
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization of multifunctional β-NaEuF4/NaGdF4 core–shell nanoparticles with narrow size distribution†
Lilli
Schneider
*a,
Thorben
Rinkel
b,
Benjamin
Voß
b,
Artur
Chrobak
c,
Johann P.
Klare
a,
Jan
Neethling
d,
Jaco
Olivier
d,
Dominik
Schaniel
e,
El-Eulmi
Bendeif
e,
Federica
Bondino
f,
Elena
Magnano
f,
Igor
Píš
fg,
Kamil
Balinski
a,
Joachim
Wollschläger
a,
Heinz-Jürgen
Steinhoff
a,
Markus
Haase
b and
Karsten
Kuepper
*a
aDepartment of Physics, Center of Physics and Chemistry of New Materials, Universität Osnabrück, Barbarastr. 7, 49076 Osnabrück, Germany. E-mail: lischroe@uos.de; kkuepper@uos.de
bInstitute of Chemistry of New Materials, Center of Physics and Chemistry of New Materials, Universität Osnabrück, Barbarastr. 7, 49076 Osnabrück, Germany
cInstitute of Physics, Silesian University, Uniwersytecka 4, 40-007 Katowice, Poland
dCentre for HRTEM, Nelson Mandela Metropolitan University, P.O. Box 77000, Port Elizabeth, South Africa
eUniversité de Lorraine, CRM2, UMR 7036, Vandoeuvre-les-Nancy, F-54506, France
fIOM CNR, Laboratorio TASC, S.S. 14 km 163.5, 34149 Basovizza, Trieste, Italy
gElettra-Sincrotrone Trieste S.C.p.A., S.S. 14 km 163.5, 34149 Basovizza, Trieste, Italy
First published on 28th December 2015
Abstract
The properties of β-NaEuF4/NaGdF4 core–shell nanocrystals have been thoroughly investigated. Nanoparticles with narrow size distribution and an overall diameter of ∼22 nm have been produced with either small β-NaEuF4 cores (∼3 nm diameter) or large β-NaEuF4 cores (∼18 nm diameter). The structural properties and core–shell formation are investigated by X-ray diffraction, transmission electron microscopy and electron paramagnetic resonance, respectively. Optical luminescence measurements and X-ray photoelectron spectroscopy are employed to gain information about the optical emission bands and valence states of the rare earth constituents. Magnetic characterization is performed by SQUID and X-ray magnetic circular dichroism measurements at the rare earth M4,5 edges. The characterization of the core–shell nanoparticles by means of these complementary techniques demonstrates that partial intermixing of core and shell materials takes place, and a significant fraction of europium is present in the divalent state which has significant influence on the magnetic properties. Hence, we obtained a combination of red emitting Eu3+ ions and paramagnetic Gd3+ ions, which may be highly valuable for potential future applications.
1 Introduction
NaREF4 nanoparticles (RE: rare earth) have attracted increasing interest in recent years for applications in biological labeling and imaging,1–20 photodynamic therapy and drug delivery,21–26 photovoltaics,27–30 photonics31–33 and security labeling.34–36 Frequently, nanocrystals with core–shell structure are synthesized, since a shell of inert material increases the quantum yield of a luminescent particle core by decreasing energy losses on the particle surface.37–39 Particles containing one or more shells of the doped material furthermore allow us to spatially separate the dopant ions with different functionalities and to control the migration of energy within such structures.40–49 For lanthanide ions emitting in the red and near infrared spectral regions, fluoride host materials are of particular interest because the very low phonon frequencies of fluoride lattices decrease the probabilities of multiphonon relaxation processes, resulting in high quantum yields. One example is the Eu3+ ion, displaying pure red emission in many host materials, because all transitions except those from the 5D0 level are quenched due to multiphonon relaxation. In the fluoride host, however, also transitions from the higher 5D1, 5D2 and 5D3 levels are observed provided that the concentration of the Eu3+ dopant is low. At higher concentrations, however, cross-relaxation between adjacent Eu3+ in the lattice leads to quenching of the 5D1, 5D2 and 5D3 transitions in these materials, too. Trivalent gadolinium, on the other hand, is characterized by a relatively large energy gap between the ground state (8S7/2) and the lowest excited level (6P7/2).21 Importantly, this excited level is situated in the ultraviolet spectral region, i.e. well above the emitting 5D0, 5D1, 5D2 and 5D3 levels of Eu3+. A NaGdF4 shell around a NaEuF4 core can therefore improve the luminescence efficiency of the core. Moreover, the lattice mismatch between the core and the shell is very low due the very similar lattice constants of NaGdF4 and NaEuF4. In addition to their interesting optical properties, europium and gadolinium have remarkable magnetic properties. Owing to their seven unpaired electrons, Gd3+ ions in the ground state can, for instance, efficiently alter the relaxation times of the surrounding water protons and are therefore the basis of the most commonly used contrast agents in magneto resonance imaging.50 While gadolinium ions usually form the oxidation state III+, europium compounds can contain either Eu3+ or Eu2+ ions, or both. Trivalent europium in the ground state (7F0, S = 3, L = 3, J = 0) has no magnetic moment while divalent europium has seven unpaired electrons in the ground state (8S7/2, S = 8, L = 0, J = 7/2). Interestingly in some trivalent compounds formation of divalent europium states on the surface was observed.51,52 Moreover, Kachkanov et al. detected an optically induced magnetic moment of Eu3+ ions in GdN.53In this work, we synthesized core–shell systems composed of a β-NaEuF4 core and a β-NaGdF4 shell. This structure has two advantages. Firstly, the influence of different surrounding media on the β-NaEuF4 core can be minimized or entirely removed. Secondly, the combination of red emitting Eu3+ ions and paramagnetic Gd3+ ions is highly valuable.
