
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multifunctional clickable and protein-repellent magnetic silica nanoparticles†
Diego
Estupiñán
a,
Markus B.
Bannwarth
a,
Steven E.
Mylon
b,
Katharina
Landfester
a,
Rafael
Muñoz-Espí
*ac and
Daniel
Crespy
*ad
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
bLafayette College, Department of Chemistry, Easton, PA 18042, USA
cInstitute of Materials Science (ICMUV), University of Valencia, PO Box 22085, 46071 Valencia, Spain. E-mail: rafael.munoz@uv.es
dVidyasirimedhi Institute of Science and Technology (VISTEC), School of Molecular Science and Engineering, Department of Material Science and Engineering, Rayong, Thailand. E-mail: crespy@mpip-mainz.mpg.de
First published on 12th January 2016
Abstract
Silica nanoparticles are versatile materials whose physicochemical surface properties can be precisely adjusted. Because it is possible to combine several functionalities in a single carrier, silica-based materials are excellent candidates for biomedical applications. However, the functionality of the nanoparticles can get lost upon exposure to biological media due to uncontrolled biomolecule adsorption. Therefore, it is important to develop strategies that reduce non-specific protein–particle interactions without losing the introduced surface functionality. Herein, organosilane chemistry is employed to produce magnetic silica nanoparticles bearing differing amounts of amino and alkene functional groups on their surface as orthogonally addressable chemical functionalities. Simultaneously, a short-chain zwitterion is added to decrease the non-specific adsorption of biomolecules on the nanoparticles surface. The multifunctional particles display reduced protein adsorption after incubation in undiluted fetal bovine serum as well as in single protein solutions (serum albumin and lysozyme). Besides, the particles retain their capacity to selectively react with biomolecules. Thus, they can be covalently bio-functionalized with an antibody by means of orthogonal click reactions. These features make the described multifunctional silica nanoparticles a promising system for the study of surface interactions with biomolecules, targeting, and bio-sensing.
Introduction
Nanoparticles are promising materials in the biomedical field as biosensors,1 therapeutic agents,2,3 drug delivery systems and molecular imaging devices.4,5 The designed nanoparticles for biomedical applications have usually relatively complex structure and chemistry so that they can perform multiple tasks.6 Silica is a suitable primary matrix to prepare nanoparticles owing to its robustness, biocompatibility, and versatility for chemical functionalization.7 The diversity of organosilane chemistry allows the controlled introduction of many different functional groups, from which the physicochemical characteristics of the nanomaterial can be carefully adjusted.8–11 Therefore, silica-based biomaterials are widely studied either as nanoparticulate carriers or as inorganic coating for nanostructures.12–15 Notwithstanding, a major issue encountered in the bio-application of these materials is the non-specific protein adsorption elicited when the nanocarriers come into contact with biological media. A dynamic “protein corona” is thus unavoidably formed over the nanoparticles16 altering their size, surface properties, and interaction with cell membranes and receptors.17–19 Moreover, the previously introduced surface functionality becomes cloaked by this protein layer, reducing its accessibility. Consequently, in order to decrease the rate and/or extent of non-specific protein adsorption and concomitant “biomolecular corona” formation, several strategies have been adopted. The most widespread method to passivate surfaces against non-specific interactions is the surface attachment of poly(ethylene glycol) (PEG) chains.20–22 However, a major drawback derived from this approach is the sensitivity of PEG to temperature and autooxidation.23,24 Therefore, alternative methodologies to attain adsorption-resistant surfaces have been proposed, including the use of poly(ethylene imine), mannitol or cellulose derivatives,24 hyperbranched polyglycerol, several co-polymers of PEG,25 starch derivatives26 and zwitterions.23,27,28 Polymeric zwitterionic coatings have been reported to dramatically reduce protein adsorption and biofilm formation.29,30 Interestingly, short-chain zwitterions as well as mixed-charged surfaces also display excellent protein-repellent properties.23,31–34 Moreover, in combination with magneto-responsive materials, zwitterionic coatings offer the possibility to obtain high-performing devices for selective recognition of biomolecules.35,36 Nevertheless, besides remarkable adsorption resistance, a manifold functionalizable surface is often required for bio-recognition and targeting purposes. Recent alternatives to address this issue encompass the direct modification of the protein-repellent agent by coupling chemistries.30,37 Although high functionalization density can be attained by these techniques, it is still challenging to selectively and orthogonally modify nanocarriers. Additionally, uncontrolled modification of the passivating agent can have deleterious effects on its biomolecule-repellency properties. Thus, another useful strategy is the introduction of reactive groups amidst the repelling layer.38,39 Herein, we report the synthesis of magneto-responsive multifunctional silica particles displaying both low non-specific protein adsorption and selective functionalization ability.Experimental
Materials
Styrene (>99%, Sigma-Aldrich) was passed through an aluminum oxide-loaded column prior to use. Ammonium hydroxide solution (28%–30%), potassium persulfate (KPS, 99%), polyoxyethylene (20) sorbitan monolaurate (Tween20, BioXtra), n-hexadecane (99%), fluorescamine (>98%), cysteamine hydrochloride (>98%), urea (99–100%), thiourea (>99.0%), and (3-aminopropyl)triethoxysilane (APTES, 99%) were purchased from Sigma-Aldrich. Sodium n-dodecyl sulfate (SDS, 99%), oleic acid (58%), and iron(II) chloride tetrahydrate (FeCl2·4H2O, 99%) were purchased from Merck. Iron(III) chloride hexahydrate (FeCl3·6H2O, 99%) and anhydrous dimethyl sulfoxide (DMSO, 99.7%) were purchased from Acros Organics. Polyvinylpyrrolidone (PVP, K 25, M 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was purchased from Carl Roth. 3-[(3-Cholamidopropyl)dimethyl ammonio]-1-propanesulfonate (CHAPS, >97%) was purchased from Serva. Isopropanol (i-PrOH, >99%) was purchased from Fisher Chemical. Tetraethoxysilane (TEOS, 99.9%) was purchased from Alfa Aesar, 3-(triethoxysillyl)propyl methacrylate (TPM, >98%) was purchased from TCI Chemicals, and 3-{[dimethyl(3-trimethoxysilyl)propyl]ammonio}-1-propanesulfonate (betaine, 95%) was purchased from Gelest Inc. Dulbecco's phosphate-buffered saline (PBS, without calcium and magnesium, pH 7.2) was purchased from Gibco. The N-hydroxysuccinimidyl ester (NHS) derivative connected to a dibenzocyclooctyne (DBCO) moiety by a PEG chain (DBCO–PEG4–NHS, >95%, Jena Bioscience), fetal bovine serum (FBS, Gibco), and lysozyme (from chicken egg white, protein >90%, Sigma-Aldrich) were stored at −20 °C. Bovine serum albumin (BSA, Sigma-Aldrich, 99%) was stored at 4 °C. Sterile water was used unless otherwise specified. The synthesis of the fluorescent BODIPY dye (Fig. S1,†λex = 523 nm, λem = 536 nm) was carried out following literature procedures.40,41 All other chemicals and solvents were purchased from Sigma-Aldrich and used as received.
000 g mol−1) was purchased from Carl Roth. 3-[(3-Cholamidopropyl)dimethyl ammonio]-1-propanesulfonate (CHAPS, >97%) was purchased from Serva. Isopropanol (i-PrOH, >99%) was purchased from Fisher Chemical. Tetraethoxysilane (TEOS, 99.9%) was purchased from Alfa Aesar, 3-(triethoxysillyl)propyl methacrylate (TPM, >98%) was purchased from TCI Chemicals, and 3-{[dimethyl(3-trimethoxysilyl)propyl]ammonio}-1-propanesulfonate (betaine, 95%) was purchased from Gelest Inc. Dulbecco's phosphate-buffered saline (PBS, without calcium and magnesium, pH 7.2) was purchased from Gibco. The N-hydroxysuccinimidyl ester (NHS) derivative connected to a dibenzocyclooctyne (DBCO) moiety by a PEG chain (DBCO–PEG4–NHS, >95%, Jena Bioscience), fetal bovine serum (FBS, Gibco), and lysozyme (from chicken egg white, protein >90%, Sigma-Aldrich) were stored at −20 °C. Bovine serum albumin (BSA, Sigma-Aldrich, 99%) was stored at 4 °C. Sterile water was used unless otherwise specified. The synthesis of the fluorescent BODIPY dye (Fig. S1,†λex = 523 nm, λem = 536 nm) was carried out following literature procedures.40,41 All other chemicals and solvents were purchased from Sigma-Aldrich and used as received.
Experimental methods
| Entry | Functionalizationa |
D
hb [nm] |
Specific surface areac [m2 g−1] | |||
|---|---|---|---|---|---|---|
| TEOS | APTES | TPM | Betaine | |||
| a Initial amounts of organosilanes used for the preparation of the silica particles. b Hydrodynamic diameter and standard deviation determined by DLS. c Specific surface area calculated from nitrogen adsorption using the BET model. | ||||||
| 1a | 20 μmol | — | — | — | 230 ± 100 | N.D. |
| 1b | 40 μmol | — | — | — | 270 ± 95 | N.D. |
| 1c | 80 μmol | — | — | — | 440 ± 210 | N.D. |
| 2a | 40 μmol | 0.4 μmol | — | — | 330 ± 140 | N.D. |
| 2b | 40 μmol | 0.8 μmol | — | — | 360 ± 160 | N.D. |
| 2c | 40 μmol | 1.2 μmol | — | — | 320 ± 130 | N.D. |
| 3a | 40 μmol | 0.8 μmol | 4 μmol | — | 270 ± 105 | 18 |
| 4a | 40 μmol | 0.8 μmol | 4 μmol | 6 μmol | 280 ± 130 | 12 |
| 4b | 40 μmol | 0.8 μmol | 4 μmol | 15 μmol | 245 ± 100 | 15 |
| 5a | 40 μmol | — | — | 15 μmol | 250 ± 75 | 22 |
The modified particles bearing the IgG primary antibody (adsorbed and covalently attached) and the unmodified dye labeled 4b particles (negative control) were incubated with a fluorescently labeled secondary antibody F(ab′)2 goat anti-mouse IgG, Alexa Fluor 633 (Life Technologies GmbH), for 30 min at room temperature. Afterwards, the fluorescence intensity of the nanoparticles was measured by flow cytometry in a Cyflow ML (Partec GmbH), using a laser power of 200 mW. Nanoparticles were displayed via SCC and FL1 channel dot plot, which both axes were scaled logarithmically.
000g. Then, the particles were washed with 1 mL of PBS and centrifuged again under the same conditions. This purification step was repeated 3 times. The adsorbed proteins were eluted from the particles surface by re-suspending the particles in 300 μL of thiourea/urea lysis buffer (thiourea 2 M, urea 7 M, CHAPS 4% w/v in H2O). After a final centrifugation step (1 h, 20000g), the supernatant was recovered and used for SDS-PAGE analysis and protein quantification. Each experiment was performed 4 times and analyzed separately. Alternatively, the same procedure was repeated replacing the centrifugation steps by magnetic purification. The samples were placed over a strong magnet for 10 min and the supernatant removed. The amount of washing steps was kept constant and the elution of the proteins was carried out as described above.
For the measurements of nanoparticle–serum mixtures, 1 mL of concentrated FBS was filtered into the cuvette through a 0.2 μm pore size Millex-GS filter (Merck Millipore). To this solution were added 5 μL of each nanoparticle dispersion (solid content 0.5 wt%) and the mixtures where incubated at 37 °C for 1 h, followed by multi-angle DLS measurements at the same temperature. The pure FBS and the particle dispersions were measured separately under the same conditions and taken as references.
For the mixtures of particles and single protein solutions (BSA and lysozyme, dissolved in PBS), time resolved DLS experiments were performed. After addition of the appropriate concentration of protein to each nanoparticle suspension in PBS (ca. 104 protein molecules per particle) at room temperature, the solution was given a brief shake and quickly inserted into the index matching vat. Dynamic light scattering measurements were started immediately. The time required being the first DLS measurement was generally less than 20 seconds. The intensity of the auto-correlation function of dilute nanoparticle suspensions over time was measured in the presence and absence of protein. All light scattering measurements were conducted by employing the detector positioned at a scattering angle of 90° from the incident laser beam. The detector signal was fed into the correlator, which accumulated each auto-correlation function for between 10 s and 20 s. The intensity-weighted hydrodynamic radius of the nanoparticle aggregates was calculated by non-linear least squares fitting of the second order cumulants (ALV software). The measurements were performed over time periods of 10 min up to 1 h. Aggregation rate constants were calculated from the slope of the curve hydrodynamic radius vs. time. When no aggregation was observed, those suspensions were characterized as kinetically inert.
The estimation of the accessible alkene groups was carried out by a thiol–ene reaction with cyestamine (Fig. S3†), catalyzed with borax. First, 200 μL of the particle dispersion (3a, 4a–b) were mixed with 200 μL of an aqueous solution of cysteamine hydrochloride (1.0 mg mL−1), 60 μL of an aqueous borax solution (0.7 mg mL−1) and 540 μL of water. The mixture was shaken overnight at room temperature and washed with water three times by magnetic purification, to remove unreacted material. The dispersions were diluted to a final volume of 200 μL with water. This procedure renders an amino-functionalized surface, which can be titrated by the fluorescamine method, as described above.
The morphology of the particles was characterized by transmission electron microscopy (TEM), on a Jeol 1400 operating at 120 kV. For sample preparation, the particle dispersions were diluted with demineralized water, drop-casted on 300-mesh carbon-coated copper grids, and dried at room temperature. The thickness of the silica shell (and standard error) was assessed for ca. 100 particles, from micrographs taken in different areas of the samples.
The specific surface area of the particles was determined by nitrogen adsorption and desorption at 77.3 K using Autosorb-1 (Quantachrome Instruments), and calculated according to the BET model. Data were evaluated using QuadraWin software from Quantachrome Instruments. Samples were degassed at 150 °C for 24 h under vacuum before analysis.
Zeta potential measurements were performed on a Malvern Instruments Zeta Nanosizer. All samples were first diluted in aqueous 1.0 mM KCl solution. For the measurements at different pH values, aqueous 1 M solutions of NaOH and HCl were used to adjust the pH.
Average hydrodynamic diameters of the particles were determined by dynamic light scattering (DLS) at 20 °C at a fixed angle of 90° with a PSS Nicomp particle sizer 380. Thermogravimetric analysis (TGA) were performed on a Mettler-Toledo 851 thermobalance with temperatures ranging from 35 °C to 1000 °C at a heating rate of 10 °C min−1 under nitrogen flow (30 mL min−1).
The solid content of the dispersions was determined gravimetrically after freeze-drying of the dispersions. Gel permeation chromatography (GPC) was used to determine the molecular weights distributions with a PSS-SECcurity (Agilent Technologies 1260 Infinity). The freeze-dried samples were dissolved in tetrahydrofuran (THF) and eluted at a flow rate of 1.0 mL min−1 at 30 °C. Polystyrene standards were used for calibration.
