
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultra-fast stem cell labelling using cationised magnetoferritin†
S.
Correia Carreira‡
*ab,
J. P. K.
Armstrong‡
c,
A. M.
Seddon
ab,
A. W.
Perriman
c,
R.
Hartley-Davies
d and
W.
Schwarzacher
*b
aBristol Centre for Functional Nanomaterials, University of Bristol, Tyndall Avenue, Bristol, BS8 1FD, UK. E-mail: S.Carreira@bristol.ac.uk
bH.H. Wills Physics Laboratory, University of Bristol, Tyndall Avenue, Bristol, BS8 1TL, UK. E-mail: W.Schwarzacher@bristol.ac.uk
cSchool of Cellular and Molecular Medicine, University of Bristol, Medical Sciences Building, University Walk, Bristol, BS8 1TD, UK
dBioengineering, Innovation, and Research Hub, University Hospitals Bristol NHS Foundation Trust, St. Michael's Hospital, Southwell Street, Bristol, BS2 8EG, UK
First published on 20th January 2016
Abstract
Magnetic cell labelling with superparamagnetic iron oxide nanoparticles (SPIONs) facilitates many important biotechnological applications, such as cell imaging and remote manipulation. However, to achieve adequate cellular loading of SPIONs, long incubation times (24 hours and more) or laborious surface functionalisation are often employed, which can adversely affect cell function. Here, we demonstrate that chemical cationisation of magnetoferritin produces a highly membrane-active nanoparticle that can magnetise human mesenchymal stem cells (hMSCs) using incubation times as short as one minute. Magnetisation persisted for several weeks in culture and provided significant T2* contrast enhancement during magnetic resonance imaging. Exposure to cationised magnetoferritin did not adversely affect the membrane integrity, proliferation and multi-lineage differentiation capacity of hMSCs, which provides the first detailed evidence for the biocompatibility of magnetoferritin. The combination of synthetic ease and flexibility, the rapidity of labelling and absence of cytotoxicity make this novel nanoparticle system an easily accessible and versatile platform for a range of cell-based therapies in regenerative medicine.
Introduction
Cell labelling with magnetic nanoparticles has enabled a host of platform biotechnologies for remotely manipulating, analysing and visualising cells.1 Magnetic cell labelling typically relies on endocytosis of superparamagnetic iron oxide nanoparticles (SPIONs), however, weak interactions between SPIONs and the cell membrane make this a highly inefficient process.2,3 As a result, lengthy incubation times (24 hours and more)4,5 and elevated SPION concentrations are required to attain sufficient cell magnetisation. These conditions can adversely affect cell viability,6,7 moreover, cell types with low internalisation capacity, such as lymphocytes, exhibit poor SPION uptake even after 40 hours.8 There has been limited success in enhancing cellular uptake by functionalising the surface of SPIONs with antibodies,9,10 transfection agents,11,12 or cell-penetrating peptides,13,14 however, these approaches are restricted by complex synthesis, expensive or cytotoxic reagents,15 and problems such as nanoparticle aggregation and precipitation.16 Accordingly, there is an unmet need for a simple surface functionalisation methodology that can produce a biocompatible SPION capable of rapidly magnetising a variety of cell types.Here, we sought to develop a facile surface functionalisation route to rapidly magnetise cells, thus avoiding lengthy exposure times. Our approach includes the synthesis of a novel cationised SPION derived from magnetoferritin, a protein-based nanoparticle comprising a SPION core mineralised inside the demineralised ferritin cavity.17 A major advantage of magnetoferritin is the ease with which its magnetic properties can be controlled, for instance by tuning the size of the mineralised nanoparticle18,19 or by doping the core with metals such as cobalt.20,21 Furthermore, the surrounding protein shell affords aqueous solubility and a large canvas of addressable amino acids that can be chemically22–24 or genetically modified.25–27 We hypothesised that cationisation of acidic amino acid residues on the magnetoferritin surface would generate a magnetic bionanomaterial that would readily interact with anionic domains present on the surface of a variety of cell types (Scheme 1). In this study, we demonstrate that cationised magnetoferritin can rapidly and persistently magnetise stem cells using incubation times as short as one minute without any adverse effects on cell viability, self-renewal and differentiation capacity. Our results represent a facile, rapid and versatile platform technology based on a non-specific labelling mechanism applicable to a variety of cell types. Furthermore, as it is possible to load the ferritin cavity with alternative functional materials, such as radioisotopes,28 quantum dots29 or anti-cancer drugs,30 this technology presents new opportunities for a host of cationised ferritin-based cell therapies.
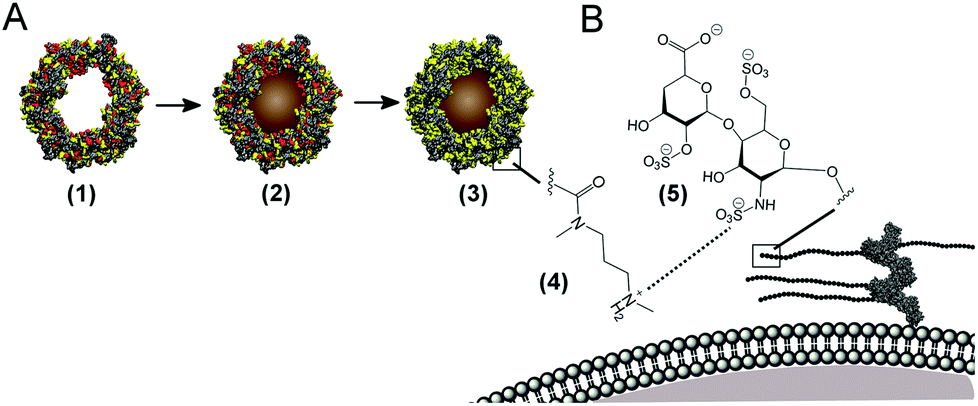 | ||
| Scheme 1 Magnetoferritin synthesis, cationisation and magnetic cell labelling (not to scale). (A) Apoferritin (1) is re-mineralised with iron and cobalt salts to produce magnetoferritin (2). Carbodiimide-mediated crosslinking of N,N′-dimethyl-1,3-propanediamine (DMPA) to surface carboxyl groups yields cationised magnetoferritin (3). Solvent accessible surface area representations show the distribution of acidic (red) and basic (yellow) amino acid residues present on the protein surface. (B) Rapid cell labelling is mediated by electrostatic interactions (dashed line) between basic residues (4) present at the cationised protein surface and anionic groups on proteoglycans, such as heparan sulfate residues (5) within the glycocalyx of the cytoplasmic membrane. | ||
Experimental section
Synthesis and cationisation of magnetoferritin
Cobalt-doped magnetoferritin was synthesised from commercially available horse spleen apoferritin (ApoF) (Sigma Aldrich, A3641, LOT 081M7011V) using an established protocol.17,31 Synthesis was performed in a double-jacketed reaction vessel at 65 °C containing 75 mL of deoxygenated 50 mM HEPES-NaOH (pH 8.6) with 3 mg mL−1 ApoF. The vessel was kept under a nitrogen atmosphere to restrict oxidation of the metal precursors. 30.3 mL of 25 mM ammonium iron sulfate hexahydrate (Sigma Aldrich), containing 2.5% (w/v) cobalt sulfate heptahydrate (Sigma Aldrich) was added at a rate of 0.15 mL min−1. Controlled oxidation of the metal precursors inside the apoferritin cavity was mediated by adding an equivalent volume of an 8.33 mM hydrogen peroxide solution at the same rate. Fresh metal precursor and hydrogen peroxide solutions were injected three times at 65 minute intervals. The solution was left to mature for 15 minutes prior to the addition of 1.5 mL of 1 M sodium citrate to chelate free metal ions in the solution.Large precipitates were removed by centrifuging the sample for 30 minutes at 4350g and then passing the supernatant through a 220 nm syringe filter. The protein was purified by anion-exchange chromatography (ANX Sepharose 4 Fast Flow, GE Healthcare, Life Sciences) to remove nanoparticles not enclosed in the protein cavity, followed by size-exclusion chromatography (HiPrep 26/60 Sephacryl S-300 HR column, GE Healthcare, Life Sciences) to isolate protein monomers.
For cationisation, N,N-dimethyl-1,3-propanediamine (DMPA, Sigma Aldrich) was coupled to aspartic and glutamic acid residues on the MF surface using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, Sigma Aldrich) as described previously.24,32 Solutions of DMPA were adjusted to pH 7 using HCl, before drop-wise addition of MF or ApoF. After an equilibration period of three hours, the coupling reaction was initiated by adding EDC and adjusting the pH to 5 using HCl. The solution was continuously stirred for four hours and then filtered through a 220 nm syringe filter to remove any precipitates. The solution was extensively dialysed (Medicell dialysis tubing, 12–14 kDa MWCO) against 50 mM phosphate buffer (pH 7) containing 50 mM NaCl to yield stable solutions of cationised MF and cationised apoferritin. Protein concentration was determined using a Bradford assay (BioRad) using a calibration curve prepared from horse spleen ferritin (Sigma Aldrich). In the Bradford assay, Coomassie Brilliant Blue G-250 dye binds to aromatic and basic amino acid residues on proteins, and the resulting complex results in a shift in absorbance maximum from 465 nm to 595 nm. Interference of the assay with the iron core was evaluated by measuring the absorbance spectrum of 1 mg mL−1 cat-MF from 250 to 750 nm. The contribution of the iron core to the absorbance at 595 nm (the wavelength at which the Bradford assay is measured) was 7% of the total absorbance measured at 595 nm after incubation with the Bradford reagent (Fig. S2 and S3†). Effects of cationisation were evaluated by preparing a range of concentrations of MF and cat-MF and determining their absorbance using the Bradford assay. Absorbance values and slopes of the linear fit were similar and variations between MF and cat-MF were within error (Fig. S3†).
Characterisation of magnetoferritin (MF) and cationised magnetoferritin (cat-MF)
Transmission electron microscopy (TEM) with a JEOL JEM 1200 EX was used to confirm mineralisation and determine MF core size. Samples were observed both unstained or stained with 2% (w/v) phosphotungstic acid (Sigma Aldrich). Iron content in MF samples digested with 50% (v/v) nitric acid was determined by inductively-coupled plasma optical emission spectrometry (710 ICP-OES, Agilent). Zeta potential and dynamic light scattering measurements of MF and cat-MF were performed on a Malvern ZetaSizer Nano-ZS at a protein concentration of approximately 1 mg mL−1 in 20 mM phosphate buffer at pH 7. Matrix-assisted laser desorption ionisation time-of-flight (MALDI-TOF) mass spectrometry was performed on ApoF and cat-ApoF samples dissolved in an equivalent volume of 20 mg mL−1 of 2,5-dihydroxybenzoic acid in methanol.Magnetic saturation and susceptibility of MF and cat-MF were measured by performing a field sweep over ±2 Tesla at 300 K using a superconducting quantum interference device (SQUID) magnetometer (Quantum Design). The diamagnetic background of the sample tube (0.2 mL PCR tube, Corning) and aqueous solvent were measured separately and subtracted from the magnetic moment of the sample. Susceptibility was calculated by determining the slope of the linear part of the magnetisation curve. Molar T1 and T2 relaxivity was assessed using a Siemens Magnetom Skyra 3 T MRI system. Samples were serially diluted from 3 μM to 0.01 μM protein concentration in Dulbecco's Phosphate Buffered Saline (PBS, Sigma Aldrich) and 200 μL of each solution was placed in tubes of a PCR plate embedded in a 1.5% (w/v) carrageenan (Sigma Aldrich) gel phantom. Longitudinal relaxation times (T1) were determined using an inversion recovery spin echo sequence (repetition time TR: 6000 ms, echo time TE: 9.6 ms, inversion times TI: 50, 150, 250, 350, 450, 650, 850, 1250, and 2700 ms, slice thickness: 3 mm, field of view: 12 cm). To measure transverse relaxation times (T2), a spin-echo sequence with different echo times was used (TR: 3500 ms, TE: 10, 20, 30, 50, 80, 100, 160, 240, 480 ms, slice thickness: 3 mm, field of view: 12 cm). T1 and T2 were determined by fitting an exponential curve to the signal intensities at each inversion (TI) or echo time (TE), respectively. Transverse and longitudinal relaxivity of each sample was calculated using the T1 and T2 data along with the MF iron content determined by ICP-OES.
Stem cell culture
Human mesenchymal stem cells (hMSC) were harvested from the proximal femur bone marrow of osteoarthritic patients undergoing total hip replacement surgery, in full accordance with Bristol Southmead Hospital Research Ethics Committee guidelines (reference #078/01) and after patient consent was obtained. hMSCs were cultured at 37 °C and 5% CO2 atmosphere in Dulbecco's Modified Eagle's Medium (DMEM), 1000 mg per L glucose (Sigma Aldrich), containing 10% (v/v) foetal bovine serum (FBS, Sigma Aldrich), 1% (v/v) penicillin/streptomycin (Sigma Aldrich), 1% (v/v) glutamax solution (Life Technologies) and 5 ng mL−1 freshly supplemented human fibroblast growth factor (PeproTech).Magnetic stem cell labelling
For magnetic labelling, 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hMSCs from three different patients were seeded into 25 cm2 tissue culture flasks and left to adhere over night. Cells were washed with PBS and exposed to 1 mL of MF or cat-MF in PBS for 1 minute to 6 hours with concentrations ranging from 0.01–3 μM. After labelling, cells were washed with PBS and harvested from the flasks using trypsin/EDTA (Sigma Aldrich). Cell pellets were re-suspended in 500 μL magnetic separation buffer (0.5% (w/v) FBS and 2 mM EDTA in PBS) and magnetic-activated cell separation (MACS) was performed using MACS MS columns (Miltenyi BIOTEC). Magnetic and non-magnetic fractions were collected and the number of cells was determined using an Improved Neubauer hemocytometer (Hawksley BS778). To assess magnetisation efficiency, the fraction of magnetised cells was determined using the following equation:
000 hMSCs from three different patients were seeded into 25 cm2 tissue culture flasks and left to adhere over night. Cells were washed with PBS and exposed to 1 mL of MF or cat-MF in PBS for 1 minute to 6 hours with concentrations ranging from 0.01–3 μM. After labelling, cells were washed with PBS and harvested from the flasks using trypsin/EDTA (Sigma Aldrich). Cell pellets were re-suspended in 500 μL magnetic separation buffer (0.5% (w/v) FBS and 2 mM EDTA in PBS) and magnetic-activated cell separation (MACS) was performed using MACS MS columns (Miltenyi BIOTEC). Magnetic and non-magnetic fractions were collected and the number of cells was determined using an Improved Neubauer hemocytometer (Hawksley BS778). To assess magnetisation efficiency, the fraction of magnetised cells was determined using the following equation:| Magnetised cell fraction = 100 × n(M)/n(M + NM) |
For the time course study, 150000 cells were seeded into 75 cm2 tissue culture flasks and left to adhere over night. The cells were washed with PBS and exposed to 1 mL of 0.5 μM cat-MF for 30 minutes. The cat-MF supernatant was removed, the cells washed with PBS and then cultured for up to five weeks. Magnetisation and iron content of the cells were analysed at weekly intervals using MACS and ICP-OES.
MRI imaging of labelled cells
800000 hMSCs were seeded in 75 cm2 tissue culture flasks and left to adhere over night. The cells were labelled with 3 mL of 0.5 μM and 1 μM of MF or cat-MF for 30 minutes. The MF or cat-MF supernatant was removed and cells were washed with PBS, harvested with trypsin/EDTA and counted. 750000 labelled cells were suspended in 200 μL of PBS and transferred into tubes of a PCR plate embedded in a 1.5% (w/v) carrageenan gel phantom and left to settle by gravity for four hours prior to MRI imaging. A gradient echo sequence was used with TR = 100 ms, TE = 10 ms, and a flip angle of 30°.
To assess the long term retention of cat-MF 800000 cells were seeded into 75 cm2 flasks, left to adhere over night, and then labelled with 3 mL of 0.5 μM of cat-MF for 30 minutes. The cat-MF supernatant was removed, the cells were washed with PBS, and cultured in DMEM for either four days or one week before being harvested. 750000 labelled hMSCs were suspended in 200 μL of PBS and transferred into tubes of a PCR plate embedded in a 1.5% (w/v) carrageenan gel phantom and left to settle by gravity for four hours prior to MRI imaging using a gradient echo sequence with TR = 100 ms, TE = 10 ms, and a flip angle of 30°.
TEM imaging of labelled cells
50000 hMSCs were seeded into a 6 well plate and left to adhere over night. The cells were washed with PBS and exposed to 1 mL of 0.5 μM cat-MF for 30 minutes. The cat-MF supernatant was removed and the cells were washed with PBS. Here, the cells were either immediately fixed and stained for TEM imaging following established procedures33 or cultured for one week before fixing and staining. Localisation of cat-MF on labelled cells was investigated using a FEI Tecnai T12 transmission electron microscope.
Prussian blue staining of labelled cells
1000000 hMSCs were seeded in culture medium into 75 cm2 tissue culture flasks and left to adhere overnight. The cells were exposed to 3 mL of 0.5 μM of cat-MF for 30 minutes. The cat-MF supernatant was removed, cells were washed with PBS and left in culture medium for 24 hours. hMSCs were harvested with trypsin/EDTA and counted. 300000 cells were suspended in 30 μL of culture medium and loaded onto fibronectin-coated polyglycolic acid (PGA) tissue engineering scaffolds of 5 mm diameter and 2 mm thickness (Biomedical Structures, USA) and placed into the inner, agarose-coated wells of a 24 well plate. Cells were left to adhere to the scaffold over night, and then cultured for six hours in culture medium. The medium was then removed and scaffolds fixed in 4% (w/v) paraformaldehyde over night at room temperature, dehydrated in 70% ethanol for two hours and then submitted to the Histology Services Unit (Medical Sciences, University of Bristol). Histological sections of 10 μm thickness were immersed for 20 minutes at room temperature in a 10% (w/v) potassium ferrocyanide solution (Sigma Aldrich, UK) containing an equal volume of 2 M hydrochloric acid. Slides were washed in dH2O three times for five minutes. Cell nuclei were counter stained with a Nuclear Fast Red (Sigma Aldrich) for five minutes and the excess stain was removed by rinsing with dH2O. Stained sections were observed using an inverted light microscope at 40× magnification (Leica DM IRB).
Labelling mechanism of cat-MF
To assess the contribution of active internalisation processes on cellular iron content 150000 hMSCs were seeded into 25 cm2 tissue culture flasks and left to adhere over night. hMSCs were exposed to 1 mL of 0.5 μM cat-MF for 5 or 30 minutes at 37 °C or 4 °C. Prior to incubation with cat-MF at 4 °C, cells were pre-cooled at 4 °C for 30 minutes in DMEM supplemented with 20 mM HEPES buffer. After each incubation period, the cat-MF supernatant was removed and the cells were washed, harvested and analysed using ICP-OES.
To test the hypothesis that anionic proteoglycans in the glycocalyx facilitate cat-MF uptake 150000 cells were cultured for four days in 25 cm2 tissue culture flasks using DMEM supplemented with 80 mM sodium chlorate. hMSCs were labelled with 1 mL of 0.5 μM cat-MF for 5 or 30 minutes at 37 °C before the cat-MF supernatant was removed and the cells were washed, harvested and analysed using ICP-OES.
The cellular iron content values after a 5 or 30 minute incubation period were compared between groups labelled in different conditions using the Friedman test in IBM SPSS Statistics version 21.
Cytotoxicity assays
For the MTS assay, 5000 cells from three different patients were seeded in quadruplicate into wells of a 96 well plate, left to adhere over night, and exposed for 30 minutes to 125 μL of MF or cat-MF at concentrations ranging from 0.01–3 μM, or PBS only (untreated control). The supernatant was removed, cells washed with PBS and cultured in DMEM. After 24 hours, DMEM was removed, cells washed with PBS and incubated for one hour at 37 °C with phenol-free DMEM containing 20% (v/v) of MTS solution (CellTiter 96, Promega). Formation of the reduced formazan product was measured at 490 nm using a spectrophotometer (SpectraMax M5e, Molecular Devices). Cell viability values were normalised with respect to the untreated control.For the LDH assay (LDH Cytotoxicity Assay Kit, Pierce), 5000 cells from three different patients were seeded in quadruplicate into wells of a 96 well plate, left to adhere over night and exposed for 30 minutes to 125 μL of MF or cat-MF at concentrations ranging from 0.01 to 3 μM, PBS only (untreated control), or a Lysis Buffer provided by the manufacturer as a positive control. The supernatant was removed, cells washed with PBS and cultured in DMEM with reduced serum content (5% FBS) for 24 h after exposure to native MF or cat-MF, because higher serum concentrations are known to interfere with the assay. The assay was performed as per manufacturer's instructions using the cell culture supernatants. Percentage (%) cytotoxicity was calculated using the following equation:
| % cytotoxicity = 100 × (LDHtreat − LDHUC)/(LDHmax − LDHUC) |
For statistical analysis of the MTS and LDH assays, data were presented as mean ± standard deviation of three biological repeats. Statistical analysis was performed using IBM SPSS Statistics version 21. The means of groups treated with MF, cat-MF, and PBS (untreated control) were compared using Two-Way Analysis of Variance (ANOVA), in which effects of nanoparticle concentration as well as surface functionalisation (un-functionalised vs. cationised) were investigated.
Cell proliferation assay
Proliferation of hMSCs was assessed using a cell counting assay. 150000 cells from three different patients were seeded into 75 cm2 tissue culture flasks and left to adhere over night. hMSCs were exposed for 30 minutes to 3 mL of 0.5 μM cat-MF or PBS (untreated cell control). The supernatant was removed, cells washed with PBS and cultured in DMEM for up to three weeks. At weekly intervals, cells were harvested, counted, and population doublings were calculated as follows:| Population doublings = log(n/150000)/log2 |
000 denotes the number of cells initially seeded. Population doublings of the cat-MF treated cells were normalised to the untreated cell control. The population doubling values of cat-MF labelled and untreated hMSCs were compared at each interval using the Wilcoxon Signed-Rank test in IBM SPSS Statistics version 21.
Differentiation capacity of hMSCs after cat-MF labelling
The multi-lineage differentiation capacity of hMSCs was investigated using osteogenesis and adipogenesis in monolayer, and chondrogenesis in a 3D cartilage engineering model. For monolayer differentiation, 37000 and 7400 hMSCs were seeded into 24 well plates for adipogenesis and osteogenesis, respectively, and left to adhere over night. Cells were exposed to 0.75 mL of 1 μM of cat-MF or PBS (untreated control) for 30 minutes. The supernatant was removed, cells washed in PBS and cultured in αMEM medium (Sigma Aldrich) with 10% (v/v) FBS, 1% (v/v) penicillin/streptomycin and 1% (v/v) glutamax solution, containing either 50 μl mL−1 human osteogenic supplement (StemXVivo, R&D Systems) or 10 μl mL−1 human adipogenic supplement (StemXVivo, R&D Systems) for three weeks with media changes performed twice a week. Osteoblast formation was visualised using Alizarin Red staining of calcium phosphate deposits. Cells were washed with PBS and fixed for one hour at 4 °C using 0.5 mL ice-cold 70% (v/v) ethanol. The fixative was removed and the cells were incubated with 0.5 mL alizarin red solution (Sigma Aldrich) for five minutes at room temperature. The cells were then washed five times with PBS.
Adipocyte formation was assessed using Oil Red staining of intracellular lipid vacuoles. Cells were washed with PBS and fixed with 4% (w/v) paraformaldehyde for 30 minutes at room temperature. The fixative was removed and the cells washed first with PBS followed by 60% (v/v) isopropanol. Cells were incubated with 0.5 mL of Oil Red stain (Sigma Aldrich) for 30 minutes at room temperature. The stain was aspirated and cells washed with 60% (v/v) isopropanol. Stained monolayers were observed using an inverted light microscope at 10× magnification (Leica DM IRB).
For tissue engineering of cartilage, 1000000 hMSCs were seeded in 75 cm2 tissue culture flasks and left to adhere over night. The cells were exposed to 3 mL of 0.5 μM of cat-MF or PBS for 30 minutes. The cat-MF or PBS supernatant was removed and cells were washed with PBS, harvested with trypsin/EDTA and counted. 300000 hMSCs were suspended in 30 μL of DMEM medium and loaded onto fibronectin-coated PGA tissue engineering scaffolds of 5 mm diameter (Biomedical Structures, USA) and placed in the inner agarose-coated wells of a 24 well plate. Cells were left to adhere to the scaffold over night, and then cultured for one week in DMEM containing 4500 mg glucose per L, 1% (v/v) penicillin/streptomycin, 1% (v/v) glutamax, 1% (v/v) sodium pyruvate, and 1% (v/v) insulin-transferrin-sodium selenite, supplemented with 100 nM dexamethasone, 80 nM ascorbic acid and 10 ng ml−1 TGFβ3. Three media changes were performed during this first week, after which the medium was additionally supplemented with 10 ng per ml insulin and scaffolds were cultured for a further four weeks with media changes three times a week. After five weeks in total, half of the scaffolds were stored at −80 °C until biochemical analysis, and the other half was prepared for histology. Histochemical staining of proteoglycans was performed by incubating 5 μm sections of the tissue engineered construct with a 5 mg mL−1 Safranin O solution (Sigma Aldrich) for six minutes. Immunohistochemical staining was used to visualise type II collagen content. Goat Anti-Type II Collagen (Cambridge Biosciences, UK, 1320-1) was the primary antibody and Biotinylated Anti-Goat IgG (VECTASTAIN Elite ABC Kit (Goat IgG), Vector Laboratories, UK, PK6105) was the secondary antibody. Avidin and biotinylated horseradish peroxidase macromolecular complex (VECTASTAIN Elite ABC Kit) was used to bind to the secondary antibody and provide an enzymatic base for the immunoperoxidase stain diaminobenzidine tetrahydrochloride (ImmPACT DAB Substrate Kit, Vectro Laboratories, UK), producing a brown stain. Sections were also stained with Prussian Blue and Nuclear Fast Red to assess the presence of cat-MF.
Biochemical analysis was performed to quantify the amount of type II collagen and GAG in the engineered cartilage constructs. First, the constructs were weighed, and then digested in a 2 mg mL−1 solution of TPCK-treated trypsin supplemented with 200 mM iodoacetamide, 200 mM EDTA and 2 mg ml−1 pepstatin A (all Sigma Aldrich), first at 37 °C over night, then for two hours at 65 °C. Samples were boiled for 15 minutes to inactivate trypsin and then centrifuged. The supernatant containing the digested cartilage matrix components was removed and the undigested scaffold freeze-dried and weighed again. The dry weight of the extracellular matrix was calculated by subtracting the dry weight of the undigested scaffold from the dry weight of the whole cartilage construct.
Type II collagen was quantified by performing an enzyme-linked immunosorbent assay (ELISA) developed by Hollander et al.34 Briefly, samples along with calibration standards were first incubated with a mouse-derived type II collagen antibody (Bioiberica, Spain) over night, and then the supernatant was transferred into plates coated with type II collagen and incubated for 30 minutes at room temperature. The plate was then washed and incubated with an anti-mouse secondary antibody (Cambridge Biosciences, UK) for two hours at 37 °C. An alkaline phosphatase solution was added to the plate and incubated for 20 minutes at 37 °C before the absorbance was measured at 405 nm. The amount of type II collagen was calculated from the calibration curve and expressed as a percentage of the extracellular matrix dry weight.
Glycosaminoglycan (GAG) content was quantified using a 160 μg ml−1 dimethylmethylene blue solution, which was added to samples and calibration standards of chondroitin sulfate (Sigma Aldrich) and measured immediately at 530 nm. The amount of GAG was calculated from the calibration curve and expressed as a percentage of the extracellular matrix dry weight.
The percentage of GAG or type II collagen in cat-MF treated and untreated cartilage constructs was compared using the Mann-Whitney U-test in IBM SPSS Statistics version 21.
Results and discussion
Synthesis and characterisation of cationised magnetoferritin
Cationised magnetoferritin (cat-MF) was prepared using a facile two step synthesis (Scheme 1). First, cobalt-doped iron oxide nanoparticles were mineralised within the apoferritin cavity to produce magnetoferritin (MF), which was subsequently cationised using carbodiimide coupling of N,N′-dimethyl-1,3-propanediamine (DMPA) to acidic residues on the protein surface (Fig. S1†). Transmission electron microscopy (TEM) confirmed the presence of nanoparticles within the protein cage (Fig. 1A), and image analysis of unstained MF samples gave an average core diameter of 5.3 ± 1.1 nm (Fig. 1B and S4†). Using inductively-coupled plasma optical emission spectrometry (ICP-OES), we measured an average of 45 ± 8 μg of iron and 0.43 ± 0.17 μg of cobalt per milligram of MF, indicating a 1% (w/w) cobalt doping of the iron oxide-based nanoparticle core. The nanoparticle size was measured using dynamic light scattering, which gave a hydrodynamic diameter of 11.8 ± 1.1 nm for MF and 12.5 ± 1.4 nm for cat-MF (Fig. S5A†), with the size increase attributed to the steric bulk of coupled DMPA. Mass spectrometry revealed a subunit molecular weight of 20.1 kDa for native apoferritin and 21.1 kDa for cationised apoferritin (Fig. S5B†). This mass increase corresponded to the cationisation of 288 residues on the entire 24-subunit protein, which is consistent with previous cationisation efficiencies obtained using horse spleen ferritin.32 Further evidence for cationisation was provided by zeta potentiometry (Table 1), and time course cationisation studies using ferritin showed that maximum zeta potential was reached after two hours crosslinking time (Fig. S6†). Magnetic saturation, susceptibility and relaxivity were similar for MF and cat-MF, indicating that cationisation had negligible impact on the magnetic properties of the enclosed SPION (Table 1, Fig. S7 and S8†). Although the relaxivity values of MF and cat-MF were lower than those of commercially available SPION-based contrast agents,35 higher iron loadings (and thus relaxivity values) can be achieved by varying the mineralisation protocol.18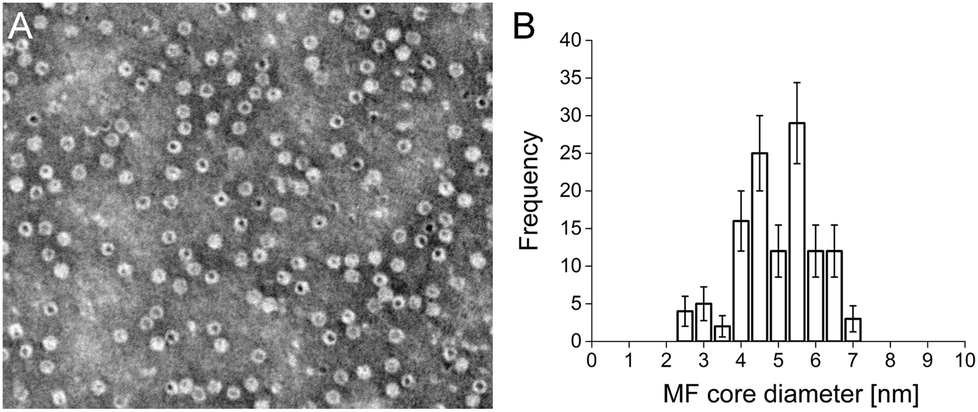 | ||
| Fig. 1 TEM analysis of MF. (A) Negative stain with 2% (w/v) phosphotungstic acid showing that the majority of protein shells (white) contain a mineralised nanoparticle core (black). Scale bar: 50 nm. (B) Image analysis performed on an unstained TEM image of MF found a distribution of core sizes with an average nanoparticle diameter of 5.2 ± 1.0 nm. Error bars represent the Poisson counting error. | ||
| MF | cat-MF | |
|---|---|---|
| Hydrodynamic diameter [nm] | 11.8 ± 1.1 | 12.5 ± 1.4 |
| Zeta potential [mV] | −10.4 ± 0.2 | 8.3 ± 0.7 |
| Magnetic saturation [emu g−1] | 48.6 ± 1.8 | 47.5 ± 1.0 |
| Magnetic susceptibility [×10−2 emu g−1 Oe−1] | 1.3 ± 0.1 | 1.3 ± 0.1 |
| Longitudinal relaxivity r1 [mM−1 s−1] | 2.6 ± 0.1 | 2.3 ± 0.0 |
| Transverse relaxivity r2 [mM−1 s−1] | 44.6 ± 1.0 | 52.8 ± 0.8 |
Magnetic stem cell labelling with cationised magnetoferritin
Magnetic-activated cell separation (MACS) and ICP-OES revealed that cat-MF was remarkably effective at magnetically labelling hMSCs (Fig. 2A). Notably, a one minute exposure to 0.5 μM cat-MF resulted in the magnetisation of 92% of the cell population and the delivery of approximately 3.6 pg of iron, or 5 × 107 cat-MF nanoparticles, per cell. Remarkably, given the extreme brevity of exposure, this cellular iron content is well within the reported range required to influence T2 and T2* contrast for MRI.36,37 Increasing the incubation time to six hours resulted in saturation of cellular iron content at a level of 20 pg iron per cell (Fig. S9†). The iron uptake rate between 1 and 30 minutes was 20.5 pg Fe per cell per hour, which slowed significantly between 30 minutes and 6 hours to 1.2 pg Fe per cell per h. A 30 minute incubation was used thereafter, because this effectively magnetised the entire cell population and resulted in high cellular iron content within a reasonably short time period. The cell magnetisation efficiency was also dependent on incubation concentration, with more concentrated solutions of cat-MF magnetising a greater proportion of hMSCs and resulting in a higher cellular iron content (Fig. 2B). In contrast, all tested concentrations of un-functionalised MF magnetised less than 3% of the cell population and cellular iron content was below the ICP-OES detection limit (Fig. S10†). These results demonstrate the direct impact of cationisation on the efficiency of the magnetic labelling process.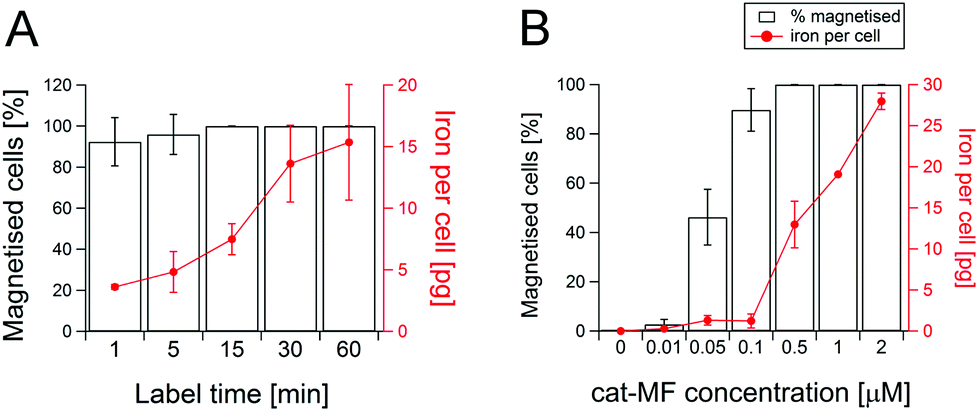 | ||
| Fig. 2 MACS and ICP-OES analysis of magnetised hMSCs. (A) Effect of labelling time on the percentage of magnetised cells and cellular iron content in hMSCs exposed to 0.5 μM cat-MF. 92% of the cell population was magnetised after just one minute, while the entire cell population was magnetised in 15 minutes. Average and standard deviation of three biological replicates are shown. (B) Effect of cat-MF concentration on magnetisation efficiency after a 30 minute incubation. Labelling efficiency and cellular iron content were found to be concentration dependent. Average and standard deviation of three biological replicates are shown. | ||
Although other charge functionalisation strategies can improve non-specific nanoparticle–cell interactions, cat-MF labelling achieves greater labelling efficiency with lower incubation concentrations. For instance, anionisation of SPIONs is known to enhance cell labelling.38 However, this approach required a 30 minute incubation with 5 mM iron to deliver 10 pg per cell, whereas cat-MF labelling attained the same cellular iron loading with an incubation concentration of 0.2 mM iron (this is the amount of iron contained in 0.5 μM cat-MF). Cellular iron content after a one hour incubation with cat-MF (15 pg) was also comparable to iron loadings achieved with TAT-functionalised SPIONs (18 pg).39 Taken together, the results presented in this work show for the first time that efficient magnetic labelling can be achieved within a one minute incubation period, and that this efficiency can also be achieved using relatively low extracellular iron concentrations. Long-term studies using hMSCs labelled with 0.5 μM cat-MF for 30 minutes revealed that 60% of the cell population remained magnetised after one week in culture, and 10% after five weeks. Cellular iron content decreased by approximately 60% per week (Fig. 3A), which is in agreement with the observation of a weekly population doubling in the magnetic cell fraction (Fig. S11†). The cellular iron content dropped below the detection limit of ICP-OES after four weeks in culture, which is accompanied by a drop in the number of magnetised cells. This reduction in iron content can be attributed to a combination of dilution through transfer to daughter cells during cell division,40 and lysosomal breakdown of the nanoparticles over time.41,42
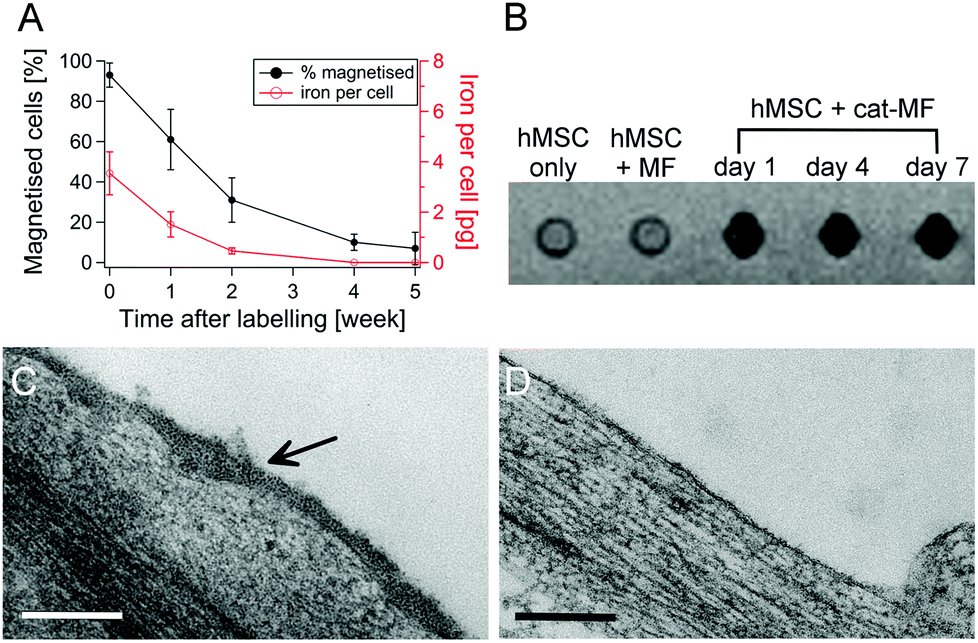 | ||
| Fig. 3 Long-term fate of cat-MF. (A) The percentage of magnetised cells and cellular iron content of hMSCs labelled with cat-MF over five weeks in culture. Average and standard deviation of three biological replicates are shown. (B) MRI of 750000 hMSCs labelled with MF and cat-MF (3 T scanner, gradient echo, TR = 100 ms, TE = 10 ms, flip angle 30°). Persistent contrast enhancement was observed in hMSCs labelled with 0.5 μM cat-MF. (C) A TEM image of magnetised hMSCs immediately after labelling with 0.5 mM cat-MF, revealing coverage of the cell surface with nanoparticles. (D) A TEM image of magnetised hMSCs one week after labelling, showing no nanoparticles at the cell surface. All scale bars: 200 nm. | ||
MRI revealed a large T2* signal loss for hMSCs labelled with cat-MF, compared to a control of unlabelled cells or cells labelled with the equivalent concentration of un-functionalised MF (Fig. 3B). In addition, the T2 relaxation rate increased from 0.9 s−1 in unlabelled hMSC to 2 s−1 after cat-MF exposure (Fig. S12A†). The contrast enhancement persisted for at least one week, after which the T2 relaxation rate was 1.4 s−1 (Fig. S12B†). The r1 and r2 relaxivity values in cat-MF labelled cells were 0.2 and 7.7 mM−1 s−1, respectively, and thus much lower compared to the relaxivity values measured for free cat-MF (Table 1 and Fig. S13†). A marked reduction in relaxivity has previously been observed in SPIONs internalised by cells, which was attributed to limited water and/or SPION diffusion inside endocytotic vesicles.35,37 Taken together, these results demonstrate that cat-MF labelling could be used to monitor stem cell therapies over time using MRI.
TEM performed immediately after cell magnetisation revealed widespread coverage of cat-MF across the cell surface (Fig. 3C), however, no nanoparticles were detected at the cell membrane after one week in culture (Fig. 3D). Given that the majority of cells was still magnetised at this time point, it follows that internalisation of cat-MF must have occurred. Evidence for this hypothesis was provided by Prussian Blue staining, which revealed extensive iron deposits present within sectioned hMSCs 48 hours after exposure to 0.5 μM cat-MF (Fig. S14†).
The mechanism underlying rapid magnetic labelling with cationised magnetoferritin
The incubation conditions were systematically varied to understand the different contributions of electrostatic interaction and internalisation in cat-MF labelling (Fig. 4). Labelling at 37 °C and 4 °C for five minutes resulted in similar cellular iron content, indicating that initial adsorption of cat-MF to the cell surface was similar in both groups. However, after 30 minutes the iron content was significantly higher in cells labelled at 37 °C compared to cells labelled at 4 °C. Low temperatures are an efficient method of non-invasively inhibiting many active internalisation pathways.43 Therefore, these results show that interactions between cat-MF and the cell surface mediate rapid magnetic labelling during the first few minutes of exposure, whilst active internalisation processes contribute to increased cellular iron content during the later stages of the labelling process. Furthermore, culturing hMSCs in medium supplemented with 80 mM sodium chlorate (NaClO3) significantly reduced the cellular iron content after a five minute incubation with cat-MF (Fig. 4). NaClO3 inhibits the synthesis of anionic functional groups on proteoglycans,44 which suggests that these glycocalyx species act as binding sites for cat-MF during the initial labelling period, as has been demonstrated for other cationic ligands.45 The observation that NaClO3-treated cells still contained a relatively large amount of iron suggests that other anionic moieties, such as phospholipid head groups, may also contribute to electrostatic binding. After 30 minutes, the cellular iron content in cells cultured in NaClO3 was still much reduced compared to cells cultured in untreated medium. This indicates that initial electrostatic adsorption of cat-MF to the cell surface is important for high internalisation rates, which is in agreement with previously calculated models that predict a higher rate of endocytosis with increased ligand–receptor interactions.46 These results are also consistent with models showing that non-specific interactions are as important as specific interactions during endocytosis, and even favour nanoparticle uptake.2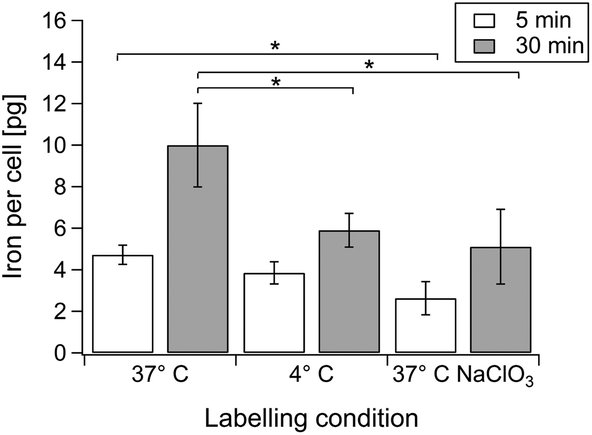 | ||
| Fig. 4 Mechanistic study of cat-MF uptake. ICP-OES was used to analyse the cellular iron content of hMSCs incubated with 0.5 μM cat-MF for either 5 or 30 minutes at 37 °C, 4 °C, or 37 °C after NaClO3-treatment. Average and standard deviation of three biological replicates are shown. For each incubation period, cellular iron content values were compared using the Friedman test, with significant differences indicated with an asterisk (p < 0.05). | ||
Toxicological evaluation of magnetoferritin and cationised magnetoferritin
Although magnetoferritin has previously been explored as a cellular contrast agent,18,47 no detailed toxicological evaluation of this nanoparticle has been undertaken. Here, we investigated acute effects on cell viability and membrane integrity using MTS and LDH assays performed 24 hours after exposure to 0.01–3 μM solutions of MF or cat-MF. The MTS assay, which uses cellular respiration as a measure of viability, showed no significant effects on cell viability after exposure to both MF and cat-MF across this concentration range (Fig. 5A). This was confirmed by an LDH assay, which showed that neither MF nor cat-MF led to cytotoxic effects affecting cell membrane integrity (Fig. 5B). The observation that similar viability levels were measured for MF and cat-MF indicates that cationisation did not confer additional cytotoxicity. This is a promising result, because cationic nanoparticles are often more cytotoxic than their anionic or neutral counterparts.48,49 This can be attributed to the relatively low zeta potential of cat-MF, which should avoid the hole formation observed in membranes exposed to nanoparticles with excessively high cationic charge density.50 The rapidity of cat-MF labelling also avoids the cytotoxic effects associated with prolonged incubation time.6,7 Furthermore, the efficiency of cat-MF labelling allows the use incubation concentrations several orders of magnitude lower than many competing SPION systems.51 Finally, the biocompatible protein shell of cat-MF circumvents the cytotoxic effects that can arise from coating agents used to synthesise functionalised SPIONs.52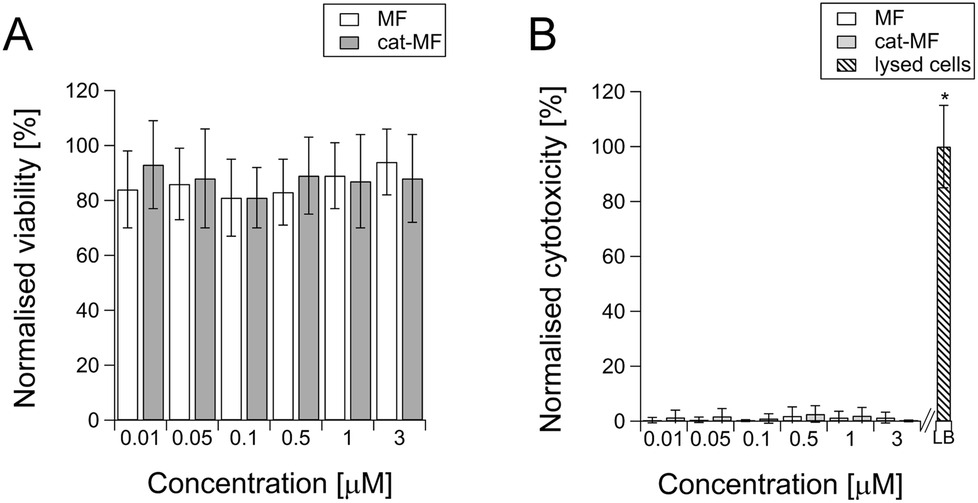 | ||
| Fig. 5 Acute effects of MF and cat-MF exposure in hMSCs. (A) Viability and (B) cytotoxicity were assessed using an MTS and LDH assay, respectively, 24 hours after exposure to MF and cat-MF for 30 minutes. Average and standard deviation of three biological replicates are shown. All values were normalised to an untreated hMSC control. No significant effects of concentration or surface functionalisation on cell viability or cytotoxicity were found using Two-Way Analysis of Variance. LB: lysis buffer used to disrupt the cell membrane and release intracellular LDH into the media (positive control). | ||
The long term effect of magnetising hMSCs with cat-MF was assessed using established proliferation and differentiation assays. hMSCs incubated with 0.5 μM cat-MF proliferated to the same extent as untreated cells (Fig. S15†), while monolayer differentiation into adipocytes and osteoblasts was observed in hMSCs exposed to elevated concentrations (1 μM) of cat-MF (Fig. 6A and B; see Fig. S16 and S17† for controls). Furthermore, hMSCs labelled with 0.5 μM cat-MF were used to grow cartilage constructs in a 35 day course of tissue engineering. Histochemical and immunohistochemical staining revealed that proteoglycan and type II collagen production and distribution was unaffected by cat-MF exposure (Fig. 6C and D; see Fig. S17† for images of the untreated control). This was confirmed by biochemical analysis of digested cartilage constructs, which showed that the level of glycosaminoglycan and type II collagen was similar when magnetised and untreated cells were used (Fig. S18†). Cartilage sections were also stained with Prussian Blue to investigate whether cat-MF was still present in the labelled cells after five weeks in culture. Blue staining indicative of the presence of iron deposits was observed in cartilage sections engineered from hMSC labelled with cat-MF, but not in cartilage grown from untreated hMSCs (Fig. S19†). Higher magnification imaging revealed that the blue stain was co-localised with the cell matrix, indicating that cat-MF was indeed still present within some cells. This is in good agreement with the results reported above, which showed that 10% of hMSCs still contained sufficient amounts of cat-MF to be retained in a MACS column after five weeks in culture.
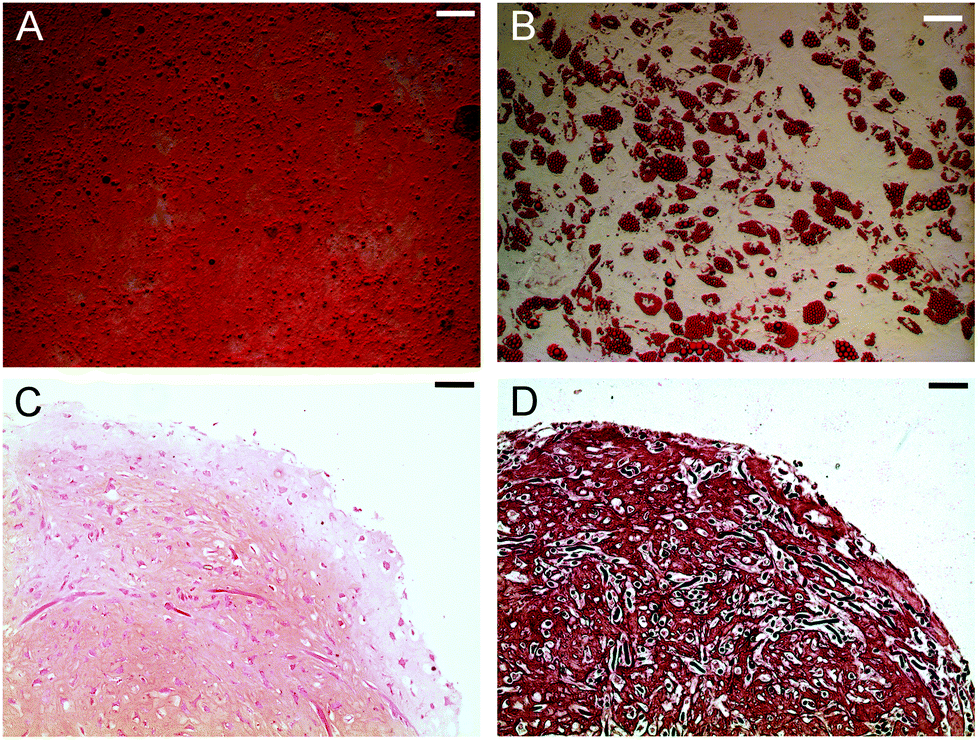 | ||
| Fig. 6 Differentiation capacity hMSCs after labelling with cat-MF. Representative bright field microscopy images of (A) hMSC-derived osteoblasts with calcium phosphate deposits stained with Alizarin Red. (B) hMSC-derived adipocytes with fatty vacuoles stained using Oil Red. (C) Engineered cartilage tissue stained for proteoglycans with Safranin O and (D) type II collagen using an immunohistochemical staining procedure. All scale bars: 100 μm. | ||
Taken together, these results indicate that the two hallmarks of viable stem cells, proliferation and differentiation, were unaffected by exposure to cat-MF. The finding that cat-MF labelling did not inhibit chondrogenesis is highly significant, given that previous studies have shown that this differentiation pathway can be adversely affected by SPION exposure.53,54 For example, Kostura et al. found that chondrogenesis was inhibited after exposure to Feridex, which resulted in cellular iron levels similar to ours (approximately 13 pg per cell).54 However, the exposure concentration and incubation time used here were much lower compared to the study by Kostura et al. Moreover, there is evidence that regulation of iron metabolism is linked to chondrogenesis,55 which has been suggested as the cause for the observed inhibition of this differentiation pathway after SPION exposure. Here, labelling with cat-MF introduced SPIONs encapsulated inside ferritin shells. It is possible that internalised cat-MF may have been able to withstand acidic environments of lysosomes better because of the relatively acid-resilient protein cover compared to the dextran coating of Feridex. Thus, iron ions might have been released more slowly, avoiding sudden disruption of the iron homeostasis.
Conclusions
We have demonstrated the synthesis of cationised magnetoferritin, a novel magnetic nanoparticle that rapidly and persistently magnetises stem cells in just one minute, which is several orders of magnitude shorter than most conventional SPION systems. The magnetised hMSCs exhibited lasting MRI contrast and retained capacity for self-renewal and differentiation, which makes this novel SPION system an attractive candidate for tracking stem cell therapy using MRI without impairing the regenerative capacity of the labelled cells. Significantly, chondrogenesis was not inhibited after cat-MF exposure, which is a differentiation pathway that is often affected by SPION labelling. The facile nature of the cationisation procedure eliminates the need for laborious functionalisation chemistry, and the non-specific labelling mechanism makes this a versatile technology that should find wide-spread application in a range of different cell types. Furthermore, the apoferritin cage represents a highly flexible vector that can be loaded with alternative functional molecules, thereby extending the presented concepts to applications beyond magnetic labelling.Acknowledgements
This work was financed through the Bristol Centre for Functional Nanomaterials, sponsored by the Engineering and Physical Sciences Research Council (EPSRC grant code EP/G036780/1). We are grateful to the staff of the Wolfson Bioimaging Facility (Paul Verkade, Judith Mantell, Gini Tilly) and Monika Jakimowicz for electron microscopy support at the University of Bristol, and to the Clinical Research and Imaging Centre (CRIC) at University Hospitals Bristol NHS Foundation Trust, particularly Sian Curtis, for MRI support.References
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- P. Decuzzi and M. Ferrari, Biomaterials, 2007, 28, 2915–2922 CrossRef CAS PubMed.
- S. Mukherjee, R. N. Ghosh and F. R. Maxfield, Physiol. Rev., 1997, 77, 759–803 CAS.
- C. H. Cunningham, T. Arai, P. C. Yang, M. V. McConnell, J. M. Pauly and S. M. Conolly, Magn. Reson. Med., 2005, 53, 999–1005 CrossRef CAS PubMed.
- H. T. Song, J. S. Choi, Y. M. Huh, S. Kim, Y. W. Jun, J. S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 9992–9993 CrossRef CAS PubMed.
- H. E. Daldrup-Link, M. Rudelius, R. A. J. Oostendorp, M. Settles, G. Piontek, S. Metz, H. Rosenbrock, U. Keller, U. Heinzmann, E. J. Rummeny and T. M. Link, Radiology, 2003, 228, 760–767 CrossRef PubMed.
- N. Singh, G. J. Jenkins, R. Asadi and S. H. Doak, Nano Rev., 2010, 1 Search PubMed.
- S. J. Dodd, M. Williams, J. P. Suhan, D. S. Williams, A. P. Koretsky and C. Ho, Biophys. J., 1999, 76, 103–109 CrossRef CAS PubMed.
- E. T. Ahrens, M. Feili-Hariri, H. Xu, G. Genove and P. A. Morel, Magn. Reson. Med., 2003, 49, 1006–1013 CrossRef CAS PubMed.
- S. Miltenyi, W. Muller, W. Weichel and A. Radbruch, Cytometry, 1990, 11, 231–238 CrossRef CAS PubMed.
- A. S. Arbab, G. T. Yocum, H. Kalish, E. K. Jordan, S. A. Anderson, A. Y. Khakoo, E. J. Read and J. A. Frank, Blood, 2004, 104, 1217–1223 CrossRef CAS PubMed.
- J. W. Bulte, T. Douglas, B. Witwer, S. C. Zhang, E. Strable, B. K. Lewis, H. Zywicke, B. Miller, P. van Gelderen, B. M. Moskowitz, I. D. Duncan and J. A. Frank, Nat. Biotechnol., 2001, 19, 1141–1147 CrossRef CAS PubMed.
- L. Josephson, C. H. Tung, A. Moore and R. Weissleder, Bioconjugate Chem., 1999, 10, 186–191 CrossRef CAS PubMed.
- M. Lewin, N. Carlesso, C. H. Tung, X. W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nat. Biotechnol., 2000, 18, 410–414 CrossRef CAS PubMed.
- A. C. Hunter, Adv. Drug Delivery Rev., 2006, 58, 1523–1531 CrossRef CAS PubMed.
- K. Montet-Abou, X. Montet, R. Weissleder and L. Josephson, Mol. Imaging, 2007, 6, 1–9 CAS.
- F. C. Meldrum, B. R. Heywood and S. Mann, Science, 1992, 257, 522–523 CAS.
- M. Uchida, M. Terashima, C. H. Cunningham, Y. Suzuki, D. A. Willits, A. F. Willis, P. C. Yang, P. S. Tsao, M. V. McConnell, M. J. Young and T. Douglas, Magn. Reson. Med., 2008, 60, 1073–1081 CrossRef CAS PubMed.
- M. J. Martinez-Perez, R. de Miguel, C. Carbonera, M. Martinez-Julvez, A. Lostao, C. Piquer, C. Gomez-Moreno, J. Bartolome and F. Luis, Nanotechnology, 2010, 21 Search PubMed.
- M. Okuda, J. C. Eloi, A. Sarua, S. E. W. Jones and W. Schwarzacher, J. Appl. Phys., 2012, 111 Search PubMed.
- M. T. Klem, D. A. Resnick, K. Gilmore, M. Young, Y. U. Idzerda and T. Douglas, J. Am. Chem. Soc., 2007, 129, 197–201 CrossRef CAS PubMed.
- M. Zborowski, C. B. Fuh, R. Green, N. J. Baldwin, S. Reddy, T. Douglas, S. Mann and J. J. Chalmers, Cytometry, 1996, 24, 251–259 CrossRef CAS PubMed.
- E. Valero, S. Tambalo, P. Marzola, M. Ortega-Munoz, F. J. Lopez-Jaramillo, F. Santoyo-Gonzalez, J. D. Lopez, J. J. Delgado, J. J. Calvino, R. Cuesta, J. M. Dominguez-Vera and N. Galvez, J. Am. Chem. Soc., 2011, 133, 4889–4895 CrossRef CAS PubMed.
- D. Danon, L. Goldstein, Y. Marikovski and E. Skutelsky, J. Ultrastruct. Res., 1972, 38, 500–510 CrossRef CAS PubMed.
- M. Uchida, M. L. Flenniken, M. Allen, D. A. Willits, B. E. Crowley, S. Brumfield, A. F. Willis, L. Jackiw, M. Jutila, M. J. Young and T. Douglas, J. Am. Chem. Soc., 2006, 128, 16626–16633 CrossRef CAS PubMed.
- Y. Ikezoe, Y. Kumashiro, K. Tamada, T. Matsui, I. Yamashita, K. Shiba and M. Hara, Langmuir, 2008, 24, 12836–12841 CrossRef CAS PubMed.
- K. Yamashita, H. Kirimura, M. Okuda, K. Nishio, K. I. Sano, K. Shiba, T. Hayashi, M. Hara and Y. Mishima, Small, 2006, 2, 1148–1152 CrossRef PubMed.
- X. Lin, J. Xie, G. Niu, F. Zhang, H. K. Gao, M. Yang, Q. M. Quan, M. A. Aronova, G. F. Zhang, S. Lee, R. Leaprnan and X. Y. Chen, Nano Lett., 2011, 11, 814–819 CrossRef CAS PubMed.
- K. K. W. Wong and S. Mann, Adv. Mater., 1996, 8, 928–932 CrossRef CAS.
- R. M. Xing, X. Y. Wang, C. L. Zhang, Y. M. Zhang, Q. Wang, Z. Yang and Z. J. Guo, J. Inorg. Biochem., 2009, 103, 1039–1044 CrossRef CAS PubMed.
- M. Okuda, J.-C. Eloi, S. E. Ward Jones, A. Sarua, R. M. Richardson and W. Schwarzacher, Nanotechnology, 2012, 23, 415601 CrossRef PubMed.
- A. W. Perriman, H. Colfen, R. W. Hughes, C. L. Barrie and S. Mann, Angew. Chem., Int. Ed., 2009, 48, 6242–6246 CrossRef CAS PubMed.
- J. R. T. van Weering, E. Brown, T. H. Sharp, J. Mantell, P. J. Cullen and P. Verkade, Methods Cell Biol., 2010, 96, 619–648 CAS.
- A. P. Hollander, T. F. Heathfield, C. Webber, Y. Iwata, R. Bourne, C. Rorabeck and A. R. Poole, J. Clin. Invest., 1994, 93, 1722 CrossRef CAS PubMed.
- G. H. Simon, J. Bauer, O. Saborovski, Y. Fu, C. Corot, M. F. Wendland and H. E. Daldrup-Link, Eur. Radiol., 2006, 16, 738–745 CrossRef PubMed.
- P. Smirnov, E. Lavergne, F. Gazeau, M. Lewin, A. Boissonnas, B. T. Doan, B. Gillet, C. Combadiere, B. Combadiere and O. Clement, Magn. Reson. Med., 2006, 56, 498–508 CrossRef CAS PubMed.
- C. Billotey, C. Aspord, O. Beuf, E. Piaggio, F. Gazeau, M. F. Janier and C. Thivolet, Radiology, 2005, 236, 579–587 CrossRef PubMed.
- C. Wilhelm, L. Bal, P. Smirnov, I. Galy-Fauroux, O. Clement, F. Gazeau and J. Emmerich, Biomaterials, 2007, 28, 3797–3806 CrossRef CAS PubMed.
- M. Lewin, N. Carlesso, C. H. Tung, X. W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nat. Biotechnol., 2000, 18, 410–414 CrossRef CAS PubMed.
- C. Wilhelm and F. Gazeau, Biomaterials, 2008, 29, 3161–3174 CrossRef CAS PubMed.
- A. S. Arbab, L. B. Wilson, P. Ashari, E. K. Jordan, B. K. Lewis and J. A. Frank, NMR Biomed., 2005, 18, 383–389 CrossRef CAS PubMed.
- S. J. Soenen, U. Himmelreich, N. Nuytten, T. R. Pisanic 2nd, A. Ferrari and M. De Cuyper, Small, 2010, 6, 2136–2145 CrossRef CAS PubMed.
- R. M. Steinman, I. S. Mellman, W. A. Muller and Z. A. Cohn, J. Cell Biol., 1983, 96, 1–27 CrossRef CAS PubMed.
- P. A. Baeuerle and W. B. Huttner, Biochem. Biophys. Res. Commun., 1986, 141, 870–877 CrossRef CAS PubMed.
- C. K. Payne, S. A. Jones, C. Chen and X. W. Zhuang, Traffic, 2007, 8, 389–401 CrossRef CAS PubMed.
- R. Vacha, F. J. Martinez-Veracoechea and D. Frenkel, Nano Lett., 2011, 11, 5391–5395 CrossRef CAS PubMed.
- J. W. Bulte, T. Douglas, S. Mann, R. B. Frankel, B. M. Moskowitz, R. A. Brooks, C. D. Baumgarner, J. Vymazal, M. P. Strub and J. A. Frank, J. Magn. Reson. Imaging, 1994, 4, 497–505 CrossRef CAS PubMed.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS PubMed.
- E. Frohlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS PubMed.
- U. O. Hafeli, J. S. Riffle, L. Harris-Shekhawat, A. Carmichael-Baranauskas, F. Mark, J. P. Dailey and D. Bardenstein, Mol. Pharm., 2009, 6, 1417–1428 CrossRef CAS PubMed.
- T. D. Henning, E. J. Sutton, A. Kim, D. Golovko, A. Horvai, L. Ackerman, B. Sennino, D. McDonald, J. Lotz and H. E. Daldrup-Link, Contrast Media Mol. Imaging, 2009, 4, 165–173 CrossRef CAS PubMed.
- L. Kostura, D. L. Kraitchman, A. M. Mackay, M. F. Pittenger and J. W. M. Bulte, NMR Biomed., 2004, 17, 513–517 CrossRef PubMed.
- K. Nakamasu, T. Kawamoto, M. Shen, O. Gotoh, M. Teramoto, M. Noshiro and Y. Kato, Biochim. Biophys. Acta, Gene Struct. Expression, 1999, 1447, 258–264 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed characterisation data, and controls of the magnetisation and toxicological profiling studies. See DOI: 10.1039/c5nr07144e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |