values can also be presented as the ratio of the total number of polymer amines and the total number of origami phosphates (nN+/nP−). ABA has two cationic blocks, and indeed by looking at the nN+/nP− ratio (Fig. 4B, lower values), the results show that all of the polymers prevent the electrophoretic mobility of DNA origamis with similar nN+/nP− ratios. Therefore, the PDMAEMA-block content in all of the immobilized samples is roughly equal and indicates that increasing the number of cationic blocks does not improve the relative binding affinity.
A similar type of binding behavior can also be observed by using the ethidium bromide (EthBr) displacement assay (see the ESI†). The ABA-type copolymer is able to decrease the EthBr fluorescence slightly more efficiently than the homopolymer (HP) or the AB-type copolymer, whereas the difference between the latter two is less prominent.
Cell viability with the polymers and polymer–origami complexes
The possible toxicity of the prepared polymer–60HB complexes and bare polymers was studied using a colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye assay. We used adenocarcinomic human alveolar basal epithelial cells (A549) and incubated them (1 h or 9 h) with polymers and polymer–origami complexes at physiologically relevant concentrations (DNA origami concentration 0.5 nM, polymers were added 10×, 100×, 1000× and 10000× excess; bare polymer concentrations 5 nM, 50 nM, 500 nM and 5 μM). In addition, we used polyethylenimine (PEI, ∼75 kDa) and PEG (6 kDa) as positive and negative controls, respectively. The cell viability can be seen in Fig. 5. During the one-hour incubation with the polymers and complexes, the cells retained their viability throughout the concentration range, whereas PEI as a positive control showed high activity. For the nine-hour incubation, PEI showed again the highest activity, and also the incubation with bare ABA polymers at high concentrations started to cause cell deaths, mainly due to their large cationic content. However, ABA–60HB complexes (and all HP and AB containing samples; both bare polymers and complexes) showed only insignificant activity.
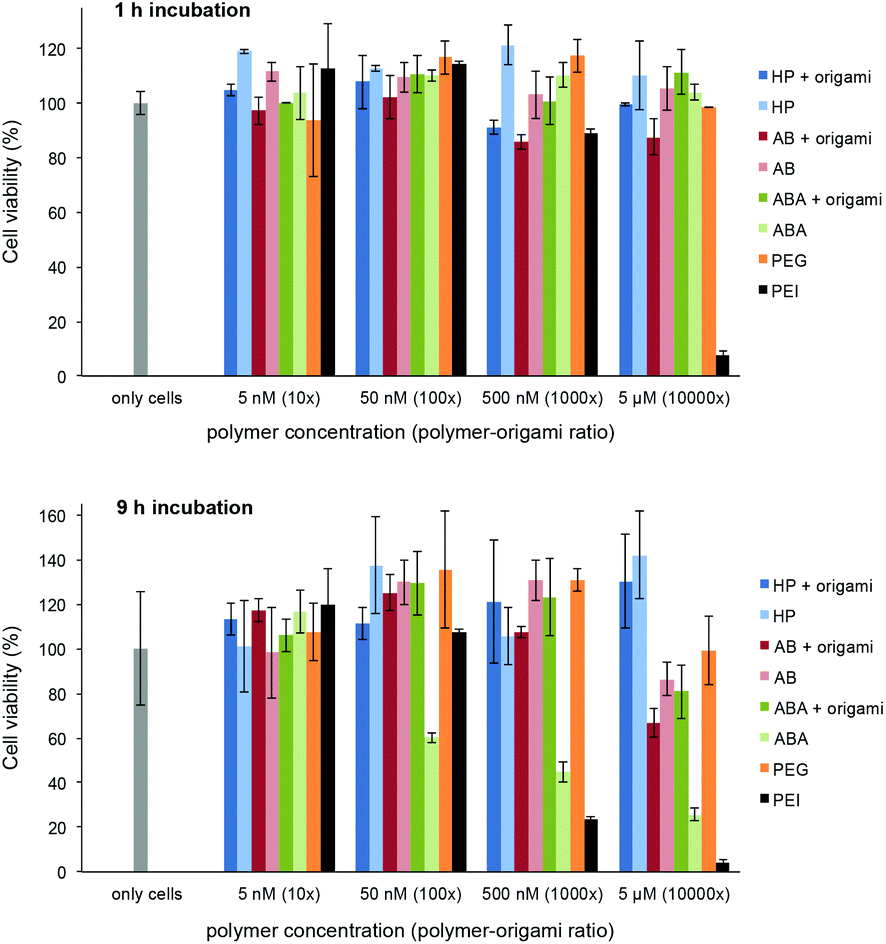 | ||
| Fig. 5 MTT assay of the polymers and polymer–60HB complexes. DNA origami concentration in all complexes is 0.5 nM (polymer concentration varies between 5 nM–5 μM). A549 human epithelial cells were incubated with the polymers for 1 h or 9 h before measuring the cell viability. The data consists of the average values measured from two independent sample sets and the error bars represent the data range. | ||
Tuning enzyme reaction rates with polymer coating
To further study the functionality of the DNA structures coated with the polymers, we used a tubular DNA origami, dubbed hexagonal tube (HT), which is reported and characterized in a previous work.49 A HT origami was loaded with streptavidin-Lucia enzymes (InvivoGen) through three biotinylated binding sites on its inner surface (Fig. 6A inset). The excess amount of added enzyme was removed using the spin-filtering procedure (see the ESI†). The Lucia enzyme (LUC) provides a highly sensitive and flash-like bioluminescence reaction when combined with a coelenterazine-based substrate (InvivoGen). With these structures we were able to systematically study the effect of different (HP, AB and ABA) polymer coatings on the luminescence decay rates of luciferase-equipped DNA origamis (LUC-origami).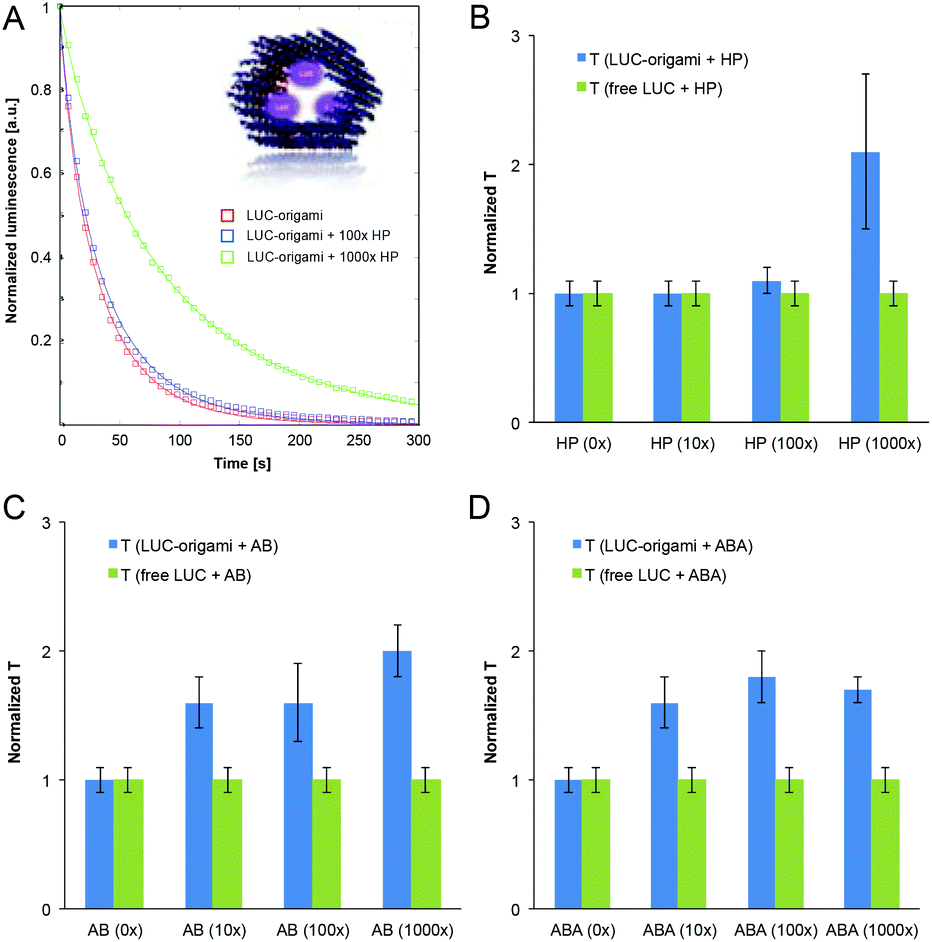 | ||
| Fig. 6 (A) Luminescence decay assay for the hexagonal tube origami equipped with streptavidin-Lucia (LUC) enzymes (inset). Homopolymer (HP) is added 100× and 1000× excess to DNA origami (10× omitted for clarity), which causes the change in the reaction rate (time constant is prolonged). Red, blue and green lines are fits to the data (stretched exponential functions). (B)–(D) Fitted normalized time constants of the luminescence decay for LUC-origami (blue) and free LUC enzymes (control assay without origamis, green) complexed with HP, AB and ABA, respectively. Time constants were determined with 4 different polymer concentrations (0×, 10×, 100× and 1000× with respect to DNA origami). For each case, the sample without added polymer has been normalized to 1. The data consists of the average values measured from two independent sample sets and the error bars represent the data range. | ||
LUC-origami decay assays (35 nM origami) and control assays of free enzymes (with and without the added polymers) with the coelenterazine-based substrate in HEPES/NaOH-based buffer (pH 6.8) were analyzed using the standard stretched exponential law,50
| I(t) = Aexp(−(t/T)β), | (1) |
The enzyme concentrations in the control assays (without origamis) were tailored to match the decay kinetics of the “(0×) LUC-origami” sample, i.e. the luminescence intensity levels, time constant (∼40–50 s) and the stretching exponent (∼0.85) were adjusted to be similar. It was noticed, that all the samples (polymer concentration varied between 0–1000× with respect to origamis) obeyed stretched exponential behaviour (fitted β varied between 0.73–0.87). An example of the normalized decay assay (A = 1) and the data fits can be seen in Fig. 6A (LUC-origami complexed with HP). Time constants (from the fits) for all the polymer–origami complexes (HP, AB and ABA-coated) are shown in Fig. 6B–D. The fitting parameters T and β for all the samples are listed in the ESI.†
LUC-enzymes showed appropriate catalytic activity in all samples (0–1000× polymer coating) indicating that the added polymer is not capable of blocking the enzyme activity completely. Importantly, the characteristic reaction time constant (T) was gradually prolonged for LUC-origamis, but not for the free enzymes when the polymers were incorporated. The trend is clearly seen in Fig. 6. The increase in the T value was most pronounced when the thickest coating (1000× polymer concentration) was used. For 1000× HP, 1000× AB and 1000× ABA, the prolongation factors of the T value were 2.1, 2.0 and 1.7, respectively. As mentioned above, the reaction time constant remained constant in all the control samples (free enzyme with polymers), thus indicating that the enzymes need to be encapsulated into/attached to origamis in order to achieve the prolongation. A plausible explanation is that the polymer coating of the origami limits the accessibility of the enzymes and restricts the diffusion rate of the substrate. This observation could lead to interesting applications, where the cationic polymer coatings could be utilized as protective layers for origami containers loaded with molecular cargos. Besides shielding of the origami, the tunable coating would also allow control over the kinetics of its enzymatic payload.
Conclusions
In this article we have verified that the PDMAEMA-based homo- and block copolymers are readily synthesizable via the ATRP; appropriate control over molecular weight and polydispersity indices was obtained. Moreover, the synthesis is fast and can be done under relatively mild conditions. To further demonstrate the feasibility of the polymers in possible bioapplications, we have systematically shown that the polymers can bind efficiently to brick-like and tubular DNA origami nanostructures through electrostatic interactions. However, it should be emphasized here that the efficiency of the polymer binding to two different origamis could not be directly compared with each other. The origamis are fundamentally distinct in structure; for 60HB, only the outer surface is used for binding, whereas there are both inner and outer surfaces available in the tubular origami design. Thus, the polymer size and charge can play a different role in these cases. For example, small HPs can bind more efficiently to the inner surface of the tube and restrict the accessibility of the enzymes, which then results in the prolongation of the characteristic time constant. Moreover, the surface of the tubular origamis contains bound enzymes that may also conjugate with the polymers to some extent (compared to bare 60HB).In the case of 60HB, the most efficient binding (in terms of molar ratios) was achieved with the ABA-type block copolymer PDMAEMA-PEG-PDMAEMA. The advantage of the PEG-containing polymers is that besides the cationic binding properties, they could simultaneously provide the protective features (PEG-moiety). Moreover, controllable polymer coatings could find uses in improving the transfection rates of DNA origami shapes and concurrently enhancing the stability of origamis in biologically relevant environments, although it was not directly demonstrated in this work. However, here we have already shown that these cationic polymers possess a great potential in possible delivery applications. We have proven that different polymer coatings could be used in controlling the catalytic activity of tubular enzyme-loaded DNA origami nanocontainers. Moreover, the polymer–origami complexes are insignificantly toxic at the biologically relevant concentration levels used in this work (polymer concentration varied between 5 nM–5 μM in the MTT assay). Therefore, we believe that the presented features could find intriguing uses in various drug-delivery approaches in the near future.
Experimental
Syntheses
For the luminescence assay, the spin-filtered origamis were incubated at least 6 h with the bioluminescent streptavidin-Lucia (LUC) luciferase enzymes (InvivoGen). The enzymes were added to the origamis in excess amounts, and thus the unbound enzymes were removed by spin-filtering (HEPES/NaOH buffer, 6.5 mM HEPES, pH 6.8) (see the ESI† for the efficiency of purification).
For more detailed description of the macroinitiator and polymer syntheses as well as DNA origami and complex preparation, see the ESI.†
Characterization
Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker Avance 400 MHz spectrometer. The chemical shifts were calibrated using residual CHCl3 peaks. The IR spectra were recorded with a Nicolet 380 FT-IR spectrometer. Gel permeation chromatography was performed with a setup consisting of a Waters 515 HPLC-pump, Waters Styragel-columns and a Waters 2410 refractive index detector. The SEC measurements were run in DMF containing 1% of LiBr and calibrated with poly(methyl methacrylate) standards. The transmission electron microscope images were obtained with a Tecnai 12 Bio-Twin and JEM 3200FSC field emission microscope (JEOL, 300 kV) in bright field mode. The gel electrophoresis tests were run in 0.8% agarose gel with EthBr (80 μl, 0.625 mg ml−1) and MgCl2 (11 mM) using 1× TAE (40 mM tris(hydroxymethyl)aminomethane (Tris), 1 mM ethylenediamine tetra acetic acid (EDTA) and acetic acid, pH 8.3) running buffer with 11 mM MgCl2. The fluorescence spectra were recorded with an Agilent Technologies Cary Eclipse Fluorescence Spectrometer. MALDI-ToF analyses were carried out with an UltrafleXtreme 2000 Hz instrument (Bruker Daltonics, Bremen Germany) equipped with a SmartBeam II laser (355 nm), operated in positive mode. Typically, mass spectra were acquired by the accumulating spectra of 10000 laser shots. FlexAnalysis v3.4 was used to assign molecular isotopic masses for polymers. The luminescence decay assay was prepared by mixing 50 μl of the ready coelenterazine-based substrate, Quanti-Luc (InvivoGen), with 10 μl of the ready sample and measuring the luminescence immediately (10 s delay due to the measurement setup). A BioTek Cytation 3 Cell Imaging Multi-Mode Reader was employed in the luminescence decay assay and in the MTT assay.
Acknowledgements
This work was supported by the Academy of Finland (263504, 267497, 273645, 286845) Emil Aaltonen Foundation, Biocentrum Helsinki, Aalto University School of Chemical Technology and Tekniikan Edistämissäätiö. This work was carried out under the Academy of Finland's Centres of Excellence Programme (2014-2019) and made use of the Aalto University Nanomicroscopy Centre (Aalto-NMC) and Meilahti Clinical & Basic Proteomics, Core Facility, Biomedicum-Helsinki premises.Notes and references
- V. Linko and H. Dietz, Curr. Opin. Biotechnol., 2013, 24, 555–561 CrossRef CAS PubMed.
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS PubMed.
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- E. S. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, W. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. P. Oliveira, J. S. Pedersen, V. Birkedal, F. Besenbacher, K. V. Gothelf and J. Kjems, Nature, 2009, 459, 73–76 CrossRef CAS PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS PubMed.
- C. E. Castro, F. Kilchherr, D.-N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221–229 CrossRef CAS PubMed.
- S. M. Douglas, J. J. Chou and W. M. Shih, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6644–6648 CrossRef CAS PubMed.
- A. Rajendran, M. Endo and H. Sugiyama, Angew. Chem., Int. Ed., 2012, 51, 874–890 CrossRef CAS PubMed.
- Y. Sannohe, M. Endo, Y. Katsuda, K. Hidaka and H. Sugiyama, J. Am. Chem. Soc., 2010, 132, 16311–16313 CrossRef CAS PubMed.
- C. Steinhauer, R. Jungmann, T. Sobey, F. Simmel and P. Tinnefeld, Angew. Chem., Int. Ed., 2009, 48, 8870–8873 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E.-M. Roller, A. Högele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed.
- Y. Krishnan and F. C. Simmel, Angew. Chem., Int. Ed., 2011, 50, 3124–3156 CrossRef CAS PubMed.
- S. F. J. Wickham, M. Endo, Y. Katsuda, K. Hidaka, J. Bath, H. Sugiyama and A. J. Turberfield, Nat. Nanotechnol., 2011, 6, 166–169 CrossRef CAS PubMed.
- R. J. Kershner, L. D. Bozano, C. M. Micheel, A. M. Hung, A. R. Fornof, J. N. Cha, C. T. Rettner, M. Bersani, J. Frommer, P. W. K. Rothemund and G. M. Wallraff, Nat. Nanotechnol., 2009, 4, 557–561 CrossRef CAS PubMed.
- B. Shen, V. Linko, K. Tapio, M. A. Kostiainen and J. J. Toppari, Nanoscale, 2015, 7, 11267–11272 RSC.
- J. B. Knudsen, L. Liu, A. L. B. Kodal, M. Madsen, Q. Li, J. Song, J. B. Woehrstein, S. F. J. Wickham, M. T. Strauss, F. Schueder, J. Vinther, A. Krissanaprasit, D. Gudnason, A. A. A. Smith, R. Ogaki, A. N. Zelikin, F. Besenbacher, V. Birkedal, P. Yin, W. M. Shih, R. Jungmann, M. Dong and K. V. Gothelf, Nat. Nanotechnol., 2015, 10, 892–898 CrossRef CAS PubMed.
- S. Surana, A. R. Shenoy and Y. Krishnan, Nat. Nanotechnol., 2015, 10, 741–747 CrossRef CAS PubMed.
- Y.-J. Chen, B. Groves, R. A. Muscat and G. Seelig, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS PubMed.
- V. Linko, A. Ora and M. A. Kostiainen, Trends Biotechnol., 2015, 33, 586–594 CrossRef CAS PubMed.
- S. H. Ko, H. Liu, Y. Chen and C. Mao, Biomacromolecules, 2008, 9, 3039–3043 CrossRef CAS PubMed.
- Q. Jiang, C. Song, J. Nangreave, X. Liu, L. Lin, D. Qiu, Z. Wang, G. Zou, X. Liang, H. Yan and B. Ding, J. Am. Chem. Soc., 2012, 134, 13396–13403 CrossRef CAS PubMed.
- Q. Zhang, Q. Jiang, N. Li, L. Dai, Q. Liu, L. Song, J. Wang, Y. Li, J. Tian, B. Ding and Y. Du, ACS Nano, 2014, 8, 6633–6643 CrossRef CAS PubMed.
- V. J. Schüller, S. Heidegger, N. Sandholzer, P. C. Nickels, N. A. Suhartha, S. Endres, C. Bourquin and T. Liedl, ACS Nano, 2011, 5, 9696–9702 CrossRef PubMed.
- A. S. Walsh, H. Yin, C. M. Erben, M. J. A. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS PubMed.
- G. D. Hamblin, K. M. M. Carneiro, J. F. Fakhoury, K. E. Bujold and H. F. Sleiman, J. Am. Chem. Soc., 2012, 134, 2888–2891 CrossRef CAS PubMed.
- Q. Mei, X. Wei, F. Su, Y. Liu, C. Youngbull, R. Johnson, S. Lindsay, H. Yan and D. Meldrum, Nano Lett., 2011, 11, 1477–1482 CrossRef CAS PubMed.
- A. H. Okholm, J. S. Nielsen, M. Vinther, R. S. Sørensen, D. Schaffert and J. Kjems, Methods, 2014, 67, 193–197 CrossRef CAS PubMed.
- J. Mikkilä, A.-P. Eskelinen, E. H. Niemelä, V. Linko, M. J. Frilander, P. Törmä and M. A. Kostiainen, Nano Lett., 2014, 14, 2196–2200 CrossRef PubMed.
- S. D. Perrault and W. M. Shih, ACS Nano, 2014, 8, 5132–5140 CrossRef CAS PubMed.
- J. Li, C. Fan, H. Pei, J. Shi and Q. Huang, Adv. Mater., 2013, 25, 4386–4396 CrossRef CAS PubMed.
- D. Ibraheem, A. Elaissari and H. Fessi, Int. J. Pharm., 2014, 459, 70–83 CrossRef CAS PubMed.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2009, 5, 2513–2533 RSC.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- S. Agarwal, Y. Zhang, S. Maji and A. Greiner, Mater. Today, 2012, 15, 388–393 CrossRef CAS.
- D. Fischer, T. Bieber, Y. Li, H. Elsässer and T. Kissel, Pharm. Res., 1999, 16, 1273–1279 CrossRef CAS.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS PubMed.
- B. I. Cerda-Cristerna, H. Flores, A. Pozos-Guillén, E. Pérez, C. Sevrin and C. Grandfils, J. Controlled Release, 2011, 153, 269–277 CrossRef CAS PubMed.
- P. Dubruel, B. Christiaens, B. Vanloo, K. Bracke, M. Rosseneu, J. Vandekerckhove and E. Schacht, Eur. J. Pharm. Sci., 2003, 18, 211–220 CrossRef CAS PubMed.
- J. Robbens, C. Vanparys, I. Nobels, R. Blust, K. Van Hoecke, C. Janssen, K. De Schamphelaere, K. Roland, G. Blanchard, F. Silvestre, V. Gillardin, P. Kestemont, R. Anthonissen, O. Toussaint, S. Vankoningsloo, C. Saout, E. Alfaro-Moreno, P. Hoet, L. Gonzalez, P. Dubruel and P. Troisfontaines, Toxicology, 2010, 269, 170–181 CrossRef CAS PubMed.
- L. van Vlerken, T. Vyas and M. Amiji, Pharm. Res., 2007, 24, 1405–1414 CrossRef CAS PubMed.
- V. Bulmus, Polym. Chem., 2011, 2, 1463–1472 RSC.
- M. E. Fox, F. C. Szoka and J. M. J. Fréchet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS PubMed.
- T. Yamaoka, Y. Tabata and Y. Ikada, J. Pharm. Sci., 1994, 83, 601–606 CrossRef CAS PubMed.
- V. Linko, B. Shen, K. Tapio, J. J. Toppari, M. A. Kostiainen and S. Tuukkanen, Sci. Rep., 2015, 5, 15634 CrossRef CAS PubMed.
- V. Linko, M. Eerikäinen and M. A. Kostiainen, Chem. Commun., 2015, 51, 5351–5354 RSC.
- M. N. Berberan-Santos, E. N. Bodunov and B. Valeur, Chem. Phys., 2005, 315, 171–182 CrossRef CAS.
- X. Zhang and K. Matyjaszewski, Macromolecules, 1999, 32, 1763–1766 CrossRef CAS.
- M. Even, D. M. Haddleton and D. Kukulj, Eur. Polym. J., 2003, 39, 633–639 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of materials, syntheses of the polymers, fabrication and purification of DNA origamis, luminescence decay assays, agarose gel electrophoresis, ethidium bromide displacement assay, MTT assay and TEM characterization. See DOI: 10.1039/c5nr08355a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2016 |