Results and discussion
Silica-coating on magnetic Fe3O4/PS nanoparticles
Nanobiotechnology applications require the quantitative integration of multiple functionalities on a single nanocarrier.6 Consequently, a coating process is commonly used to give hydrophilicity to the particles and to introduce chemical functionality to their surfaces. Nanoparticles coated with silica present many advantages because the formed shell imparts water dispersibility and it acts as functionalizable protective layer for the core material. Herein, we have chosen a strongly magneto-responsive core because magnetic nanoparticles are used in magnetic resonance imaging and as biosensors.35,36,44 Additionally, higher versatility is attained by modulating the physicochemical surface properties of the nanocarriers after functionalization by silica coating. Strongly magneto-responsive polystyrene/iron oxide hybrid nanoparticles (hydrodynamic diameter Dh = 170 nm, standard deviation 55 nm), were obtained by miniemulsion polymerization, as reported elsewhere.42 Indeed, the miniemulsion process is a suitable technique for preparing nanocapsules by using droplets as templates.45,46 This technique allows the immobilization of a high content of magnetite (inorganic content 75% as determined by TGA) into the polymer matrix. To form a homogeneous silica shell around the magnetic nanospheres, their surfaces were modified with the amphiphilic non-ionic polymer polyvinylpyrrolidone. The adsorbed polymer layer enhanced the stability of the dispersion in non-aqueous solvents47 and promoted the formation of an uniform silica coating over the particles surface, acting as structure directing agent.48 Additionally, the use of polyoxyethylene (20) sorbitan monolaurate (Tween20) as surfactant ensured that the hydrolysis and condensation of TEOS took place over the surface of the particles.49 Therefore, secondary nucleation of silica was reduced, affording magnetic silica nanoparticles with core–shell morphology and homogeneous size distribution (Dh = 270 ± 95 nm). The composition of the particles before and after silica coating was estimated from TGA measurements. An increase of 26% in the inorganic content of the sample due to the introduction of the silica shell was detected. Furthermore, the shell thickness could be controlled, from 7 ± 3 nm (sample 1a) to 85 ± 5 nm (sample 1c), by changing the initial amount of TEOS and the pH of the reaction medium. For the subsequent experiments, the coating thickness was adjusted to 38 ± 2 nm (sample 1b) as determined by TEM (Fig. 1).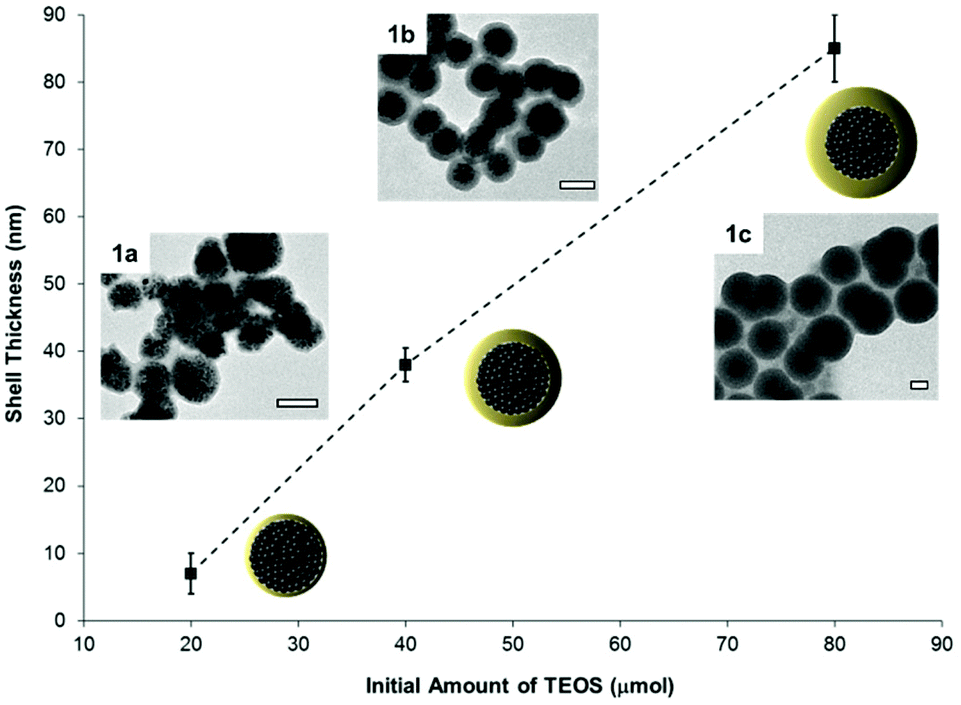 | ||
| Fig. 1 The thickness of the silica shell of the magnetic hybrid particles is controlled by the amount of TEOS for the synthesis. The insets show particles with shell thicknesses of 7 ± 3 nm (sample 1a), 38 ± 2 nm (sample 1b), and 85 ± 5 nm (sample 1c) as determined by TEM (scale bars 100 nm). | ||
Surface functionalization of the magnetic silica nanoparticles
The formed silica shell allows the introduction of several functional groups and therefore the subsequent grafting of different (bio)molecules onto the nanocarriers. The different functional groups were introduced on the surface of the magnetic carriers via hydrolysis and condensation of functionalized alkoxysilanes, yielding single- or multifunctionalized silica nanoparticles. The selected groups were amino, alkene, betaine (zwitterion), and a mixture of these functionalities (see Fig. 2 and Table 1).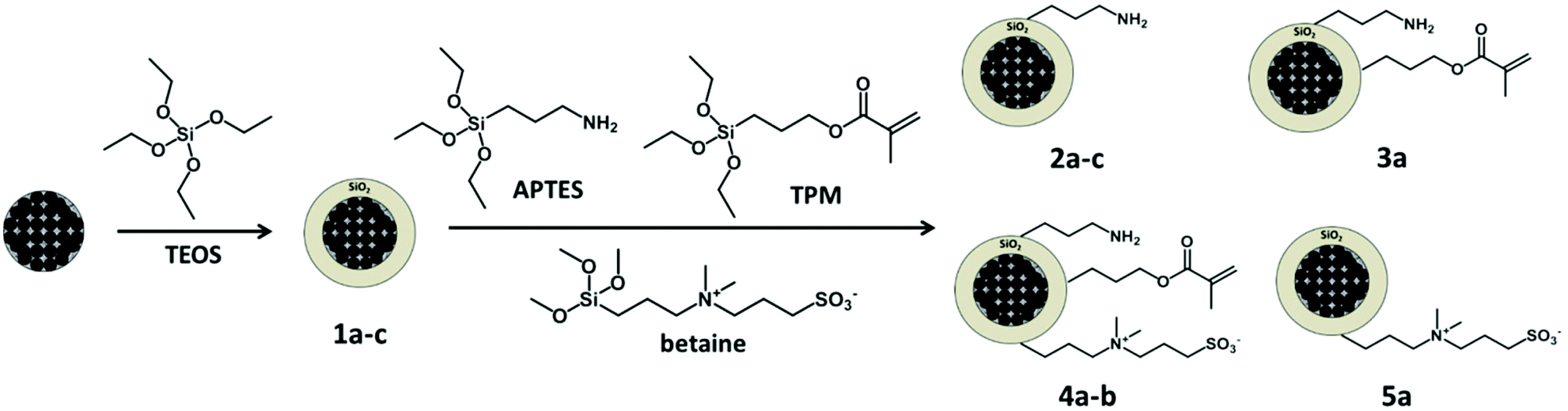 | ||
| Fig. 2 An initial silica shell is formed from TEOS around the hybrid particles. Afterwards, the amino, alkene and zwitterion functional groups are introduced into the silica surface using the APTES, TPM, and betaine organosilanes, respectively. Monofunctional (amino 2a–c, zwitterion 5a), bifunctional (amino/alkene 3a) and trifunctional (amino/alkene/zwitterion 4a–b) nanoparticles were synthesized. | ||
The functionalization of silica surfaces with these chemical groups is well documented in recent literature.8,9,32,48,50 However, the combination of these functionalities to yield a protein-repellent and yet orthogonally reactive surface has not been studied so far.
To confer a multiple but controlled functionalization ability to the nanoparticles, the chemical groups should fulfill several requirements. Ideally, they should not present cross reactivity with each other or interfere with the hydrolysis and condensation of the alkoxysilanes. Moreover, the functional groups must be prone to a wide range of orthogonal chemical modifications. We selected amino and alkene groups for model dual-functionalized particles (sample 3a). These chemical groups are not mutually reactive and allow post-functionalization by thiol–ene reactions8 and EDC coupling.29 Simultaneously, a sulfonate betaine derivative was chosen as alkoxysilane to introduce protein-repellent properties to the particles (samples 4a–b).
A post-grafting strategy was adopted to introduce the different chemical groups on the magnetic particles coated with silica 1b (Fig. 2). This functionalization ensures that the majority of the groups will be on the surface and thus accessible for chemical modification.
High reproducibility, as well as an effective modulation of the degree of functionalization was attained by this approach. Indeed, the surface charge of the amino functionalized particles 2a–c, determined by zeta potential, becomes more positive when a higher concentration of APTES is used for the grafting procedure (see Fig. S5†). As shown in Table 1, the degree of mono-, bi-, and tri-functionalization was varied without significant change in the physical characteristics of the particles such as morphology, size, and specific surface area. However, a precise quantification of the concentration of functional groups present on the surface of the nanoparticles is limited by the sensitivity of the spectrophotometric methods available for aqueous colloidal dispersions. Therefore, to have an estimation of the amount of surface-available amino groups on the nanoparticles, a titration with fluorescamine was performed (Fig. S3†). This methodology has been employed for the determination of low concentrations of amino acids and proteins which present primary amino groups.51 Some reports also describe the use of fluorescamine to estimate the number of amino functionality within aqueous colloidal dispersions.10,52–55 Fluorescamine is a non-fluorescent spiro-derivative that reacts selectively with primary amines under mild conditions. The product of the reaction is a stable highly fluorescent compound that offers a linear response that allows for the determination of the initial amino groups present in the sample.54 This strategy provided a rough assessment of reactive amino groups grafted to the surface of the particles. It is not possible to give absolute values from this titration since several factors such as different reactivity within polyamine species or quenching effects of the fluorophore cannot easily be accounted for. Nonetheless, we obtained semi-quantitative trends which could later be compared between the samples (Table S1†). These results were in good agreement with previously reported values of amino functional density on silica nanoparticles.54,55 On the other hand, the reactive alkene groups were determined with a borax-catalyzed thiol–ene reaction in water,56 in order to give an estimation of the available methacrylate groups on the particles surface. The alkene moieties were coupled with cysteamine to yield amino functional groups that can in turn, be titrated by the fluorescamine assay as described before (see Fig. S3 and Table S1†).
To prevent non-specific adsorption of biomolecules on the nanoparticles, their surfaces were functionalized with a short-chain zwitterion (see Fig. 2). Mixed multifunctional surfaces with (samples 4a–b, Table 1) and without (sample 5a, Table 1) reactive amino and alkene groups were prepared. It has been reported that zwitterionic coatings yield protein-repellent properties to the particles and also increase their colloidal stability in salt media.32–34 The resistance to colloidal aggregation, induced by the presence of salt, has been attributed to a tightly bound hydration layer formed by ionic solvation of the zwitterionic structure.34,57 Therefore, the effect of the ionic strength of the continuous phase of the dispersion on the particle size was investigated to corroborate the successful modification of the particles with the zwitterion. Samples 3a, 4a–b and 5a were suspended in different salt solutions and the evolution of the hydrodynamic diameter in time was monitored by dynamic light scattering (DLS). As shown in Fig. 3, the presence of the betaine group yielded stable particle dispersions in saline media. The salt present in the medium screened the electrostatic repulsion among the silica surfaces leading to aggregation of the nanoparticles of sample 3a. Conversely, the particles bearing zwitterion groups 4a–b and 5a did not aggregate when incubated in 100 mM NaCl and CaCl2 solutions (Fig. 3).
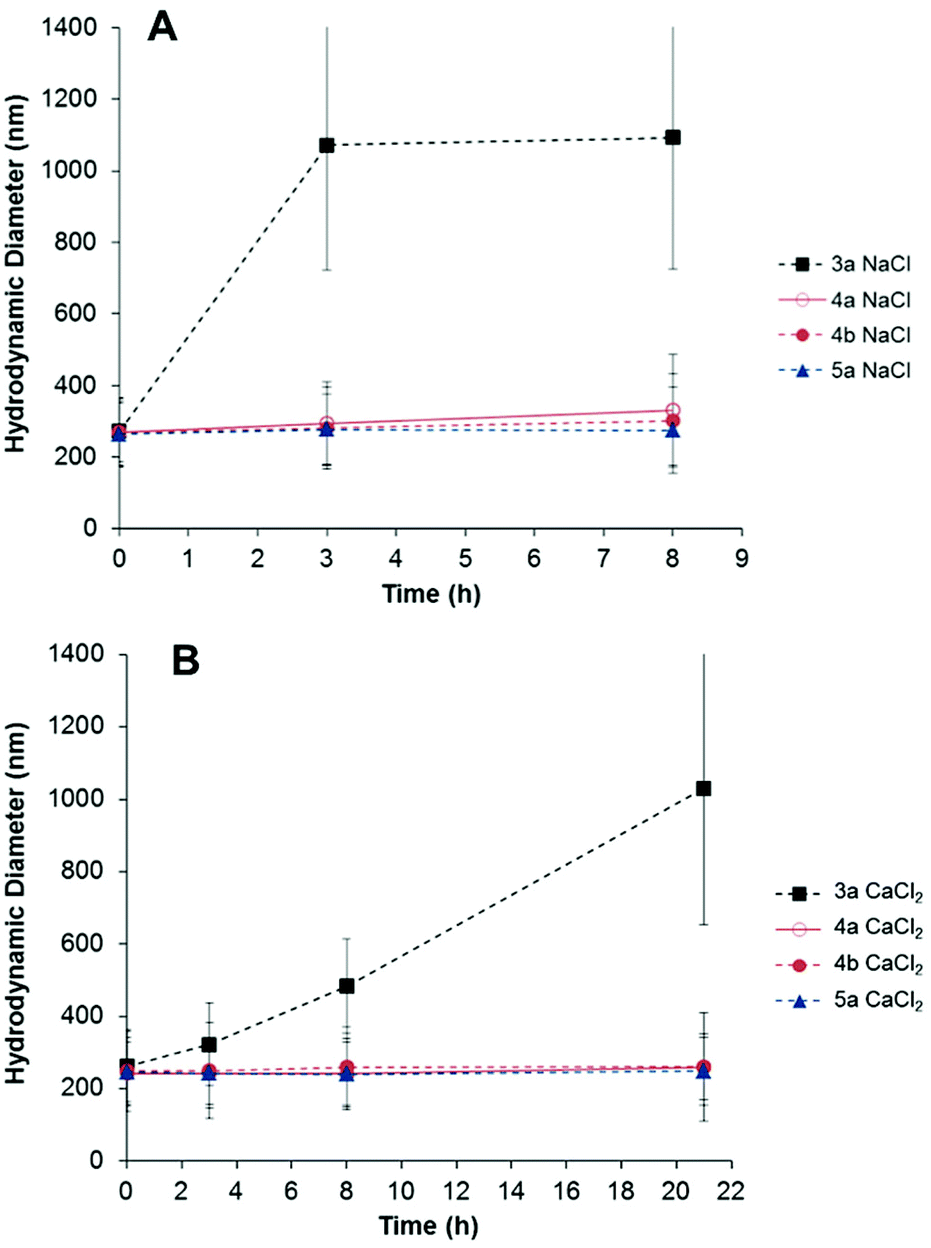 | ||
| Fig. 3 Temporal evolution of the hydrodynamic diameter of multifunctional particles with (4a–b, 5a) and without zwitterion (3a), dispersed in 100 mM NaCl (A) and CaCl2 (B) solutions. | ||
Prevention of non-specific protein adsorption
It is suggested that the protein-repellent properties of materials coated with zwitterions originates from the large number of tightly bound water molecules around the zwitterions. On the contrary to PEG that displays a hydration layer through hydrogen bonding, the zwitterionic chains are strongly hydrated via ionic solvation.57 A mechanism based on prevention of ion pairing between protein and zwitterionic surface, avoiding the release of counterions and concomitant gain of entropy, has been hypothesized.32 The multifunctional particles were incubated in a complex mixture of proteins (FBS) and in single protein solutions (BSA and lysozyme), in order to gain insights into the protein-repellency properties of mixed passivated/reactive surfaces.The multifunctional particles 3a, 4a–b and 5a (Table 1) were exposed to undiluted fetal bovine serum (FBS) and the quantification of the adsorbed proteins was carried out. Extensive washing by centrifugation and magnetic separation was applied to remove the excess of unbound proteins, as described in the Experimental section. Afterwards, the amount of strongly adsorbed proteins (hard protein corona) was determined spectro-photometrically (Pierce 660 nm assay) after eluting the proteins from the particles surface with urea/thiourea buffer. After normalizing the total amount of adsorbed protein to the nanoparticle surface area, the effect of the surface functionalization on the interaction between the serum proteins and the particles was investigated (Fig. 4A). The modification of the nanoparticles with zwitterionic structures decreased the affinity of the serum proteins. A clear reduction in the non-specific protein adsorption (ca. 65%) was reached for the particles 5a solely functionalized with the betaine, in comparison to the amino/alkene particles 3a. The samples with mixed functionality 4a and 4b displayed intermediate protein-repellent properties, which increased along with the amount of zwitterion grafted on the nanoparticles surface (see Fig. 4 and Table 1). These results demonstrate the possibility of decreasing the extent of non-specific protein adsorption onto a surface chemically functionalized with reactive amino and alkene groups. A slightly larger amount of adsorbed proteins was found for all the tested samples when magnetic purification was carried out in comparison with purification by centrifugation. Possibly, owing to the milder processing conditions exerted during the magnetic purification, some loosely bound proteins were not removed, yielding a higher amount of quantified proteins.
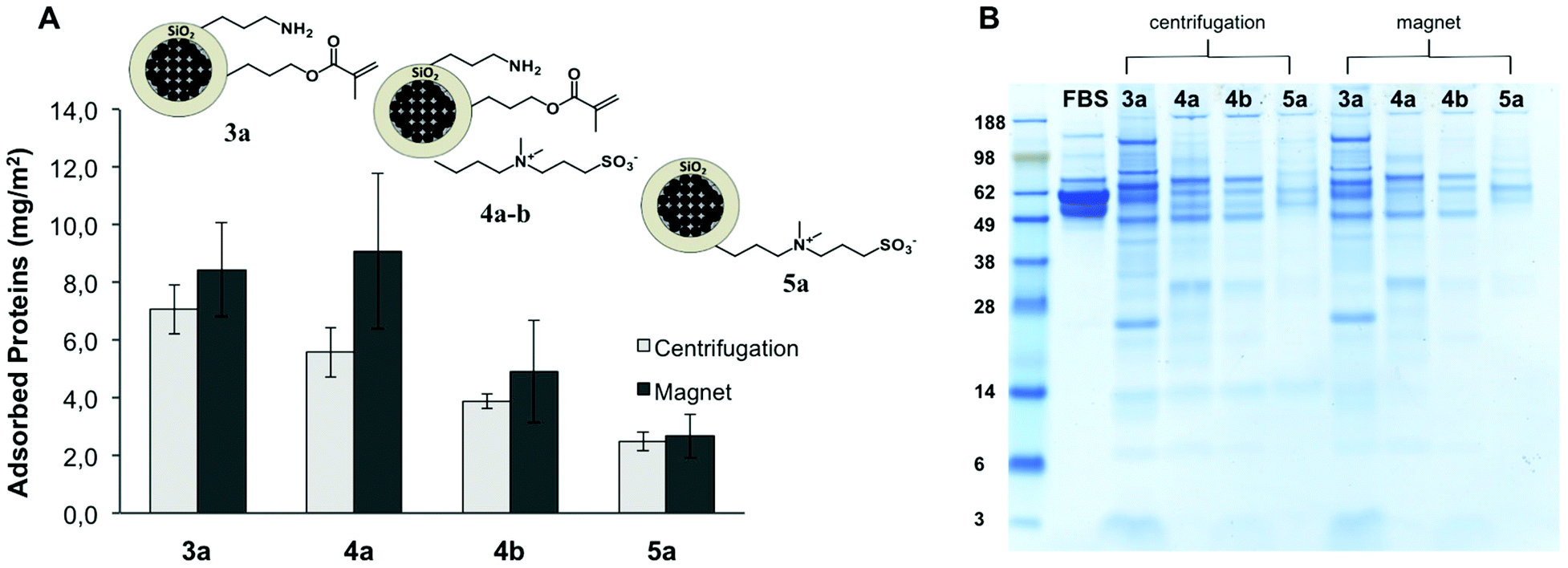 | ||
| Fig. 4 (A) Quantification of total amount of proteins from FBS adsorbed on the nanoparticles surface per area unit. (B) The profile of the composition of the adsorbed proteins was assessed by SDS-PAGE. The molecular weights (in kDa) of the standard marker on the left lane are reported for reference. | ||
The pattern of adsorbed proteins in the samples 3a, 4a–b, and 5a was analyzed by SDS-PAGE (Fig. 4B). With this technique, it is possible to determine a profile of the composition of the hard protein corona attached to the nanoparticles surface. From the amount and relative intensity of the observed bands in the gel after staining, it can be inferred that the particles having a higher amount of grafted zwitterion (4b, 5a) adsorbed not only less but also different kind of proteins, in comparison with the amino/alkene functional particles 3a. For all the studied samples, the most intense bands correspond to low molecular weight proteins (26–30 kDa) and some larger proteins (60–120 kDa). The determined protein patterns were in agreement with the reported composition of the hard protein corona formed on 200 nm silica particles incubated in concentrated plasma.58 By analogy with this previous study, the proteins that contributed more to the overall corona in our case probably corresponded to apolipoprotein A-I, fibrinogen, and histidine-rich glycoproteins.58 It is noteworthy to mention that the profile of the most abundant proteins in FBS (albumin, immunoglobulin) is significantly different from the distribution of proteins recovered from the particles (Fig. 4B). In fact, upon exposure of the nanoparticles to the serum, a dynamic process was elicited in which the different proteins competed for the available surface.17 Thus, the composition of the protein corona depends not only on the relative abundance of the proteins present in the biological medium, but also on their affinity for a certain surface. In accordance with other publications,16,59 we have found in our study that the pattern of adsorbed proteins was dictated primarily by the surface functionality, i.e. the physicochemical identity of the surface of the nanoparticle.
The behavior of the particles after incubation in FBS was also studied by DLS. The auto-correlation functions of the scattering intensity for the dispersion of particles, the serum, and the mixture of dispersion and serum were analyzed to check for the presence of aggregates upon incubation. When no aggregation takes place, the autocorrelation function of the mixture can be described by the sum of each individual auto-correlation functions.60
The results of these experiments revealed that no large aggregates of particles were formed upon incubation of any of the samples in undiluted FBS. Surprisingly, for the samples without (3a) and with a low amount of zwitterion (4a), no structures larger than the largest particle or serum component were formed after incubation (Fig. S6†). Although we observed previously the formation of aggregates larger than 1 μm when the particles were dispersed in saline media (see Fig. 3), the presence of proteins changes the aggregation behavior of the particles. A possible scenario can be described in terms of steric stabilization by the adsorbed protein corona, which prevents the aggregation of the particles. Previous studies have reported this phenomenon for functionalized silica particles.9,61 Nevertheless, the aggregation of plain silica particles in presence of FBS has been also observed,62 suggesting again that the specific physicochemical surface properties of the nanoparticles have different influence on their stability and the formation of the protein corona, after exposure to biological media.
BSA and lysozyme were chosen as model proteins for the adsorption studies from buffer on the multifunctional nanoparticles. These two biomolecules present opposite net charge at physiological pH with the reported isoelectric point (IEP) values of 4.8 and 11.1 for BSA and lysozyme, respectively.63 After incubation, the amount of adsorbed protein on the different functionalized particles 3a, 4a–b, and 5a was quantified spectro-photometrically. In the case of BSA, an evident reduction in the total protein adsorption by increasing the zwitterion content on the particles surface was observed, whereas the effect of the zwitterion was less pronounced with lysozyme (Fig. 5). This phenomenon can be explained by favorable electrostatic interactions between the negatively charged silica surface and the positively charged lysozyme, which is not the case for BSA.63
 | ||
| Fig. 5 Quantification of adsorption of BSA (A) and lysozyme (B) from PBS buffer per area unit of the different multifunctional nanoparticles. | ||
However, the overall protein adsorption is low (ca. five-fold less compared to FBS) for both cases. zeta-potential values were obtained for the nanoparticles before and after incubation with BSA and lysozyme. For the samples with and without zwitterion, this data show that the surface charges become more negative when BSA is present in the media, and more positive when lysozyme is used, (Fig. S7†). This corroborates that the proteins adsorb to the surface of the particles, changing their surface charge, as reported in recent literature.9,63 The net variation of the zeta-potential is less pronounced when the zwitterion is present on the particles surface for both cases indicating a barrier to protein adsorption.
Moreover, the functionalization of the silica surface with the betaine decreased the extent of total non-specific adsorption from both serum and single protein solution. This analysis confirms the observations from the SDS-PAGE experiments in which no significant amount of albumin was present in the hard corona. Although BSA was the most abundant protein present in serum, other proteins with higher affinity attached preferentially to the particles surface. When different proteins were present simultaneously in solution, the final composition of the protein layer was mainly comprised of proteins capable of irreversible adsorption. The population of the proteins in solution changed with time until those interacting irreversibly with the surface replaced the weakly adsorbed ones.58
Finally, the stability of the particles in both protein solutions was assessed by DLS. The intensity-weighted hydrodynamic radius (Rh) of the particles was determined in the presence and absence of protein (in phosphate buffer). The aggregation kinetics in the selected media was determined by measuring changes in Rh with time. As discussed before, the zwitterion modification over the particles enhanced their stability in saline media in comparison to the ones without zwitterion 3a (see Fig. 3). However, the presence of proteins prevented the aggregation of those particles in serum (see Fig. S6†). Therefore, it was interesting to study if also single protein solutions changed the aggregation behavior of the particles in PBS.
As shown in Fig. 6 (red plot), the sample 3a displayed an initial aggregation rate constant of 2.4 × 10−18 m3 s−1, calculated from the slope of the curve, in the phosphate buffer (ionic strength 166 mM). However, when BSA was present in the medium, the slope decreased (black symbols in Fig. 6), indicating a lower initial rate constant of aggregation (1.3 × 10−18 m3 s−1). Most probably, the BSA adsorbed to the particles surface provides a measure of steric stabilization, as already observed in serum.
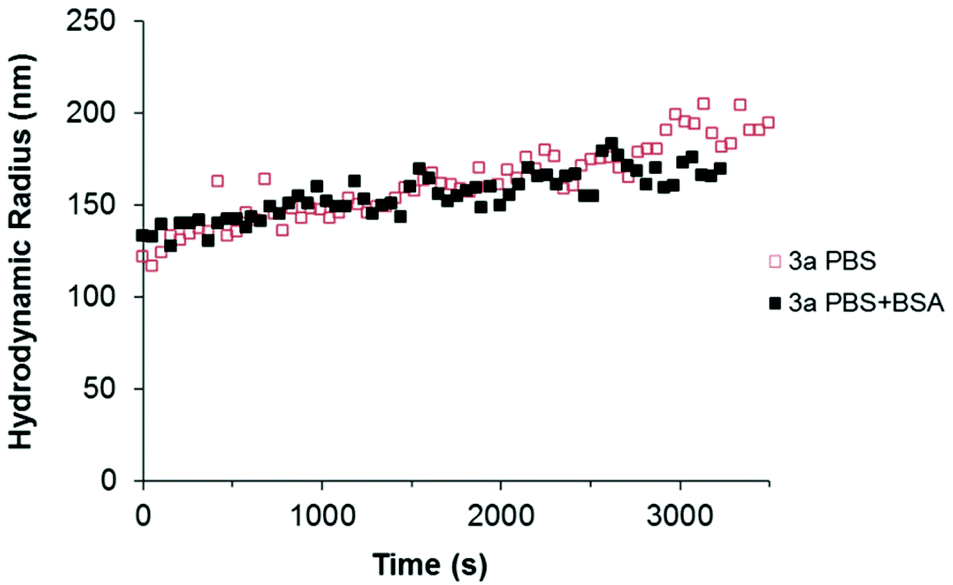 | ||
| Fig. 6 Time resolved dynamic light scattering of dilute 3a nanoparticle suspensions in the presence (solid black symbols) and absence (open red symbols) of BSA. The intensity-weighted RH of the nanoparticle aggregates was calculated by non-linear least squares fitting of the second order cumulants (ALV software). | ||
None of the particles 4a–b and 5a showed evident aggregation in the buffer solution or in the presence of protein (BSA and lysozyme). These samples were characterized as kinetically inert (Fig. S8†). Remarkably, the aggregation of the particles 3a in the buffer was completely suppressed by the adsorption of lysozyme. A steric stabilization due to the adsorbed lysozyme can be suggested in order to explain this phenomenon.
Bio-functionalization of the zwitterion modified particles
Although protein-repellent properties were suitable and achieved in our study, it is important that the functionalization ability of the particles for bio-sensing, recognition and targeting purposes is retained.5 Therefore, bio-orthogonally addressable reactions are convenient strategies for affording nanoparticle bioconjugates in a controlled manner.7 To demonstrate the accessibility of the reactive orthogonal groups present in the mixed particles functionalized with the zwitterions, a biomolecule was covalently attached to the surface by click chemistry. As proof of principle, mouse IgG antibody was selected as target biomolecule to be chemically immobilized on the surface of the multifunctional particles 4b. This sample was chosen due to the protein repellency displayed by the particles (see Fig. 4 and 5) along with the presence of reactive amino and alkene functional groups on their surface (see Table S1†).Measurements on temporal evolution of the hydrodynamic diameter of the particles upon incubation with IgG show that the zwitterionic multifunctional colloids 4b remain stable in the presence of the antibody. Conversely, the particles without zwitterion 3a aggregate over time as displayed in Fig. 7A. A comparison between the aggregation rate of the sample 3a in phosphate buffer and in buffer plus antibody reveals that the IgG delays the rate in which the mean diameter of the particles increase (Fig. 7A), suggesting the adsorption of the antibody on their surface which results in steric stabilization of the colloid suspension. Moreover, the strong variation on the Z-potential values obtained after incubation with IgG, support the scenario of adsorption of the antibody on the surface of the particles 3a but not onto the zwitterionic particles 4b (Fig. 7B).
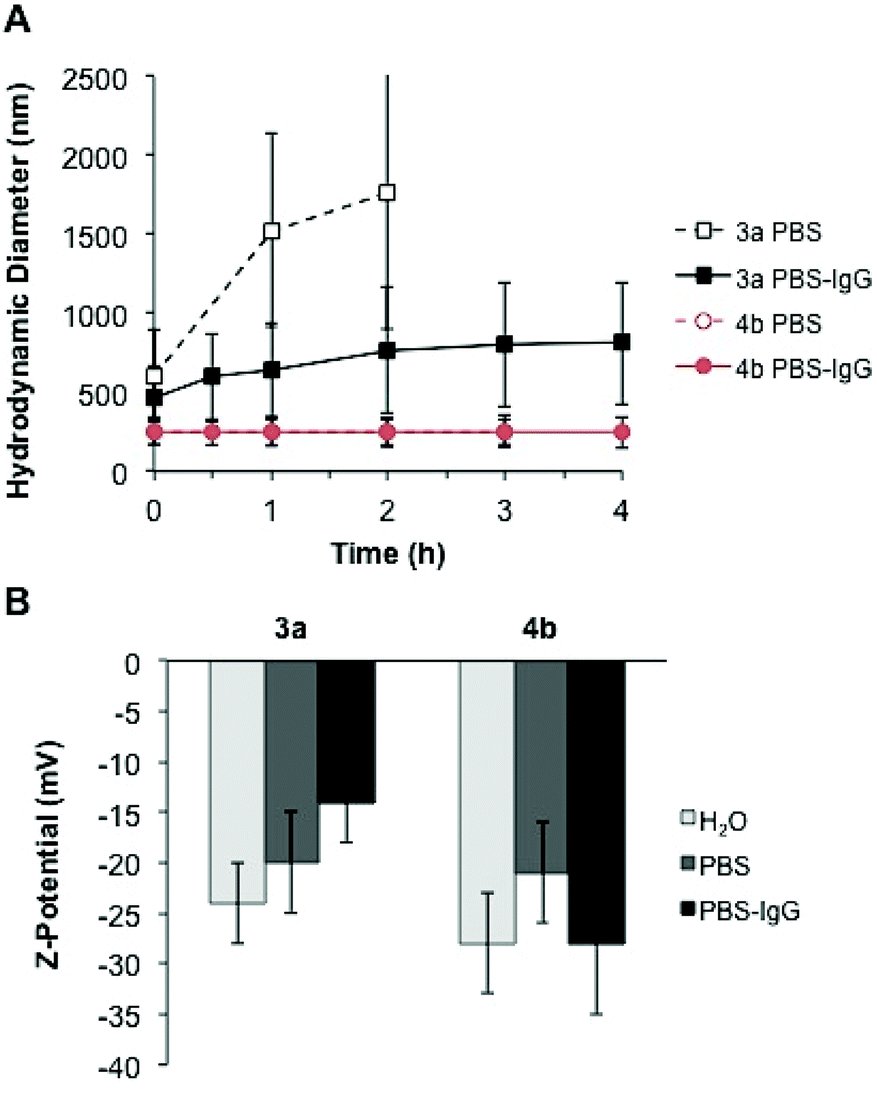 | ||
| Fig. 7 (A) Temporal evolution of the hydrodynamic diameter of particles 3a and 4b, dispersed in phosphate buffer (open symbols) and phosphate buffer plus IgG (solid symbols). (B) Z-potential values of the particles before and after incubation with IgG. | ||
The adopted strategy for the covalent bio-functionalization of these particles includes (i) condensation of a NHS ester derivative of DBCO (strained alkyne) to the amino groups, and (ii) copper free alkyne–azide cycloaddition to the introduced DBCO units using an azide-modified mouse IgG antibody (Fig. 8). This methodology allows orthogonal bio-functionalization of the particles and leaves available the alkene moiety of the methacrylate group for further modification via thiol–ene chemistry.
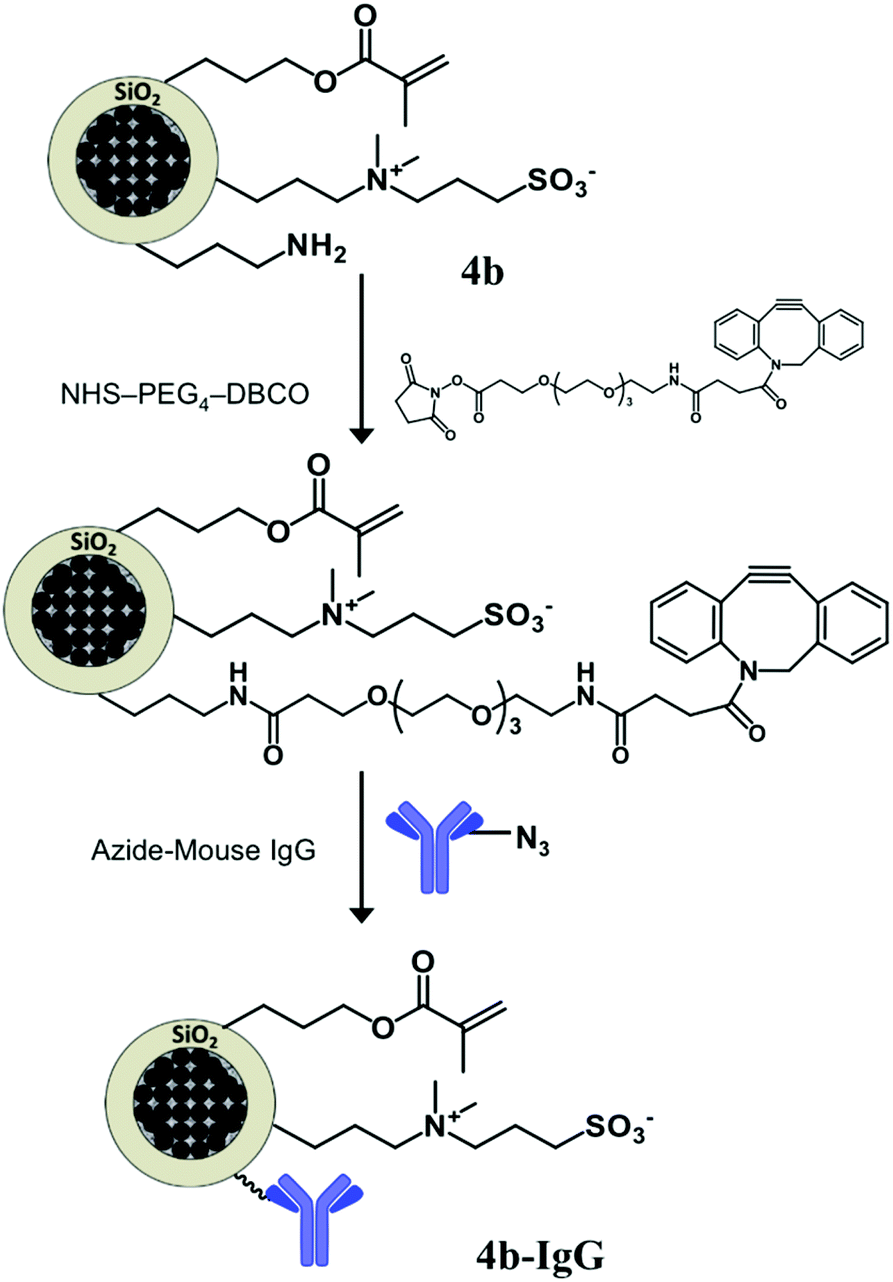 | ||
| Fig. 8 Modification of the multifunctional zwitterion-bearing particles 4b with a strained alkyne (DBCO) that allows the covalent immobilization of an antibody (mouse IgG) by means of alkyne–azide click chemistry, along with a reduced non-specific adsorption over the particles surface. | ||
The effective functionalization with the antibody was assessed by flow cytometry measurements. The dye-labeled particles 4b and 4b-IgG (see Fig. 8) were incubated with a fluorescent secondary antibody, F(ab′)2 goat anti-mouse IgG (H + L), which was attached specifically to the mouse IgG macromolecule.
By measuring the fluorescence intensity of the particles, it is possible to recognize if the antibody is present on the particles surface. The median fluorescence intensity determined for the bio-functionalized 4b-IgG particles was higher than for the control sample 4b, indicating the presence of the IgG antibody on the surface of the particles (Fig. 9).
 | ||
| Fig. 9 Fluorescence intensity of the particles functionalized covalently with mouse IgG (4b-IgG) and with physically adsorbed IgG, determined by flow cytometry. Unmodified 4b particles were employed as negative control. | ||
Additionally, the particles 4b were incubated in mouse IgG antibody (physical adsorption) and analyzed by flow cytometry. The measured fluorescence intensity for this sample was also lower than the one for the covalently attached antibody. These results demonstrated a reduction in the non-specific protein adsorption, preserving at the same time the functionalization ability of the multifunctional nanoparticles. Nonetheless, future work should be devoted to a detailed study of the properties and performance of the bio-modified particles, to assess their potential application in bio-sensing and molecular recognition.
Conclusions
Highly magnetic iron oxide/polystyrene nanoparticles were synthesized and coated with a silica shell. Subsequently, by surface grafting of organosilanes, the particles were simultaneously functionalized with amino, alkene, and betaine (zwitterion) groups. The presence of the zwitterion on the surface of the nanoparticles played an important role in their stability in salt media and their interaction with biomolecules. After incubation in fetal bovine serum and solutions of serum albumin and lysozyme, the particles bearing higher amounts of zwitterion on the surface displayed low non-specific protein adsorption (up to 65% less in comparison with the particles without zwitterion). Remarkably, the introduction of the protein-repellent moieties on the particles surface did not hinder the accessibility of the reactive amino and alkene groups. Indeed, the particles could be selectively bio-functionalized with a mouse IgG antibody by click chemistry. Flow cytometry measurements revealed that the biomolecule was covalently attached to the surface of the nanoparticles, and the extent of physical adsorption was reduced. Therefore, the presented methodology comprised the parallel introduction of protein-repellant groups and orthogonally addressable moieties within the same surface. In this way, the physical and chemical identity of the nanocarriers can be carefully modulated, allowing us to assess the impact of surface functionalization on protein binding.Acknowledgements
The authors gratefully thank Manuel Tonigold, Johanna Simon and Christine Rosenauer for the flow cytometry, SDS-PAGE and light scattering analyses, respectively. RME acknowledges the financial support from the Spanish Ministry of Economy and Competitiveness through a Ramón y Cajal grant (grant no. RYC-2013-13451).Notes and references
- P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 1247390 CrossRef PubMed.
- S. Kroll, R. Macrez, F. Docagne, G. Defer, S. Laurent, M. Rahman, M. J. Hajipour, P. G. Kehoe and M. Mahmoudi, Chem. Rev., 2013, 113, 1877 CrossRef PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS PubMed.
- G. Bao, S. Mitragotri and S. Tong, Annu. Rev. Biomed. Eng., 2013, 15, 253 CrossRef CAS PubMed.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028 RSC.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825 CrossRef CAS PubMed.
- A. T. Dickschat, F. Behrends, M. Bühner, J. Ren, M. Weiß, H. Eckert and A. Studer, Chem. – Eur. J., 2012, 18, 16689 CrossRef CAS PubMed.
- S. Shahabi, L. Treccani, R. Dringen and K. Rezwan, ACS Appl. Mater. Interfaces, 2015, 7, 13821 CAS.
- J. Fickert, P. Rupper, R. Graf, K. Landfester and D. Crespy, J. Mater. Chem., 2012, 22, 2286 RSC.
- J. Fickert, D. Schaeffel, K. Koynov, K. Landfester and D. Crespy, Colloid Polym. Sci., 2014, 292, 251 CAS.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S.-Y. Lin, Adv. Funct. Mater., 2007, 17, 1225 CrossRef CAS.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590 RSC.
- T. Kim, E. Momin, J. Choi, K. Yuan, H. Zaidi, J. Kim, M. Park, N. Lee, M. T. McMahon, A. Quinones-Hinojosa, J. W. M. Bulte, T. Hyeon and A. A. Gilad, J. Am. Chem. Soc., 2011, 133, 2955 CrossRef CAS PubMed.
- D. K. Yi, T. Selvan, S. S. Lee, G. C. Papaefthymiou, D. Kundaliya and J. Y. Ying, J. Am. Chem. Soc., 2005, 127, 4990 CrossRef CAS PubMed.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772 CrossRef CAS PubMed.
- V. Mirshafiee, M. Mahmoudi, K. Lou, J. Cheng and M. L. Kraft, Chem. Commun., 2013, 49, 2557 RSC.
- Z. W. Lai, Y. Yan, F. Caruso and E. C. Nice, ACS Nano, 2012, 6, 10438 CrossRef CAS PubMed.
- K. Feldman, G. Hähner, N. D. Spencer, P. Harder and M. Grunze, J. Am. Chem. Soc., 1999, 121, 10134 CrossRef CAS.
- K. L. Prime and G. M. Whitesides, Science, 1991, 252, 1164 CrossRef CAS PubMed.
- P. Kingshott and H. J. Griesser, Curr. Opin. Solid State Mater. Sci., 1999, 4, 403 CrossRef CAS.
- K. Pombo García, K. Zarschler, L. Barbaro, J. A. Barreto, W. O'Malley, L. Spiccia, H. Stephan and B. Graham, Small, 2014, 10, 2516 CrossRef PubMed.
- R. S. Kane, P. Deschatelets and G. M. Whitesides, Langmuir, 2003, 19, 2388 CrossRef CAS.
- K. G. Neoh and E. T. Kang, Polym. Chem., 2011, 2, 747 RSC.
- M. Noga, D. Edinger, R. Kläger, S. V. Wegner, J. P. Spatz, E. Wagner, G. Winter and A. Besheer, Biomaterials, 2013, 34, 2530 CrossRef CAS PubMed.
- S. Chen and S. Jiang, Adv. Mater., 2008, 20, 335 CrossRef CAS.
- S. Chen, J. Zheng, L. Li and S. Jiang, J. Am. Chem. Soc., 2005, 127, 14473 CrossRef CAS PubMed.
- Y. Zhu, H. S. Sundaram, S. Liu, L. Zhang, X. Xu, Q. Yu, J. Xu and S. Jiang, Biomacromolecules, 2014, 15, 1845 CrossRef CAS PubMed.
- G. Chang, H. Xue, Z. Zhang, S. Chen and S. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8831 CrossRef PubMed.
- A. K. Murthy, R. J. Stover, W. G. Hardin, R. Schramm, G. D. Nie, S. Gourisankar, T. M. Truskett, K. V. Sokolov and K. P. Johnston, J. Am. Chem. Soc., 2013, 135, 7799 CrossRef CAS PubMed.
- Z. G. Estephan, P. S. Schlenoff and J. B. Schlenoff, Langmuir, 2011, 27, 6794 CrossRef CAS PubMed.
- J. E. Rosen and F. X. Gu, Langmuir, 2011, 27, 10507 CrossRef CAS PubMed.
- F. Hu, K. Chen, H. Xu and H. Gu, Colloids Surf., B, 2015, 126, 251 CrossRef CAS PubMed.
- X. Li, B. Zhang, L. Tian, W. Li, H. Zhang and Q. Zhang, Sens. Actuators, B, 2015, 208, 559 CrossRef CAS.
- Z.-A. Lin, J.-N. Zheng, F. Lin, L. Zhang, Z. Cai and G.-N. Chen, J. Mater. Chem., 2011, 21, 518 RSC.
- S. Jiang and Z. Cao, Adv. Mater., 2010, 22, 920 CrossRef CAS PubMed.
- T. L. Lasseter, B. H. Clare, N. L. Abbott and R. J. Hamers, J. Am. Chem. Soc., 2004, 126, 10220 CrossRef CAS PubMed.
- I. M. Rio-Echevarria, F. Selvestrel, D. Segat, G. Guarino, R. Tavano, V. Causin, E. Reddi, E. Papini and F. Mancin, J. Mater. Chem., 2010, 20, 2780 RSC.
- I. García-Moreno, A. Costela, L. Campo, R. Sastre, F. Amat-Guerri, M. Liras, F. López Arbeloa, J. Bañuelos Prieto and I. López Arbeloa, J. Phys. Chem. A, 2004, 108, 3315 CrossRef.
- D. Schaeffel, R. H. Staff, H.-J. Butt, K. Landfester, D. Crespy and K. Koynov, Nano Lett., 2012, 12, 6012 CrossRef CAS PubMed.
- M. B. Bannwarth, S. Utech, S. Ebert, D. A. Weitz, D. Crespy and K. Landfester, ACS Nano, 2015, 9, 2720 CrossRef CAS PubMed.
- M. B. Bannwarth, S. W. Kazer, S. Ulrich, G. Glasser, D. Crespy and K. Landfester, Angew. Chem., Int. Ed., 2013, 52, 10107 CrossRef CAS PubMed.
- J. Thevenot, H. Oliveira, O. Sandre and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 7099 RSC.
- L.-P. Lv, Y. Zhao, N. Vilbrandt, M. Gallei, A. Vimalanandan, M. Rohwerder, K. Landfester and D. Crespy, J. Am. Chem. Soc., 2013, 135, 14198 CrossRef CAS PubMed.
- J. Fickert, C. Wohnhaas, A. Turshatov, K. Landfester and D. Crespy, Macromolecules, 2013, 46, 573 CrossRef CAS.
- C. Graf, D. L. J. Vossen, A. Imhof and A. van Blaaderen, Langmuir, 2003, 19, 6693 CrossRef CAS.
- Y. Piao, A. Burns, J. Kim, U. Wiesner and T. Hyeon, Adv. Funct. Mater., 2008, 18, 3745 CrossRef CAS.
- H. Xu, L. Cui, N. Tong and H. Gu, J. Am. Chem. Soc., 2006, 128, 15582 CrossRef CAS PubMed.
- Y. Chen, M. Wu, K. Wang, B. Chen, S. Yao, H. Zou and L. Nie, J. Chromatogr., A, 2011, 1218, 7982 CrossRef CAS PubMed.
- S. Udenfriend, S. Stein, P. Böhlen, W. Dairman, W. Leimgruber and M. Weigele, Science, 1972, 178, 871 CAS.
- F. Ganachaud, G. Mouterde, T. Delair, A. Elaissari and C. Pichot, Polym. Adv. Technol., 1995, 6, 480 CrossRef CAS.
- G. Deng, M. A. Markowitz, P. R. Kust and B. P. Gaber, Mater. Sci. Eng., C, 2000, 11, 165 CrossRef.
- Y. Cheng and Y. Zhang, Anal. Bioanal. Chem., 2011, 399, 2503 CrossRef PubMed.
- O. M. Evbuomwan, M. E. Merritt, G. E. Kiefer and D. Sherry, Contrast Media Mol. Imaging, 2012, 7, 19 CrossRef CAS PubMed.
- S. Hussain, S. K. Bharadwaj, M. K. Chaudhuri and H. Kalita, Eur. J. Org. Chem., 2007, 374 CrossRef CAS.
- Y. He, J. Hower, S. Chen, M. T. Bernards, Y. Chang and S. Jiang, Langmuir, 2008, 24, 10358 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525 CrossRef CAS PubMed.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailänder, Biomacromolecules, 2015, 16, 1311 CrossRef CAS PubMed.
- K. Rausch, A. Reuter, K. Fischer and M. Schmidt, Biomacromolecules, 2010, 11, 2836 CrossRef CAS PubMed.
- F. Catalano, L. Accomasso, G. Alberto, C. Gallina, S. Raimondo, S. Geuna, C. Giachino and G. Martra, Small, 2015, 11, 2919 CrossRef CAS PubMed.
- B. Díaz, C. Sánchez-Espinel, M. Arruebo, J. Faro, E. de Miguel, S. Magadán, C. Yagüe, R. Fernández-Pacheco, M. R. Ibarra, J. Santamaría and Á. González-Fernández, Small, 2008, 4, 2025 CrossRef PubMed.
- F. Turci, E. Ghibaudi, M. Colonna, B. Boscolo, I. Fenoglio and B. Fubini, Langmuir, 2010, 26, 8336 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and additional experimental light scattering and zeta-potential data. See DOI: 10.1039/c5nr08258g |
| This journal is © The Royal Society of Chemistry 2016 |