
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonocyte cell membrane-derived nanoghosts for targeted cancer therapy†
S.
Krishnamurthy
a,
M. K.
Gnanasammandhan
b,
C.
Xie
a,
K.
Huang
b,
M. Y.
Cui
a and
J. M.
Chan
*a
aSchool of Chemical and Biomedical Engineering and Lee Kong Chian School of Medicine, Nanyang Technological University, Block N1.3, #B4-10, 70 Nanyang Drive, Singapore 637457. E-mail: julianachan@ntu.edu.sg
bDepartment of Biomedical Engineering, National University of Singapore, 9 Engineering Drive 1, Block EA, #03-12, Singapore 117575
First published on 3rd March 2016
Abstract
Core–shell type ‘nanoghosts’ were synthesized with a drug-loaded biodegradable PLGA core and a monocyte cell membrane-derived shell. The nanoghosts were monodisperse with an average size <200 nm, and showed good serum stability for 120 h. Doxorubicin-loaded nanoghosts showed greater cellular uptake and cytotoxicity compared to non-coated nanoparticle controls in metastatic MCF-7 breast cancer cell lines.
Nanoparticle (NP)-mediated drug delivery systems have contributed significantly to the advancement of clinical therapy.1,2 Liposomes and polymeric NPs are the most commonly-used NP drug delivery platforms,3 and many of which have entered clinical trials and received US Food and Drug Administration (FDA) approval for a variety of clinical indications.2
In recent years, however, researchers have explored newer modalities of drug delivery, such as the use of whole biological cells as targeted delivery systems. These whole cell carriers, as they are commonly referred to, have intact cytoplasm and organelles, and are typically chosen for their ability to home towards a site of interest or release a therapeutic payload.4 To date, these carriers include red blood cells (RBCs),5–7 exosomes,8,9 mesenchymal stem cells,10,11 macrophages,12 lymphocytes,13 and cancer cells,14–16 among others.
Although whole cell carriers have been used successfully in pre-clinical studies, there are a number of limitations to their use. Therapeutic or diagnostic molecules, which are typically loaded by electroporation or osmosis, leak from the cells during and after loading.17 As these are viable cells, the whole cell carriers have to remain biologically active upon administration.5
To retain the biological targeting properties of the whole cell carriers and concomitantly overcome cell viability and drug loading issues, researchers have designed cell ghosts, which are mammalian cell-surface membranes in the nano- to micro-range devoid of cytoplasm and organelles.18,19 This approach of coating naturally-functionalized cell membranes onto preformed NPs greatly simplifies the current methods of NP functionalization.20 Moreover, the presence of an additional lipid (cell membrane, in the case of cell ghosts) layer has been shown to provide greater NP stability and improved drug release profiles,1 which are advantageous for in vivo drug delivery applications. The cell ghosts are produced by cell lysis and serial ultracentrifugation and have a biologically intact bilayer membrane that can be used for both active and passive targeting. Additionally, serial extrusion can give rise to cell ghosts in the 100–200 nm range (‘nanoghosts’)21 that can be used to exploit the enhanced permeation and retention (EPR) effect observed in tumors.22
Typically, drugs are conjugated to or encapsulated within the cell ghosts, with glutaraldehyde commonly used as a common chemical cross linker to conjugate drugs such as daunorubicin23 and doxorubicin (Dox)24,25 to RBC ghosts. To increase the drug loading capacity of the nanoghosts and prevent drug leakage, researchers have encapsulated drug-loaded NPs into the nanoghosts instead of free or conjugated drugs.26–28 Zhang et al. encapsulated poly(lactic-co-glycolic acid) (PLGA) NPs within RBC ghosts by serial extrusion, for use as nanosponges that absorb pore forming toxins29 and for antitoxin vaccination.29 Other NPs that have been encapsulated include gold nanocages for photothermal cancer therapy30,31 and gelatin NPs containing vancomycin as a broad-spectrum antibiotic.32 Besides RBCs, researchers have used cancer cells and leukocytes to encapsulate NPs. Zhang et al. used B16-F10 mouse melanoma cell coated-polymeric NPs to deliver adjuvants for dendritic cell maturation and elicit a tumor-specific immune response.18,33 Leukocyte cell carriers have also been used to evade the immune system and for payload delivery of nanoporous silicon microparticles34 and Dox-encapsulated silica NPs.35
We have previously designed hybrid NP systems comprising a polymeric core and a lipid shell.36,37 These hybrid NP systems typically contain a PLGA core surrounded by a lipid monolayer shell consisting of either synthetic lipids or purified lipid populations extracted from soybean. In this study, we design monocyte-derived cell membrane-coated PLGA NPs to actively target cancers that recruit circulating monocytes as a supply of cell-proliferating growth factors.38 Previously, breast cancer cell lines were shown to overexpress cell adhesion molecules such as VCAM-1 that bind to circulating monocytes expressing cell adhesion molecules such as α4β1 integrin.39 PLGA was selected for the NP core due to its excellent biocompatibility and prior FDA approval for clinical use.40 Dox was chosen for its anti-proliferative properties and loaded into the PLGA core by nanoprecipitation. Meanwhile, cell membranes from U937 monocytes were purified by hypotonic lysis of the cells, homogenization, and subsequent isolation of the membrane fraction by serial ultracentrifugation (Fig. 1A). The purified cell membranes were coated onto Dox-loaded PLGA NPs by serial extrusion through polycarbonate membranes (pore size 400 nm and 200 nm) to form nanoghosts as shown in Fig. 1B.
 | ||
| Fig. 1 (A) Illustration of cell membrane purification and (B) nanoghost preparation. (A) U937 monocyte cell membranes were extracted by hypotonic lysis, homogenization and serial ultracentrifugation. (B) Subsequently, nanoghosts were prepared by serial extrusion with pre-formed PLGA NPs and U937 cell membrane fractions. | ||
Before cell membrane coating, dynamic light scattering studies showed that the Dox-loaded PLGA NPs were monodisperse with an average hydrodynamic diameter of 157 nm and a zeta potential of −8.3 mV. The size and zeta potential of the pure membrane fraction were 28 nm and −13.6 mV, respectively. After coating, the average diameter of the nanoghosts increased by ∼20–40 nm and the zeta potential became more negative at −16.5 mV. Transmission electron microscopy (TEM) images showed similar size profiles before and after cell membrane coating (Fig. 2A and B). The nanoghosts and PLGA NPs exhibited good serum stability over a period of 120 h (Fig. 2C). Dox loading of PLGA NPs was high at 21% (drug to polymer weight) and remained at similar levels after extrusion to form the nanoghosts. The nanoghosts released Dox in a sustained fashion over 72 h, similar to PLGA NP controls, but drug release was slower initially (<30 h) and plateaued at 60 h as shown in Fig. 2D. The slower release from nanoghosts could be due to an additional cell membrane bilayer that possesses a diffusional barrier.
 | ||
| Fig. 2 (A) TEM characterization of PLGA NPs and (B) nanoghosts. Scale bar: 200 nm. (C) Dynamic light scattering studies on nanoghosts and PLGA NPs. (D) Cumulative drug release from nanoghosts and PLGA NPs in PBS at 37 °C. | ||
To confirm the post-purification retention and integrity of CD49d, which is a part of the heterodimeric integrin α4β1 that binds to VCAM-1 on target cells, the nanoghost membrane protein profile was compared to the U937 cell membrane extract profile by SDS-PAGE and western blotting. As shown in Fig. 3A, protein expression patterns were similar between U937 cell membranes and nanoghosts, including integrin α4β1 and control cell membrane marker proteins sodium–potassium ATPase and GAPDH, suggesting that the U937 cell membrane coated onto the nanoghosts are biologically relevant.
 | ||
| Fig. 3 (A) Western blot studies on the U937 cell membrane extract and nanoghosts. Percentage of FITC-positive MCF-7 (B) and mouse skeletal myocyte (C) cells in cell populations treated with either FITC-loaded nanoghosts or PLGA NPs for 2, 4 or 6 h. Statistical analysis was performed using Student's t-test. Differences are considered statistically significant when probability P < 0.05. *, P < 0.05 vs. PLGA. | ||
The intracellular uptake of nanoghosts into metastatic breast cancer cells was tested using MCF-7 cell lines. MCF-7 cells were treated with FITC-loaded nanoghosts or FITC-loaded PLGA NP controls for 2, 4 and 6 h and NP uptake was quantified by flow cytometry. At each time point, the percentage of FITC-positive cells in the overall cell population was analyzed. The number of FITC-positive cells increased over time in both samples, and the percentage of FITC-positive cells was significantly higher at all timepoints in the nanoghost samples, in comparison with PLGA NP control samples (Fig. 3B). Similarly, the uptake of these NPs in primary mouse skeletal myocytes that have negligible expression of cell adhesion molecules like VCAM-1, ICAM-1 and ELAM-1 was carried out as a control (Fig. 3C). NP uptake was similar for both the nanoghosts and PLGA NP samples at all time points and reached a maximum of ∼10%. We hypothesize that the higher cellular uptake observed with the nanoghosts in cancer cells is due to the monocyte cell membrane coating that provides enhanced cancer cell targeting, cell receptor-specific binding and intracellular uptake.
Finally, the cytotoxicity of Dox-loaded nanoghosts was evaluated using MCF-7 cells. The cytotoxicity of blank nanoghosts and PLGA NPs was assessed to rule out non-specific toxicity. Both formulations showed no observable toxicity towards MCF-7 cells up to the tested concentrations of 2 mg mL−1 and for a treatment period of 72 h (Fig. 4A). Next, MCF-7 cells were treated with free Dox, Dox-loaded nanoghosts or Dox-loaded PLGA NPs for 6 h, followed by 66 hours of incubation. Using a standard MTS cytotoxicity assay, free Dox was shown to have a half maximal inhibitory concentration (IC50) of 0.5 μM, while nanoghosts and PLGA NPs had respective IC50 values of 4 μM and 12 μM (Fig. 4B).
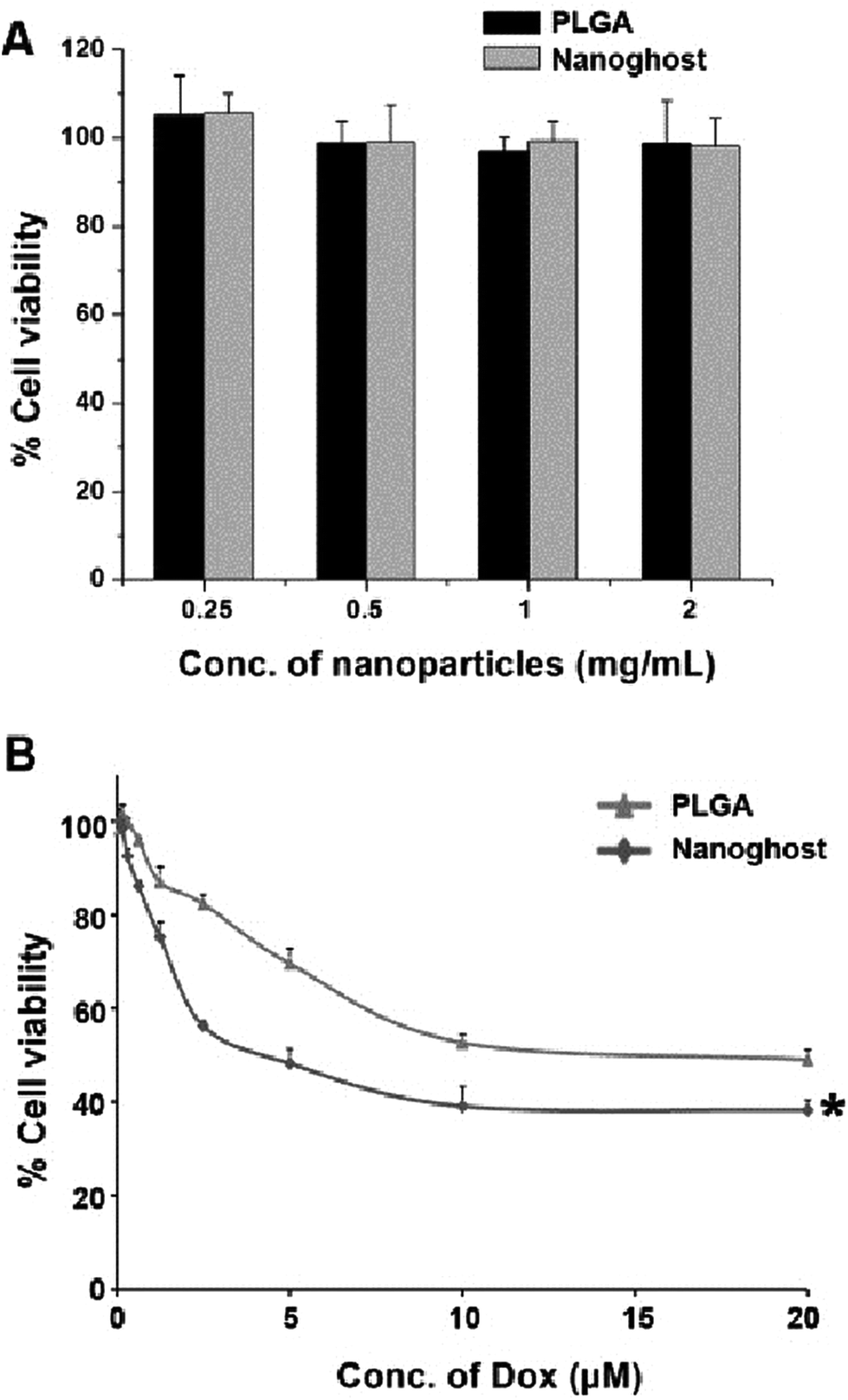 | ||
| Fig. 4 (A) Cytotoxicity of ‘blank’ nanoghosts and PLGA NPs against MCF-7 cells. (B) Cytotoxicity of Dox-loaded nanoghosts and PLGA NPs against MCF-7 cells. Statistical analysis was performed using Student's t-test. Differences are considered statistically significant when probability P < 0.05. *, P < 0.05 vs. PLGA at all Dox concentrations from 2 μM. | ||
The difference in cytotoxicity between the free drug and NP samples could be because of the fundamentally different routes of uptake. Comparing the two NP formulations, however, it can be seen that the nanoghosts had a significantly lower IC50 value compared to PLGA NPs. Therefore, these results demonstrate the significance of using biological cell membrane coatings for efficient NP-mediated drug delivery applications.
In summary, nanoghosts combine both synthetic and biological features for enhanced target specificity and efficacy. Further studies planned include optimizing the ratio of PLGA NPs and cell membranes to minimize the presence of free cell membranes that might cause physical aggregation. In vivo experiments aimed at investigating the pharmacokinetic and pharmacodynamic profiles, tumor-specific accumulation and tumor reduction of the nanoghosts will be performed on MCF-7 xenograft mice models. We believe that cell membrane-coated NP systems such as our monocyte cell membrane-derived nanoghosts hold great promise in biomimetic applications for targeted cancer therapy, and that further research will only help advance their clinical relevance.
Acknowledgements
JMC acknowledges start-up grant funding from Nanyang Technological University (NTU) and the Lee Kong Chian School of Medicine (LKCMedicine). We thank Ronnie Fang and Professor Liangfang Zhang of UCSD for experimental advice.References
- S. Krishnamurthy, R. Vaiyapuri, L. Zhang and J. M. Chan, Biomater. Sci., 2015, 3, 923–936 RSC.
- L. Zhang, F. Gu, J. Chan, A. Wang, R. Langer and O. Farokhzad, Clin. Pharmacol. Ther., 2007, 83, 761–769 CrossRef PubMed.
- S. Krishnamurthy, X. Ke and Y. Y. Yang, Nanomedicine, 2015, 10, 143–160 CrossRef CAS PubMed.
- C. Latorre-Romero, M. R. Marin-Yaseli, C. Belmar-Lopez, R. del Moral, P. C. Marijuan, M. Quintanilla and P. Martin-Duque, Clin. Transl. Oncol., 2011, 13, 10–17 CrossRef PubMed.
- M. Hamidi, A. Zarrin, M. Foroozesh and S. Mohammadi-Samani, J. Controlled Release, 2007, 118, 145–160 CrossRef CAS PubMed.
- A. Fraternale, A. Casabianca, C. Orlandi, A. Cerasi, L. Chiarantini, G. Brandi and M. Magnani, Antiviral Res., 2002, 56, 263–272 CrossRef CAS PubMed.
- R. Luo, S. Mutukumaraswamy, S. S. Venkatraman and B. Neu, J. Mater. Sci. Mater. Med., 2012, 23, 63–71 CrossRef CAS PubMed.
- C. Théry, M. Ostrowski and E. Segura, Nat. Rev. Immunol., 2009, 9, 581–593 CrossRef PubMed.
- A. J. O'Loughlin, C. A. Woffindale and M. J. Wood, Curr. Gene Ther., 2012, 12, 262–274 CrossRef.
- C. D. Porada and G. Almeida-Porada, Adv. Drug Delivery Rev., 2010, 62, 1156–1166 CrossRef CAS PubMed.
- N. E. Toledano Furman, Y. Lupu-Haber, T. Bronshtein, L. Kaneti, N. Letko, E. Weinstein, L. Baruch and M. Machluf, Nano Lett., 2013, 13, 3248–3255 CrossRef CAS PubMed.
- N. Doshi, A. J. Swiston, J. B. Gilbert, M. L. Alcaraz, R. E. Cohen, M. F. Rubner and S. Mitragotri, Adv. Mater., 2011, 23, H105–H109 CrossRef CAS PubMed.
- C. H. June, B. R. Blazar and J. L. Riley, Nat. Rev. Immunol., 2009, 9, 704–716 CrossRef CAS PubMed.
- K. Iguchi, F. Sakurai, K. Tomita, K. Katayama, T. Yamaguchi, K. Kawabata, M. Tagawa, M. Kawabata, T. Shirakawa and H. Mizuguchi, Cancer Gene Ther., 2012, 19, 118–125 CrossRef CAS PubMed.
- T. Zhang, K. Hamada, M. Hyodo, H. Itoh, K. Tani, H. Goda, K. I. Nakashiro and H. Hamakawa, Oncol. Rep., 2011, 25, 795–802 CAS.
- A. T. Power, J. H. Wang, T. J. Falls, J. M. Paterson, K. A. Parato, B. D. Lichty, D. F. Stojdl, P. A. J. Forsyth, H. Atkins and J. C. Bell, Mol. Ther., 2007, 15, 123–130 CrossRef CAS PubMed.
- S. Ravilla, B. R. Chandu, S. Nama and B. Nagaveni, J. Appl. Pharm. Sci., 2012, 2, 166–176 CrossRef.
- R. H. Fang, C. M. J. Hu, B. T. Luk, W. W. Gao, J. A. Copp, Y. Y. Tai, D. E. O'Connor and L. F. Zhang, Nano Lett., 2014, 14, 2181–2188 CrossRef CAS PubMed.
- C.-M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- S. A. McCarthy, G.-L. Davies and Y. K. Gun'ko, Nat. Protoc., 2012, 7, 1677–1693 CrossRef CAS PubMed.
- C. M. J. Hu, R. H. Fang and L. F. Zhang, Adv. Healthcare Mater., 2012, 1, 537–547 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- A. Lejeune, M. Moorjani, C. Gicquaud, J. Lacroix, P. Poyet and R. C. Gaudreault, Anticancer Res., 1994, 14, 915–919 CAS.
- A. Alachi and M. Boroujerdi, Drug Dev. Ind. Pharm., 1990, 16, 2199–2219 CrossRef CAS.
- A. Alachi and M. Boroujerdi, Drug Dev. Ind. Pharm., 1990, 16, 1325–1338 CrossRef CAS.
- A. C. Anselmo, V. Gupta, B. J. Zern, D. Pan, M. Zakrewsky, V. Muzykantov and S. Mitragotri, ACS Nano, 2013, 7, 11129–11137 CrossRef CAS PubMed.
- E. Chambers and S. Mitragotri, J. Controlled Release, 2004, 100, 111–119 CrossRef CAS PubMed.
- E. Chambers and S. Mitragotri, Exp. Biol. Med., 2007, 232, 958–966 CAS.
- C. M. J. Hu, R. H. Fang, B. T. Luk and L. F. Zhang, Nat. Nanotechnol., 2013, 8, 933–938 CrossRef CAS PubMed.
- W. Gao, C. M. J. Hu, R. H. Fang, B. T. Luk, J. Su and L. Zhang, Adv. Mater., 2013, 25, 3549–3553 CrossRef CAS PubMed.
- J.-G. Piao, L. Wang, F. Gao, Y.-Z. You, Y. Xiong and L. Yang, ACS Nano, 2014, 8, 10414–10425 CrossRef CAS PubMed.
- L. L. Li, J. H. Xu, G. B. Qi, X. Z. Zhao, F. Q. Yu and H. Wang, ACS Nano, 2014, 8, 4975–4983 CrossRef CAS PubMed.
- D. J. Schwartzentruber, D. H. Lawson, J. M. Richards, R. M. Conry, D. M. Miller, J. Treisman, F. Gailani, L. Riley, K. Conlon, B. Pockaj, K. L. Kendra, R. L. White, R. Gonzalez, T. M. Kuzel, B. Curti, P. D. Leming, E. D. Whitman, J. Balkissoon, D. S. Reintgen, H. Kaufman, F. M. Marincola, M. J. Merino, S. A. Rosenberg, P. Choyke, D. Vena and P. Hwu, N. Engl. J. Med., 2011, 364, 2119–2127 CrossRef CAS PubMed.
- A. Parodi, N. Quattrocchi, A. L. van de Ven, C. Chiappini, M. Evangelopoulos, J. O. Martinez, B. S. Brown, S. Z. Khaled, I. K. Yazdi and M. V. Enzo, Nat. Nanotechnol., 2013, 8, 61–68 CrossRef CAS PubMed.
- M. Xuan, J. Shao, L. Dai, Q. He and J. Li, Adv. Healthcare Mater., 2015, 4(11), 1645–1652 CrossRef CAS PubMed.
- L. Zhang, J. M. Chan, F. X. Gu, J.-W. Rhee, A. Z. Wang, A. F. Radovic-Moreno, F. Alexis, R. Langer and O. C. Farokhzad, ACS Nano, 2008, 2, 1696–1702 CrossRef CAS PubMed.
- J. M. Chan, L. Zhang, R. Tong, D. Ghosh, W. Gao, G. Liao, K. P. Yuet, D. Gray, J.-W. Rhee and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2213–2218 CrossRef CAS PubMed.
- D. Laoui, E. Van Overmeire, P. De Baetselier, J. A. Van Ginderachter and G. Raes, Front. Immunol., 2014, 5, 1–5 CAS , part of eCollection.
- Q. Chen, X. H.-F. Zhang and J. Massagué, Cancer Cell, 2011, 20, 538–549 CrossRef CAS PubMed.
- R. A. Jain, Biomaterials, 2000, 21, 2475–2490 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr07588b |
| This journal is © The Royal Society of Chemistry 2016 |