DOI:
10.1039/C5NR08163G
(Paper)
Nanoscale, 2016,
8, 8101-8110
Genetically designed biomolecular capping system for mesoporous silica nanoparticles enables receptor-mediated cell uptake and controlled drug release†
Received
18th November 2015
, Accepted 14th February 2016
First published on 17th February 2016
Abstract
Effective and controlled drug delivery systems with on-demand release and targeting abilities have received enormous attention for biomedical applications. Here, we describe a novel enzyme-based cap system for mesoporous silica nanoparticles (MSNs) that is directly combined with a targeting ligand via bio-orthogonal click chemistry. The capping system is based on the pH-responsive binding of an aryl-sulfonamide-functionalized MSN and the enzyme carbonic anhydrase (CA). An unnatural amino acid (UAA) containing a norbornene moiety was genetically incorporated into CA. This UAA allowed for the site-specific bio-orthogonal attachment of even very sensitive targeting ligands such as folic acid and anandamide. This leads to specific receptor-mediated cell and stem cell uptake. We demonstrate the successful delivery and release of the chemotherapeutic agent Actinomycin D to KB cells. This novel nanocarrier concept provides a promising platform for the development of precisely controllable and highly modular theranostic systems.
Introduction
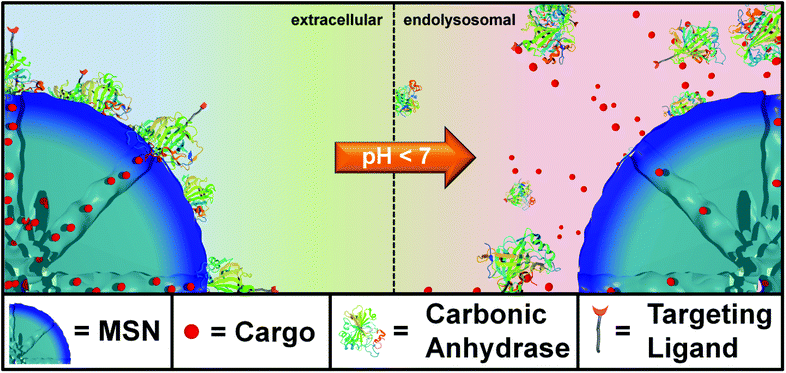
The development of effective systems for targeted drug delivery combined with on demand release behavior can be considered one of the grand challenges in nanoscience. In particular, porous nanocarriers with high drug loading capacity, immunological stealth behavior and tunable surface properties are promising candidates for biomedical applications such as cancer therapy and bioimaging.1–5 Specifically, multifunctional mesoporous silica nanoparticles (MSNs) have great potential in drug delivery applications due to their attractive porosity parameters and the possibility to conjugate release mechanisms for diverse cargos6,7 including gold nanoparticles,8,9 iron oxide nanocrystals,10 bio-macromolecules,11,12 enzymes,13 and polymers.14 Control over a stimuli-responsive cargo release can be achieved via different trigger mechanisms such as redox reactions,15 pH changes,16 light-activation,6,17 or change in temperature.7 Drug delivery vehicles equipped with acid-sensitive capping mechanisms are highly desirable for acidified target environments such as the transition from early to late endosomes, tumors, or inflammatory tissues.
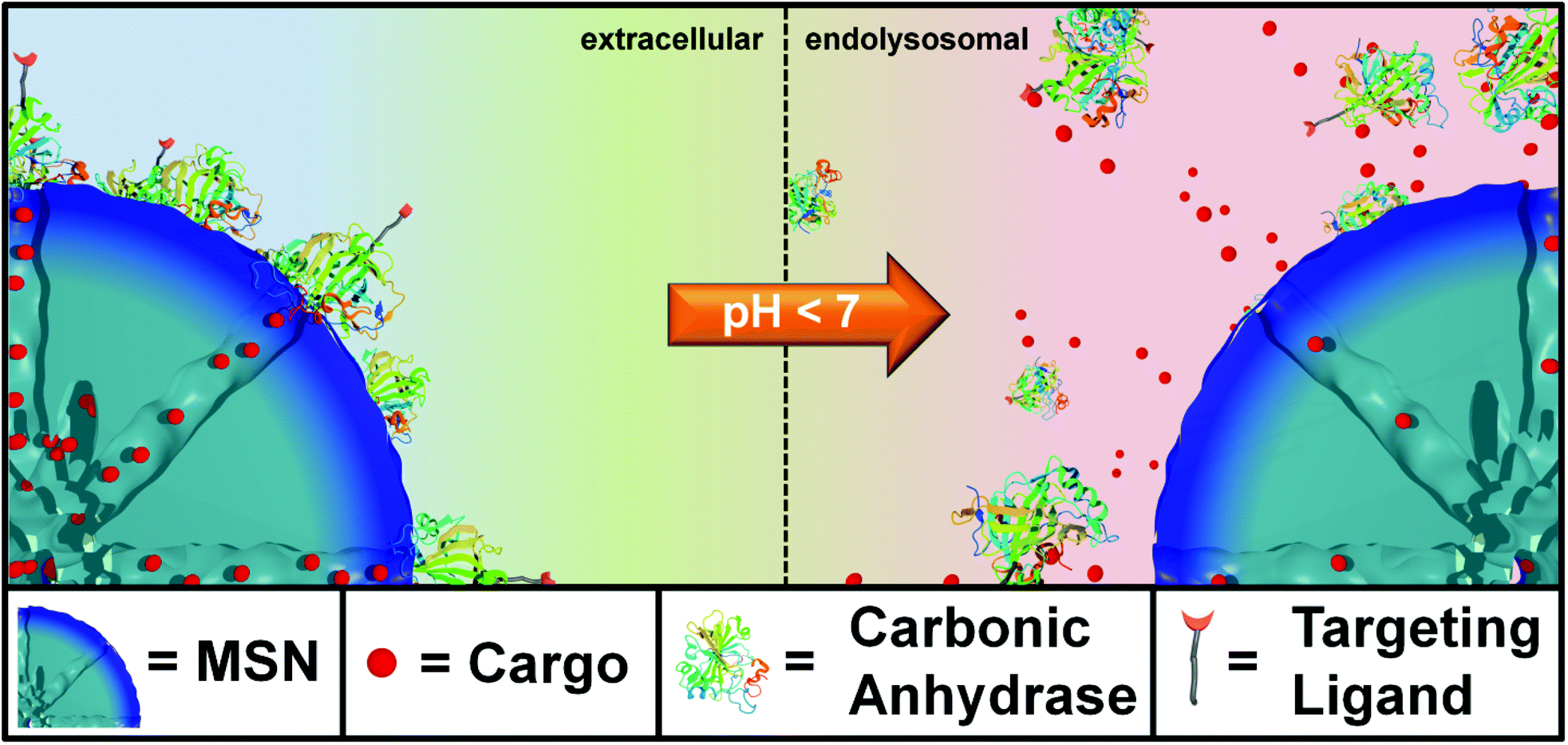
Here, we present genetically designed enzyme-capped MSNs that combine two important prerequisites for advances in drug delivery, namely stimuli-responsive drug release and specific cell targeting (Scheme 1). Specifically, these pH-responsive MSNs consist of a capping structure based on carbonic anhydrase (CA). CA is a model enzyme abundant in humans and animals and generally catalyzes the hydration of carbon dioxide and the dehydration of bicarbonate.18 It is attached to the silica nanoparticle surface via aryl sulfonamide groups. Acting as inhibitors, sulfonamide groups strongly bind to the active site of the CA. This enzyme-sulfonamide binding is reversible depending on the pH, where an acidic medium causes protonation of the sulfonamide, resulting in cleavage of the coordination bond and access to the porous system.19 The CA gatekeepers were used to exploit the endosomal pH change as an internal cellular trigger and to gain control over the release of cargo molecules from the mesoporous system.