The nominal size is the expected size based on the used materials respectively precursor particles. We use a number of complementary methods to evaluate the structure of β-NaEuF4/NaGdF4 core–shell particles and to study their electronic and magnetic properties. Detailed knowledge of these properties is indispensable to compare the results with those of Eu3+ doped β-NaGdF4 nanoparticles and to further optimize the core–shell system in question. Structural properties and core–shell formation are determined by means of X-ray powder diffraction (XRD) and high resolution transmission electron microscopy (HR-TEM). Core–shell formation is further investigated by electron paramagnetic resonance (EPR) and optical luminescence spectroscopy. Chemical and electronic properties with special emphasis on the Eu and Gd valence states are determined employing X-ray photoelectron spectroscopy (XPS), whereas magnetic characterization is performed with a superconducting quantum interference device (SQUID) magnetometer. The measured magnetization curves are analyzed by means of Monte Carlo based Heisenberg simulations. Finally we performed element specific X-ray magnetic circular dichroism (XMCD) at the Eu M4,5 and Gd M4,5 edges to gain a complete picture of the internal magnetic structure of the multifunctional β-NaEuF4/NaGdF4 core–shell nanoparticles.
2 Results and discussion
2.1 Total scattering measurement and PDF analysis
Fig. 1 shows the pair distribution functions (PDFs) for both core–shell samples as well as the 22 nm β-NaGdF4 nanocrystals doped with 1% Eu. The two samples containing low Eu and high Gd concentration (20 nm β-NaEuF4/NaGdF4 core–shell nanoparticles (NPs) with a 3 nm β-NaEuF4 core, 22 nm β-NaGdF4 nanoparticle (NP) doped with 1% Eu) exhibit very similar PDFs that differ significantly from the core–shell sample with the 18 nm β-NaEuF4 core. Comparing the two core–shell samples (20 nm β-NaEuF4/NaGdF4 core–shell nanoparticles with a 3 nm β-NaEuF4 core, 22 nm β-NaEuF4/NaGdF4 core–shell nanoparticles with an 18 nm β-NaEuF4 core) we can clearly identify shifts of the peak positions in the PDF (corresponding to atom–atom distances). Note that the structures of bulk β-NaEuF4 and β-NaGdF4 are quite similar and the differences in bond distances between the different atoms are thus expected to be small. In order to enhance the visibility of the structural differences we calculated the differences between the corresponding PDFs, given as ΔG(r) in Fig. 1. One can clearly see that the difference between the PDF of the 20 nm β-NaEuF4/NaGdF4 core–shell nanoparticles with a 3 nm β-NaEuF4 core and the 22 nm β-NaGdF4 NP doped with 1% Eu is very small, resulting in very weak features in the ΔG(r). A core–shell nanoparticle with a small β-NaEuF4 core thus exhibits almost the same structure as a nanoparticle doped with 1% Eu, indicating that the structural relaxation is similar for these two cases. In other words, from the PDF analysis alone it is impossible to distinguish between the structure of the core–shell NP and a homogeneously doped NP. In contrast, the difference of the two PDFs (ΔG(r)) of the two core–shell samples is much more pronounced. Almost all peak positions are affected and the shifts are more pronounced for larger atom–atom distances r. In order to investigate this result in more detail we performed simulations of the PDFs based on pure crystalline, spherical 22 nm β-NaGdF4 and β-NaEuF4 nanocrystals. The results are shown in Fig. 2. The distances are slightly shorter in the β-NaGdF4 nanoparticle as expected from the smaller lattice parameters. We observe a good agreement between the simulated peak positions in the PDF for the two pure nanoparticles and the experimentally determined PDF. The peaks are broader in the experimental PDF, which is due to a lower resolution of the experiment, but also due to the fact that in the core–shell nanoparticles we expect some strain and defects as well as a not perfectly spherical shape. We don't expect a significant impact of the particle-size distribution on the PDF since the size distributions are rather narrow, e.g., the β-NaEuF4/NaGdF4 (3 nm core) nanoparticles exhibit a size distribution of 16.8 nm ± 5% (see Table 1), corresponding to particles ranging from 16 nm to 17.7 nm. If the structural difference (e.g. distances between first neighbours) between two such nanoparticles is well below the resolution of 0.138 Å, there will be no visible effect on the experimental PDF. For illustration one can compare the two simulated PDFs for the 22 nm β-NaGdF4 and β-NaEuF4 particles given in Fig. 2, where the difference in the first neighbour distances between the pure Gd and the pure Eu sample is of the order of 0.02 Å, which is at the limit of visibility in the G(r). Structural differences between two identical nanoparticles differing only by 5% in size would therefore not be detected in the experiment. As a first conclusion we can state that a β-NaEuF4/NaGdF4 core–shell nanoparticle with a 3 nm β-NaEuF4 core exhibits approximately the same structure as a slightly Eu-doped β-NaGdF4 nanoparticle and its main structural parameters are those of the β-NaGdF4 structure. β-NaEuF4/NaGdF4 core–shell nanoparticles with an 18 nm β-NaEuF4 core on the other hand resemble the corresponding pure β-NaEuF4 structure. Due to the limited resolution of the experiment a more detailed modelling, e.g. to investigate the core–shell interface, is currently not possible. High-resolution electron microscopy investigations were carried out for further characterisation of the nanoparticles. | ||
| Fig. 1 Comparison of atomic PDFs in real space, G(r) for 20 nm β-NaEuF4/NaGdF4 core–shell nanoparticles with a 3 nm β-NaEuF4 core and ∼8.5 nm β-NaGdF4 shell, 22 nm β-NaEuF4/NaGdF4 core–shell nanoparticles (NPs) with an 18 nm β-NaEuF4 core and 22 nm β-NaGdF4 nanoparticle (NP) doped with 1% Eu and their differential PDFs. | ||
 | ||
| Fig. 2 Comparison of observed PDFs for 20 nm β-NaEuF4/NaGdF4 core–shell nanoparticles with a 3 nm β-NaEuF4 core and ∼8.5 nm β-NaGdF4 shell and 22 nm β-NaEuF4/NaGdF4 core–shell NPs with an 18 nm β-NaEuF4 core and calculated PDFs of 22 nm β-NaGdF4 and β-NaEuF4 spheres with the corresponding differential PDFs (spectra are normalized to the first peak). | ||
| Samples | Nominal size | Size (from TEM) |
|---|---|---|
| NaEuF4/NaGdF4 (3 nm core) | 20 nm | 16.8 nm ± 5% |
| NaEuF4/NaGdF4 (18 nm core) | 22 nm | 23 nm ± 3.8% |
| NaGdF4:1% Eu | 22 nm | 25.4 nm ± 4% |
| NaEuF4 (3 nm) | 3 nm | Not evaluable |
| NaEuF4 (18 nm) | 18 nm | 19.7 nm ± 4.2% |
| NaGdF4/NaEuF4 (3 nm core) | 20 nm | 20.2 nm ± 3.9% |
| NaEuF4:1% Gd | 22 nm | 24.5 nm ± 5.4% |
| NaGdF4 (3 nm) | 3 nm | Not evaluable |
| NaGdF4 (22 nm) | 22 nm | 25.6 nm ± 5.8% |
2.2 TEM-HRTEM
The morphology of the nanoparticles was characterized by transmission electron microscopy (TEM). It can be seen that the particles are nearly spherical or slightly elongated and retain the initial morphology of the precursor core particles (Fig. 3). Inter-planar spacings were measured using fast Fourier transform (FFT) analysis of the HRSTEM images. Inter-planar spacings with distances 0.29 nm, 0.18 nm and 0.16 nm were measured. These match very closely to the theoretical values for the (1,1,0), (0,0,2) and (1,1,2) planes respectively. Because the lattice constants between the core and the shell are similar, no significant differences are observed. Fig. 4(a) shows the high-resolution bright field scanning transmission electron microscopy (BF STEM) images of the core–shell nanoparticles with an 18 nm β-NaEuF4 core and a 2 nm β-NaGdF4 shell and (b) the corresponding high-angle annular dark field (HAADF) image. | ||
| Fig. 3 TEM images showing (top) general morphology and (bottom) high-resolution images of the (a) 18 nm β-NaEuF4 NP and (b) 22 nm β-NaEuF4/NaGdF4 NP with a 2 nm β-NaGdF4 shell around the 18 nm β-NaEuF4 core. The insets show the fast Fourier transforms (FFTs) of HR-TEM images. | ||
 | ||
| Fig. 4 High resolution scanning transmission electron microscopy bright field (HRSTEM-BF) image of (a) 22 nm β-NaEuF4/NaGdF4 core–shell NPs with an 18 nm β-NaEuF4 core and 2 nm β-NaGdF4 shell and (b) the corresponding high-angle annular dark field (HAADF) high resolution scanning transmission electron microscope (HRSTEM) image. And (c) scanning transmission electron microscope-electron energy loss spectroscopy (STEM-EELS) survey image and colour map of Eu (green) and Gd (red). | ||
The HAADF HRSTEM image of the 22 nm β-NaEuF4/NaGdF4 core–shell NPs with an 18 nm β-NaEuF4 core and 2 nm β-NaGdF4 shell in Fig. 4(b) does not show significant variation within the nanoparticle.
The STEM-EELS mapping of the 22 nm β-NaEuF4/NaGdF4 core–shell NPs with an 18 nm β-NaEuF4 core and 2 nm β-NaGdF4 shell (Fig. 4(c)) shows according to the colour map (inset) evidence of a core shell structure with Eu (green) in the center and Gd (red) on the surface.
2.3 Luminescence
The optical properties of the β-NaEuF4 core and β-NaEuF4/NaGdF4 core–shell nanocrystals were studied by photoluminescence spectroscopy. As expected, excitation at the 7F0,1 → 5L6 transition of Eu3+ at ∼394 nm results in Eu3+ emission for all nanoparticles containing Eu3+ ions (Fig. 5). In all cases the highest emission intensities are recorded at ∼591, 614 and 696 nm corresponding to the 5D0 → 7F1, 5D0 → 7F2 and 5D0 → 7F4 intra-4f shell transitions of Eu3+ respectively.54 These transitions are characteristic for luminescent materials containing Eu3+ ions and are, for instance, also observed for the β-NaGdF4 nanocrystals doped with 1 mol% of Eu3+. Fig. 5 shows, however, that the emission spectrum of such weakly doped particles displays additional lines between 450 nm and 580 nm, caused by transitions from the higher excited 5D3, 5D2 and 5D1 levels of Eu3+ to its 7FJ ground state multiplet. These emission lines are very weak in materials containing Eu3+ in high concentrations, since cross-relaxation between adjacent Eu3+ ions is known to quench the luminescence from these states. The 5D3, 5D2 and 5D1 emission lines are therefore characteristic for Eu3+ ions having no Eu3+ neighbors in the crystal lattice and display negligible intensity, for instance, in the case of β-NaEuF4 core particles (Fig. 5).55,56Fig. 5 shows, however, that after the formation of the NaGdF4 shell, the 5D3, 5D2 and 5D1 emission lines appear in the spectrum of the core–shell particles. This indicates that some Eu3+ ions are released from the β-NaEuF4 core and incorporate into NaGdF4 during formation of the shell. The figure also shows that the transitions from the higher 5D states are much less intense for core–shell particles with 18 nm cores indicating that the number of Eu3+ ions released decreases with increasing core size. This observation is in accord with the lower surface-to-bulk ratio of 18 nm core particles compared to 3 nm core particles, since the release of Eu3+ ions should be proportional to the total surface area of all core particles. To further substantiate this result, we have investigated the core–shell particles containing Gd3+ by EPR spectroscopy. | ||
| Fig. 5 Normalized emission spectra of 3 nm β-NaEuF4, 20 nm β-NaEuF4/NaGdF4 NPs with a 3 nm core, 22 β-NaGdF4 NP doped with 1% Eu, 18 nm β-NaEuF4 and 22 nm β-NaEuF4/NaGdF4 NPs with an 18 nm β-NaEuF4 core. The spectra were recorded under 394 nm excitation of the Eu3+ ions (7F0,1 → 5L6). All particles consist of hexagonal phase (β-phase). | ||
2.4 Electron paramagnetic resonance spectroscopy
In general, the EPR spectrum of Gd3+ ions (S = 7/2) is characterized by the Zeeman interaction with a g-value ≈2 and exhibits significant contributions from zero-field splitting. The mean distance between two adjacent Gd3+ ions in the crystal lattice is reflected in the broadness of the EPR spectrum that increases with increasing Gd concentration due to dipolar and isotropic exchange interactions between the ions, as observed in Fig. 6 for 3 nm β-NaGdF4 particles (green spectrum).57 A similar broadening due to Gd3+–Gd3+ interactions has been observed for NaYF4/NaGdF4 core–shell nanocrystals, even if the shell of NaGdF4 is as thin as 1 nm.58 In the study presented here we investigated core–shell particles with significantly thicker shells, consisting of a small 3 nm β-NaEuF4 core and an ∼8.5 nm thick β-NaGdF4 shell. Since the EPR signal of such particles would be fully dominated by the broad spectrum of the undiluted Gd3+ ions in the shell, we used core–shell nanocrystals with inverted composition in the EPR measurements, i.e., 3 nm β-NaGdF4 core particles with an ∼8.5 nm β-NaEuF4 shell. In Fig. 6, the EPR spectrum of these core–shell particles (red line) is compared with the EPR spectrum of β-NaEuF4 nanocrystals doped with 1% Gd3+ (blue line) and the EPR spectrum of 3 nm β-NaGdF4 core particles (green line). Note that all spectra are normalized at maximum signal height as indicated by the scaling factors given in the figure. The highest scaling factor was used for the NaGdF4 core particles as the strong interaction of the Gd3+ ions in these particles leads to strong broadening of their EPR signal (green line). The lowest scaling factor was used in the spectrum of the weakly doped β-NaEuF4:1% Gd particles. This spectrum exhibits a dominating narrow EPR signal (peak-to-peak width, ΔBpp ≈ 10 mT) in the g = 2 region and additional contributions at higher g-values (lower magnetic fields), the latter indicating the presence of Gd3+ ions at sites with different symmetries and/or crystal fields compared to the bulk sites. Since we used rather large NaEuF4:1% Gd particles with a diameter of 22 nm, the contribution from surface Gd3+ sites, however, is almost negligible in this case. The additional contributions therefore indicate that the Gd3+ dopant ions are not homogeneously distributed inside the particles as observed before.58 Finally, the high scaling factor used for the β-NaGdF4/NaEuF4 core–shell nanocrystals shows that their EPR signal is much weaker than the signal of the doped β-NaEuF4:1% Gd particles, although both samples contain europium and gadolinium in the same molar ratio of 99 to 1. The much lower intensity is in accord with the core shell structure, since the signal of the Gd3+ ions in the core is expected to be strongly broadened. Similar to the doped β-NaEuF4:1% Gd particles, however, the normalized EPR spectrum of the core–shell particles also displays a narrow signal at g ≈ 2.0 characteristic for isolated or weakly interacting Gd3+ ions. Since this signal is even narrower (ΔBpp ≈ 3 mT) and of higher relative intensity compared to the doped β-NaEuF4:1% Gd particles, it indicates the presence of highly diluted (≪1%) Gd3+ ions in the β-NaEuF4 shell. Komban et al. concluded from the EPR data for NaYF4:Gd core–shell particles that the shell structure grows from dissolved precursor particles and that most likely also Y3+ ions are released from the surface of the core-precursors during the reaction.58 The EPR data for the β-NaGdF4:Eu core–shell particles shown in Fig. 6 indicates that these nanocrystals are formed in a similar way and therefore support the conclusions drawn above from luminescence spectroscopy. | ||
| Fig. 6 Normalized EPR spectra of 20 nm core–shell particles composed of a 3 nm β-NaGdF4 core and an ∼8.5 nm β-NaEuF4 shell (red), 22 nm β-NaEuF4 NPs doped with 1% Gd (blue) and 3 nm β-NaGdF4 precursor particles (green). Due to the large width of the EPR spectrum of the precursor particles it has a significantly lower intensity compared to the other spectra and consequently displays more noise when the spectra are normalized to the same signal height (scaling factors: blue, ×1; red, ×10; green, ×11). The asterisk denotes an impurity signal of the microwave cavity present in all spectra at 175 mT. | ||
2.5 Chemical properties, core level XPS
We employed core level XPS to investigate the chemical states of the Eu and Gd ions of the nanoparticles and core–shell systems in question. Fig. 7(a) displays the Gd 3d X-ray photoelectron spectra of the core–shell and pure β-NaGdF4 (3 nm diameter) nanoparticles. The corresponding spectrum of a GdF3 single crystal52 is also shown for comparison. For all samples the Gd 3d5/2 and 3d3/2 core level binding energies are located at 1187.5 eV and 1219.5 eV, respectively. All the spectra show no distinct satellite features between the 3d5/2 and 3d3/2 main peaks indicating the absence of any metallic Gd contributions.59 | ||
| Fig. 7 Normalized XPS spectra of: (a) Gd 3d-core level of β-NaEuF4/NaGdF4 core–shell nanoparticles with 18 nm and 3 nm β-NaEuF4 cores, 3 nm pure β-NaGdF4 nanoparticles, and a GdF3 single crystal52 as a reference. (b) Eu 4d-core level of pure 3 nm β-NaEuF4 nanoparticles, de-convoluted into Eu2+ and Eu3+ fractions. (c) Eu 4d-core level of pure 18 nm β-NaEuF4 nanoparticles, de-convoluted into Eu2+ and Eu3+ fractions. (d) Eu 4d-core level of β-NaEuF4/NaGdF4 core–shell nanoparticles with an 18 nm β-NaEuF4 core. Eu and Gd spectra are partly overlapping, and are de-convoluted into Eu2+, Eu3+ and Gd3+ contributions. | ||
Fig. 7(b)–(d) show the Eu 4d (and Gd 4d (Fig. 7(d))) spectra of pure β-NaEuF4 (3 nm and 18 nm (Fig. 7(b) and (c))) and the β-NaEuF4/NaGdF4 core–shell nanoparticles with an 18 nm β-NaEuF4 core (Fig. 7(d)). The Eu 4d XPS of the 3 nm β-NaEuF4 nanoparticles comprises three distinct features located at 129.9 eV (A), 137.4 eV (B), and 143.4 eV (C). Features B and C can be attributed to the Eu3+ 4d5/2 and Eu3+ 4d3/2 states, whereas the lower binding energy peak A represents the Eu2+ 4d5/2 states,52 whereas the Eu2+ 4d3/2 states are overlapping with the Eu3+ 4d5/2 states and not visible as distinct features in the corresponding Eu 4d spectra. We de-convolute these spectra using Gaussian/Lorentzian line profiles. In the case of the β-NaEuF4/NaGdF4 core–shell nanoparticles (18 nm β-NaEuF4 core) this de-convolution approach is extended to the Gd3+ 4d5/2 and Gd3+ 4d3/2 states, assuming a pure trivalent Gd valence state in accordance with the results presented in Fig. 7(a). With the fitting approach described we are able to determine the Eu cation distribution of the nanoparticles investigated here. One can already see by the naked eye that the 3 nm β-NaEuF4 nanoparticles comprise the highest amount of Eu2+ ions, we find the Eu valence state in this sample to be composed out of 30% Eu2+ and 70% Eu3+ ions. The Eu2+ fraction is significantly reduced for the other two samples, we find fractions of 20% Eu2+ for the 18 nm β-NaEuF4 nanoparticles and 18% Eu2+ for the β-NaEuF4/NaGdF4 core–shell nanoparticles, respectively. We assume that this result can be likely due to the different surface to bulk ratios of the samples as the formation of divalent Eu should predominantly occur at the surface (interface) of the sample, since a similar effect has been reported by Burian et al. on different EuF3 surfaces and interfaces.52
2.6 Electronic and magnetic properties
In order to elucidate further the magnetic properties all over the nanocrystals superconducting quantum interference device (SQUID) magnetometry measurements were performed.Fig. 8(a) and (b) show thermomagnetic curves M(T) of the precursor particles of pure β-NaGdF4 and β-NaEuF4 respectively. In the case of β-NaGdF4 particles, the M(T) dependences fulfill the Curie law but with small deviation that is clearly demonstrated in the M × T(T) curves. For pure Curie law (i.e. M ∼ 1/T), the M × T(T) dependence should be a flat line, while the observed linear deviation indicates temperature independent component of magnetization which, in our case, can be attributed to some blocked magnetic moments of Gd. In the case of β-NaEuF4 particles, the thermomagnetic curves are typical as for Eu3+ ions, showing the so-called van Vleck paramagnetism which is relatively weak and temperature independent. At low temperatures, below 20 K, traces of classical paramagnetic behavior are observed, i.e. sharp increase in magnetization with decreasing temperature. This effect is caused by an expected contribution of Eu2+ ions that pose magnetic moment and follow the Curie law. The possible origin of the Eu2+ presence is a surface valence transition, typical for nanoparticles containing Eu.
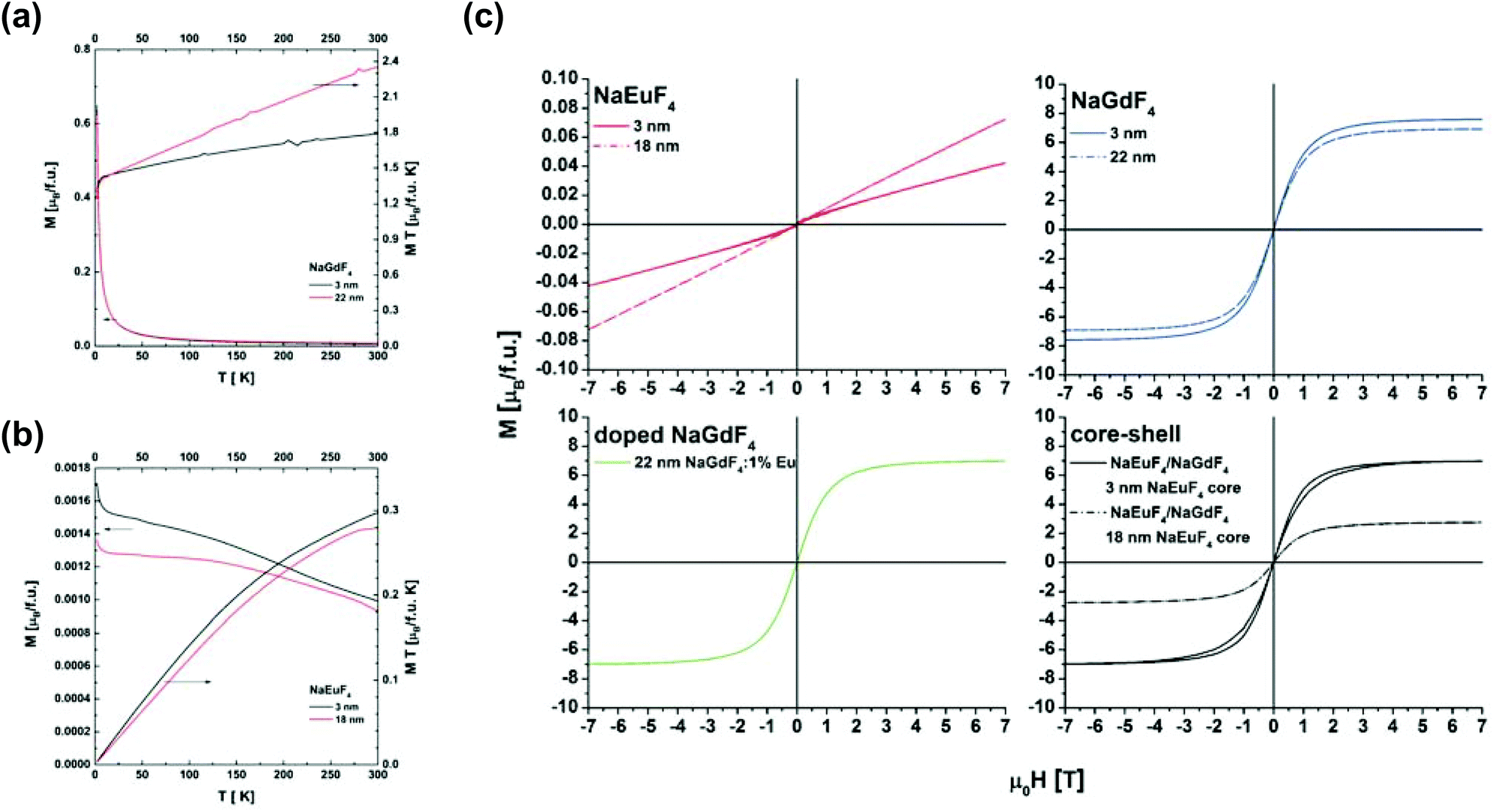 | ||
| Fig. 8 Thermomagnetic M(T) curves measured in μ0H = 0.1 T of (a) β-NaGdF4 and (b) β-NaEuF4 particles and (c) hysteresis loops of pure β-NaEuF4 and β-NaGdF4 precursor particles, 22 nm β-NaGdF4 nanocrystals doped with 1% Eu and β-NaEuF4/NaGdF4 core–shell particles at 2 K. | ||
Fig. 8(c) shows hysteresis loops, determined at 2 K, for (i) pure β-NaEuF4 and β-NaGdF4 precursor particles, (ii) 22 nm β-NaGdF4 nanocrystals doped with 1% of Eu and (iii) β-NaEuF4/NaGdF4 core–shell particles. Generally, the β-NaEuF4 samples (3 nm and 18 nm) are paramagnetic. However, for the β-NaEuF4 3 nm nanocrystals some saturated component is detected which confirms the presence of magnetic Eu2+ ions. This answers the expectation of Eu2+ formation on the surface, consequently due to the large bulk to surface ratio in the 18 nm nanocrystals no signal is detectable. The pure β-NaGdF4 precursor particles, β-NaEuF4/NaGdF4 core–shell particles with the 18 nm β-NaEuF4 core and 22 nm β-NaGdF4 nanocrystals doped with 1% Eu are paramagnetic with saturation magnetization as expected from the literature.60 Surprisingly the β-NaEuF4/NaGdF4 core–shell nanocrystals with the 3 nm core show a butterfly shaped hysteresis loop. A similar behavior was recently found in rare earth based single molecular magnets.61 The explanation of the hysteresis loop for the β-NaEuF4/NaGdF4 core–shell particles with the 3 nm β-NaEuF4 core can be some magnetic anisotropy. Accounting the fact that for 22 nm β-NaGdF4 and 3 nm β-NaGdF4 samples the effect of hysteresis broadening was not observed one may propose a model in which the anisotropy exists at the interface between a magnetic shell and a non-magnetic core.
In order to confirm this hypothesis Monte Carlo simulations were performed. The simulations are based on the Heisenberg model and the so-called simulated annealing, as described by Ziółkowski and Chrobak.62 The analyzed particle consists of a spherical magnetic shell (7 nm in diameter) and a nonmagnetic core (3 nm). In the magnetic net, spin S = 3.5, exchange integral parameter J = 0 and radial magnetic anisotropy coefficient K = kBT. Fig. 9 depicts the spin configurations for different applied magnetic fields from 0 T to 10 T.
 | ||
| Fig. 9 Spin configurations (central section) for the simulated particles in the fields of 0 T, 1 T, 2 T, 6 T and 10 T (scales in nm) and the simulated hysteresis loops (the first quadrant) for the particle. | ||
The resulting average spin in the direction of magnetic field (i.e. magnetization of the particle) as a function of increasing and decreasing field is presented in Fig. 9. As shown, this part of simulated hysteresis loop also reveals the butterfly shape. It should be underlined that good qualitative agreement with experimental data could be obtained only with the assumption of the surface anisotropy. The performed simulations including an influence of dipolar interactions, magnetic frustrations, weak ferro and antiferro couplings do not bring satisfactory results. Finally, one can conclude that the magnetization processes of β-NaGdF4 nanoparticles can be affected by anisotropy that leads to the appearing of butterfly shaped hysteresis loops and deviation from the Curie law (see Fig. 8(a)).
In order to tackle the internal magnetic structure of selected β-NaEuF4 and the β-NaEuF4/NaGdF4 core–shell nanocrystals with an 18 nm β-NaEuF4 core in detail we performed X-ray magnetic circular dichroism (XMCD) at the Gd and Eu M4,5-edges. XMCD is a very powerful tool to determine the magnetic properties of a compound in question valence and element specific.
Fig. 10 presents the Eu M4,5-edge X-ray absorption spectroscopy (XAS) and XMCD spectra of the β-NaEuF4/NaGdF4 core–shell nanocrystals with an 18 nm β-NaEuF4 core along with the Gd M5-edge of the 2 nm β-NaGdF4 shell. The Gd M5-edge exhibits a large dichroic signal (∼20%) of negative sign and a typical Gd3+ multiplet structure.63
 | ||
| Fig. 10 (a) Gd M5 and Eu M4,5-edge XAS and XMCD of the β-NaEuF4/NaGdF4 core–shell nanocrystals with an 18 nm β-NaEuF4 core. (b) Eu M4,5-edge XAS and XMCD of the β-NaEuF4/NaGdF4 core–shell nanocrystals with an 18 nm β-NaEuF4 core and the pure 3 nm β-NaEuF4 nanoparticles. (c) Eu M4,5-edge XMCD spectra in comparison with the corresponding atomic multiplet calculations of the 7F2 Eu3+ and the 8S7 Eu2+ final states. The simulated spectra have been extracted from Kachkanov et al.53 The experiments have been performed under an external magnetic field of B = 6 T and with the samples at a temperature of 6 K. Spectra are offset vertically for clarity. | ||
In contrast the Eu M4,5-edge XMCD shows a rather weak negative dichroism of ∼2% at the M5 edge. However, this measured dichroic signal is significantly stronger than expected according to the SQUID data of the pure β-NaEuF4 and β-NaGdF4 nanoparticles. Here the maximum recorded magnetization (see Fig. 8(c)) of the β-NaEuF4 nanocrystals is less than 1% compared to the β-NaGdF4 nanocrystals of same size. Since both, Eu and Gd M5 exhibit negative signs the net moments are parallelly aligned to each other. Fig. 10(b) displays the Eu M4,5-edge XA- and XMCD spectra of the core–shell nanoparticles with those of pure 3 nm β-NaEuF4 nanoparticles. The XPS spectra (see section 2.5) reveal an increased amount of divalent Eu ions which is also reflected in the X-ray absorption- and XMCD spectra of this sample. The M5 edge XMCD of the 3 nm β-NaEuF4 nanoparticles is dominated by two distinct features located at 1128 eV and 1131 eV, which can be attributed to Eu2+ and Eu3+ contributions to the XMCD signal.64 In the case of the core–shell nanocrystals the intensity of the XMCD at 1128 eV is much less pronounced, which is in accordance with the XPS results indicating a significantly lower Eu2+ fraction compared to the 3 nm β-NaEuF4 nanoparticles. The XMCD peak at 1132 eV can be presumed as being due to Eu3+ ions with a total magnetic quantum number J ≠ 0, since Eu3+ in its magnetic ground state with J = 0 would produce no dichroic signal at all. On the other hand, Eu2+ ions are in a J = 7/2 ground state, leading to a “spin only” moment of 7μB per Eu ion. This means that already quite a small fraction of uncompensated moments originating from Eu2+ ions may lead to a significant contribution to the overall Eu M4,5-edge XCMD signal. As we have already pointed out the overall dichroic Eu signal is much higher than expected from the SQUID magnetometry data. One potential reason is that the Eu3+-signal stems from ions in the 7F2 (J = 1) state. This is likely populated via the excitation of the probing X-ray beam during the XMCD measurement. Note that 4f intrashell transitions into the 7F2 also dominate the luminescence spectra presented in Fig. 5. A similar effect has been reported by Kachkanov et al.53 for Eu3+ doped GaN layers. It seems that β-NaEuF4 based nanocrystals and core–shell systems could be another example of a material in which the magnetic state can be altered by light excitation. In order to investigate this highly interesting effect in more detail future experiments by means of magneto optical SQUID and/or XMCD measurements in luminescence yield would be desirable.
3 Conclusions
We synthesized β-NaEuF4/NaGdF4 core–shell nanoparticles with narrow size distribution and an overall size of around 22 nm diameter. The particles contained either a small β-NaEuF4 core with a diameter of only ∼3 nm or a large β-NaEuF4 core with a diameter of ∼18 nm. Structural PDF analysis of the XRD measurements shows differences in peak positions between the both core–shell samples, although the differences in bond distance between the different atoms of bulk β-NaEuF4 and β-NaGdF4 are small. Comparison with simulation based on pure crystalline, spherical β-NaEuF4 and β-NaGdF4 nanoparticles point out that the β-NaEuF4/NaGdF4 core–shell nanoparticle with a 3 nm β-NaEuF4 core exhibits structural parameters of β-NaGdF4 structure, whereas β-NaEuF4/NaGdF4 core–shell nanoparticles with an 18 nm β-NaEuF4 core resemble the β-NaEuF4 structure. HR-TEM images reveal core–shell formation for the β-NaEuF4/NaGdF4 core–shell nanoparticles with an 18 nm β-NaEuF4 core. The Eu3+ photoluminescence and Gd3+ ESR spectra both indicate that rare earth ions of the small 3 nm particle cores were released during shell growth and incorporate into the shell material. In contrast the system with the large β-NaEuF4 core does not exhibit additional peaks in the Eu3+ luminescence spectra indicating that only a small number of Eu3+ ions are present in the NaGdF4 shell. As to the Eu and Gd valance states, X-ray photoelectron-core level spectroscopy reveals significant fractions of Eu2+ ions in the β-NaEuF4 precursors as well as the β-NaEuF4/NaGdF4 core–shell nanoparticles with an 18 nm β-NaEuF4 core. Larger β-NaEuF4 nanoparticles and surrounded by the β-NaGdF4 shell reduce the Eu2+ fraction in agreement with results reported for EuF3 thin film surfaces and interfaces.52 The presence of Eu2+ in the β-NaEuF4 nanoparticles as well as in the core–shell systems is confirmed by SQUID magnetometry. M(T) curves of the β-NaEuF4 precursors exhibit a sharp increase in magnetization below 20 K stemming from Eu2+ ions since only the Eu3+ ions contribute a weak van Vleck paramagnetism. In the case of the core–shell system with 3 nm β-NaEuF4 core we find a butterfly-shaped hysteresis loop which is known for single molecular magnets, for instance. This result could be reproduced by Heisenberg model simulations of a magnetic sphere comprising a non-magnetic core. XMCD at the Eu and M4,5 edges demonstrates the parallel alignment of Gd and Eu moments in the core–shell nanoparticles with a large β-NaEuF4 core. The magnetic properties are dictated by the paramagnetic Gd3+ ions in the surrounding β-NaGdF4 shell. However, both the Eu2+ and Eu3+ ions contribute to the overall magnetic properties of the system. In particular the Eu3+ contribution is much higher than expected from the weak van Vleck paramagnetism. This might be due to the fact that the exciting X-rays populate the J = 1 state of Eu3+via 4f transitions which can be also seen in the optical spectra. In future studies the core–shell formation with small β-NaEuF4 cores will be further optimized, also to investigate the interesting butterfly shaped magnetic hysteresis in more detail. The fact that the magnetic state of Eu3+ might be altered by external light excitation is another interesting aspect which may be investigated by SQUID experiments under optical light excitation or XMCD using luminescence yield as the detection mode. Finally the multifunctionality of the β-NaEuF4/NaGdF4 core–shell nanoparticles could be further improved, e.g. by transition metal doping into the β-NaEuF4 core and/or the β-NaGdF4 shell. In summary we have successfully synthesized and carefully characterized multifunctional β-NaEuF4/NaGdF4 core–shell nanoparticles combining intense red optical emission from the Eu3+ ions with the paramagnetic properties stemming mainly from the Gd3+ ions.4 Experimental
4.1 Materials
Sodium oleate (82%, Sigma-Aldrich), sodium fluoride (99%, Sigma-Aldrich), ammonium fluoride (98%, Sigma-Aldrich), oleic acid (90%, Alfa Aesar), 1-octadecene (90%, Alfa Aesar) and hydrated rare-earth chlorides of EuCl3 and GdCl3 (99.9%, Treibacher Industrie AG) were used as received.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 in oleic acid and 1-octadecene (10 ml of each solvent per 1 mmol of rare-earth oleate) followed by degassing at 100 °C for 1 h under vacuum (100 Pa) in a Schlenk-line. Subsequently, 4 mmol ammonium fluoride per 1 mmol of rare-earth oleate was added at 100 °C to the clear yellowish solution under nitrogen flow. The vessel was three times subjected to vacuum and then refilled with nitrogen (vacuum applied for 5 seconds only) and the stirred solution was subsequently heated at 200 °C for 60 min under slow nitrogen flow. After cooling to room temperature an equal volume of ethanol was added to the solution, resulting in precipitation of the precursor particles. The particles were separated by centrifugation and purified as given above for the cubic phase precursor nanocrystals.
:2 mixture of hexane and ethanol, the precipitate was separated from the solution by centrifugation. The precipitate was purified two times as given above for the precursor particles. The synthesis procedure for all NaREF4 product particles was the same.
:2.5 in oleic acid and 1-octadecene (10 ml of each solvent per 1 mmol of rare-earth oleate) followed by degassing at 100 °C for 1 h under vacuum (100 Pa) in a Schlenk-line. Subsequently, 4 mmol ammonium fluoride per 1 mmol of rare-earth oleate was added at 100 °C to the clear yellowish solution under nitrogen flow. The vessel was three times subjected to vacuum and then refilled with nitrogen (vacuum applied for 5 seconds only) and the stirred solution was subsequently heated at 200 °C for 60 min under slow nitrogen flow. After cooling to room temperature an equal volume of ethanol was added to the solution, resulting in precipitation of the precursor particles. The particles were separated by centrifugation and purified as given above for the cubic phase precursor nanocrystals.
:2 mixture of hexane and ethanol, the precipitate was separated from the solution by centrifugation. The precipitate was purified two times as given above for the precursor particles. The synthesis procedure for all NaREF4 product particles was the same.
4.2 Experimental methods
The total scattering X-ray measurements were performed on the high energy scattering beamline P02.1 at PETRA III (DESY, Hamburg, Germany). The data sets were collected using a high energy monochromatic beam (59.875 keV, λ = 0.20707 Å) and a Perkin Elmer flat panel detector placed at 303 mm from the samples. The wavelength and the sample to detector distance have been determined by using a LaB6 NIST standard. The used experimental setup allows to collect total scattering data over a sufficiently high momentum transfer (Q ∼ 22.72 Å−1) corresponding to a d spacing of 0.138 Å. We note here that the total scattering measurements have been conducted under the same experimental conditions for all the studied samples. The collected data were then corrected for experimental effects (absorption, multiple scattering, polarization, Compton scattering and Laue diffuse scattering) and the scattering signals from the air and the experimental set up were measured independently under exactly the same conditions as the samples and subtracted as a background in the data reduction procedure. For obtaining the experimental atomic pair distribution function G(r) by a direct sine Fourier transformation of the resulting total scattering structure function S(Q), the data were truncated at a finite maximum value of Qmax = 18.13 Å−1 beyond which the signal-to-noise ratio became unfavourable. All data processing was done using Fit-2D68 and PDFgetX369 software.A double Cs corrected JEOL ARM 200F transmission electron microscope operated at 200 kV was used for the analysis in the study. High angle annular dark field (HAADF) scanning transmission electron microscopy (STEM) was used along with electron energy loss spectroscopy (EELS) Spectrum Imaging (SI) using the DualEELS™ mode on the Gatan GIF Quantum 965 ERS™ spectrometer fitted on the microscope as well as EDS using an Oxford XMax 80 SDD.
Fluorescence spectra of the nanocrystals were recorded with a Fluorolog 3-22 spectrometer (Jobin Yvon) equipped with double monochromators for both channels.
The EPR spectra of the sample were recorded with a home made X-band EPR spectrometer (9 GHz) equipped with a dielectric resonator (DM5) (Bruker Biospin, Rheinstetten, Germany). The microwave power was set to 1.0 mW, the B-field modulation amplitude was 0.3 mT.
The XPS measurements were performed using a PHI 5600CI multitechnique spectrometer with monochromatic Al Kα = 1486.6 eV radiation of 0.3 eV at full width at half-maximum. The overall resolution of the spectrometer is 1.5% of the pass energy of the analyzer, 0.88 eV in the present case. The XPS measurements were performed at room temperature.
The XAS and XMCD measurements were performed at the Beamline for Advanced Dichroism (BACH) at the Elettra Synchrotron Facility.70 The samples were mounted into a cryostat equipped with a 6.5 T superconducting magnet, the sample stage was connected to a pumped helium cryostat, reaching a base temperature of around 6.0 K during the experiments presented here. The measurements at the Gd and Eu M4,5 edges have been recorded under external magnetic fields of 6 T in the total electron yield mode.
The FC–ZFC and hysteresis loop measurements were performed at the A. Chełkowski Institute of Physics, University of Silesia, Katowice, Poland, with the use of SQUID magnetometer (MPMS XL7 Quantum Design). All measurements have been performed in the 2 K–300 K temperature range up to 7 T magnetic field.
Acknowledgements
Parts of this research were carried out at the light source PETRA III at DESY, a member of the Helmholtz Association (HGF). We would like to thank J. Gamcová for assistance in using beamline P02.1. We thank all Beamline scientists for their excellent and generous technical support.References
- S. Gai, C. Li, P. Yang and J. Lin, Chem. Rev., 2014, 114, 2343–2389 CrossRef CAS.
- C. Liu, Y. Hou and M. Gao, Adv. Mater., 2014, 26, 6922–6932 CrossRef CAS PubMed.
- D. K. