
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystal phase-controlled synthesis of rod-shaped AgInTe2 nanocrystals for in vivo imaging in the near-infrared wavelength region†
Tatsuya
Kameyama
a,
Yujiro
Ishigami
a,
Hiroshi
Yukawa
ab,
Taisuke
Shimada
a,
Yoshinobu
Baba
abc,
Tetsuya
Ishikawa
d,
Susumu
Kuwabata
e and
Tsukasa
Torimoto
*a
aGraduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: torimoto@apchem.nagoya-u.ac.jp
bImPACT Research Center for Advanced Nanobiodevices, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan
cInstitute of Innovation for Future Society, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan
dDepartment of Medical Laboratory Sciences, Graduate School of Medicine, Nagoya University, Daikominami, Higashi-ku, Nagoya 461-8673, Japan
eGraduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan
First published on 10th February 2016
Abstract
Rod-shaped AgInTe2 nanocrystals (NCs) exhibiting intense near-band edge photoluminescence in the near-infrared (NIR) wavelength region, were successfully prepared by the thermal reaction of metal acetates and Te precursors in 1-dodecanethiol. Increasing the reaction temperature resulted in the formation of larger AgInTe2 NCs with crystal structures varying from hexagonal to tetragonal at reaction temperatures of 280 °C or higher. The energy gap was increased from 1.13 to 1.20 eV with a decrease in rod width from 8.3 to 5.6 nm, accompanied by a blue shift in the photoluminescence (PL) peak wavelength from 1097 to 1033 nm. The optimal PL quantum yield was approximately 18% for AgInTe2 NCs with rod widths of 5.6 nm. The applicability of AgInTe2 NCs as a NIR-emitting material for in vivo biological imaging was examined by injecting AgInTe2 NC-incorporated liposomes into the back of a C57BL/6 mouse, followed by in vivo photoluminescence imaging in the NIR region.
Colloidal semiconductor nanocrystals (NCs) with absorbing and emitting properties in the near-infrared (NIR) light region have attracted considerable attention because of their potential applications in biological imaging,1–7 sensors,8,9 and solar energy conversion systems.4–6,9–14 NIR light penetrates more efficiently into biological tissues such as skin and blood than light in the ultraviolet, visible, and IR wavelength regions; thus making them promising probes for biological applications, particularly for in vivo imaging.15–18 To this end, many studies have attempted to develop NIR light-emitting semiconductor nanocrystals as photoluminescent markers, because they exhibited unique and excellent physicochemical properties, including a broad absorption band, size-tunable photoluminescence (PL) peak, and high PL quantum yield (QY), in comparison with other NIR light-emitting materials such as organic dyes or lanthanide-based upconversion nanoparticles.1–3,7
To date, high-quality semiconductor NCs that exhibit NIR photoluminescence, such as CdTe,19,20 PbS,21 and PbSe,22 have been prepared via solution syntheses. In vivo imaging has been successfully conducted by the surface modification of NCs with bifunctional agents containing hydrophilic functional groups or by the incorporation of NCs into liposomes or micelles. Furthermore, nanocrystals not containing toxic metal elements such as Ag2S,20,23 Ag2Se,24 AgInS2,20,25 and CuInS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5,26,27 have been reported to show intense PL in the NIR region. For example, binary semiconductors of Ag2S and Ag2Se with band gaps of 1.0 and 0.15 eV, respectively, have been utilized as NIR-emitting markers in vivo. Hocaoglu et al. successfully prepared highly photoluminescent Ag2S NCs (PL QY < 39%) that showed good cytocompatibility using NIH/3T3 cell lines.23 Wang et al. demonstrated the in vivo imaging of vascular structures of mice using Ag2Se NCs with a PL QY of 29%.24 Since an AgInTe2 semiconductor was reported to have a bulk band gap of approximately 1.0 eV,28 size-controlled AgInTe2 NCs show promise as NIR light-emitting markers in the second NIR window at wavelengths of 1000–1350 nm, in which the signal-to-noise ratios of in vivo PL imaging are much more improved than those in the first NIR window with wavelengths from 700 to 900 nm, because of reduced photon absorption and lower autofluorescence by biological tissues.16,29 However, with the exception of the recent preparation of spherical, approximately 10.6 nm AgInTe2 NCs that exhibited a low PL QY of 0.06%,30 no attempts have been made to fabricate highly photoluminescent AgInTe2 NCs with sizes less than several tens of nanometers. Moreover, such AgInTe2 NCs have never been successfully applied to in vivo biological imaging because of their low QY of NIR photoluminescence.