 |
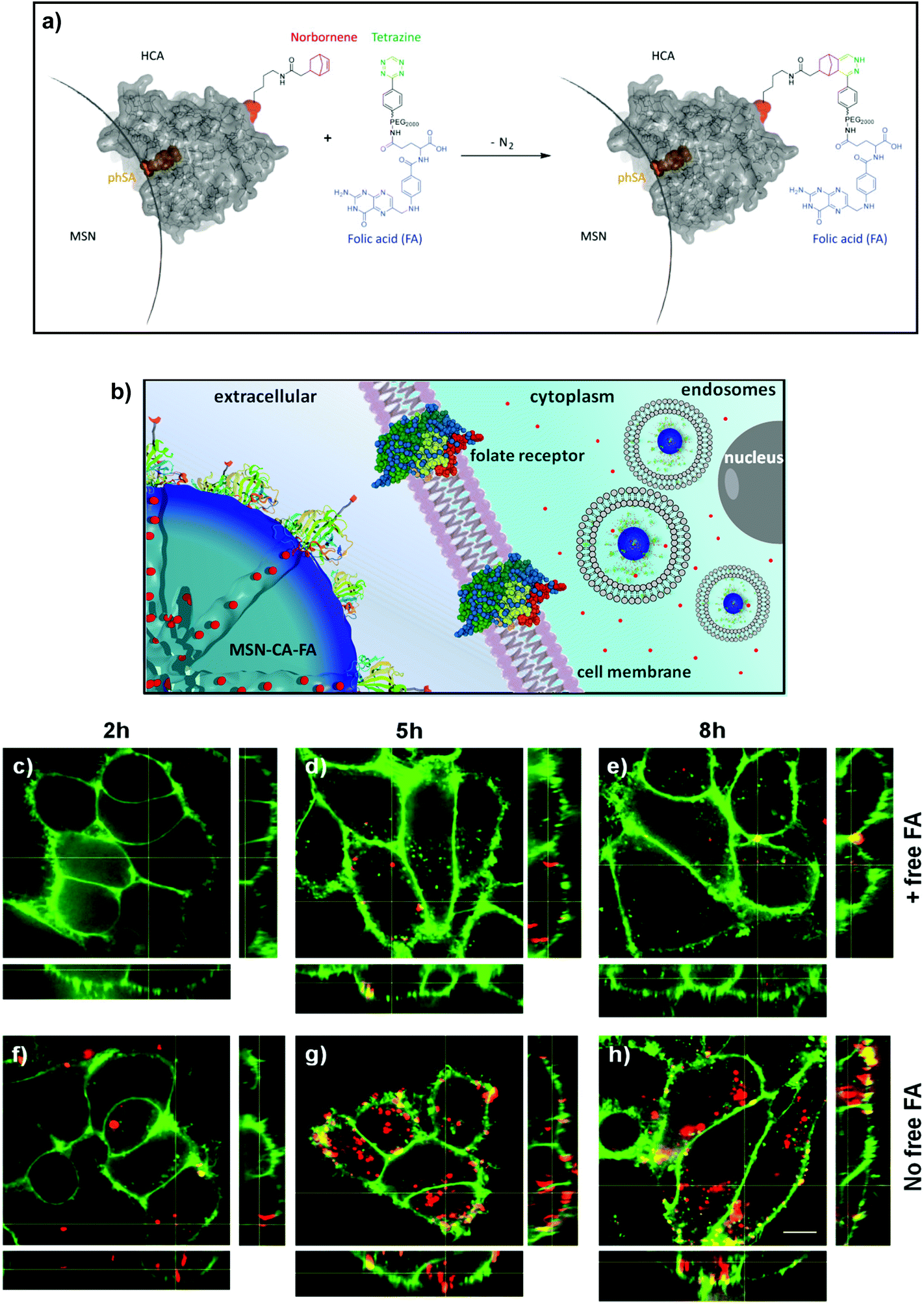
| | Scheme 1 Schematic illustration of the genetically designed biomolecular pore gating system providing a pH-responsive drug release from mesoporous silica nanoparticles (MSNs). Aryl sulfonamide functionalized MSNs offer pH-dependent reversible attachment of the bulky enzyme carbonic anhydrase, which efficiently blocks the pore entrances to prevent premature cargo release. Furthermore, specific cancer cell targeting can be achieved via site-specific modification of a genetically incorporated norbornene amino acid in the biomolecular gatekeepers. | |
This stimuli-responsive capping system on MSNs was combined with cell targeting specificity via a bio-orthogonal click chemistry approach. Targeting ligands provide specific binding to certain cell membrane receptors allowing for an enhanced and distinctive cellular uptake of such modified nanocarriers. For example, various cell receptors are overexpressed on cancer cells, which can lead to a preferential receptor-mediated endocytosis of modified MSNs. For the attachment of such targeting ligands exclusively to the outer periphery of the enzyme gatekeepers, we exploited a recently developed method that takes advantage of the pyrrolysine amber suppression system followed by bio-orthogonal copper-free click chemistry.20–22 This system has already been utilized in applications such as optical gene control.23 To the best of our knowledge, this is the first time the pyrrolysine amber suppression system is used in a combination with porous nanocarriers for specific cell recognition and drug delivery. The incorporation of an unnatural amino acid (UAA) containing a norbornene moiety into CA provides a bio-orthogonal reaction pathway by covalently attaching tetrazine-modified targeting ligands.24,25 It has recently been demonstrated that norbornene–tetrazine click chemistry is a favorable synthesis strategy over various other methods including thiol–maleimide reaction and amide formation due to extremely mild and biocompatible reaction conditions and higher selectivity.26 Here, copper-free click chemistry of norbornene-modified human carbonic anhydrase II with targeting ligands was performed to prepare folate- and anandamide-modified multifunctional mesoporous silica nanocarriers.27 The anandamide is, due to the cis-configured double bonds, a particularly sensitive receptor ligand that requires extremely mild coupling conditions. The targeting system based on folate-modified silica nanocarriers was studied on KB cancer cells, which are known to overexpress the folate receptor FR-α.6,28 The targeting system based on anandamide-modified particles was tested on neural stem cells and A431 cells. The combination of on-demand release and specific receptor-mediated cell uptake properties within one multifunctional mesoporous silica nanocarrier system, containing biomolecular valves based on carbonic anhydrase, is anticipated to offer promising potential for controlled drug delivery applications including cancer therapy.
Experimental part
Synthesis of thiol-functionalized MSNs (MSN-SH)
A mixture of TEOS (1.92 g, 9.22 mol) and TEA (14.3 g, 95.6 mmol) was heated to 90 °C for 20 min under static conditions in a polypropylene reactor. Then, a preheated (60 °C) mixture of CTAC (2.41 mL, 1.83 mmol, 25% in H2O) and NH4F (100 mg, 0.37 mmol) in bidistilled H2O (21.7 g, 1.21 mol) was added and the resulting reaction mixture was stirred vigorously (700 rpm) for 30 min while cooling down to room temperature. Afterwards, TEOS (18.2 mg, 92 μmol) and MPTES (18.1 mg, 92 μmol) were premixed briefly before addition to the reaction mixture. The final reaction mixture was stirred over night at room temperature. After dilution with absolute ethanol (100 mL), the nanoparticles were collected by centrifugation (19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 43146 rcf, 20 min) and redispersed in absolute ethanol. Template extraction was performed in an ethanolic solution of MSNs (100 mL) containing NH4NO3 (2 g) which was heated at reflux conditions (90 °C oil bath) for 45 min. This was followed by a second extraction step (90 mL absolute ethanol and 10 mL hydrochloric acid (37%)) under reflux conditions for 45 min (the material was washed with absolute ethanol after each extraction step and collected by centrifugation); finally the particles were redispersed in absolute ethanol and stored as colloidal suspension.
000 rpm, 43146 rcf, 20 min) and redispersed in absolute ethanol. Template extraction was performed in an ethanolic solution of MSNs (100 mL) containing NH4NO3 (2 g) which was heated at reflux conditions (90 °C oil bath) for 45 min. This was followed by a second extraction step (90 mL absolute ethanol and 10 mL hydrochloric acid (37%)) under reflux conditions for 45 min (the material was washed with absolute ethanol after each extraction step and collected by centrifugation); finally the particles were redispersed in absolute ethanol and stored as colloidal suspension.
Synthesis of sulfonamide-functionalized MSNs (MSN-phSA)
For the covalent attachment of a sulfonamide derivative to the external particle surface, a thiol-reactive linker was synthesized. 6-Maleimidohexanoic acid N-hydroxysuccinimide ester (mal-C6-NHS, 10 mg, 33 μmol) was dissolved in DMF (500 μL, dry) and was added to an ethanolic solution (15 mL) containing 4-(2-aminoethyl)benzene sulfonamide (6.7 mg, 33 μmol). The resulting reaction mixture was stirred for 1 h at room temperature. Afterwards, thiol-functionalized silica nanoparticles (MSN-SH, 100 mg) in absolute ethanol (10 mL) were added and the mixture was stirred over night at room temperature. Subsequently, the particles were collected by centrifugation (19000 rpm, 41146 rcf, 20 min), washed twice with absolute ethanol and were finally redispersed in ethanol (15 mL) to obtain a colloidal suspension.
Cargo loading and particle capping
MSNs (MSN-phSA, 1 mg) were immersed in an aqueous solution of fluorescein (1 mL, 1 mM), DAPI (500 μL, 14.3 mM) or Actinomycin D (500 μL [14 v% DMSO], 140 μM) and stirred over night or for 1 h, respectively. After collection by centrifugation (14000 rpm, 16837 rcf, 4 min), the loaded particles were redispersed in a HBSS buffer solution (1 mL) containing carbonic anhydrase (1 mg) and the resulting mixture was allowed to react for 1 h at room temperature under static conditions. The particles were thoroughly washed with HBSS buffer (4 times), collected by centrifugation (5000 rpm, 2200 rcf, 4 min, 15 °C), and finally redispersed in HBSS buffered solution.
Click chemistry of norbornene-containing hCA
MSNs (MSN-phSA, 0.5 mg) were immersed in 500 μL HBSS buffer solution and 0.5 mg norbornene-containing hCA was added. In the meantime, 2.5 μg tetrazine p-benzylamine (DMSO stock solution, 0.92 mg mL−1) and 0.41 mg NHS-PEG2000-FA were mixed in 100 μL HBSS and stirred overnight in the dark at room temperature. The solutions were mixed afterwards and stirred for two hours, washed several times and redispersed in 1 mL HBSS buffer. Subsequently, 1 μL Atto633mal (DMF stock solution, 0.5 mg mL−1) was added and the mixture was stirred for 1 hour. The particles were thoroughly washed with HBSS buffer (4 times), collected by centrifugation (5000 rpm, 2200 rcf, 4 min, 15 °C), and finally redispersed in HBSS buffered solution.
Synthesis of Knorb
The norbornene containing amino acid Knorb was synthesized as described in ref. 29.
Mutagenesis of pACA_HCA H36amber
Adapted from ref. 30 with permission from The Royal Society of Chemistry. The amber codon (TAG) was introduced into the expression vector pACA_HCA31 at position His36 of the human carbonic anhydrase II gene by blunt end site directed mutagenesis using the primers forward HCA H36amber and reverse HCA H36amber (see Table 1).
Table 1 Sequences of the used primers for the generation of expression vector pACA_HCA H36amber. The introduced Amber codon is shown in bold
| Name |
Sequence |
| Forward HCA H36amber |
5′phosph GTT GAC ATC GAC ACT TAG ACA GCC AAG TAT GAC |
| Reverse HCA H36amber |
5′phosph AGG GGA CTG GCG CTC TCC CTT GG |
Expression of norbornene-containing HCA
Adapted from ref. 30 with permission from The Royal Society of Chemistry. The expression vector pACA_HCA H36amber was transformed together with pACyc_pylRS Norb, 3xpylT29 which contains the genes of the triple mutant of PylRS and three copies of pylT in E. coli BL21(DE3) cells (NEB). 1 L of LB medium containing 34 mg L−1 chloramphenicol, 100 mg L−1 carbenicillin and 2 mM norbornene amino acid Knorb was inoculated with 10 mL of an overnight culture. The cells were stirred at 37 °C until an OD600 of 0.9. At this optical density 1 mM ZnSO4 and 0.1 mM IPTG were added to induce the expression of the HCA H36amber gene. After further 10 h at 37 °C the cells were harvested and stored at −20 °C until further use. The harvested cells were resuspended in washing buffer (25 mM tris; 50 mM Na2SO4; 50 mM NaClO4; pH 8.8) and disrupted by French Press procedure. The supernatant of the centrifuged lysate was used for sulfonamide affinity protein purification using an ÄKTA purifier system. The self-packed 3 mL column of p-aminomethylbenzenesulfonamide–agarose resin (Sigma-Aldrich, A0796) was equilibrated with washing buffer. After binding (0.75 mL min−1) of the protein solution, the column was washed with 7 column volumes of washing buffer. HCA was eluted by lowering the pH by elution buffer (100 mM NaOAc; 200 mM NaClO4; pH 5.6). The protein containing fractions were combined, analyzed by SDS-PAGE, dialyzed against water and lyophilized. Typical yields of the pure norbornene amino acid Knorb containing protein HCA H36Knorb were 20 mg L−1 expression medium.
Tryptic digestion and MS/MS of norbornene-containing HCA
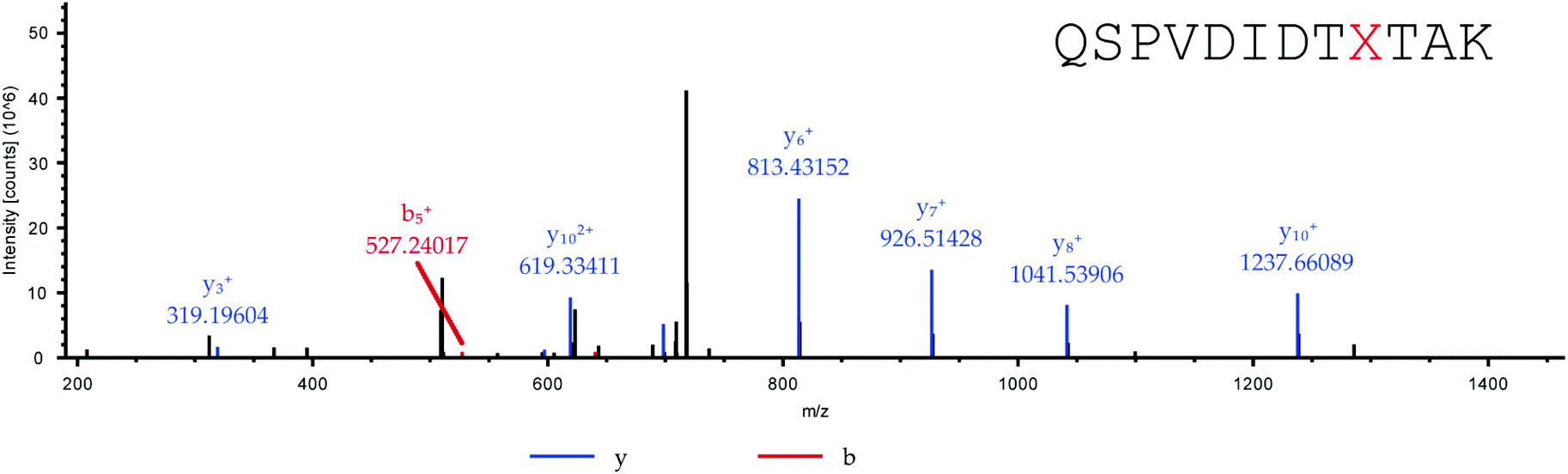
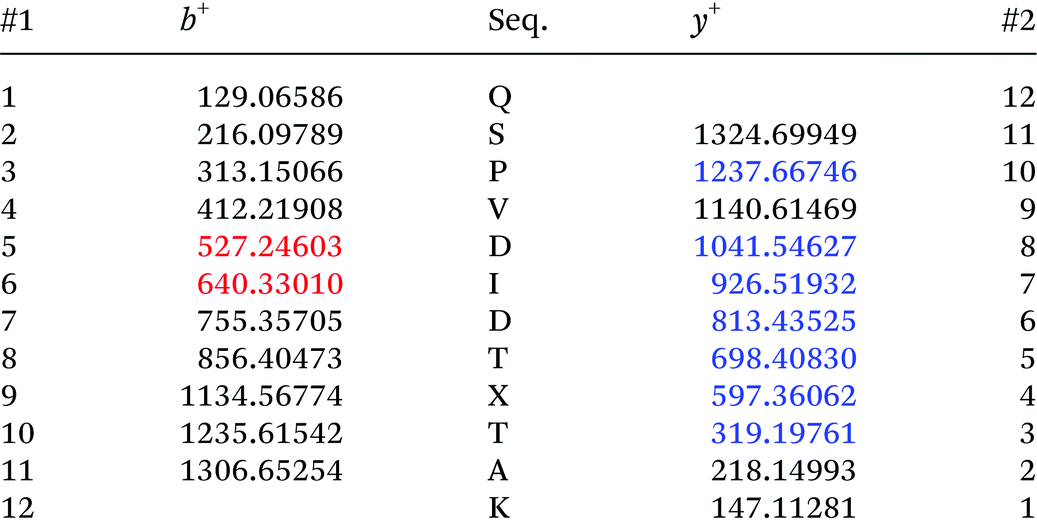
Adapted from ref. 30 with permission from The Royal Society of Chemistry. The sequence of HCA II is shown in Table 2. Position His36, which was chosen for the incorporation of amino acid Knorb, is shown in red. The peptide generated after tryptic digestion is emphasized in bold letters. Fig. 1 shows the corresponding MS/MS spectrum. Table 3 shows the expected and identified MS/MS fragments of the relevant tryptic peptide.
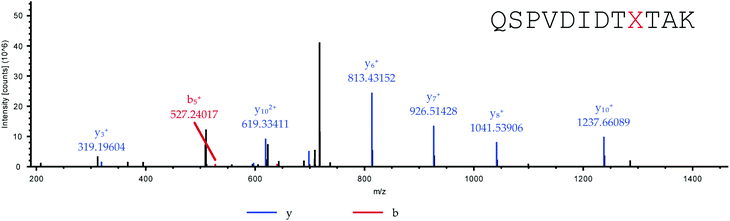 |
| | Fig. 1 MS/MS spectrum of the tryptic peptide QSPVDIDTXTAK (X = 4). Parent ion: [M + 2H]2+calc. = 726.8829, [M + 2H]2+obs. = 726.8807 (ΔM = 3 ppm). | |
Table 2 Amino acid sequence of HCA II
Table 3 Expected and identified MS/MS fragments of the tryptic peptide QSPVDIDTXTAK (X = Knorb). Identified fragments are shown in red for b ions and blue for y ions
Cell culture
HeLa and A431 cells were grown in Dulbecco's modified Eagle's medium (DMEM):F12 (1:1) (lifeTechnologies) with Glutamax I medium and KB cells in folic acid deficient Roswell Park Memorial Institute 1640 medium (RPMI 1640, lifeTechnologies), both supplemented with 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 humidified atmosphere. The cells were seeded on collagen A-coated LabTek chambered cover glass (Nunc). For live cell imaging the cells were seeded 24 or 48 h before measuring, at a cell density of 2 × 104 or 1 × 104 cells per cm2.
The FGF-2 and EGF dependant neural stem cell line ENC1 was derived from E14 mouse embryonic stem cells and cultured as described.32 ENC1 cells were maintained in gelatine coated flasks and propagated in a 1:1 mixture of Knockout-DMEM (Life Technologies) and Ham's F-12 (Sigma) supplemented with 2 mM GlutaMAX-I (Life Technologies), 100 U ml−1 penicillin (Sigma), 100 μg ml−1 streptomycin (Sigma) 1% N2 and 20 ng ml−1 each of mouse recombinant FGF-2 and EGF (Peprotech). N2 supplement was produced in house as described, with the exception that insulin was of human origin (Sigma I9278) instead of bovine. Stem cells and A431 cells were seeded on ibidi 8-well μ-slides.
In vitro cargo release
Cells were incubated 7–24 h prior to the measurements at 37 °C under a 5% CO2 humidified atmosphere. Shortly before imaging, the medium was replaced by CO2-independent medium (Invitrogen). During the measurements all cells were kept on a heated microscope stage at 37 °C. The subsequent imaging was performed as described in the spinning disk confocal microscopy section.
Endosomal compartment staining
To stain the early/late endosome or the lysosome with GFP, commercially available CellLight© staining from lifeTechnologies was used. The cells were simultaneously incubated with MSNs and the BacMam 2.0 reagent. The concentration of the labeling reagent was 25 particles per cell (PCP) of the BacMam 2.0 reagent (cf. staining protocol33). For incubation, the cells stayed at 37 °C under 5% CO2 humidified atmosphere for 21–24 h before the measurement.
Caspase-3/7 staining
For apoptosis detection commercially available CellEvent™ caspase-3/7 Green Detection Reagent was used. A final concentration of 2.5 μM caspase-3/7 reagent and 0.5 μg mL−1 Hoechst 33342 were added to the cells for 30 min and imaging was performed without further washing steps.
Uptake studies
The functionality of the folic acid targeting ligand was evaluated in a receptor competition experiment. For this purpose, one part of the KB cells was pre-incubated with 3 mM folic acid, to block the receptors, for 2 h at 37 °C under a 5% CO2 humidified atmosphere. Then the KB cells were incubated with particles for 2/5/8 h at 37 °C under a 5% CO2 humidified atmosphere. For staining the cell membrane, the cells were incubated with a final concentration of 10 μg mL−1 wheat germ agglutinin Alexa Fluor 488 conjugate for one minute. The cells were washed once with CO2-independent medium and imaged. For stem cell uptake studies cells were seeded the day prior to incubation. They were incubated for 2 h with free anandamide–tetrazine at a final concentration of 10 μg ml−1. After 2 h 15 μg of particles were added and incubated for another 2 h. Then, the cells were washed 3× with medium containing growth factors and if preincubated free anandamide–tetrazine and incubated until imaging. Immediately before imaging, cell membranes were stained using cell mask deep red (lifetechnologies) and washed with medium.
Spinning disc confocal microscopy
Confocal microscopy for live-cell imaging was performed on a setup based on the Zeiss Cell Observer SD utilizing a Yokogawa spinning disk unit CSU-X1. The system was equipped with a 1.40 NA 100× Plan apochromat oil immersion objective or a 0.45 NA 10× air objective from Zeiss. For all experiments the exposure time was 0.1 s and z-stacks were recorded. DAPI and Hoechst 33342 were imaged with approximately 0.16 W mm−2 of 405 nm light, GFP and the caspase-3/7 reagent were imaged with approximately 0.48 W mm−2 of 488 nm excitation light. Atto 633 was excited with 11 mW mm−2 of 639 nm light. In the excitation path a quad-edge dichroic beamsplitter (FF410/504/582/669-Di01-25 × 36, Semrock) was used. For two color detection of GFP/caspase-3/7 reagent or DAPI/Hoechst 33342 and Atto 633, a dichroic mirror (560 nm, Semrock) and band-pass filters 525/50 and 690/60 (both Semrock) were used in the detection path. Separate images for each fluorescence channel were acquired using two separate electron multiplier charge coupled device (EMCCD) cameras (PhotometricsEvolve™).
Results and discussion
pH-Responsive MSNs with an average particle size of 150 nm (average pore diameter: 3.8 nm) containing biomolecular valves based on the enzyme carbonic anhydrase (CA, hydrodynamic diameter: 5.5 nm) were synthesized via a delayed co-condensation approach.34 An outer functional shell consisting of benzene sulfonamide groups (pHSA) acts as an anchor point for the enzymatic gatekeepers. The formation of the inhibitor–enzyme complex (phSA-CA) leads to a dense coating at the external particle surface (MSN-phSA-CA). The characterization of the particle system by dynamic light scattering (DLS), zeta potential measurements, transmission electron microscopy (TEM), nitrogen sorption isotherms, infrared and Raman spectroscopy confirms the successful synthesis of carbonic anhydrase-coated MSNs. In vial release experiments demonstrate efficient sealing of the pores with carbonic anhydrase acting as a bulky gatekeeper, preventing premature cargo release and allowing for release upon acid-induced detachment of the capping system (for detailed information about synthesis and characterization see ESI†).
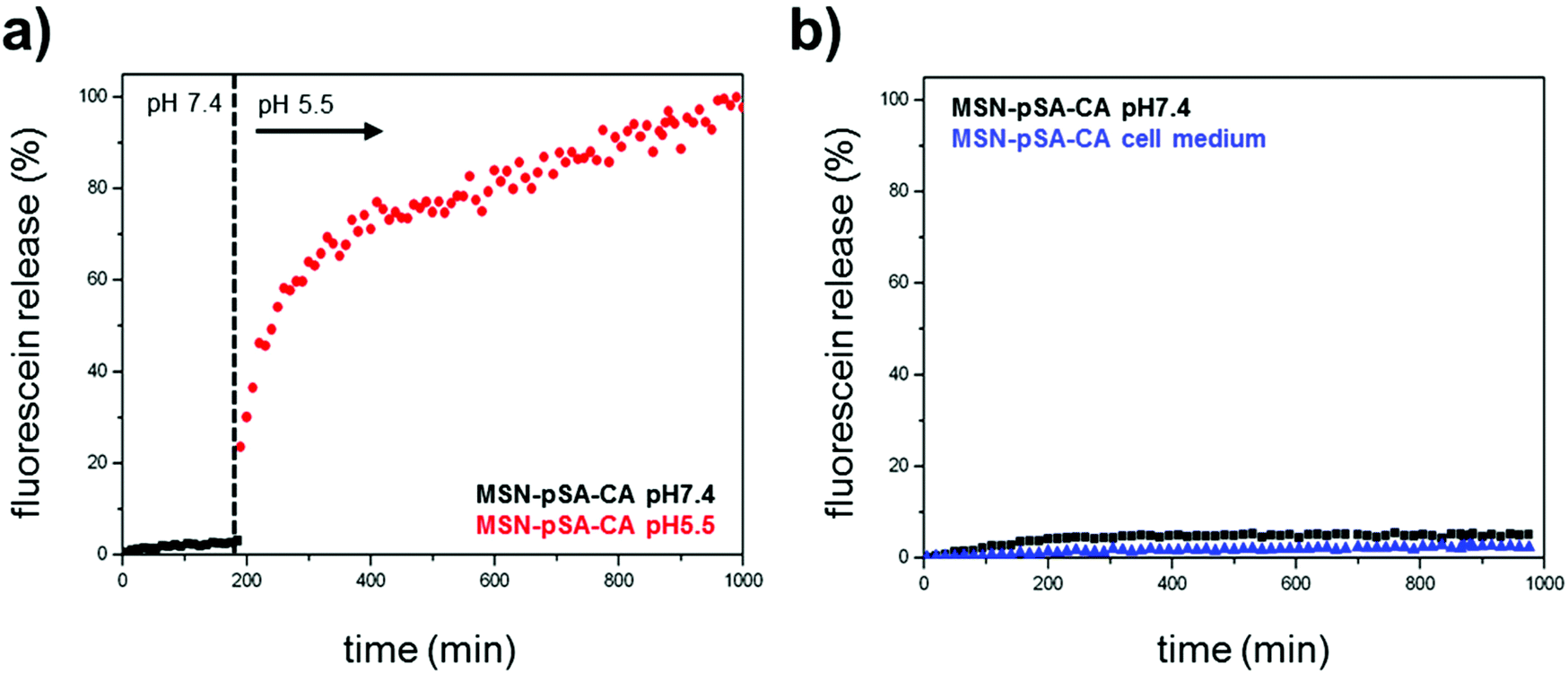
In order to investigate the pH-responsive removal of the bulky gatekeepers from the particles, in vial cargo release experiments were performed. We used a custom-made two-compartment system to analyze the time-based release of the fluorescent model cargo fluorescein.12 After incorporation of fluorescein molecules into the mesoporous system, carbonic anhydrase was added to block the pore entrances. An efficient sealing of the pores and no premature release of the cargo was observed for the sample MSN-phSA-CA dispersed in HBSS buffer (pH 7.4) at 37 °C (Fig. 2a, closed state, black curve). After 3 h the solution was exchanged and the particles were dispersed in citric-acid phosphate buffer (CAP buffer, pH 5.5). The change to acidic milieu, which simulates the acidification of endosomes, causes a significant increase in fluorescence intensity over time (open state, red curve). Furthermore, we could show the long-term stability of the capping system for more than 16 hours in HBSS buffer and cell medium at pH 7.4 (Fig. 2b). These in vial release experiments demonstrate efficient sealing of the pores with carbonic anhydrase acting as a bulky gatekeeper, preventing premature cargo release and allowing for release upon acid-induced detachment of the capping system.
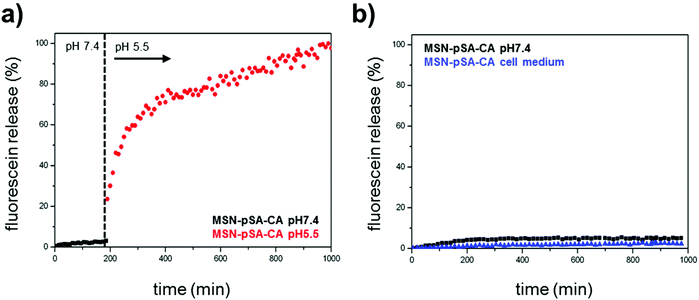 |
| | Fig. 2
In vial release kinetics of fluorescein molecules from the enzyme-coated MSNs at different pH values. (a) Sample MSN-pSA-CA features no premature release of the fluorescent cargo molecules in HBSS buffer solution at pH 7.4 (closed state, black curve). After 3 h the medium was changed to slightly acidic milieu (CAP buffer, pH 5.5, red curve) resulting in a significant increase in fluorescence intensity. The gatekeepers are detached from the particle surface upon acidification, causing an efficient and precisely controllable release of fluorescein from the mesoporous system. (b) Long-term stability of the capping system was investigated in HBSS buffer (pH 7.4, black curve) and cell medium (blue curve). No unintended cargo release was observed within about 16 h. | |
For efficient receptor-mediated cancer cell uptake and selective drug delivery a targeting ligand needs to be implemented. Since the particle surface is covered with bulky enzymes (CA), we aimed for the attachment of the targeting moieties directly to the outer periphery of the enzyme, in order to be accessible for cell receptors. For this approach to be successful, the site of targeting ligand attachment on the enzyme is of key importance. Ideally it should be positioned opposite of the binding site of the enzyme, to prevent blocking of the active site and thus leakage of the capping system. However, site-specific chemical modifications of proteins are highly challenging. Several methods, such as the reaction of thiol groups with maleimide or of lysine chains with activated esters, lack specificity. A more specific method is the incorporation of unnatural amino acids into the protein.20,35,36 Among others, the genetic incorporation of UAAs bearing side chains with alkyne,37,38trans-cyclooctene,39 cyclooctyne40 or norbornene20,24 functionalities has been reported previously. Subsequently these residues can be modified specifically and bio-orthogonally, for example by reverse electron-demanding Diels–Alder reactions with tetrazines.25,26,39 The natural PylRS/tRNAPyl pair is perfectly suitable to genetically incorporate UAAs due to its orthogonality to common expression strains. Recently, a norbornene-containing Pyl analogue (Knorb) has been developed by some of us.20,21 Here, the synthesis of norbornene-functionalized human carbonic anhydrase II (HCA) was accomplished similar to a previously described procedure yielding HCA H36Knorb.41 The correct position of the UAA was confirmed by tryptic digestion of the protein followed by HPLC-MS/MS analysis (see ESI†). HCA H36Knorb carrying norbornene on the opposite face of its phSA-binding site was bound to phSA-MSN and then treated with an excess of folate–PEG2000–tetrazine (Fig. 3) or anandamide–tetrazine. The excess of the tetrazine reagent could be easily removed by centrifugation of the nanoparticles followed by washing. The efficiency of the folate-targeting system was examined on KB-cells presenting either free or blocked FA-receptors (Fig. 3). For visualization, the cell membrane of the KB cells was stained with WGA488 (green), and the particles were labeled with Atto633 (red). In Fig. 3c–e we present the folic acid receptor blocked cells that were incubated with particles between 2 and 8 h. With increasing incubation time, only a few particles were internalized and unspecific cell uptake was observed only to a minor degree. In contrast, the cells with available folic acid receptor on their surface (Fig. 3f–h) exhibit a significant and increasing uptake behavior and a considerably higher degree of internalized particles. Thus we could confirm the successful application of bioorthogonal modification of a capping enzyme to act as targeting ligand. We also proved that the genetically modified enzyme capping strategy described here can be used to attach even sensitive ligands like arachidonic acid via mild click-chemistry conditions, e.g. for the site-specific targeting of neural stem cells and different cancer cells.32 We tested the anandamide-targeting system on neural stem cells and A431 cells. Neural stem cells have anandamide receptors and successfully internalize the anandamide-particles (see ESI, Fig. S9†). A431 cells (epidermoid carcinoma) are also known to over-express the G-protein coupled cannabinoid-based receptor CB2. These receptors can interact with anandamide-functionalized MSNs. Similar to the folate-based targeting experiment, the cannabinoid receptors on the A431 cells were either blocked or free. After 3 h of incubation the receptor-blocked cells internalized just a few anandamide-functionalized MSNs. In contrast, the amount of intracellular particles is clearly much larger in the case of non-blocked cannabinoid receptors (Fig. S10†). The successful experiments with different cell lines and targeting ligands show that the investigated bio-orthogonal attachment concept could be expanded to a variety of enzymes and ligands.
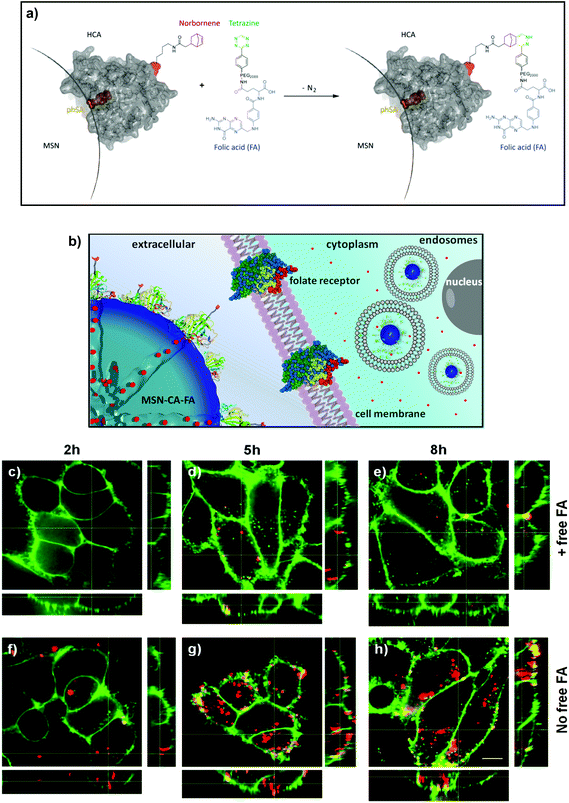 |
| | Fig. 3 (a) Norbornene-functionalized carbonic anhydrase (HCA H36Norb) with indicated functionalization site (red) and active site (blue) is able to react in a reversed-electron-demand Diels–Alder reaction with a folate–PEG2000–tetrazine derivative to give HCA-FA. (b) Schematic receptor-mediated uptake of folate-functionalized MSN-CA nanoparticles. (c–e) Nonspecific and (f–h) receptor-mediated endocytosis of MSN-phSA-CA-FA (red) by KB cells (WGA488 membrane staining, green). A specific receptor-mediated cell uptake was observed for MSN-phSA-CA-FA with KB cells (not pre-incubated with FA) after 5 and 8 h incubation at 37 °C (g/h). Incubation of MSN-phSA-CA-FA with FA-pre-incubated KB cells for 2, 5, 8 h at 37 °C showed only minor unspecific cellular uptake over all incubation times (c–e). The scale bar represents 10 μm. | |
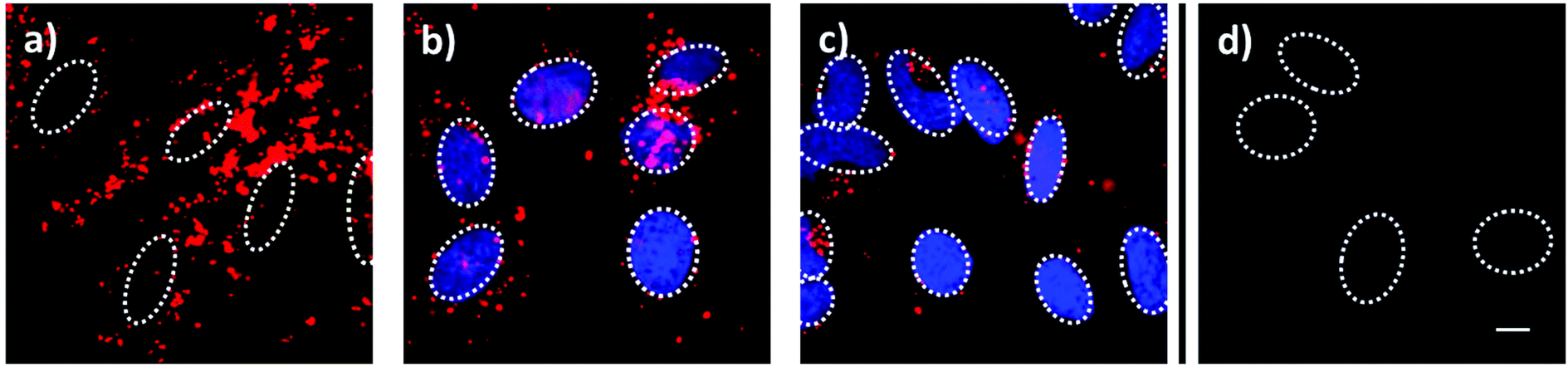
Employing fluorescent live-cell imaging, we investigated the in vitro release behavior of encapsulated 4′,6-diamidino-2-phenylindole (DAPI) in HeLa cancer cells. The molecular size of DAPI is similar to fluorescein. It was therefore expected to efficiently enter the mesoporous system of the silica nanoparticle. Due to its effective turn-on fluorescence upon intercalation into DNA double strands, DAPI is commonly used as nuclei counterstain in cell imaging (about 20 fold enhancement in fluorescence intensity).42 Since DAPI is cell membrane permeable, free fluorescent dye molecules are able to stain the nucleus within very short time periods (1–5 min), as described in several staining protocols.43 After incorporation of DAPI into the mesoporous system of the silica nanocarriers, the pores were sealed by addition of carbonic anhydrase. The HeLa cells were incubated for a total time period of 24 h with the loaded particles, which were additionally labeled with Atto 633 dye (red), as depicted in Fig. 4. After 7 h of incubation, MSNs were efficiently taken up by the cells and were found to be located in endosomes. Importantly, almost no staining of the nuclei with DAPI (blue) could be observed at this time point. Only after 15 h, blue fluorescence (even more intensive after 24 h) provided evidence of efficiently released DAPI from the MSNs. Control experiments in which the sample supernatant after particle separation (centrifugation) was added to the HeLa cells showed no significant nuclei staining even after 24 h (Fig. 4d). These cell experiments prove a substantial time-dependent release of DAPI from the mesopores of our nanocarrier system and also show that no free dye molecules were present in the solution. We suggest that the observed delayed nuclei staining results from a cascaded release mechanism. First, acidification throughout the endosomal pathway to late endosomes or endolysosomes is of key importance. Only the pH change to mildly acidic values (about 5.5) makes the detachment of the bulky gatekeepers from the MSN hosts possible. Subsequent opening of the pores leads to an efficient cargo release.
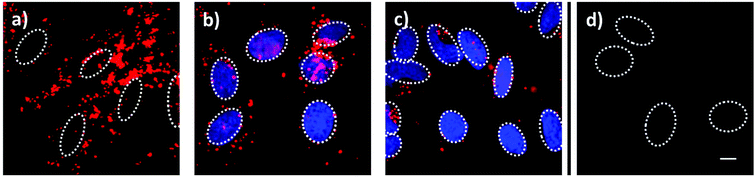 |
| | Fig. 4 Fluorescence microscopy of HeLa cells incubated with MSN-phSA-CA nanoparticles loaded with DAPI and labeled with Atto 633 (red) after (a) 7 h, (b) 12 h and (c) 24 h of incubation. The delayed nuclei staining with DAPI (blue) is caused by a time-dependent release of DAPI based on the need for an acidic environment. (d) In a control experiment, the incubation with the supernatant solution (without MSNs) showed no staining of the nuclei with DAPI after 24 h, suggesting that no free DAPI molecules were present in the particle solution. The nuclei are indicated with dashed circles. The scale bar represents 10 μm. | |
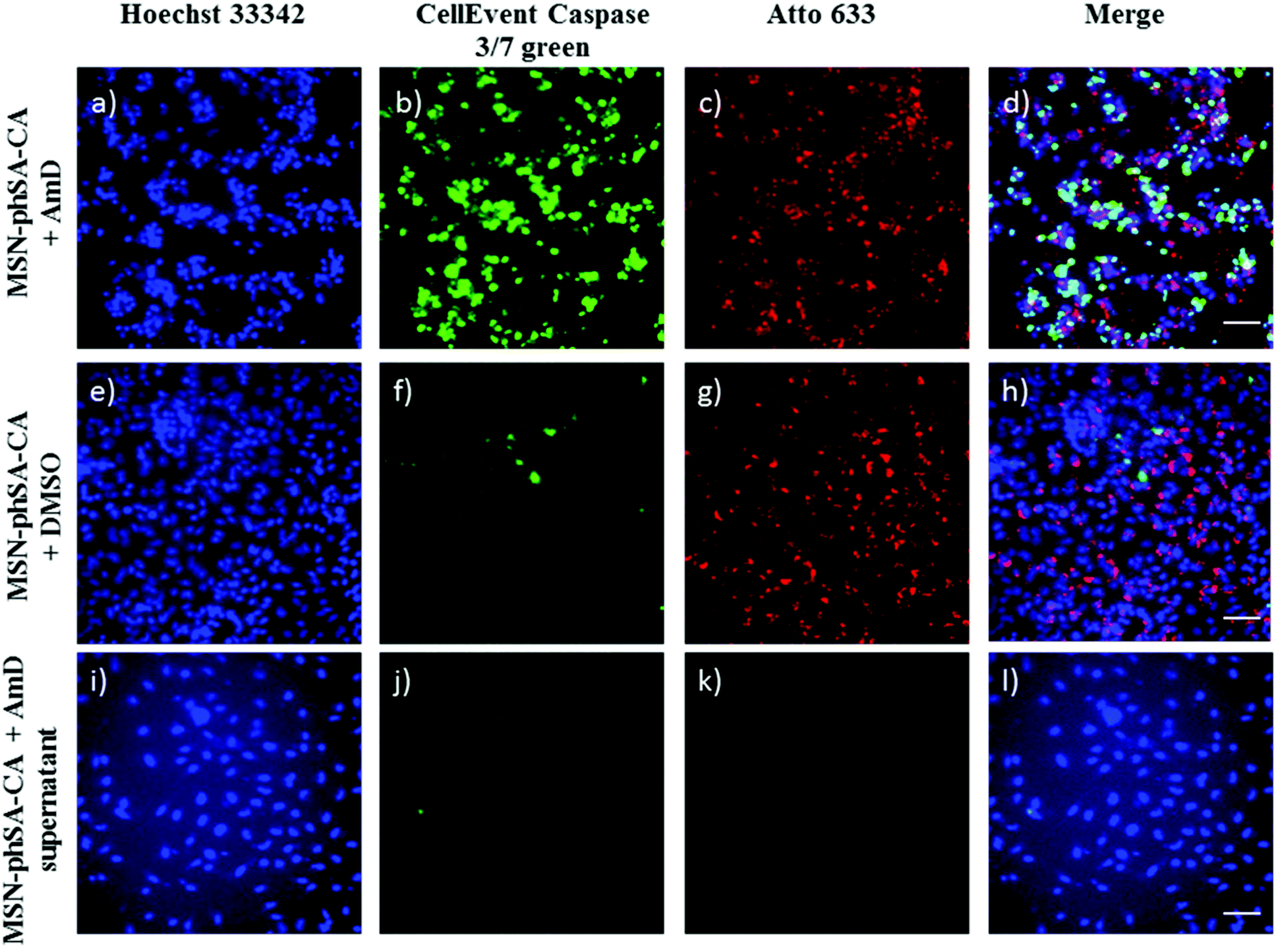
Additional co-localization experiments showed the localization of CA-capped nanoparticles in acidic cell compartments after endocytosis (Fig. S8†). To examine the ability of our newly developed MSN drug delivery system to transport chemotherapeutics and to affect cells with their cargo, we incorporated Actinomycin D (AmD), a cytostatic antibiotic, dissolved in DMSO. Free AmD is membrane permeable and induces an uncontrolled cell death within a few hours. MSN-phSA-CA provided intracellular AmD release and caused efficient cell death after 24 h. The delayed reaction demonstrates that AmD was delivered in a controlled manner via the particles and released only after acidification of the endosome and subsequent de-capping of the gate-keeper CA. In Fig. 5 cell death is visualized by a caspase 3/7 stain – a marker for apoptotic/dead cells. Control particles loaded with pure DMSO did not induce significant cell death at all, nor did the supernatant solution after particle separation via centrifugation (Fig. 5i–l). The results are in good accordance with dose-dependent cell viability studies (Fig. S11†) where the AmD-loaded particles killed HeLa cells effectively after 24 h of incubation (IC50,rel = 8.3 μg mL−1). This experiment shows the great potential of the MSN-phSA-CA system to efficiently deliver chemotherapeutics to cancer cells. The pH-responsive genetically modified capping system provides the ability to act as a general platform for different targeting ligands and cargos.
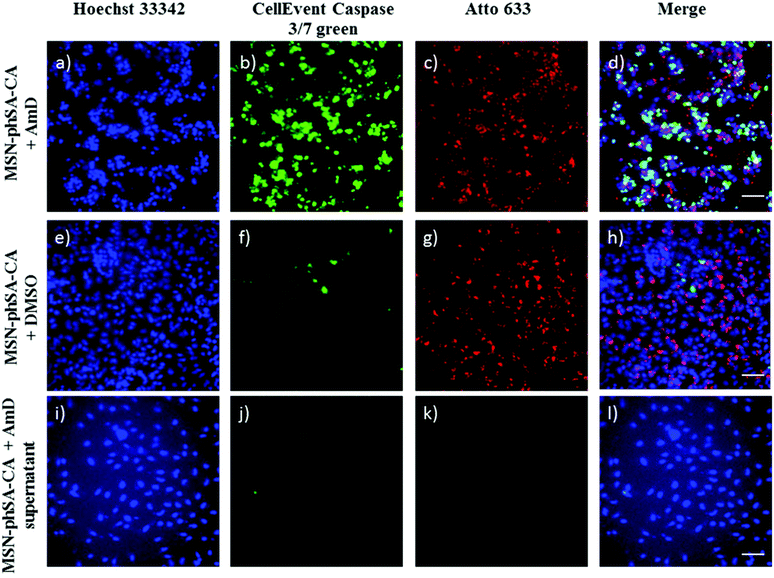 |
| | Fig. 5 Representative fluorescence microscopy images of HeLa cells incubated with MSN-phSA-CA nanoparticles loaded with Actinomycin D (AmD; a–d) or DMSO (e–h) and labeled with Atto 633 (red) after 24 h of incubation. As a control, the supernatant of AmD loaded particles after particle separation was incubated with the cells (i–l). Cell nuclei were stained with Hoechst 33342 (blue). For live/dead discrimination CellEvent caspase 3/7 (green) was used. Due to activation of caspase-3/7 in apoptotic cells, DNA can be stained after cleavage of the DNA-binding dye from a binding-inhibiting peptide. MSNs were efficiently taken up by cells (c/d and g/h). Cell death can only be observed for cells treated with AmD loaded MSN-phSA-CA after 24 h of incubation (increased DNA staining in green) (b). In contrast, nanoparticles loaded with DMSO or the sample supernatant do not induce significant apoptosis (almost no DNA-staining) (f and j). The scale bars represent 50 μm. | |
Conclusions
We conclude that the novel capping system concept based on pH-responsive detachment of carbonic anhydrase combined with folic acid as targeting ligand allows for highly controllable drug release from porous nanocarriers. Our drug delivery system provides an on-demand release mechanism shown by in vial and in vitro cargo release experiments. The multifunctional MSNs were efficiently endocytosed in cancer cells and could be located in acidic cell compartments where they released their cargo. Furthermore, the system has an on-board targeting mechanism as demonstrated in additional in vitro experiments. The targeting mechanism is attached at a specific site of the capping enzyme preventing interference with the closure mechanism. Our newly developed pH-responsive gatekeepers with genetically designed targeting functions provide a promising platform for the design of versatile and modular drug delivery systems.
Acknowledgements
The authors acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG) in the context of SFB 749 and SFB 1032, the Excellence Clusters Nanosystems Initiative Munich (NIM) and Center for Integrated Protein Science Munich (CIPSM), and the Center for Nano Science (CeNS). S. D. thanks the “Dr Klaus-Römer-Stiftung” for financial support. We thank Dr Bastian Rühle for 3D graphics design.
References
- Z. X. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Y. Lin, Small, 2010, 6, 1952–1967 CrossRef CAS PubMed.
- J. M. Rosenholm, V. Mamaeva, C. Sahlgren and M. Linden, Nanomedicine, 2012, 7, 111–120 CrossRef CAS PubMed.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- A. Ott, X. Yu, R. Hartmann, J. Rejman, A. Schutz, M. Ochs, W. J. Parak and S. Carregal-Romero, Chem. Mater., 2015, 27, 1929–1942 CrossRef CAS.
- S. A. Mackowiak, A. Schmidt, V. Weiss, C. Argyo, C. von Schirnding, T. Bein and C. Bräuchle, Nano Lett., 2013, 13, 2576–2583 CrossRef CAS PubMed.
- A. Schlossbauer, S. Warncke, P. M. E. Gramlich, J. Kecht, A. Manetto, T. Carell and T. Bein, Angew. Chem., Int. Ed., 2010, 49, 4734–4737 CrossRef CAS PubMed.
- E. Aznar, L. Mondragon, J. V. Ros-Lis, F. Sancenon, M. Dolores Marcos, R. Martinez-Manez, J. Soto, E. Perez-Paya and P. Amoros, Angew. Chem., Int. Ed., 2011, 50, 11172–11175 CrossRef CAS PubMed.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S. Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS PubMed.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S. Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038–5044 CrossRef CAS PubMed.
- Z. Luo, K. Cai, Y. Hu, L. Zhao, P. Liu, L. Duan and W. Yang, Angew. Chem., Int. Ed., 2011, 50, 640–643 CrossRef CAS PubMed.
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef CAS PubMed.
- W. Zhao, H. Zhang, Q. He, Y. Li, J. Gu, L. Li, H. Li and J. Shi, Chem. Commun., 2011, 47, 9459–9461 RSC.
- R. Liu, X. Zhao, T. Wu and P. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef CAS PubMed.
- C. Wang, Z. Li, D. Cao, Y.-L. Zhao, J. W. Gaines, O. A. Bozdemir, M. W. Ambrogio, M. Frasconi, Y. Y. Botros, J. I. Zink and J. F. Stoddart, Angew. Chem., Int. Ed., 2012, 51, 5460–5465 CrossRef CAS PubMed.
- Q. Gan, X. Lu, Y. Yuan, J. Qian, H. Zhou, X. Lu, J. Shi and C. Liu, Biomaterials, 2011, 32, 1932–1942 CrossRef CAS PubMed.
- S. Carregal-Romero, M. Ochs, P. Rivera-Gil, C. Ganas, A. M. Pavlov, G. B. Sukhorukov and W. J. Parak, J. Controlled Release, 2012, 159, 120–127 CrossRef CAS PubMed.
- T. H. Maren, Phys. Rev., 1967, 47, 595–781 CAS.
- P. W. Taylor, R. W. King and A. S. V. Burgen, Biochemistry, 1970, 9, 3894–3902 CrossRef CAS PubMed.
- E. Kaya, M. Vrabel, C. Deiml, S. Prill, V. S. Fluxa and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 4466–4469 CrossRef CAS PubMed.
- S. Schneider, M. J. Gattner, M. Vrabel, V. Flügel, V. López-Carrillo, S. Prill and T. Carell, ChemBioChem, 2013, 14, 2114–2118 CrossRef CAS PubMed.
- L. Davis and J. W. Chin, Nat. Rev. Mol. Cell Biol., 2012, 13, 168–182 CAS.
- J. Hemphill, E. K. Borchardt, K. Brown, A. Asokan and A. Deiters, J. Am. Chem. Soc., 2015, 137, 5642–5645 CrossRef CAS PubMed.
- K. Lang, L. Davis, J. Torres-Kolbus, C. J. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298–304 CrossRef CAS PubMed.
- T. Plass, S. Milles, C. Koehler, J. Szymanski, R. Müller, M. Wiessler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2012, 51, 4166–4170 CrossRef CAS PubMed.
- M. Vrabel, P. Kölle, K. M. Brunner, M. J. Gattner, V. López-Carrillo, R. de Vivie-Riedle and T. Carell, Chem. – Eur. J., 2013, 19, 13309–13312 CrossRef CAS PubMed.
- J. Willibald, J. Harder, K. Sparrer, K.-K. Conzelmann and T. Carell, J. Am. Chem. Soc., 2012, 134, 12330–12333 CrossRef CAS PubMed.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Linden, ACS Nano, 2009, 3, 197–206 CrossRef CAS PubMed.
- E. Kaya, M. Vrabel, C. Deiml, S. Prill, V. S. Fluxa and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 4466–4469 CrossRef CAS PubMed.
- M. J. Gattner, M. Ehrlich and M. Vrabel, Chem. Commun., 2014, 50, 12568–12571 RSC.
- S. K. Nair, T. L. Calderone, D. W. Christianson and C. A. Fierke, J. Biol. Chem., 1991, 266, 17320–17325 CAS.
- K. Brunner, J. Harder, T. Halbach, J. Willibald, F. Spada, F. Gnerlich, K. Sparrer, A. Beil, L. Möckl, C. Bräuchle, K.-K. Conzelmann and T. Carell, Angew. Chem., Int. Ed., 2015, 54, 1946–1949 CrossRef CAS PubMed.
-
http://tools.lifetechnologies.com/content/sfs/manuals/mp10582.pdf
.
- V. Cauda, A. Schlossbauer, J. Kecht, A. Zürner and T. Bein, J. Am. Chem. Soc., 2009, 131, 11361–11370 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- T. Fekner, X. Li, M. M. Lee and M. K. Chan, Angew. Chem., Int. Ed., 2009, 48, 1633–1635 CrossRef CAS PubMed.
- E. Kaya, K. Gutsmiedl, M. Vrabel, M. Muller, P. Thumbs and T. Carell, ChemBioChem, 2009, 10, 2858–2861 CrossRef CAS PubMed.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- T. Plass, S. Milles, C. Koehler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2011, 50, 3878–3881 CrossRef CAS PubMed.
- M. J. Gattner, M. Ehrlich and M. Vrabel, Chem. Commun., 2014, 50, 12568–12571 RSC.
- M. L. Barcellona, G. Cardiel and E. Gratton, Biochem. Biophys. Res. Commun., 1990, 170, 270–280 CrossRef CAS PubMed.
-
http://tools.lifetechnologies.com/content/sfs/ma+nuals/mp01306.pdf
.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, further characterization of functionalized MSNs, synthesis of anandamide–tetrazine, results of additional cell studies, dose-dependent MTT assay. See DOI: 10.1039/c5nr08163g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence