
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembly of supramolecular triarylamine nanowires in mesoporous silica and biocompatible electrodes thereof†
Erol-Dan
Licsandru
a,
Susanne
Schneider
bc,
Sophie
Tingry
a,
Thomas
Ellis
b,
Emilie
Moulin
b,
Mounir
Maaloum
b,
Jean-Marie
Lehn
c,
Mihail
Barboiu
*a and
Nicolas
Giuseppone
*b
aAdaptative Supramolecular Nanosystems Group, Institut Européen des Membranes, ENSCM/UMII/UMR-CNRS 5635, Pl. Eugène Bataillon, CC 047, 34095, Montpellier, Cedex 5, France. E-mail: mihail-dumitru.barboiu@univ-montp2.fr
bSAMS Research Group, University of Strasbourg, Institut Charles Sadron – CNRS, 23 rue du Loess, BP 84047, 67034 Strasbourg Cedex 2, France. E-mail: giuseppone@unistra.fr
cISIS, Institut de Science et d'Ingénierie Supramoléculaires, 8, allée Gaspard Monge, 67000 Strasbourg, France
First published on 10th February 2016
Abstract
Biocompatible silica-based mesoporous materials, which present high surface areas combined with uniform distribution of nanopores, can be organized in functional nanopatterns for a number of applications. However, silica is by essence an electrically insulating material which precludes applications for electro-chemical devices. The formation of hybrid electroactive silica nanostructures is thus expected to be of great interest for the design of biocompatible conducting materials such as bioelectrodes. Here we show that we can grow supramolecular stacks of triarylamine molecules in the confined space of oriented mesopores of a silica nanolayer covering a gold electrode. This addressable bottom-up construction is triggered from solution simply by light irradiation. The resulting self-assembled nanowires act as highly conducting electronic pathways crossing the silica layer. They allow very efficient charge transfer from the redox species in solution to the gold surface. We demonstrate the potential of these hybrid constitutional materials by implementing them as biocathodes and by measuring laccase activity that reduces dioxygen to produce water.
Introduction
Nanostructured materials presenting dense arrays of electroactive domains have received growing attention in a number of applications during the last few years.1–3 In particular, the increase of nanometric contacts over large effective areas in hybrid materials can improve their performance.4 For example, it has been shown that carbon nanotube based electrodes display strongly enhanced responses when compared with amorphous carbon microelectrodes.5 One important challenge in this direction concerns the generation of high-density arrays of electrochemical contacts which can interact with metabolites or biomolecules in order to probe essential physiological processes.Although it is well-established that biocompatible silica-based mesoporous materials, which present extremely high surface areas, can be organized by orienting their nanoporous channels parallel to a surface, perpendicular orientations are more difficult to achieve.6–12 Recently, Walcarius et al. reported the electrochemically-assisted self-assembly of orthogonally oriented mesoporous silica thin films on various supports and used them for the electrochemical detection of small ionic species diffusing along the mesopores toward the electrode surface.13 However, silica is by essence an electrically insulating material14 and species bigger than the dimension of the mesopores cannot reach the electroactive surface, precluding a number of applications.
One alternative to overcome such physical restrictions at the surface would consist of using the perpendicularly aligned mesopores as a scaffolding matrix to incorporate self-assembled electroactive contacts crossing the silica layer. This strategy is of potential applicative interest for the design of conducting biocompatible materials and, in particular, for bioelectrodes. However, to the best of our knowledge, the preparation of such materials has not yet been demonstrated and implemented in devices, for instance in biofuel cell elements.
We have previously demonstrated that a large variety of molecules can be self-assembled and organized in various mesopores by using supramolecular interactions.15–20 The approach described hereafter thus consists of self-assembling electrically conducting supramolecular nanowires within the confined environment of oriented mesoporous silica. For that, we have used chemically tailored triarylamines (TAA) which self-assemble in chloroform and under light irradiation to produce monodimensional supramolecular polymers with metal-like conductivity.21–27 The detailed mechanism of this unique supramolecular polymerization is very complex,28,29 but it can be summarized for TAA molecules as follows (Fig. 1): (i) oxidation of a catalytic quantity of triarylamines to their radical cations TAA˙+, with concomitant reduction of the chlorinated solvent producing chloride counterions; (ii) formation (above a critical concentration of 10 nM) of a nucleus of at least 6 triarylammonium radicals in a double columnar arrangement involving hydrogen bonds; (iii) stacking of neutral triarylamines onto the nucleus and subsequent growth of the primary fibril. In addition to this unique supramolecular polymerization process, ohmic conductivities higher than 5 × 103 S m−1 were determined for these nanostructured fibers over 100 nm distances.
 | ||
| Fig. 1 Simplified supramolecular polymerization process of conducting triarylamine nanowires. The catalytic oxidation of C12-TAA molecules by light irradiation in chlorinated solvents leads to a nucleation/growth mechanism of the self-assembly in which TAA radical cations stack with their neutral counterparts to produce double columnar (i.e. zipped by hydrogen bonds) primary fibrils that contain charge transfer complexes. A top view of the characteristic molecular packing between 4 TAA molecules is represented to illustrate the dimensions of a fibril section (not including the C12 lateral chains).28 | ||
The physical reasons for such conductivity in these wires were fully explained by the formation of charge transfer complexes delocalized along the stacked structure (i.e. supramolecular polarons).30 These functional molecules thus appeared very promising for implementing them in the confined space of mesoporous silica by using a light-triggered in situ self-assembly.
Here we show that we can grow supramolecular stacks of triarylamine molecules in the confined space of oriented mesopores of a silica nanolayer covering a gold electrode.
Results and discussion
Fabrication and characterization of the functional electrodes
Functional electrodes were first constructed on a gold surface by electrodeposition of silica using a mixture of tetraethylsiloxane (TEOS) in the presence of a surfactant (Fig. 2a and b). We also used (3-mercaptopropyl)-trimethoxysilane (MPTMS) in order to provide conditions to tightly attach the silica layer to the gold surface. After washing the nanostructure, the silica surface and pores were made hydrophobic by the attachment of C6-alkyl chains (Fig. 2c). The electrodes were then immersed in a chloroform solution of C12-TAA molecules which were self-assembled in situ by visible light irradiation for one hour with a halogen lamp (10 W cm−2), and with the aim to non-covalently confine the resulting nanowires within the hydrophobic mesopores (i.e. by van der Waals interactions between alkyl chains) (Fig. 2d). To first probe the presence of organic TAA molecules in the mesopores, X-ray photoelectron spectrometry (XPS) experiments were conducted on a series of electrodes after each modification (Table S1 in the ESI†). The porous electrode showed the typical silicon oxygen signal for the silica, as well as some nitrogen, sulfur, and chlorine atom contamination originating from the electrolyte and remaining surfactant. The C6-modified electrode showed an increased concentration of carbon and a full disappearance of the nitrogen, sulfur, and chlorine contaminants ensuring that the surfactant is completely removed. After the addition of C12-TAA into the pores of the silica, a corresponding new nitrogen peak appeared as expected. By using the nitrogen concentration and considering the area of a self-assembled fibril section (Fig. 1), one can estimate that C12-TAA molecules have filled about one third of pores in the silica thin film electrode (see the ESI† section 2). | ||
| Fig. 2 Fabrication of the functional electrodes. (a) Electrodeposition of the silica matrix on the gold surface in the presence of surfactant produces the mesoporous structure depicted in (b). (c) The silica surface is then functionalized with alkyl chains, allowing the non-covalent confinement and self-assembly of conducting C12-TAA nanowires in the hydrophobic pores (d). It is shown in (e) that each step of the fabrication process (empty pores (1), pores blocked with hydrophobic alkyl chains (2), and TAA doped nanopores (3)) should lead to a specific electrochemical behavior when using a Ru3+/ Ru2+ redox probe. | ||
Microscopy techniques were then used to confirm the fabrication of the functional electrodes. Scanning electron microscopy (SEM) micrographs show that the silica layer is attached to the gold surface and has a regular thickness of about 100 nm (Fig. 3a). Further analyses using atomic force microscopy (AFM) – before nanowire formation – confirm the presence of empty pores with diameters in the range of 10 nm (Fig. 3b). After in situ supramolecular polymerization of C12-TAA under light illumination, the nanowires’ tips can be observed by AFM when protruding out of the pores (Fig. 3c and d). The diameter of these tips is less than 5 nm, a dimension in agreement with the expected diameter of a TAA primary fibril considering a core of 2.3 nm (ref. 28) which is extended by lateral C12 alkyl chains (Fig. 1). Conducting AFM finally confirmed the electroactive nature of the C12-TAA-filled nanopores over a large surface covering about 50% of the total area (bright zones in Fig. 3e as compared with the empty nanoporous electrode as a reference in Fig. 3f). One can notice qualitatively that not all the pores present a similar conductivity, with a color range from yellow to white, probably indicating the presence of pores partially filled with conducting wires. This result is coherent with the total quantity of TAA previously determined by XPS (Table S1 in the ESI†).
 | ||
| Fig. 3 (a) SEM micrograph of the cross section of a gold electrode covered by a silica film obtained by electrodeposition (−1.4 V, 10 s) and showing the structure consisting of a 91 nm silica layer on the top of a 400 nm gold layer. (b) Typical AFM topography images of the empty mesoporous silica layer, and (c) of the corresponding C12-TAA doped layer showing several tips of fibrils within nanopores. (d) High resolution AFM image of single pore after the silica layer was filled with a single self-assembled fibril of TAA, in agreement with the size of their section. (e) Conducting AFM image of a large surface of the C12-TAA-functionalized electrode showing the distribution of the conducting mesopores. (f) Conducting AFM image of the reference nanoporous electrode without C12-TAA functionalization. | ||
We then investigated the electrical properties of the C12-TAA/silica hybrid films by using cyclic voltammetry with Ru(NH3)63+ as a redox probe (Fig. 2e). Fig. 4a displays the cyclic voltammograms (CVs) of Ru(NH3)63+ (1 mM in 100 mM aqueous NaNO3) using either bare gold electrodes or electrodes that were subjected to successive steps of (i) silica thin film deposition, (ii) hydrophobic modification, and (iii) doping by C12-TAA. After the removal of the surfactants, the CV curve of the thin-layer mesoporous silica gold electrode is characterized by well-defined Ru2+/Ru3+ oxidation and reduction peaks, indicating the access of the redox probe to the gold surface by diffusion within the mesoporous array (Fig. 4a, dashed blue line). By comparing the CVs of Ru(NH3)63+ at the gold electrode and at the porous electrode, the electroactive surface area of the latter was evaluated from the ip (peak current density) values at different scan rates using the Randles–Sevcik equation (see the ESI† section 3), and estimated to be 0.15 cm2 compared to a gold surface of 0.226 cm2. By performing a similar CV experiment with the mesoporous electrode blocked by the presence of alkyl chains within the pores, the weak electrochemical signal confirms the inaccessibility of the hydrophobic nanochannels for the redox probe (for some electrodes, the residual redox signal is not even detectable). However, in the presence of the self-assembled triarylamines in the pores, the intensity of the signal corresponding to the redox probe is increased and nearly equals the one recorded for the empty porous system (Fig. 4a, black line). Regarding the previous structural data (Fig. 3), and because the hydrophobicity of the electrodes is not changed significantly when functionalized with C12-TAA (see contact angle measurements in the ESI† section 4), this current density recovery suggests that the electron transfer process is mainly supported by the self-assembled wires. The matching of the current peak heights for both filled and empty pores indicates that C12-TAA can conduct the current from the redox probe with remarkable efficiency. Importantly, the structure of the material is stable for at least one month without isolation from atmospheric oxygen (see the ESI† section 5).
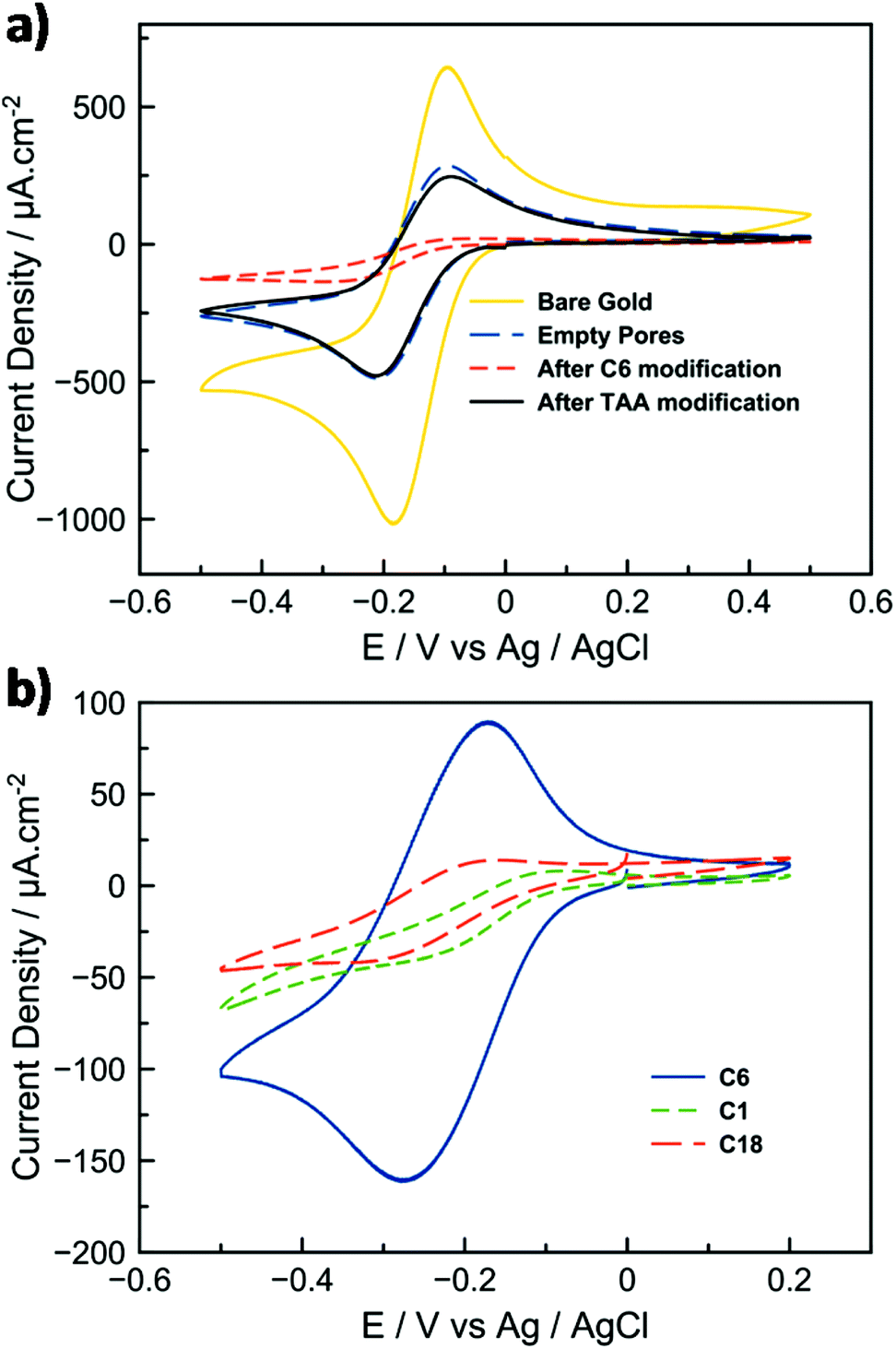 | ||
| Fig. 4 (a) Cyclic voltammetry curves of Ru(NH3)63+ (c = 1 mM , ν = 20 mV s−1, in a 0.1 M aqueous solution of NaNO3) at bare gold electrode (yellow), after mesoporous silica thin film electrodes were prepared by electrogeneration (dashed blue), after hydrophobic modification (dashed red), and after doping with C12-TAA (black). (b) Cyclic voltammetry of the redox probe after C12-TAA doping using different initial hydrophobic modifications (i.e. chain lengths of methyl (C1), hexyl (C6), octyldecyl (C18)). | ||
The effect of varying alkyl chain lengths of the modified silica mesopores (C1, C6 or C18) was tested towards its efficiency to incorporate the triarylamine within the pores. CVs show a much better confinement of the self-assembly in the presence of C6 alkyl chains which fit better the size of the C12-TAA fibrils for optimal confinement (Fig. 4b).
The importance of light irradiation on the device was also investigated. Comparison of porous electrodes immersed in non-irradiated and irradiated solutions respectively confirmed the appearance of a current for the latter only. The importance of the doped supramolecular nanowire to transfer electrons is also evident when looking at data obtained by electrochemical impedance spectroscopy (see the ESI† section 6). The illuminated C12-TAA electrode has twice the charge transfer conductivity of the empty pores, and nearly five times the conductivity of the non-irradiated C12-TAA electrode. For both porous and doped electrodes, the linear dependence of ip with the square root of the scan rate (ν1/2) supports the view that the electron-transfer process is coupled to the diffusion of the redox probes (Fig. S2†). A supplementary control experiment using non-electroactive tridodecylamine was performed under the same conditions and did not show significant increase of current from the C6-functionalized porous layer (Fig. S3†). These results confirm that the conductivity displayed by the devices doped with C12-TAA is not just an effect of introducing a new adsorbent, or of differing ionic conditions to the surface, but is truly an effect of the highly conducting self-assembled nanowires.
Evaluation of the functional electrodes as biocathodes
The hybrid electrodes were further evaluated toward the electrocatalytic reduction of dioxygen by an enzyme. This system is inspired by biofuel cells (BFCs) which have been considered for the conversion of chemical energy into electrical energy and hold promise for numerous applications in biomedical research, environmental monitoring, and as a power source for portable electronic devices.31–35 BFC devices are built from the integration of a bioanode, which oxidizes the fuel substrate (e.g. glucose, ethanol or methanol), with a biocathode, where the electrons generated at the anode combine with an oxidant (typically oxygen) and protons to form a product (typically water).31,33,34,36 Here, the mesoporous C12-TAA electrode was covered on its surface by a mixture of laccase and ABTS entrapped in the Nafion® polymer to adhere and stabilize the species on the support. Laccase is considered as an attractive biocatalyst to reduce molecular oxygen into water at very high redox potentials. Its functioning is enhanced by the presence of ABTS as a redox mediator which transfers electrons by diffusion from the electrode to the enzymatic active site.37 The enzyme was also previously mixed with conducting carbon black nanoparticles providing an additional surface area for adsorption. The polarization curve of the resulting biocathode, shown in Fig. 5a, is characteristic of the electrochemical response (surface area), with and without C12-TAA, for enzymatic reduction of O2 in phosphate buffer at pH 5. The oxygen reduction current starts at 0.58 V vs. Ag/AgCl, and current densities feature a semi-plateau indicating a control of the electrocatalytic reaction by diffusion of O2 to the active enzymes in the film. The presence of a peak around 0.3 V is due to the reduction of some ABTS molecules weakly adsorbed at the electrode. The C12-TAA bioelectrode delivers current densities of 240 μA cm−2 and shows good stability on successive scans. No catalytic response was obtained in the absence of ABTS, suggesting that electrical communication between the redox protein and C12-TAA self-assemblies is not strong enough to avoid a chemical mediator. For comparison, the current density was evaluated for 2 biocathodes prepared from (i) the porous electrode, and (ii) the C6-functionalized electrode. In the absence of C12-TAA, the biocathodes deliver half of the current density (130 μA cm−2) compared to the triarylamine modified layer, supporting the electrical connection provided by the triarylamine nanowires.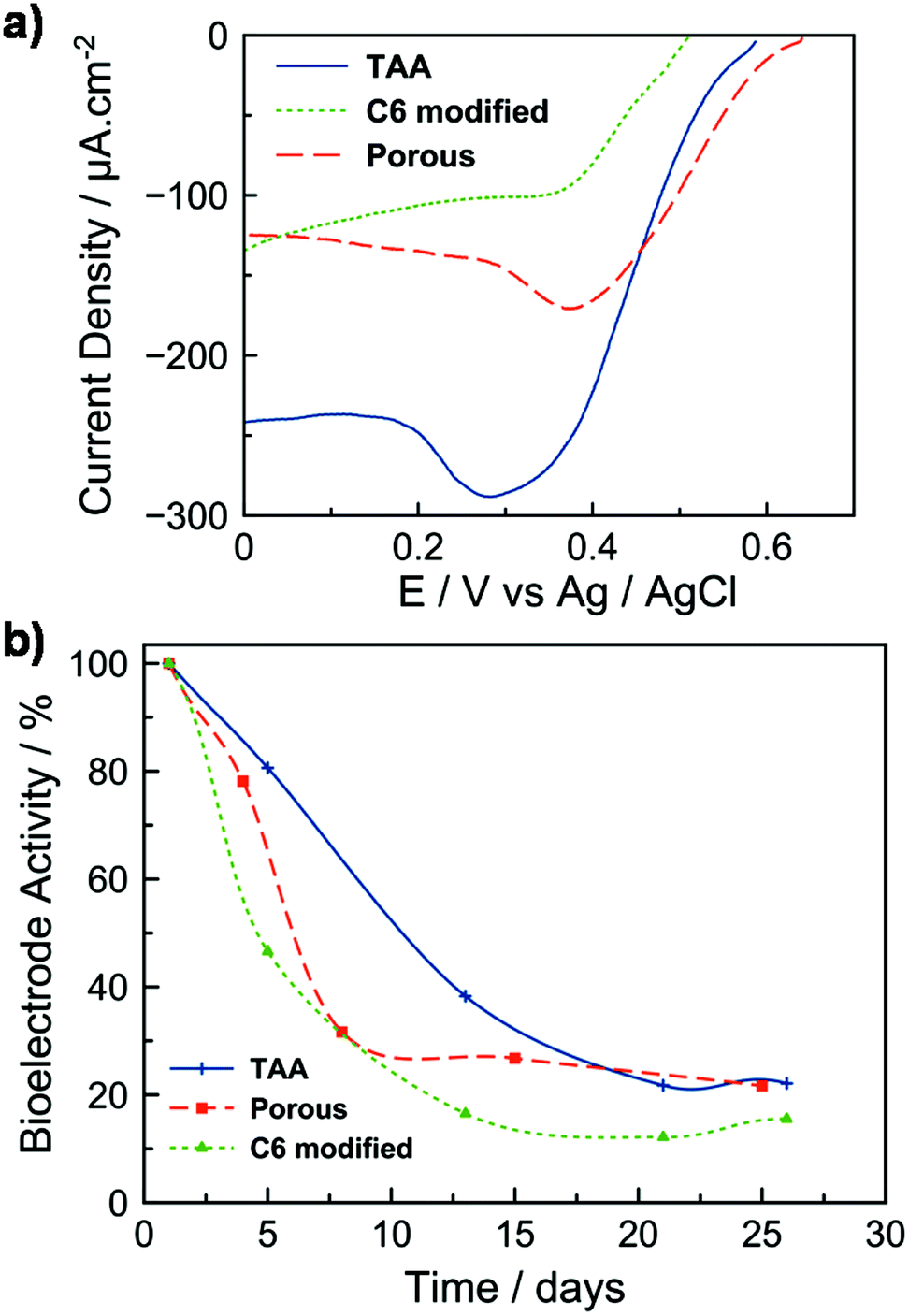 | ||
| Fig. 5 (a) Polarization curves of a laccase-based system using different mesoporous biocathodes (empty, C6-modified, and C12-TAA-hybrid) in an O2-saturated phosphate solution (pH = 5, c = 0.1 M, scan rate = 3.3 mV s−1). The current density is reported to the geometrical surface area. (b) Normalized activity evolution of the same biocathodes as a function of time. | ||
The stability of the bioelectrodes was tested over time by keeping them at 5 °C between each polarization test (Fig. 5b). The C12-TAA biocathode retained 50% of its initial electroactivity over 10 days. Although bare gold electrodes with immobilized enzymes exhibit similar performance to that of the TAA-doped mesoporous electrode, the enzyme layer does not stick properly and collapses on the gold surface with the stability of the resulting electrode restricted to one day38,39 (see also Fig. S5†). In the absence of C12-TAA and over the same period, bioelectrodes exhibited only 20% of their initial electroactivity. This decrease can be mainly attributed to the loss of entrapped ABTS from the electrodes’ surface to the solution.40 In the presence of C12-TAA nanowires, we attributed the slower release of ABTS to the π-stacking and charge interactions between ABTS2− and C12-TAA˙+. Such a phenomenon was already described by Willner et al. in the case of the confinement of CNTs in mesoporous carbon nanoparticles modified by ABTS2−.41 However, after 26 days, the denaturation of the immobilised enzymes resulted in a lower electroactivity (less than 20%) for all configurations, showing that the nanowires themselves do not affect the stability of such biocathodes. In previous studies, for systems based on laccase/ABTS entrapment in Nafion, the loss of bioelectrode stability with time was also reported to be about 50% after 5 days on a glassy carbon electrode,40 and 60% after 7 days on a carbon fiber electrode.42
Conclusions
We have shown how to confine self-assembled triarylamine nanowires in a pre-oriented scaffold of mesoporous silica. The perpendicular orientation of the wires within the silica layer is important as it optimizes the number of contacts and allows their filling by 100 nm long nanowires which present optimized conduction properties over this distance.21 One of the interesting features of this efficient process is illustrated here by associating biocompatible (but insulating) silica layers with highly conducting self-assembled nanowires. The formation of a continuous pathway of supramolecular charge transfer complexes across the hybrid silica layers allows for a highly efficient electronic conduction. The first implementation of this strategy was illustrated by fabricating a biocathode device for the reduction of O2 into water. This study takes advantage of the metallic conductivity of triarylamine nanowires, and shows how their unique light-triggered self-assembly process can be used to address their formation in pre-patterned nanostructures, thus merging several bottom-up technologies in order to produce advanced functional materials.43 Further implementations of self-assembled triarylamine organic metals as nanometer-sized electrodes thus appear promising for high-resolution chemical imaging of surfaces and interfaces, and for the development of microscopic chemical sensors.Experimental section
Imaging
Scanning electron microscopy (SEM) micrographs were obtained with a Hitachi S-4500 microscope (0.5–30 kV). Atomic force microscopy (AFM) and conducting AFM images were obtained by scanning the samples using a Nanoscope 8 (Bruker) operating in Peak-Force tapping mode. For the contact current images, the scale is 3.6 pA, the image size is 0.4 μm × 0.4 μm, taken at 1 nN PeakForce, 5 V DC bias, using Bruker's PeakForce TUNA probe (Pt/Ir coating, spring constant of 0.4 N m−1) on an Icon dimension with an electrically conductive tip having a radius of about 50–100 nm.Electrodeposition of mesoporous silica layer
The silica matrix was made through electrodeposition methods with a procedure adapted from Walcarius et al.13 A mixture of tetraethylsiloxane (TEOS) and (3-mercaptopropyl)-trimethoxysilane (MPTMS) in ethanol was mixed with CTAB (hexadecyltrimethylammonium bromide) as a template agent dissolved in a 0.1 M solution of NaNO3. The solution was aged under stirring for 2.5 h at pH 3 prior to electrodeposition. The mesoporous silica films were deposited by applying a negative potential of −1.4 V for 10 s on the gold electrode immersed in the pre-hydrolysed precursor solution. The electrode was removed from the solution and the electrodeposited surfactant-templated film was cured overnight at 60 °C and washed in ethanol.Alkylation of mesoporous silica layer and subsequent in-pores self-assembly of TAA nanowires
The resulting electrode was modified with hexyltrichlorosilane (C6) in a toluene solution at 60 °C for 6 h in order to obtain a hydrophobic surface coating covalently linked to the inner silica mesopores. Triarylamines were non-covalently confined in these hydrophobic mesopores by immersing the functionalized electrodes in a 1.5 mM chloroform solution of C12-TAA that was irradiated for one hour with a 20 W (10 W cm−2) halogen lamp.Bioelectrode preparation
15 mg of commercial carbon powder Super P (TIMCAL) were sonicated for 5 min in PBS (phosphate buffer solution) pH = 5 and stirred for 30 min. 333 μL of this mixture were pipetted over 5 mg of laccase enzyme from Trametes Versicolor (20 U per mg solid), and mixed for 2 min. Separately, a solution of ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid)) was made out of 2.7 mg of ABTS in 500 μL of PBS at pH = 5 and homogenized. 100 μL of the enzyme solution were mixed with 90 μL of the ABTS solution, and stirred for another 30 min. 10 μL of Nafion resin (5 wt%) solution were added and the solution was homogenized for another 3 min. The electrode surfaces were pre-treated with a solution of poly(acrylic acid). 15 μL of the enzyme solution was drop-cast on a 20 mm2 surface (estimated laccase loading of 560 μg cm−2). The electrodes were left to dry at room temperature and then stored in the fridge.Acknowledgements
This work was supported by funds from ITN DYNANO, PITN-GA-2011-289033 (http://www.dynano.eu). The research leading to these results has received funding from the European Research Council under the European Community's Seventh Framework Program (FP7/2007–2013)/ERC Starting Grant Agreement No. 257099 (N. G.). We acknowledge the Agence Nationale pour la Recherche (ANR-11-BS08-06 and ANR-11-EMMA-009), the COST action (CM1304), the University of Strasbourg (UdS), the Centre National de la Recherche Scientifique (CNRS), the international center for Frontier Research in Chemistry (icFRC), the Laboratory of Excellence for Complex Systems Chemistry (LabEx CSC), and the Institut Universitaire de France (IUF). We thank I. A. Dascalu (“Petru Poni” Institute of Macromolecular Chemistry of Romanian Academy, Iasi, Romania) for XPS Measurements.References
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed.
- Y.-G. Guo, J.-S. Hu and L.-J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS.
- M. Ramanathan, S. M. Kilbey II, Q. Ji, J. P. Hill and K. Ariga, J. Mater. Chem., 2012, 22, 10389 RSC.
- L. B. Sheridan, D. K. Hensley, N. V. Lavrik, S. C. Smith, V. Schwartz, C. Liang, Z. Wu, H. M. Meyer and A. J. Rondinone, J. Electrochem. Soc., 2014, 161, H558–H563 CrossRef CAS.
- A. G. Zestos, C. B. Jacobs, E. Trikantzopoulos, A. E. Ross and B. J. Venton, Anal. Chem., 2014, 86, 8568–8575 CrossRef CAS PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- S.-H. Wu, C.-Y. Mou and H.-P. Lin, Chem. Soc. Rev., 2013, 42, 3862–3875 RSC.
- K. S. Jang, H. J. Kim, J. R. Johnson, W. G. Kim, W. J. Koros, C. W. Jones and S. Nair, Chem. Mater., 2011, 23, 3025–3028 CrossRef CAS.
- X. Fang, X. Zhao, W. Fang, C. Chen and N. Zheng, Nanoscale, 2013, 5, 2205–2218 RSC.
- T. Nakamura, F. Sugihara, H. Matsushita, Y. Yoshioka, S. Mizukami and K. Kikuchi, Chem. Sci., 2015, 6, 1986–1990 RSC.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- A. Walcarius, E. Sibottier, M. Etienne and J. Ghanbaja, Nat. Mater., 2007, 6, 602–608 CrossRef CAS PubMed.
- H. Nishihara, T. Kwon, Y. Fukura, W. Nakayama, Y. Hoshikawa, S. Iwamura, N. Nishiyama, T. Itoh and T. Kyotani, Chem. Mater., 2011, 23, 3144–3151 CrossRef CAS.
- M. Barboiu, A. Cazacu, S. Mihai, Y.-M. Legrand, G. Nasr, Y. Le Duc, E. Petit and A. van der Lee, Microporous Mesoporous Mater., 2011, 140, 51–57 CrossRef CAS.
- A. Cazacu, Y.-M. Legrand, A. Pasc, G. Nasr, A. Van der Lee, E. Mahon and M. Barboiu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8117–8122 CrossRef CAS PubMed.
- S. Mihai, A. Cazacu, C. Arnal-Herault, G. Nasr, A. Meffre, A. van der Lee and M. Barboiu, New J. Chem., 2009, 33, 2335–2343 RSC.
- S. Mihai, Y. Le Duc, D. Cot and M. Barboiu, J. Mater. Chem., 2010, 20, 9443–9448 RSC.
- A. Cazacu, S. Mihai, G. Nasr, E. Mahon, A. van der Lee, A. Meffre and M. Barboiu, Inorg. Chim. Acta, 2010, 363, 4214–4219 CrossRef CAS.
- Y. Le Duc, A. Gilles, S. Mihai, V. Rouessac, S. Tingry and M. Barboiu, Chem. Commun., 2013, 49, 3667–3669 RSC.
- E. Moulin, F. Niess, M. Maaloum, E. Buhler, I. Nyrkova and N. Giuseppone, Angew. Chem., Int. Ed., 2010, 49, 6974–6978 CrossRef CAS PubMed.
- V. Faramarzi, F. Niess, E. Moulin, M. Maaloum, J.-F. Dayen, J.-B. Beaufrand, S. Zanettini, B. Doudin and N. Giuseppone, Nat. Chem., 2012, 4, 485–490 CrossRef CAS PubMed.
- E. Moulin, J.-J. Cid and N. Giuseppone, Adv. Mater., 2013, 25, 477–487 CrossRef CAS PubMed.
- E. Moulin, F. Niess, G. Fuks, N. Jouault, E. Buhler and N. Giuseppone, Nanoscale, 2012, 4, 6748–6751 RSC.
- Y. Domoto, E. Busseron, M. Maaloum, E. Moulin and N. Giuseppone, Chem. – Eur. J., 2015, 21, 1938–1948 CrossRef CAS PubMed.
- A. Wolf, E. Moulin, J. J. Cid Martín, A. Goujon, G. Du, E. Busseron, G. Fuks and N. Giuseppone, Chem. Commun., 2015, 51, 4212–4215 RSC.
- J. J. Armao, Y. Domoto, T. Umehara, M. Maaloum, C. Contal, G. Fuks, E. Moulin, G. Decher, N. Javahiraly and N. Giuseppone, ACS Nano, 2016, 10 DOI:10.1021/acsnano.5b06294.
- I. Nyrkova, E. Moulin, J. J. Armao, M. Maaloum, B. Heinrich, M. Rawiso, F. Niess, J.-J. Cid, N. Jouault, E. Buhler, A. N. Semenov and N. Giuseppone, ACS Nano, 2014, 8, 10111–10124 CrossRef CAS PubMed.
- E. Busseron, J.-J. Cid, A. Wolf, G. Du, E. Moulin, G. Fuks, M. Maaloum, P. Polavarapu, A. Ruff, A.-K. Saur, S. Ludwigs and N. Giuseppone, ACS Nano, 2015, 9, 2760–2772 CrossRef CAS PubMed.
- J. J. Armao, M. Maaloum, T. Ellis, G. Fuks, M. Rawiso, E. Moulin and N. Giuseppone, J. Am. Chem. Soc., 2014, 136, 11382–11388 CrossRef CAS PubMed.
- I. Ivanov, T. Vidaković-Koch and K. Sundmacher, Energies, 2010, 3, 803–846 CrossRef CAS.
- B. E. Logan and K. Rabaey, Science, 2012, 337, 686–690 CrossRef CAS PubMed.
- I. Willner and E. Katz, Angew. Chem., Int. Ed., 2000, 39, 1180–1218 CrossRef.
- A. Zebda, C. Gondran, A. Le Goff, M. Holzinger, P. Cinquin and S. Cosnier, Nat. Commun., 2011, 2, 370 CrossRef PubMed.
- V. Andoralov, M. Falk, D. B. Suyatin, M. Granmo, J. Sotres, R. Ludwig, V. O. Popov, J. Schouenborg, Z. Blum and S. Shleev, Sci. Rep., 2013, 3, 3270 Search PubMed.
- B. E. Logan, B. Hamelers, R. Rozendal, U. Schröder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181–5192 CrossRef CAS PubMed.
- D. Selloum, A. A. Chaaya, M. Bechelany, V. Rouessac, P. Miele and S. Tingry, J. Mater. Chem. A, 2014, 2, 2794–2800 CAS.
- M. Pita, C. Gutierrez-Sanchez, D. Olea, M. Velez, C. Garcia-Diego, S. Shleev, V. M. Fernandez and A. L. De Lacey, J. Phys. Chem. C, 2011, 115, 13420–13428 CAS.
- S. Shleev, J. Tkac, A. Christenson, T. Ruzgas, A. I. Yaropolov, J. W. Whittaker and L. Gorton, Biosens. Bioelectron., 2005, 20, 2517–2554 CrossRef CAS PubMed.
- A. Both Engel, A. Cherifi, M. Bechelany, S. Tingry and D. Cornu, ChemPlusChem, 2015, 80, 494–502 CrossRef CAS.
- A. Trifonov, K. Herkendell, R. Tel-Vered, O. Yehezkeli, M. Woerner and I. Willner, ACS Nano, 2013, 7, 11358–11368 CrossRef CAS PubMed.
- K. Karnicka, K. Miecznikowski, B. Kowalewska, M. Skunik, M. Opallo, J. Rogalski, W. Schuhmann and P. J. Kulesza, Anal. Chem., 2008, 80, 7643–7648 CrossRef CAS PubMed.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, XPS measurements, contact angle measurements, additional cyclic voltammograms and electrochemical impedance spectroscopy. See DOI: 10.1039/c5nr06977g |
| This journal is © The Royal Society of Chemistry 2016 |