5,26,27 have been reported to show intense PL in the NIR region. For example, binary semiconductors of Ag2S and Ag2Se with band gaps of 1.0 and 0.15 eV, respectively, have been utilized as NIR-emitting markers in vivo. Hocaoglu et al. successfully prepared highly photoluminescent Ag2S NCs (PL QY < 39%) that showed good cytocompatibility using NIH/3T3 cell lines.23 Wang et al. demonstrated the in vivo imaging of vascular structures of mice using Ag2Se NCs with a PL QY of 29%.24 Since an AgInTe2 semiconductor was reported to have a bulk band gap of approximately 1.0 eV,28 size-controlled AgInTe2 NCs show promise as NIR light-emitting markers in the second NIR window at wavelengths of 1000–1350 nm, in which the signal-to-noise ratios of in vivo PL imaging are much more improved than those in the first NIR window with wavelengths from 700 to 900 nm, because of reduced photon absorption and lower autofluorescence by biological tissues.16,29 However, with the exception of the recent preparation of spherical, approximately 10.6 nm AgInTe2 NCs that exhibited a low PL QY of 0.06%,30 no attempts have been made to fabricate highly photoluminescent AgInTe2 NCs with sizes less than several tens of nanometers. Moreover, such AgInTe2 NCs have never been successfully applied to in vivo biological imaging because of their low QY of NIR photoluminescence.
Here, we report rod-shaped AgInTe2 NCs with controlled widths and lengths. The obtained NCs exhibit intense PL and the peak wavelength in the NIR region is tunable by changing the rod width, enabling their use as an NIR marker for in vivo biological imaging.
AgInTe2 NCs were prepared through the thermal reaction of metal acetates and Te in a hot organic solvent. Elemental Te powder (10.7 mmol) and trioctylphosphine (30 cm3) were loaded in a three-neck flask under vacuum and then heated to 80 °C under vigorous stirring. The flask was then purged with N2 and heated to 240 °C for 3 h, yielding a clear orange solution. The obtained Te precursor solution was cooled to room temperature and stored under an N2 atmosphere. Powders of silver acetate (0.075 mmol) and indium acetate (0.075 mmol) were placed in a test tube with 3.0 cm3 of 1-dodecanethiol followed by vigorous stirring under an N2 atmosphere. A 0.42 cm3 portion of the Te precursor solution containing 0.15 mmol of Te was injected into the solution, and the mixture was then immediately heat-treated at various temperatures (180 °C–300 °C) for 5 min with vigorous stirring under an N2 atmosphere. The resulting suspension was subjected to centrifugation at 4000 rpm for 5 min to remove large particles. AgInTe2 NCs were separated from the supernatant by the addition of 5 cm3 ethanol. The obtained precipitate was isolated by centrifugation and washed twice with ethanol. Finally, the AgInTe2 NCs were dissolved in octane for various characterizations. Detailed information about the characterization of the NCs is shown in the ESI.†
Fig. 1a shows the X-ray powder diffraction (XRD) patterns of the obtained nanocrystals prepared at 180 °C–300 °C. The particles prepared at temperatures of 280 °C or higher exhibited three predominant peaks at 2θ = 24.0°, 39.8°, and 46.8°, which were respectively assigned to the diffractions of the (112), (204), and (312) planes of tetragonal AgInTe2 (Fig. 1b; PDF# 01-075-0119). In contrast, the XRD patterns of nanocrystals prepared at temperatures of 250 °C or lower did not match the previously reported patterns or standard ICDD (PDF-2) patterns of AgInTe2 or other conceivable crystal phases such as AgIn8Te5, Ag2Te, and In2Te3. The hexagonal wurtzite crystal phase has been reported in the NCs of multinary semiconductors such as CuInS2,31 CuGaS2,32 and Cu2ZnSnS432 depending on the preparation conditions, even if the bulk crystals have predominantly tetragonal or cubic zinc-blende crystal structures. Thus, we simulated the XRD pattern of cation-disordered hexagonal AgInTe2 (Fig. 1c) according to previously reported methods for CuInS2,31 in which the hexagonal crystal structure was considered to be a derivative of wurtzite ZnS in such a way that the metal ions of Cu and In randomly substituted the Zn site (see the ESI† for details). The simulated XRD pattern of cation-disordered wurtzite AgInTe2 (the bottom of Fig. 1a) matched well with the patterns observed for the nanocrystals prepared at temperatures of 250 °C or lower. It should be noted that peaks at 23°, 26°, and 43° assignable to hexagonal AgInTe2 were still detected in the XRD pattern (iv) of particles prepared at 280 °C shown in Fig. 1a but their intensities were much smaller than those assigned to tetragonal AgInTe2, indicating that the phase transition of most AgInTe2 nanoparticles from the hexagonal to tetragonal structure was completed within a relatively narrow temperature range between 250 and 280 °C.
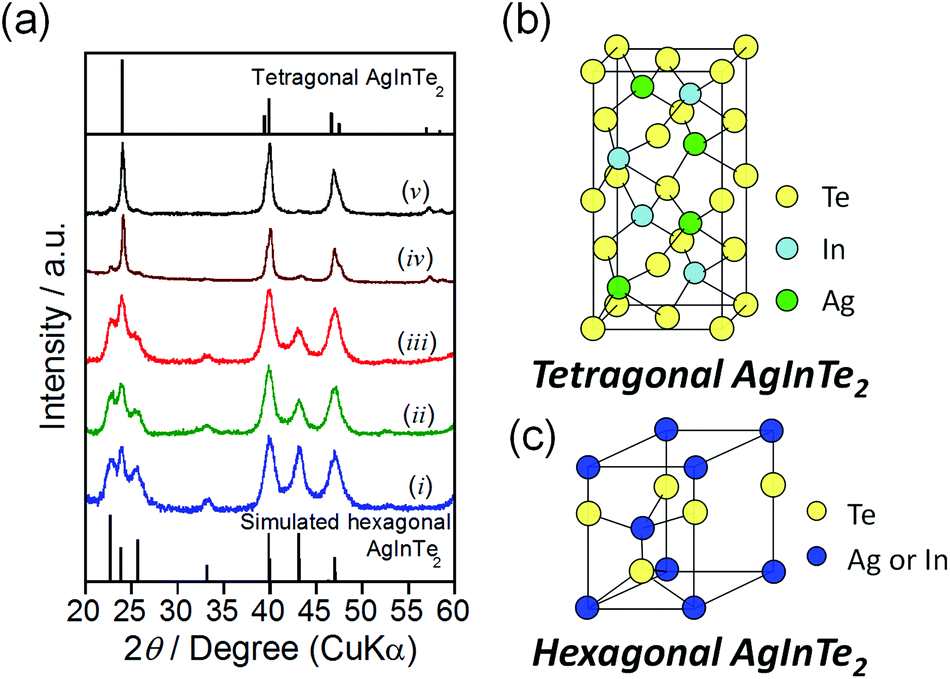 | ||
| Fig. 1 (a) XRD patterns of AgInTe2 NCs prepared at various temperatures; (i) 180 °C, (ii) 220 °C, (iii) 250 °C, (iv) 280 °C, and (v) 300 °C. Schematic illustration of unit cells of AgInTe2 with tetragonal (b) and hexagonal crystal structures (c). | ||
Elemental analysis revealed that the obtained AgInTe2 NCs had nearly stoichiometric chemical compositions, irrespective of reaction temperature (Fig. S1†), although slightly In-rich and Ag-deficient compositions were detected in the nanocrystals prepared at temperatures of 250 °C or lower. These results suggested that the AgInTe2 NCs prepared in this study contained small amounts of crystal defects such as Ag vacancies, interstitial In, and/or antisite defects of InAg.Fig. 2a–c show representative TEM images of the AgInTe2 NCs formed at 180 °C, 250 °C, and 300 °C (images of NCs obtained at other temperatures are shown in Fig. S2†). Rod-like particles were formed, and their sizes increased slightly with increasing formation temperature. As shown in Fig. 2d, the average width of the particles increased monotonically from 5.5 to 8.3 nm with increasing reaction temperature from 180 °C to 300 °C, accompanied by an increase in the average length from 11.4 to 13.5 nm. It should be noted that reaction temperatures higher than 280 °C induced the tetragonal crystal structure, produced a larger fraction of AgInTe2 NCs with irregular non-rod shapes, and resulted in a wider size distribution.
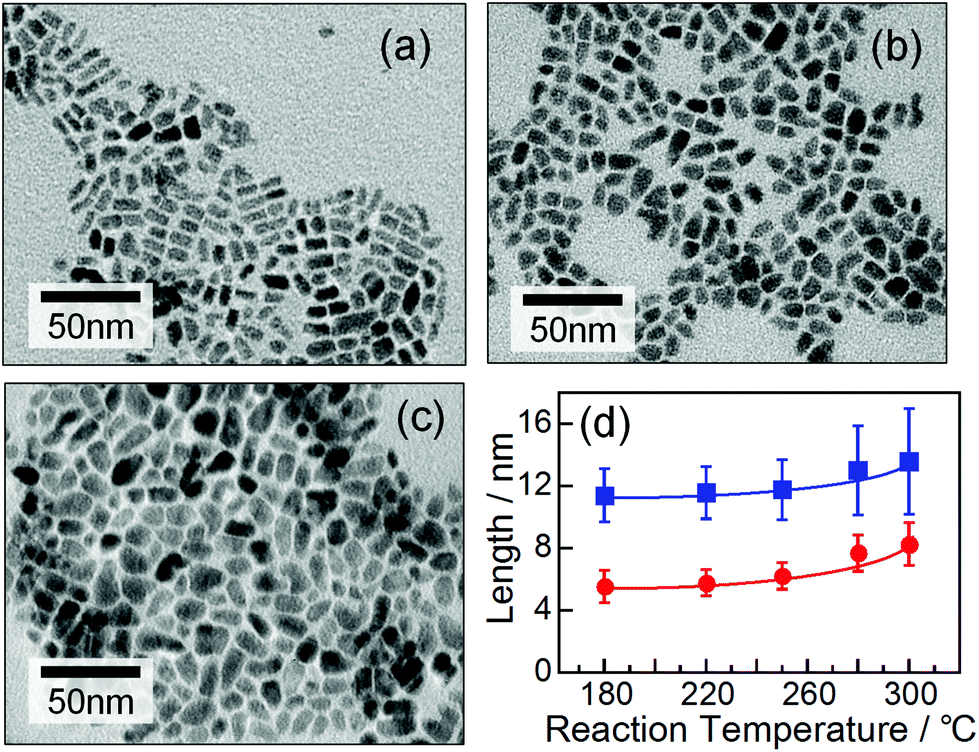 | ||
| Fig. 2 Representative TEM images of AgInTe2 NCs prepared at (a) 180 °C, (b) 250 °C, and (c) 300 °C. (d) Dependences of width (circles) and length (squares) of AgInTe2 NCs on the temperature in preparation. | ||
The solutions containing AgInTe2 NCs were deep-black in color due to strong light absorption in the visible and NIR wavelength regions, as observed in the absorption spectra shown in Fig. 3a. Irrespective of the synthetic temperature, an obscure absorption shoulder attributed to the exciton transition appeared at approximately 1000 nm with the absorption onset at approximately 1150–1200 nm. Relatively strong PL was observed in the NIR wavelength region for all AgInTe2 NCs. The PL spectra (Fig. 3a) exhibited somewhat wide FWHMs of approximately 100–150 nm, corresponding to approximately 110–180 meV. Each PL peak was located near the corresponding absorption onset, and the peak wavelength was red-shifted, as expected from the enlargement in the width of AgInTe2 NCs with increasing reaction temperature (Fig. 2d) as discussed below. Typical I–III–VI2 semiconductor nanocrystals such as AgInS2, AgInSe2, and CuInS2 have been reported to exhibit broad PL peaks (FWHM of approximately 200–400 meV)33–35 in the visible-light region with a large Stokes shifts of >100 meV; in these cases, the PL was attributed to donor–acceptor pair recombination and/or emission from trap sites. In contrast, binary, NIR-emitting, high-quality semiconductor nanocrystals, such as CdTe and PbS, exhibited narrow near-band edge emissions with FWHMs of approximately 50–100 meV.36,37 Considering these results, the PL of the AgInTe2 NCs formed in the present study can likely be attributed to radiative emission from donor–acceptor levels near the band edge.
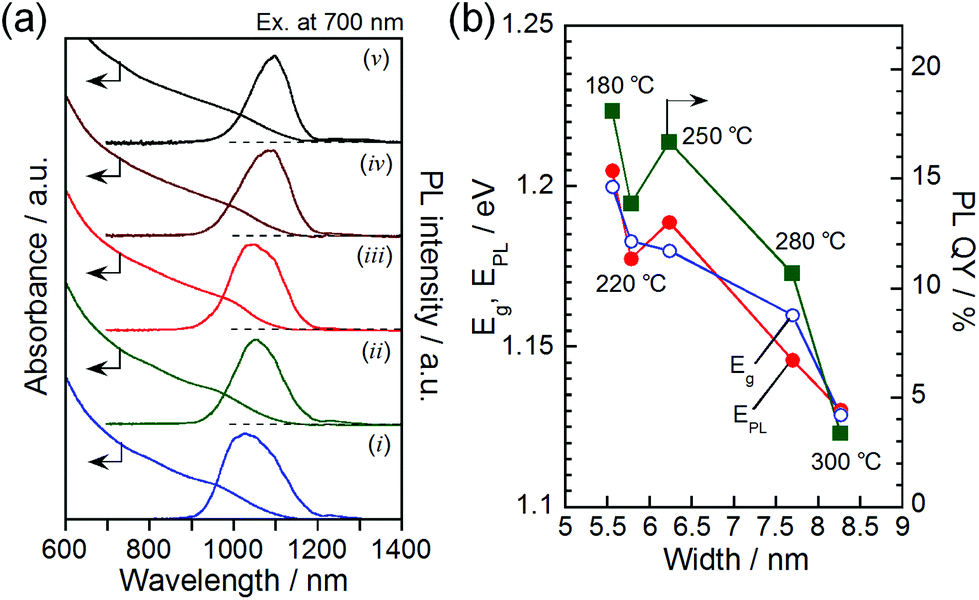 | ||
| Fig. 3 (a) Absorption and PL spectra of AgInTe2 NCs prepared at temperatures of (i) 180 °C, (ii) 220 °C, (iii) 250 °C, (iv) 280 °C, and (v) 300 °C. The wavelength of excitation light for PL measurement was 700 nm. (b) Plots of Eg, EPL, or PL QY as a function of the width of AgInTe2 NCs. | ||
Fig. 3b shows the energy gap (Eg) obtained from the absorption onset, the PL peak energy (EPL), and the PL QY as a function of AgInTe2 NC width. Since the exciton peak was not clearly observed in the individual absorption spectra, a Tauc plot was adopted to determine the value of Eg (Fig. S3 in the ESI†). Both the Eg and EPL increased with decreasing particle size. The exciton Bohr radius, aB, of AgInTe2 was calculated to be 3.5 nm from the dielectric constant of 6.48 (ref. 38) and the effective masses of electrons and holes of 0.108 and 1.012, respectively.39 Since the quantum size effect appears when the NC size becomes smaller than 2aB, it is reasonable to assume that decreasing the AgInTe2 NC width to smaller than approximately 7 nm enlarged both Eg and EPL due to the quantum size effect.
The PL QY has been reported to be enhanced with decreasing size for various types of semiconductor nanocrystals, including CuInS2,40 CdS,41 CdTe,42 and Si.43 These results were most likely due to the greater confinement of photogenerated electron–hole pairs in smaller-sized NCs, which increased the probability of radiative recombination. This can be true for the AgInTe2 NCs in the current study, in which the PL QY was improved when the widths of the rod-shaped AgInTe2 NCs decreased (Fig. 3b). The maximum PL QY of 18%, obtained in the present study for the hexagonal phase AgInTe2 NCs 5.5 nm in width, was quite larger than that previously reported for spherical AgInTe2 NCs having a relatively large diameter of 10.6 nm, 0.06%, probably because of the greater confinement of photogenerated carriers in smaller NCs. Although this value was approximately half of the PL QYs observed for conventional NIR-emissive semiconductor nanocrystals of PbS, Ag2S, or Ag2Se, it was comparable to that of indocyanine green, which is practically used as a dye for biological imaging.44 Hence, the relatively intense NIR emission of the AgInTe2 NCs demonstrates their potential use as an NIR marker for biological imaging.
The as-prepared AgInTe2 NCs could not be uniformly dispersed in aqueous solutions because of their hydrophobic surface modified with 1-dodecanethiol (used as a stabilizing agent). This makes the direct injection of these NCs in biological tissues difficult. Therefore, in this study, small unilamellar liposomes of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC; Fig. 4a) were utilized as carriers to transfer the AgInTe2 NCs into aqueous solution without aggregation. A previously reported preparation method for DSPC liposomes containing hydrophilic CdSe NCs was employed.45 The hexagonal AgInTe2 NCs with a PL QY of 18% were incorporated into DSPC liposomes [see the ESI† for the detailed preparation procedure of the DSPC–AgInTe2 NC nanocomposite (DSPC–AgInTe2)]. Dynamic light scattering measurements revealed that the DSPC–AgInTe2 composite particles had an average hydrodynamic diameter of 1.8 × 102 nm (Fig. S4†), in rough agreement with the previously reported value for DSPC–CdSe nanocomposites (approximately 1.7 × 102 nm).45 The average number of AgInTe2 NCs in an individual liposome was estimated to be 4.4 × 102 particles using the concentrations of DSPC and AgInTe2 NCs and the molar absorption coefficient (ε) of AgInTe2 NCs (Fig. S5†). We should note that the AgInTe2 NCs exhibited relatively large ε values greater than approximately 105 dm3 mol−1 (particles) cm−1 at incident photon energies greater than approximately 1.1 eV. These ε values were comparable to those of conventional NIR-emissive PbS nanocrystals of 6.5 nm in average size (ca. 6 × 106 dm3 mol−1 (particles) cm−1 at 3.1 eV).46 Both the PL peak and the absorption onset of the AgInTe2 NCs incorporated into DSPC liposomes were red shifted by 50–100 nm compared with those of the original particles (Fig. 4b); these red shifts were accompanied by a decrease in the PL QY from 18% to 2.2%. These results suggest that NC coalescence slightly occurred during the incorporation process. To utilize the thus prepared DSPC–AgInTe2 NC composites for biological imaging, it is important to evaluate the durability of photoluminescent AgInTe2 NCs under various environmental conditions.47,48 We dispersed the DSPC–AgInTe2 NC composites in 10 mmol dm−3 phosphate buffer solutions at different pH values (pH 5.4, 7.0, or 8.5) and monitored the changes in the PL intensity of AgInTe2 NCs (Fig. S6†). The AgInTe2 NCs exhibited an almost constant PL intensity for at least 24 h when a DSPC–AgInTe2 NC composite solution at pH 7.0 was stored in the dark under an N2 atmosphere. On the other hand, the PL intensities of AgInTe2 NC solutions at both pH 7.0 and 5.4 gradually decreased in air with the elapse of time and finally reached ca. 85% of the initial values after 24 h, while the deterioration of the PL intensity in air was enhanced in a basic buffer solution at pH 8.5 where AgInTe2 NCs exhibited ca. 20% of the initial PL intensity after 24 h. These results suggested that the ideal pH range of DSPC–AgInTe2 NC composites as a PL probe was between pH 7.0 and 5.4. It should be noted that the continuous irradiation to the DSPC–AgInTe2 NC composite solution (pH 7.0) in air by the excitation light with a wavelength of 700 nm (the intensity of 17.5 mW cm−2) also induced the remarkable deterioration of the PL peak probably due to the partial photooxidation of the AgInTe2 NC surface, the intensity being 20% as small as the initial value after ca. 8 h.
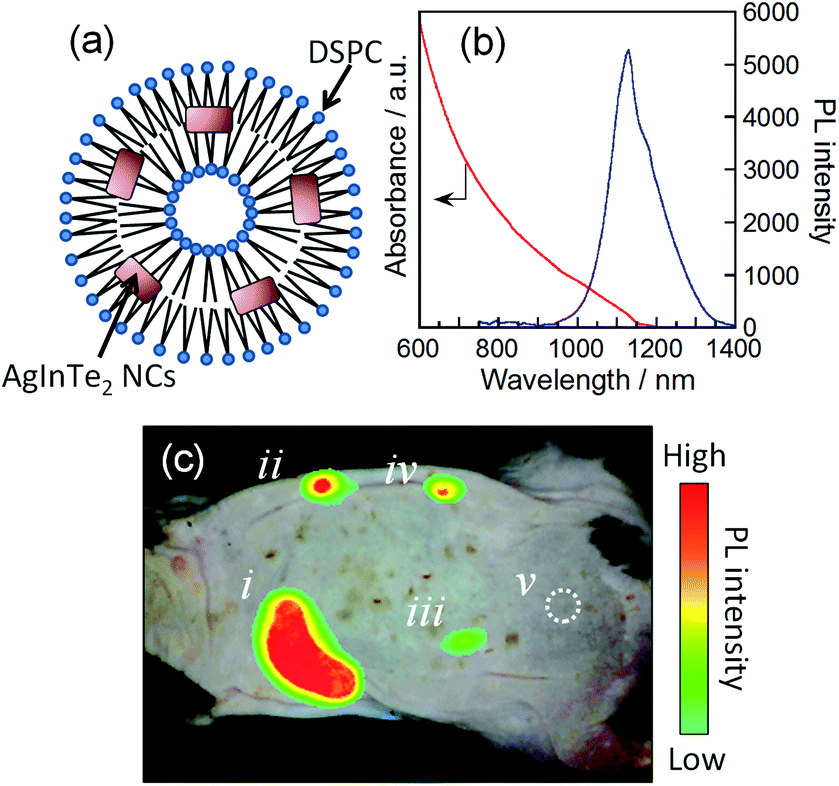 | ||
| Fig. 4 (a) Schematic illustration of DSPC–AgInTe2 NC composites and (b) their absorption and PL spectra. The wavelength of excitation light for PL measurement was 700 nm. (c) In vivo photoluminescence imaging of a mouse after injection of a 50 mm3 portion of DSPC–AgInTe2 dispersion. The concentrations of AgInTe2 NCs injected in dispersion were 50 (i), 25 (ii), 12.5 (iii), 6.25 (iv), and 0 nmol (particles) dm−3 (v). | ||
As mentioned above, the obtained water-soluble DSPC–AgInTe2 can emit in the NIR wavelength region. To explore the potential application of AgInTe2 NCs for in vivo imaging, the DSPC–AgInTe2 composites were injected into the back of a hair-removed C57BL/6 mouse after the dilution of the original composite dispersion containing 50 nmol (particles) dm−3 AgInTe2 NCs with phosphate-buffered saline (pH 7.4). Fig. 4c shows an NIR PL image obtained using an SAI-1000 portable in vivo fluorescence imaging system (SHIMADZU; excitation laser, 980 nm; emission filter, 1080 nm long-pass). In Fig. 4c, the obtained PL image was superimposed on the photograph of the mouse. The NIR PL emitted from the AgInTe2 NCs was clearly observed through the mouse skin, which was approximately 2 mm in thickness, even when the concentration of AgInTe2 NCs in dispersion was as low as 6.25 nmol (particles) dm−3. The NIR PL signals were detectable for 5 h after injection when the concentration was higher than 12.5 nmol (particles) dm−3, although the PL intensity at the site of the injection of 12.5 nmol (particles) dm−3 AgInTe2 NCs was approximately 30% as high as in the initial state (Fig. S7†).
Conclusions
In conclusion, size-quantized, rod-shaped AgInTe2 NCs were prepared via the thermal reaction of metal acetates and Te precursors in 1-dodecanethiol. The widths and lengths of the NCs increased monotonically with increasing reaction temperature. Their crystal structures, either hexagonal or tetragonal, could be controlled by choosing the reaction temperature. The energy gap and PL peak position of the rod-shaped NCs were tunable and depended on the particle width. The highest intensity of near-band edge PL in the NIR region was obtained for the particles prepared at 180 °C (PL QY = 18%), demonstrating the applicability of the AgInTe2 NCs as an efficient NIR-emitting marker for in vivo imaging. Since the surface coating of NCs, such as CdSe, CuInS2 and AgInS2, with a wider band gap semiconductor, such as ZnS and CdS, has been reported to enhance the stability of NCs and increase their PL QY several times greater than that without coating,26,34,49 the fabrication of such core–shell structures with AgInTe2 NCs as a core will be a useful strategy for further improving their performance as a biological marker. The findings of this study provide a new candidate for NIR-light-sensitive materials without highly toxic metal elements, such as Cd and Pb, for various optical applications, including in vivo biological imaging and solar energy conversion systems.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Artificial photosynthesis (AnApple)” (no. 15H00870) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, a Grant-in-Aid for Scientific Research (B) (no. 15H03876) and Grant-in-Aid for Young Scientists (B) (no. 26790006) from the Japan Society for the Promotion of Science, and the Japan Agency for Medical Research and Development (A-MED) through its “Research Center Network for Realization of Regenerative Medicine.” One of the authors (T. K.) thanks the Nippon Sheet Glass Foundation for Materials Science and Engineering for funding support.Notes and references
- S. Kim, Y. T. Lim, E. G. Soltesz, A. M. De Grand, J. Lee, A. Nakayama, J. A. Parker, T. Mihaljevic, R. G. Laurence, D. M. Dor, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2004, 22, 93–97 CrossRef CAS PubMed.
- H. Yukawa, M. Watanabe, N. Kaji, Y. Okamoto, M. Tokeshi, Y. Miyamoto, H. Noguchi, Y. Baba and S. Hayashi, Biomaterials, 2012, 33, 2177–2186 CrossRef CAS PubMed.
- S. E. Lohse and C. J. Murphy, J. Am. Chem. Soc., 2012, 134, 15607–15620 CrossRef CAS PubMed.
- H. Z. Zhong, Z. L. Bai and B. S. Zou, J. Phys. Chem. Lett., 2012, 3, 3167–3175 CrossRef CAS PubMed.
- J. Kolny-Olesiak and H. Weller, ACS Appl. Mater. Interfaces, 2013, 5, 12221–12237 CAS.
- S. V. Kershaw, A. S. Susha and A. L. Rogach, Chem. Soc. Rev., 2013, 42, 3033–3087 RSC.
- J. Niu, X. Wang, J. Lv, Y. Li and B. Tang, TrAC Trends Anal. Chem., 2014, 58, 112–119 CrossRef CAS.
- Y. S. Xia, L. Song and C. Q. Zhu, Anal. Chem., 2011, 83, 1401–1407 CrossRef CAS PubMed.
- T. Torimoto, T. Kameyama and S. Kuwabata, J. Phys. Chem. Lett., 2014, 5, 336–347 CrossRef CAS PubMed.
- J. B. Sambur, T. Novet and B. A. Parkinson, Science, 2010, 330, 63–66 CrossRef CAS PubMed.
- J. Jasieniak, B. I. MacDonald, S. E. Watkins and P. Mulvaney, Nano Lett., 2011, 11, 2856–2864 CrossRef CAS PubMed.
- P. V. Kamat, Acc. Chem. Res., 2012, 45, 1906–1915 CrossRef CAS PubMed.
- B. K. Chen, S. Chang, D. Y. Li, L. L. Chen, Y. T. Wang, T. Chen, B. S. Zou, H. Z. Zhong and A. L. Rogach, Chem. Mater., 2015, 27, 5949–5956 CrossRef CAS.
- T. Kawawaki, H. B. Wang, T. Kubo, K. Saito, J. Nakazaki, H. Segawa and T. Tatsuma, ACS Nano, 2015, 9, 4165–4172 CrossRef CAS PubMed.
- T. Niidome, Y. Akiyama, K. Shimoda, T. Kawano, T. Mori, Y. Katayama and Y. Niidome, Small, 2008, 4, 1001–1007 CrossRef CAS PubMed.
- A. M. Smith, M. C. Mancini and S. M. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- F. Wang, D. Banerjee, Y. S. Liu, X. Y. Chen and X. G. Liu, Analyst, 2010, 135, 1839–1854 RSC.
- Y. Il Park, K. T. Lee, Y. D. Suh and T. Hyeon, Chem. Soc. Rev., 2015, 44, 130s2–11317 RSC.
- Y. G. Zheng, S. J. Gao and J. Y. Ying, Adv. Mater., 2007, 19, 376–380 CrossRef CAS.
- J. van Embden, A. S. R. Chesman and J. J. Jasieniak, Chem. Mater., 2015, 27, 2246–2285 CrossRef CAS.
- D. W. Deng, J. F. Xia, J. Cao, L. Z. Qu, J. M. Tian, Z. Y. Qian, Y. Q. Gu and Z. Z. Gu, J. Colloid Interface Sci., 2012, 367, 234–240 CrossRef CAS PubMed.
- A. J. Shuhendler, P. Prasad, H. K. C. Chan, C. R. Gordijo, B. Soroushian, M. Kolios, K. Yu, P. J. O'Brien, A. M. Rauth and X. Y. Wu, ACS Nano, 2011, 5, 1958–1966 CrossRef CAS PubMed.
- I. Hocaoglu, M. N. Cizmeciyan, R. Erdem, C. Ozen, A. Kurt, A. Sennaroglu and H. Y. Acar, J. Mater. Chem., 2012, 22, 14674–14681 RSC.
- B. H. Dong, C. Y. Li, G. C. Chen, Y. J. Zhang, Y. Zhang, M. J. Deng and Q. B. Wang, Chem. Mater., 2013, 25, 2503–2509 CrossRef CAS.
- T. Torimoto, T. Adachi, K. Okazaki, M. Sakuraoka, T. Shibayama, B. Ohtani, A. Kudo and S. Kuwabata, J. Am. Chem. Soc., 2007, 129, 12388–12389 CrossRef CAS PubMed.
- L. Li, T. J. Daou, I. Texier, T. K. C. Tran, Q. L. Nguyen and P. Reiss, Chem. Mater., 2009, 21, 2422–2429 CrossRef CAS.
- M. Sakamoto, L. H. Chen, M. Okano, D. M. Tex, Y. Kanemitsu and T. Teranishi, J. Phys. Chem. C, 2015, 119, 11100–11105 CAS.
- B. Tell, J. L. Shay and H. M. Kasper, Phys. Rev. B: Solid State, 1974, 9, 5203–5208 CrossRef CAS.
- Y. T. Lim, S. Kim, A. Nakayama, N. E. Stott, M. G. Bawendi and J. V. Frangioni, Mol. Imaging, 2003, 2, 50–64 CrossRef CAS PubMed.
- M. A. Langevin, T. Pons, A. M. Ritcey and C. N. Allen, Nanoscale Res. Lett., 2015, 10, 255 CrossRef PubMed.
- D. C. Pan, L. J. An, Z. M. Sun, W. Hou, Y. Yang, Z. Z. Yang and Y. F. Lu, J. Am. Chem. Soc., 2008, 130, 5620–5621 CrossRef CAS PubMed.
- S. H. Chang, B. C. Chiu, T. L. Gao, S. L. Jheng and H. Y. Tuan, CrystEngComm, 2014, 16, 3323–3330 RSC.
- R. G. Xie, M. Rutherford and X. G. Peng, J. Am. Chem. Soc., 2009, 131, 5691–5697 CrossRef CAS PubMed.
- T. Torimoto, S. Ogawa, T. Adachi, T. Kameyama, K. I. Okazaki, T. Shibayama, A. Kudo and S. Kuwabata, Chem. Commun., 2010, 46, 2082–2084 RSC.
- D. W. Deng, L. Z. Qu and Y. Q. Gu, J. Mater. Chem. C, 2014, 2, 7077–7085 RSC.
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS.
- Y. A. Yang, H. M. Wu, K. R. Williams and Y. C. Cao, Angew. Chem., Int. Ed., 2005, 44, 6712–6715 CrossRef CAS PubMed.
- G. Kanellis and K. Kampas, Mater. Res. Bull., 1978, 13, 9–16 CrossRef CAS.
- M. Quintero, R. Tovar, C. Bellabarba and J. C. Woolley, Phys. Status Solidi B, 1990, 162, 517–521 CrossRef CAS.
- H. Z. Zhong, Y. Zhou, M. F. Ye, Y. J. He, J. P. Ye, C. He, C. H. Yang and Y. F. Li, Chem. Mater., 2008, 20, 6434–6443 CrossRef CAS.
- T. Torimoto, H. Nishiyama, T. Sakata, H. Mori and H. Yoneyama, J. Electrochem. Soc., 1998, 145, 1964–1968 CrossRef CAS.
- T. Uematsu, H. Kitajima, T. Kohma, T. Torimoto, Y. Tachibana and S. Kuwabata, Nanotechnology, 2009, 20, 215302 CrossRef CAS PubMed.
- C. M. Hessel, D. Reid, M. G. Panthani, M. R. Rasch, B. W. Goodfellow, J. W. Wei, H. Fujii, V. Akhavan and B. A. Korgel, Chem. Mater., 2012, 24, 393–401 CrossRef CAS.
- R. C. Benson and H. A. Kues, Phys. Med. Biol., 1978, 23, 159–163 CAS.
- W. W. Zheng, Y. Liu, A. West, E. E. Schuler, K. Yehl, R. B. Dyer, J. T. Kindt and K. Salaita, J. Am. Chem. Soc., 2014, 136, 1992–1999 CrossRef CAS PubMed.
- I. Moreels, K. Lambert, D. Smeets, D. De Muynck, T. Nollet, J. C. Martins, F. Vanhaecke, A. Vantomme, C. Delerue, G. Allan and Z. Hens, ACS Nano, 2009, 3, 3023–3030 CrossRef CAS PubMed.
- C. Zhao, Z. Bai, X. Liu, Y. Zhang, B. Zou and H. Zhong, ACS Appl. Mater. Interfaces, 2015, 7, 17623–17629 CAS.
- X. Liu, G. B. Braun, H. Zhong, D. J. Hall, W. Han, M. Qin, C. Zhao, M. Wang, Z.-G. She, C. Cao, M. J. Sailor, W. B. Stallcup, E. Ruoslahti and K. N. Sugahara, Adv. Funct. Mater., 2016, 26, 267–276 CrossRef CAS.
- X. G. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019–7029 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A detailed synthesis procedure of DSPC-AgInTe2 and analytical data of AgInTe2 NCs. See DOI: 10.1039/c5nr07532g |
| This journal is © The Royal Society of Chemistry 2016 |