
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Doxorubicin loaded dual pH- and thermo-responsive magnetic nanocarrier for combined magnetic hyperthermia and targeted controlled drug delivery applications†
Aziliz
Hervault
abc,
Alexander E.
Dunn
d,
May
Lim
d,
Cyrille
Boyer
e,
Derrick
Mott
c,
Shinya
Maenosono
*c and
Nguyen T. K.
Thanh
*ab
aBiophysics Group, Department of Physics & Astronomy, University College London, Gower Street, London, WC1E 6BT, UK. E-mail: ntk.thanh@ucl.ac.uk; Fax: +44 (0)2076 702920; Tel: +44 (0)2074 916509
bUCL Healthcare Biomagnetic and Nanomaterials Laboratories, 21 Albemarle Street, London W1S 4BS, UK
cSchool of Materials Science, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: shinya@jaist.ac.jp; Fax: +81 761511625; Tel: +81 761511611
dSchool of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
eAustralian Centre for Nanomedicine and Centre for Advanced Macromolecular Design, School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 10th February 2016
Abstract
Magnetic nanocarriers have attracted increasing attention for multimodal cancer therapy due to the possibility to deliver heat and drugs locally. The present study reports the development of magnetic nanocomposites (MNCs) made of an iron oxide core and a pH- and thermo-responsive polymer shell, that can be used as both hyperthermic agent and drug carrier. The conjugation of anticancer drug doxorubicin (DOX) to the pH- and thermo-responsive MNCs via acid-cleavable imine linker provides advanced features for the targeted delivery of DOX molecules via the combination of magnetic targeting, and dual pH- and thermo-responsive behaviour which offers spatial and temporal control over the release of DOX. The iron oxide cores exhibit a superparamagnetic behaviour with a saturation magnetization around 70 emu g−1. The MNCs contained 8.1 wt% of polymer and exhibit good heating properties in an alternating magnetic field. The drug release experiments confirmed that only a small amount of DOX was released at room temperature and physiological pH, while the highest drug release of 85.2% was obtained after 48 h at acidic tumour pH under hyperthermia conditions (50 °C). The drug release kinetic followed Korsmeyer–Peppas model and displayed Fickian diffusion mechanism. From the results obtained it can be concluded that this smart magnetic nanocarrier is promising for applications in multi-modal cancer therapy, to target and efficiently deliver heat and drug specifically to the tumour.
Introduction
Magnetic nanoparticles (NPs) have emerged as a promising technology for their potential use in biomedical applications, and more particularly for cancer treatment and diagnosis, such as magnetic resonance imaging (MRI),1–3 cell separation,4,5 magnetic hyperthermia6–8 or drug delivery.9–11 In particular, iron oxide NPs (IONPs), already approved by the US Food and Drug Administration (FDA), have been greatly investigated due to their good biocompatibility, low toxicity and ease of synthesis. Below a certain size (around 27 nm),12 IONPs become superparamagnetic. Superparamagnetic NPs are more suitable than ferri- or ferromagnetic NPs for hyperthermia applications as they do not retain any magnetization once the magnetic field is removed. Hyperthermia is known as a treatment where tumours are exposed to increased temperatures (40–45 °C)13 causing cancer cells to undergo apoptosis,14 while sparing normal cells, as cancer cells have greater sensitivity to heat than healthy cells.15,16 When superparamagnetic NPs are subjected to an alternating magnetic field (AMF), these NPs generate heat through Néel and Brownian relaxation losses. Magnetic hyperthermia has already shown its therapeutic efficacy in clinical trials.17,18Hyperthermia is also often used as an adjuvant therapy with radio- or chemotherapy,19 as heat makes cancer cells more sensitive to the effect of radiation15 and antineoplastic agents.16,20 An enhanced therapeutic efficacy of chemotherapy in combination with hyperthermia has been demonstrated in many clinical trials,21–24 resulting in a synergistic effect of the cancer therapy. Increased anti-cancer drug accumulation in tumour and thermal enhancement of drug cytotoxicity mainly caused by an improved intracellular uptake of drugs due to an increased cell membrane permeability, inhibition of DNA-repair of the chemically induced lethal or sublethal damages, and acceleration of the cytotoxic chemical reaction, are among the most important mechanisms behind the synergistic effect of thermo-chemotherapy.25,26 One of the major advantage of using magnetic NPs as drug carrier over conventional chemotherapy is that NPs can be potentially injected and further concentrated anywhere in the body through magnetic targeting, allowing the treatment of all kinds of tumours by delivering drugs to specific locations in the body, therefore reducing the side effects.
While relatively recent, the approach of combining magnetic hyperthermia and drug delivery features in the same formulation to effectively deliver heat and drug locally and benefit from the synergistic effect has already shown promising results.27 Several studies reported the enhanced cytotoxicity to cancer cells of drugs when combined with magnetic hyperthermia in vitro or in vivo.28–32 Moreover, combining drug delivery with magnetic hyperthermia has also proven itself to be a useful technique in overcoming multidrug resistance on drug-resistant cancer.33,34
Smart polymers, a class of materials that respond to environmental stimuli such as pH or temperature, represent a very attractive choice of polymer coating for biomedical applications and particularly drug delivery to achieve a controlled release of the drug.35 Thermo-responsive polymers can either exhibit a upper critical solution temperature (UCST) or a lower critical solution temperature (LCST). Polymers displaying a UCST become soluble upon heating while the LCST is the temperature at which polymers undergo a reversible conformational change from a swollen hydrophilic state to a shrunken hydrophobic one resulting in a volume decrease due to expelling of the aqueous content from its chains.36,37 As a consequence, the use of thermo-responsive polymers having a LCST in the hyperthermia temperature range to magnetically remotely trigger the release of the drug is desirable. At physiological temperature, the drug is retained in the composite. When heat is produced by the magnetic NPs in an AMF, the surrounding temperature rises above the LCST and triggers the release of the drug in a temporal control manner. pH-responsive nanocarriers exploit the physiological differences found between healthy and tumour tissues, as extracellular tumour environment often exhibits acidic pH (from 5.7 to mostly lower than 7.2) as compared to normal tissue (ca. 7.4).38,39 Moreover, if nanocarriers are taken up by cells through endocytosis, they will be trapped in intracellular organelles such as endosomes and lysosomes, which have even lower pH (6.0–6.5 in early endosomes, 5.0–6.0 in late endosomes and 4.5–5.0 in lysosomes).
Our work reports the development and characterization of a dual pH- and thermo-responsive MNC drug carrier suitable for multi-modal cancer therapy, by combining magnetic targeting, hyperthermia and controlled drug delivery. The dual stimuli-responsive features offer spatial and temporal control over the release of the anticancer drug, which is triggered as a consequence of hyperthermia and tumour acidic pH. The MNCs are composed of an iron oxide core, synthesized by a microwave-assisted co-precipitation method,40 and a thermo-responsive polymer shell. This polymer was previously designed and used by A. E. Dunn et al.,41 in the development of nanocomposites with an acicular magnetite core for controlled drug delivery and MRI applications. However, the magnetic hyperthermia of those nanoparticles were not reported and a dye was used as a model for the anticancer drug. While, in this work, a high heating efficiency iron oxide spherical magnetic nanoparticles synthesized in our lab using a microwave40 was conjugated with the polymer. The thermo-responsive copolymer was engineered to have its LCST in the hyperthermia temperature range. The heating performances of the MNCs were assessed and the MNCs were subsequently loaded with the anticancer drug doxorubicin (DOX) through formation of pH-sensitive imine bonds between the amine group of DOX and the aldehyde group of the copolymer (Fig. 1). The loading and in vitro release profiles under different temperatures and pH conditions were studied using UV-Vis spectroscopy.
 | ||
| Fig. 1 Synthesis of the DOX-MNCs. | ||
Experimental
Materials
Iron(II) chloride tetrahydrate (FeCl2·4H2O, purity 99%), iron(III) chloride hexahydrate (FeCl3·6H2O, purity >99%), sodium carbonate (Na2CO3, purity 99%), hydrochloric acid (HCl, 37 vol%), poly(ethylene glycol) methyl ether methacrylate (PEGMA, Mn = 300 g mol−1), di(ethylene glycol) methyl ether methacrylate (DEGMA, purity 95%), 3-(trimethoxysilyl)propyl methacrylate (TMSPMA, purity 98%), 3-vinylbenzaldehyde (VBA, purity 97%), 4,4′-azobis(4-cyanovaleric acid) (ACVA, purity ≥75%), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPPA, purity 97%), chloroform-d NMR solvent, triethylamine (TEA, purity 98%), acetonitrile (purity 99.8%), toluene (purity 99.8%) and petroleum ether were purchased from Sigma Aldrich. Tetrahydrofuran (THF, stabilizer free, purity 99.8%) was provided from Wako Chemicals. Ethanol (purity 99.5%) was obtained from Nacalai Tesque Japan. All the reagents were used as purchased without any further purification.Synthesis of P(DEGMA-co-PEGMA-b-[TMSPMA-co-VBA])
The P(DEGMA-co-PEGMA-b-[TMSPMA-co-VBA]) polymer was synthesized by reversible addition fragmentation chain transfer (RAFT) polymerization according to a previous publication with some modifications.41 The PEGMA and DEGMA monomers were mixed in a molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.2 with the CPPA as the RAFT chain transfer agent and ACVA as the initiator. Under Ar atmosphere, anhydrous acetonitrile was added to the flask and the mixture was bubbled with Ar for 30 min. After degassing, the mixture was heated to 70 °C and maintained at this temperature for 5 h. The resulting polymer was then washed using petroleum ether and dried under vacuum overnight. A chain extension was then carried out by mixing the dry P(DEGMA-co-PEGMA) polymer used as macro-RAFT agent with ACVA initiator, TMSPMA and VBA in toluene. The monomers were added in a ratio of 1:4. This mixture was then degassed under Ar and heated to 90 °C for 4 h. The resulting polymer was again purified by precipitation in petroleum ether, and dried under vacuum.
:2.2 with the CPPA as the RAFT chain transfer agent and ACVA as the initiator. Under Ar atmosphere, anhydrous acetonitrile was added to the flask and the mixture was bubbled with Ar for 30 min. After degassing, the mixture was heated to 70 °C and maintained at this temperature for 5 h. The resulting polymer was then washed using petroleum ether and dried under vacuum overnight. A chain extension was then carried out by mixing the dry P(DEGMA-co-PEGMA) polymer used as macro-RAFT agent with ACVA initiator, TMSPMA and VBA in toluene. The monomers were added in a ratio of 1:4. This mixture was then degassed under Ar and heated to 90 °C for 4 h. The resulting polymer was again purified by precipitation in petroleum ether, and dried under vacuum.
Synthesis of IONPs
IONPs were synthesized following a modified procedure consisting of microwave-assisted co-precipitation of Fe(II) and Fe(III) with Na2CO3 and the aid of a SP-Discovery Microwave (CEM, USA).40 FeCl2·4H2O (0.4 mmol) and FeCl3·6H2O (0.8 mmol) were dissolved in 20 mL of Milli-Q water (Millipore, 18.2 MΩ cm−1), transferred into a vial and sealed with a pressure cap. The solution was heated to 60 °C (300 W), and Na2CO3 aqueous solution (4 mL, 1 M) was added drop wise at a rate of 2 mL min−1 using a syringe pump (KD Scientific Inc., KDS100). The solution was kept at this temperature for 20 min before allowing to cool down to room temperature. The NPs were then washed three times with ultrapure water by magnetic separation.Grafting of the copolymer onto the IONP surface
The grafting of the polymer P(DEGMA-co-PEGMA-b-[TMSPMA-co-VBA]) onto the IONP surface was realized through a silanisation reaction between the trimethoxysilane groups of the polymer and the hydroxyl groups present on the surface of the bare IONPs, forming a covalent Si–O–Fe bond. Freshly synthesized naked IONPs (20 mg) were mixed with 60 mg of P(DEGMA-co-PEGMA-b-[TMSPMA-co-VBA]) polymer dissolved in 20 mL of ethanol. The mixture was acidified to pH = 2–3 by adding 50 μL of HCl, sonicated for 10 min and then stirred at room temperature for 24 h. The copolymer-functionalized IONPs, hereafter referred to as composite NPs (MNCs) were then washed by magnetic separation two times with ethanol and once with ultrapure water before being redispersed in ultrapure water for further use.Characterization of the MNCs and their components
1H nuclear magnetic resonance (NMR) spectroscopy was used to monitor the polymerization reaction advancement and analyze the polymer composition. 1H NMR spectra were recorded on a Bruker Avance III 400 MHz NMR spectrometer using chloroform-d as a solvent. Number average molecular weight (Mn) and polydispersity (PDI = Mw/Mn) of the synthesized polymers were measured by size exclusion chromatography (SEC) performed on a Shimadzu Prominence high-performance liquid chromatograph equipped with a LC-20AD liquid chromatograph, a RID-10A refractive index detector, a CTO-20A column oven and a DGU-20A3 degasser. THF (stabilizer free) was used as the mobile phase. Samples were prepared by dissolving 5 mg of vacuum-dried polymer in 2 mL of THF. The molecular weights of the polymers were determined by a conventional calibration using polystyrene standards ranging from 5.8 × 102 g mol−1 to 2.0 × 104 g mol−1. The LCST of the polymer in aqueous solution was determined by UV-Vis spectroscopy using a JASCO V-630 spectrophotometer equipped with an EYELA NCB-1200 temperature controller. The solution was heated from 25 °C to 47 °C whilst the transmittance was recorded at 540 nm. The LCST value was interpreted as the temperature at which the solution transmittance reached 80%.The crystalline structures of the IONPs were determined by X-ray diffraction (XRD) on a PANalytical X-ray diffractometer using Co Kα radiation (λ = 1.78901 Å). The crystallite size was extracted from the obtained XRD pattern using the Scherrer formula. Particle size, size distribution and morphology were examined with a Hitachi H-7650 transmission electron microscope (TEM) operated at an acceleration voltage of 100 kV. Samples were prepared by dropping the aqueous IONP suspension onto a carbon-coated TEM grid and air-dried. The hydrodynamic diameter of the IONPs was measured by a dynamic light scattering instrument (DLS, Zetasizer Nano, Malvern Instruments).
Colloidal stability of the MNCs in an aqueous dispersion over time was investigated by monitoring the hydrodynamic size of the MNCs after preparation and throughout 14 days. Effect of pH and different dispersion medium on the stability of the MNCs were also assessed. The magnetic properties of the NPs were evaluated with a hybrid superconducting quantum interference device-vibrating sample magnetometer (SQUID-VSM, Quantum Design). Magnetisation curves were recorded at 300 K and 5 K, with applied fields up to 5 T. Attenuated total reflectance-Fourier transformed infra-red (ATR-FTIR) spectra were taken with a Perkin Elmer Spectrum 100 spectrometer. Thermogravimetric analysis (TGA) was used to monitor the mass loss of a known quantity of MNCs as a function of temperature. Dry powder was placed on an aluminium pan and heated from 25 °C to 500 °C at a rate of 10 °C min−1 under N2 atmosphere on a Seiko EXSTAR600 TG/DTA 6200 instrument. Prior to the measurement, powder were heated to 100 °C for 30 min to remove water that could be present in the sample. In order to determine the grafting density of the polymer on the NP surface, the mass loss of bare NPs was also recorded under the same conditions and used as reference.
Drug conjugation to the MNCs
Conjugation of DOX with the MNCs was achieved through formation of imine bond, also called Schiff base bond, between the primary amine group of DOX and the aldehyde group of the P(DEGMA-co-PEGMA-b-[TMSPMA-co-VBA]) polymer. DOX and MNCs were mixed in PBS (pH = 7.4) with a ratio 1:10 w/w in the presence of TEA. The mixture was shaken gently in the dark for 24 h at room temperature, thereby leading to the conjugation of DOX via imine linkage. The DOX-loaded MNCs (DOX-MNCs) were retrieved by magnetic separation and washed thoroughly with PBS until no DOX was detected in the supernatant (at least 10 washing cycles). The drug conjugation efficiency, η (%), is calculated by measuring the absorbance of the supernatant at 480 nm of the free DOX remaining in solution, and after each washing cycle, using the following equation: | (1) |
Here Mfeed is the initial mass of DOX used for the conjugation, Mconjugated is the mass of DOX actually conjugated to the MNCs, and Mexcess is the total mass of DOX found in the supernatants after the drug loading and each washing step. The DOX content in DOX-MNCs, WDOX (%), is also evaluated, and is defined as follow:
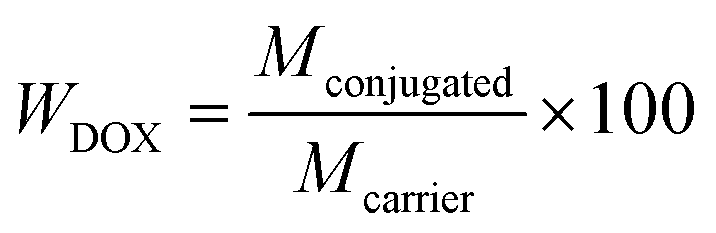 | (2) |
In vitro hyperthermia
Magnetic heating measurements were performed to assess the heating performances of the nanocomposites using a magnetic alternating current hyperthermia (MACH) system (Resonant Circuit Ltd, UK) operating at a frequency f = 960 kHz and field amplitude H = 6.88 kA m−1. Samples made of 0.5 mL of MNCs dispersed in water with a concentration of 3 mg mL−1 were placed in the middle of the coil. The increase in temperature was continuously logged using fibers optic probes centered in the suspension. Two probes were used in order to limit possible errors coming from a non-homogeneous spatial heating of the suspension. The measurement was started when the temperature of the suspension was stabilised to room temperature, and the measurement time was limited to 10 min. The specific absorption rate (SAR) is commonly employed to quantify the heat dissipation rate of a given ferrofluid, even though it is not an intrinsic property of the system as it is strongly dependent upon the frequency and field amplitude used during the measurement. | (3) |
 | (4) |
Hence one can directly compare the heating efficiency between different nanosystems measured under different experimental conditions using ILP. The heating power will therefore be characterized with the ILP value, but the SAR will be given as indication.
In vitro drug release behaviour in the absence of AMF
In order to study the pH and temperature dependence of the drug release kinetics in the absence of an AMF, 6 sets of experiments were performed. The drug release was studied at 25 °C (room temperature), 37 °C (physiological temperature, <LCST) and 50 °C (hyperthermia temperature, >LCST). For each temperature, one sample was held at pH = 7.4 (physiological pH) and another at pH = 5.7 to mimic acidic tumour pH. Typically, 3.3 mg of DOX-MNCs dispersed in 6 mL of PBS with the appropriate pH (7.4 or 5.7) were inserted in a Slyde-A-Lyzer dialysis cassette (MWCO 10 kDa) and dialyzed against 60 mL of PBS with the appropriate pH under mild stirring. A dialysate volume of 10 times that of the dialysis cassettes was chosen to insure sink conditions for the DOX. This volume was required for the dialysis cassette to be fully immersed in the medium, while the absorbance of DOX in solution is in the detectable range of UV-Vis. Dialysis set up were either maintained to room temperature, or placed into preheated water baths at 37 °C and 50 °C. At predetermined time points (i.e. 0.25, 0.5, 0.75, 1, 2, 4, 8, 24 and 48 h), 3 mL of dialysate were taken out for UV-Visible spectroscopy, and replaced with 3 mL of fresh PBS of the appropriate pH to keep the total volume constant and preserve sink conditions. The DOX content of the extracted solution was measured by UV-visible spectrophotometry and the DOX cumulative release was determined.DOX release kinetics analysis
In order to determine the mechanism of drug release and the release rate, the data obtained from the drug release studies were fitted with the most relevant kinetic models for our system, such as first order, Higuchi and Korsmeyer–Peppas models.43The first order kinetic release is concentration dependant and can be expressed as:
| Mt = M∞[1 − exp(−K1t)] | (5) |
 | (6) |
| Mt = M∞Kkptn | (7) |
Results and discussion
Characterization of the polymer
Polyethylene glycol (PEG) is FDA-approved and is widely used for biomedical applications, as it is non-toxic to cells and biocompatible with both blood and tissue. Therefore, PEG chains in the P(DEGMA-co-PEGMA-b-[TMSPMA-co-VBA]) polymer (Fig. 2) provide biocompatibility and water dispersibility to the nanosystem. Finally, PEG reduces protein adsorption and thus confers prolonged blood circulation time in the body.44 Copolymerizing two PEG chains of different length provides temperature-responsive behaviour to the polymer, with the possibility to tune the LCST by varying the molar ratio PEGMA:DEGMA (see ESI, Fig. S1†). A molar ratio of 1:2.2 was selected, as it yields a polymer with a sharp LCST at 40 °C (higher than the physiological temperature 37 °C and can be easily reached through magnetic hyperthermia). After this first step, Mn of the polymer is around 12000 g mol−1 (PDI = 1.10) as determined by SEC. Calculation of the theoretical molecular weight of the polymer by NMR was in close accordance with SEC results. Subsequently, a chain extension was carried out with the TMSPMA and VBA monomers (molar ratio TMSPMA:VBA = 1:4), using P(DEGMA-co-PEGMA) as macro-RAFT agent. TMSPMA contains a trimethoxysilane group, which will be used to chemically attach the polymer to the IONP surface via the formation of covalent Si–O–Fe bond. VBA will be used as drug storage unit. The successful chain extension was confirmed by NMR, dominantly shown by the new signal at 10 ppm corresponding to the proton of the aldehyde group (see ESI, Fig. S2†). An increase in the molecular weight of the polymer for 12000 g mol−1 to 14000 g mol−1 (PDI = 1.15) further confirms the polymer was chain extended.
 | ||
| Fig. 2 Chemical structure of P(DEGMA-co-PEGMA-b-[TMSPMA-co-VBA]) polymer. | ||
Characterization of IONPs
Fig. 3 shows a TEM image of the as-synthesized IONPs. The IONPs are spheroidal in shape with an average size of 13.3 ± 2.2 nm calculated from TEM images (n = 305). | ||
| Fig. 3 TEM image of the as-synthesized IONPs (left) and their size distribution histogram (right). | ||
Sharp and clearly defined peaks can be observed on the XRD pattern of the IONPs (Fig. 4), which indicates high degree of crystallinity of the particles. Diffraction peaks matching with a Fe3O4 magnetite phase (JCPDS PDF no. 00-019-0629) seem to indicate that the IONPs are primarily magnetite. A mean crystallite size of 11.1 nm was calculated from the XRD pattern, which is slightly lower than the particle size obtained from TEM. The magnetic properties of the IONPs were evaluated at 5 K and 300 K (Fig. 5). Superparamagnetic behaviour was observed at 300 K with a saturation magnetisation around 70 emu g−1. At 5 K, a hysteresis cycle can be observed due to the transition to a low temperature blocked state, with a saturation magnetisation of 82 emu g−1 and a coercive field around 200 Oe.
 | ||
| Fig. 4 XRD pattern of IONPs. Peaks are indexed according to the reference pattern for magnetite (JCPDS PDF no. 00-019-0629). | ||
 | ||
| Fig. 5 Magnetization curves of IONPs at 300 K (red line) and 5 K (black line). The inset shows a zoom into the low magnetic field region. | ||
Characterization of MNCs
The presence of the polymer on the NP surface was confirmed by FTIR (Fig. 6a). Absorption bands at 1728 cm−1 and 1110 cm−1 correspond to characteristic carbonyl ester bonds C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching, respectively. A small aromatic peak at 1450 cm−1 can also be observed and the small band at 1034 cm−1 is assignable to Si–O bonds.
O and C–O stretching, respectively. A small aromatic peak at 1450 cm−1 can also be observed and the small band at 1034 cm−1 is assignable to Si–O bonds.
 | ||
| Fig. 6 (a) FTIR spectra and (b) weight loss curves as a function of temperature of as-synthesized IONPs (black line) and MNCs (blue line). | ||
The average hydrodynamic diameter DH of bare IONPs was 194 nm with a polydispersity index of 0.30 while after functionalization, the DH of MNCs was reduced to 120 nm with a polydispersity index of 0.16. Introducing a hydrophilic polymer coating on the NP surface stabilizes the IONPs, preventing particle agglomeration and therefore also reducing the attractive inter-particle dipole–dipole interaction and van der Waals forces. NPs in the size range of 10–200 nm preferentially accumulate into tumour than healthy tissue owing to the enhanced permeability and retention (EPR) effect.45 Due to the defective vascular architecture of tumours and the impaired lymphatic clearance, NP extravasation and retainment into tumour tissue is favoured. The DH of the MNCs of 120 nm, is optimal for the EPR effect.
The organic content of the MNCs was determined using TGA in the temperature range from 25 °C to 500 °C. Fig. 6b shows the weight loss of the MNCs as a function of temperature. Polymer thermal decomposition occurs between 240 °C and 420 °C with a weight loss of 8.1 wt%, therefore corresponding to the polymer content of the MNCs. An average number of 24 polymer chains per NP can be calculated.
Colloidal stability of the MNCs
The colloidal stability of the MNCs over time and in different media is of significant importance for biomedical application purposes. The stability in aqueous suspension of the MNCs over time was estimated by measuring the change in DH of the MNCs. No significant changes in hydrodynamic radius was determined by DLS over two weeks, indicating that the MNCs remain well dispersed in water (see ESI, Fig. S3a†). As the pH in the human body can vary considerably, the DH of the MNCs was measured in aqueous suspension with a pH varying from 2 to 12. The DH of the MNCs remained constant at each different pH condition, suggesting that the pH does not have any impact on the colloidal stability of the MNCs (see ESI, Fig. S3b†). Finally, the DH of the MNCs was measured in biological media, i.e. Dubelcco's modified Eagles medium (DMEM) supplemented with 10% fetal bovine serum (FBS), yielding a DH of 147 nm, representing a 27 nm increase as compared to MNCs in water. However, the MNCs remain stable in DMEM + 10% FBS for several weeks before any aggregation of the MNCs could be observed. The increase in DH is most likely due to the formation of a protein corona around the MNCs.46Magnetic heating measurement
In vitro heating behaviour of an aqueous suspension of MNCs (3 mg mL−1) subjected to an AMF of frequency f = 960 kHz and field amplitude H = 6.88 kA m−1 is shown in Fig. 7. The MNCs exhibited a temperature rise of 16 °C in only 10 min, sufficient to reach hyperthermia temperatures, and yielding an ILP of 1.0 nHm2 K−1 (which translated into a SAR of 47 W g−1). This ILP value compares favourably to other ILP values obtained for iron oxide magnetic materials specially synthesized for hyperthermia applications,26 and commercially available ferrofluids with ILP values ranging from 0.15 nHm2 K−1 to 3.1 nHm2 K−1.40,41,47 | ||
| Fig. 7 Typical heating curve of the MNCs dispersed in water with a concentration of 3 mg mL−1 and subjected to an AMF (f = 960 kHz and H = 6.88 kA m−1). | ||
The concentration of MNCs of 3 mg mL−1 used to determine their heating abilities, is lower than that used by most other research groups, and the average concentration of 112 mgFe mL−1 used by Jordan et al. for clinical trials.18,48–50 Therefore, if higher temperatures needed to be reached in a shorter time, increasing the concentration of the MNCs in suspension could be feasible.
DOX conjugation efficiency
In this work, DOX was chosen as anti-cancer drug because of its primary amine group, which can react with an aldehyde to form a Schiff base bond. DOX can then be released by hydrolysis of the Schiff base bond without any damage to its chemical structure. DOX is one of the most widely used chemotherapeutic drug. It intercalates into DNA and inhibits the enzyme topoisomerase II, which is necessary for cell division and growth, therefore resulting in DNA damage and cell death. However, its use as anti-cancer agent is still limited by its detrimental side effects, and especially its cardiotoxicity.51 DOX encapsulation and targeting to the desired site, is therefore a very attractive solution to alter the pharmacokinetics and biodistribution of DOX toward its specific accumulation in tumour tissue, therefore reducing systematic effects and the therapeutic dose needed for an efficient therapy. Ideally, the drug is harmless to healthy tissue while circulating in the body because it is stored and protected in the MNC until it enters the tumour, where it is released in high concentrations. Two methods can be used to encapsulate a drug in an inorganic core/organic shell NPs: physical loading and chemical conjugation via labile bonds. The second method being more reliable, as chemical binding of the drug limit the leakage in a great extent through the polymer shell. In this study, DOX was efficiently encapsulated in the MNCs via heat- and acid-cleavable amine linkage with a conjugation efficiency of η = 82.3%. The DOX content of the nanocarrier was calculated to be WDOX = 7.6 wt%. A weight ratio of DOX to MNC of 1:10 was used, as a higher amount of DOX did not lead to significant increase in the conjugation efficiency.
Drug release profile as a function of pH and temperature
Plots for cumulative DOX release as a function of time without application of an AMF but at different temperatures, i.e. 25 °C, 37 °C and 50 °C, in PBS at pH = 7.4 or 5.7 are represented in Fig. 8. While 50 °C may seem slightly high temperature to simulate hyperthermia, it was chosen because the existence of local heating effects in the vicinity of magnetic NPs has been demonstrated, leading to high temperatures at the magnetic NP surface without necessarily observing a significant increase in the surrounding medium.52,53 For example, T. T. T. N'Guyen and coworkers showed that by subjecting magnetic NPs to an AMF, they could initiate a retro-Diels–Alder reaction in the polymer layer functionalizing the magnetic NPs, that generally requires temperatures up to 90–110 °C.54 Therefore, as the temperature was maintained via a water bath, we chose to use 50 °C to simulate magnetic hyperthermia conditions instead of the more widely used temperature of 45 °C. The drug release profiles of DOX-MNCs under each conditions show two stages: a rapid release of DOX is obtained within 8 h followed by a slower rate of release. This behaviour is more pronounced at pH = 5.7 and 50 °C with 57.8% of DOX released after 8 h and 85.2% after 48 h, while at pH = 7.4 and 25 °C, 13.6% of DOX is released after 8 h and 19.7% after 48 h. The pH and the temperature greatly influence the release behaviour of DOX. The minimal release of DOX is obtained at pH = 7.4 and 25 °C (followed by pH = 7.4 and 37 °C), because at room temperature and physiological pH the imine bond is quite stable and its hydrolysis kinetic is really slow. The initial small burst release of DOX at pH = 7.4 and 25 °C is probably due to physically adsorbed DOX on the outer shell of the polymer layer, despite the numerous washing of the DOX-MNCs. By using a pH stimulus, the amount of DOX released at 25 °C is doubled, with 26.5% of DOX released after 8 h and 39.4% after 48 h. This is because acidic pH facilitates the release of the drug by promoting the hydrolysis of the Schiff base bond (pKa of imine bond is usually around 4). When only the temperature stimulus was used (pH = 7.4 and 50 °C), 38.1% of DOX was released after 8 h and 72.3% after 48 h. The maximal release of DOX was found at pH = 5.7 and 50 °C, when the DOX-MNCs were exposed to both pH and temperature stimuli. The temperature has two different effects: first, it accelerates the rate of the hydrolysis reaction. Second, the polymer being thermo-responsive, upon reaching the LCST, the polymer becomes hydrophobic and shrinks, expelling its aqueous content and pushing the DOX molecules out at the same time. It should also be noted that at acidic pH, the primary amine group of free DOX is protonated (pKa = 8.3), therefore increasing its solubility in aqueous medium and facilitating its expulsion out of the polymer layer. Thus, by combining both the effect of the pH and the temperature, a controlled release of the DOX can be obtained. | ||
| Fig. 8 In vitro cumulative drug release profiles of DOX-MNCs dispersed in PBS at pH = 7.4 or pH = 5.7 at 25 °C, 37 °C and 50 °C. | ||
Drug release kinetics and mechanism of release
In this system, the DOX molecules are chemically bound to the polymer shell of the MNCs. The drug release is therefore mainly dependant on two mechanisms; firstly, the cleavage of the imine bond to release the DOX molecules of the bond, and secondly diffusion of DOX molecules from the polymer matrix to the surrounding dialysis medium.The use of mathematical models is necessary to predict the drug release kinetics of a particular system. In this study, different kinetic models i.e. first order kinetic, Higuchi and Korsmeyer–Peppas were used to fit the release data obtained for each pH and temperature condition applied. The release kinetic and mechanism of DOX from the DOX-MNCs nanosystem were therefore determined according to those mathematical models. The release parameters for each model (K1, Kh, Kkp and n) are shown in Table 1 as well as the correlations values (R2). The first order kinetic model does not fit well with experimental data as manifested by low R2 values, though an increasing of R2 can be observed with increasing temperature and decreasing pH, suggesting a more concentration-dependant release kinetic. Higuchi model also does not fit well with experimental data, even though it has better R2 values than the first order model. The highest R2 value is obtained with the Korsmeyer–Peppas model. Values for the release exponent n were found to be lower than 0.5 indicating a Fickian diffusion mechanism. However, an increase in the n values with the increasing temperature and decreasing pH can be observed, such as at pH 5.7 and 50 °C, the n value reached 0.475, which becomes close to the limit with anomalous diffusion. This can be attributed to the involvement of the imine bond cleavage, which becomes more and more important as the pH decreases and the temperature increases.
| Temperature (°C) | pH (−) | First order | Higuchi | Korsmeyer–Peppas | ||||
|---|---|---|---|---|---|---|---|---|
| K 1 | R 2 | K h | R 2 | K kp | n | R 2 | ||
| 25 | 7.4 | 0.794 | 0.8188 | 2.352 | 0.8523 | 8.181 | 0.277 | 0.9918 |
| 25 | 5.7 | 0.288 | 0.8861 | 5.510 | 0.9204 | 12.530 | 0.361 | 0.9893 |
| 37 | 7.4 | 0.293 | 0.8821 | 4.589 | 0.9179 | 10.548 | 0.363 | 0.9911 |
| 37 | 5.7 | 0.353 | 0.9039 | 6.232 | 0.9040 | 14.841 | 0.378 | 0.9981 |
| 50 | 7.4 | 0.127 | 0.9385 | 10.585 | 0.9844 | 14.520 | 0.451 | 0.9913 |
| 50 | 5.7 | 0.259 | 0.9536 | 12.282 | 0.9177 | 22.175 | 0.475 | 0.9875 |
Conclusions
In this study, composite magnetic NPs were developed and their suitability for multimodal cancer therapy, i.e. targeted controlled drug delivery and localized magnetic hyperthermia was evaluated. The magnetic core, made of iron oxide, was synthezised by a microwave-assisted co-precipitation method, yielding crystalline NPs with a magnetic core size around 13 nm and superparamagnetic behaviour at room temperature. The iron oxide NPs were functionalized with a thermo-responsive polymer shell [composite NPs (MNCs)], resulting in increased colloidal stability. The LCST of the polymer could be easily tuned by varying the initial monomer molar ratio. The MNCs were found to be performant nanoheaters for magnetic hyperthermia at relatively low NP concentration. High conjugation of the anticancer drug DOX to the MNCs was achieved through formation of heat- and acid-labile imine bonds. The dual temperature-and pH-responsive behaviour of the nanocarrier is an important feature of the nanosystem to control where and when the drug is released. A burst release of the drug can be achieved under hyperthermia and tumour acidic pH conditions, as high temperature and acidic pH were shown to act as triggers for the release of the drug. At physiological pH and temperature, the amount of released drug is low; that is the drug is correctly retained in the nanocarrier, which is desirable in clinical applications to limit distribution of the drug to healthy tissues and unwanted side effects. The nanosystem developed here is therefore promising for thermo-chemotherapy applications. It is necessary to investigate the behaviour of the drug release under application of an AMF at physiological pH and acidic tumour pH. Indeed, as the heat comes from the core of the nanocarrier, the release profiles may be different as when the heating is applied through a water bath. Moreover, as already mentioned earlier in the text, in magnetic hyperthermia, studies have shown the existence of local heating effects in the vicinity of magnetic NPs leading to high temperatures at the magnetic NP surface without necessarily observing a significant increase in the surrounding medium.52,53 This phenomenon could favourably influence the drug release behaviour as the heat is directly transmitted to the polymer shell. In future work, the drug release under application of an AMF will be studied, and the potential of the DOX-MNCs for thermo-chemotherapy will be tested on cells and in vivo.Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Number 26600053 (S. M.). N. T. K. Thanh thanks the EPSRC and AFOSR/AOARD for financial support.Notes and references
- J. Huang, X. D. Zhong, L. Y. Wang, L. L. Yang and H. Mao, Theranostics, 2012, 2, 86 CrossRef CAS PubMed.
- R. Hachani, M. Lowdell, M. Birchall and N. T. K. Thanh, Nanoscale, 2013, 5, 11362 RSC.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133 CrossRef CAS.
- M. Schwalbe, K. Pachmann, K. Hoffken and J. H. Clement, J. Phys.: Condens. Matter, 2006, 18, S2865 CrossRef CAS.
- H. Y. Xu, Z. P. Aguilar, L. Yang, M. Kuang, H. W. Duan, Y. H. Xiong, H. Wei and A. Wang, Biomaterials, 2011, 32, 9758 CrossRef CAS PubMed.
- I. Sharifi, H. Shokrollahi and S. Amiri, J. Magn. Magn. Mater., 2012, 324, 903 CrossRef CAS.
- I. Hilger, Int. J. Hyperthermia, 2013, 29, 828 CrossRef PubMed.
- S. Laurent, S. Dutz, U. O. Hafeli and M. Mahmoudi, Adv. Colloid Interface Sci., 2011, 166, 8 CrossRef CAS PubMed.
- M. Arruebo, R. Fernández-Pacheco, M. R. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22 CrossRef.
- M. Mahmoudi, S. Sant, B. Wang, S. Laurent and T. Sen, Adv. Drug Delivery Rev., 2011, 63, 24 CrossRef CAS PubMed.
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284 CrossRef CAS PubMed.
- K. M. Krishnan, IEEE Trans. Magn., 2010, 46, 2523–2558 CrossRef CAS PubMed.
- A. Chicheł, J. Skowronek, M. Kubaszewska and M. Kanikowski, Rep. Pract. Oncol. Radiother., 2007, 12, 267 CrossRef.
- B. V. Harmon, Y. S. Takano, C. M. Winterford and G. C. Gobe, Int. J. Radiat. Biol., 1991, 59, 489 CrossRef CAS PubMed.
- R. Cavaliere, E. C. Ciocatto, B. C. Giovanella, C. Heidelberger, R. O. Johnson, M. Margottini, B. Mondovi, G. Moricca and A. Rossi-Fanelli, Cancer, 1967, 20, 1351 CrossRef CAS PubMed.
- J. van der Zee, Ann. Oncol., 2002, 13, 1173 CrossRef CAS PubMed.
- B. Thiesen and A. Jordan, Int. J. Hyperthermia, 2008, 24, 467 CrossRef CAS PubMed.
- K. Maier-Hauff, F. Ulrich, D. Nestler, H. Niehoff, P. Wust, B. Thiesen, H. Orawa, V. Budach and A. Jordan, J. Neurooncol., 2011, 103, 317 CrossRef PubMed.
- P. Wust, B. Hildebrandt, G. Sreenivasa, B. Rau, J. Gellermann, H. Riess, R. Felix and P. M. Schlag, Lancet Oncol., 2002, 3, 487 CrossRef CAS PubMed.
- R. D. Issels, Eur. J. Cancer, 2008, 44, 2546 CrossRef CAS PubMed.
- K. Sugimachi, H. Kuwano, H. Ide, T. Toge, M. Saku and Y. Oshiumi, Int. J. Hyperthermia, 1994, 10, 485 CrossRef CAS PubMed.
- R. D. Issels, S. Abdel-Rahman, C. M. Wendtner, M. H. Falk, V. Kurze, H. Sauer, U. Aydemir and W. Hiddemann, Eur. J. Cancer, 2001, 37, 1599 CrossRef CAS PubMed.
- R. D. Issels, L. H. Lindner, J. Verweij, P. Wust, P. Reichardt, B. C. Schem, S. Abdel-Rahman, S. Daugaard, C. Salat, C. M. Wendtner, Z. Vujaskovic, R. Wessalowski, K. W. Jauch, H. R. Durr, F. Ploner, A. Baur-Melnyk, U. Mansmann, W. Hiddemann, J. Y. Blay and P. Hohenberger, Lancet Oncol., 2010, 11, 561 CrossRef CAS PubMed.
- M. K. Angele, M. Albertsmeier, N. J. Prix, P. Hohenberger, S. Abdel-Rahman, N. Dieterle, M. Schmidt, U. Mansmann, C. J. Bruns, R. D. Issels, K. W. Jauch and L. H. Lindner, Ann. Surg., 2014, 260, 749 CrossRef PubMed.
- W. Rao, Z.-S. Deng and J. Liu, Crit. Rev. Biomed. Eng., 2010, 38, 101 CrossRef PubMed.
- J. P. May and S.-D. Li, Expert Opin. Drug Delivery, 2013, 10, 511 CrossRef CAS PubMed.
- A. Hervault and N. T. K. Thanh, Nanoscale, 2014, 6, 11553 RSC.
- S. Kossatz, J. Grandke, P. Couleaud, A. Latorre, A. Aires, K. Crosbie-Staunton, R. Ludwig, H. Dahring, V. Ettelt, A. Lazaro-Carrillo, M. Calero, M. Sader, J. Courty, Y. Volkov, A. Prina-Mello, A. Villanueva, A. Somoza, A. L. Cortajarena, R. Miranda and I. Hilger, Breast Cancer Res., 2015, 17, 66 CrossRef PubMed.
- T. J. Li, C. C. Huang, P. W. Ruan, K. Y. Chuang, K. J. Huang, D. B. Shieh and C. S. Yeh, Biomaterials, 2013, 34, 7873 CrossRef CAS PubMed.
- K. Hayashi, M. Nakamura, H. Miki, S. Ozaki, M. Abe, T. Matsumoto, W. Sakamoto, T. Yogo and K. Ishimura, Theranostics, 2014, 4, 834 CrossRef PubMed.
- J. S. Lee, H. L. Rodriguez-Luccioni, J. Mendez, A. K. Sood, G. Lpez-Berestein, C. Rinaldi and M. Torres-Lugo, J. Nanosci. Nanotechnol., 2011, 11, 4153 CrossRef CAS PubMed.
- P. Kulshrestha, M. Gogoi, D. Bahadur and R. Banerjee, Colloids Surf., B, 2012, 96, 1 CrossRef CAS PubMed.
- Y. Ren, H. Zhang, B. Chen, J. Cheng, X. Cai, R. Liu, G. Xia, W. Wu, S. Wang, J. Ding, C. Gao, J. Wang, W. Bao, L. Wang, L. Tian, H. Song and X. Wang, Int. J. Nanomedicine, 2012, 7, 2261 CAS.
- O. Taratula, R. K. Dani, C. Schumann, H. Xu, A. Wang, H. Song, P. Dhagat and O. Taratula, Int. J. Pharm., 2013, 458, 169 CrossRef CAS PubMed.
- P. Bawa, V. Pillay, Y. E. Choonara and L. C. du Toit, Biomed. Mater., 2009, 4, 022001 CrossRef PubMed.
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276 RSC.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215 CrossRef CAS.
- X. He, J. Li, S. An and C. Jiang, Ther. Delivery, 2013, 4, 1499 CrossRef CAS PubMed.
- J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X. J. Liang, Biotechnol. Adv., 2014, 32, 693 CrossRef CAS PubMed.
- C. Blanco-Andujar, D. Ortega, P. Southern, Q. A. Pankhurst and N. T. K. Thanh, Nanoscale, 2015, 7, 1768 RSC.
- A. E. Dunn, D. J. Dunn, A. Macmillan, R. Whan, T. Stait-Gardner, W. S. Price, M. Lim and C. Boyer, Polym. Chem., 2014, 5, 3311 RSC.
- M. Kallumadil, M. Tada, T. Nakagawa, M. Abe, P. Southern and Q. A. Pankhurst, J. Magn. Magn. Mater., 2009, 321, 1509 CrossRef CAS.
- S. Dash, P. N. Murthy, L. Nath and P. Chowdhury, Acta Pol. Pharm., 2010, 67, 217–223 CAS.
- R. Gref, A. Domb, P. Quellec, T. Blunk, R. H. Muller, J. M. Verbavatz and R. Langer, Adv. Drug Delivery Rev., 1995, 16, 215 CrossRef CAS PubMed.
- H. Kobayashi, R. Watanabe and P. L. Choyke, Theranostics, 2014, 4, 81 CrossRef CAS PubMed.
- S. R. Saptarshi, A. Duschl and A. L. Lopata, J. Nanobiotechnol., 2013, 11, 26 CrossRef CAS PubMed.
- C. Blanco-Andujar, D. Ortega, P. Southern, S. A. Nesbitt, N. T. K. Thanh and Q. A. Pankhurst, Nanomedicine, 2016, 11, 121–136 CrossRef CAS PubMed.
- F. K. van Landeghem, K. Maier-Hauff, A. Jordan, K. T. Hoffmann, U. Gneveckow, R. Scholz, B. Thiesen, W. Bruck and A. von Deimling, Biomaterials, 2009, 30, 52 CrossRef CAS PubMed.
- K. Maier-Hauff, R. Rothe, R. Scholz, U. Gneveckow, P. Wust, B. Thiesen, A. Feussner, A. von Deimling, N. Waldoefner, R. Felix and A. Jordan, J. Neurooncol., 2007, 81, 53 CrossRef CAS PubMed.
- M. Johannsen, U. Gneveckow, B. Thiesen, K. Taymoorian, C. H. Cho, N. Waldöfner, R. Scholz, A. Jordan, S. A. Loening and P. Wust, Eur. Urol., 2007, 52, 1653 CrossRef PubMed.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein and R. B. Altman, Pharmacogenet. Genomics, 2011, 21, 440 CrossRef CAS PubMed.
- M. Creixell, A. C. Bohorquez, M. Torres-Lugo and C. Rinaldi, ACS Nano, 2011, 5, 7124–7129 CrossRef CAS PubMed.
- B. Kozissnik, A. C. Bohorquez, J. Dobson and C. Rinaldi, Int. J. Hyperthermia, 2013, 29, 706–714 CrossRef PubMed.
- T. T. T. N'Guyen, H. T. T. Duong, J. Basuki, V. Montembault, S. Pascual, C. Guibert, J. Fresnais, C. Boyer, M. R. Whittaker, T. P. Davis and L. Fontaine, Angew. Chem., Int. Ed., 2013, 52, 14152–14156 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr07773g |
| This journal is © The Royal Society of Chemistry 2016 |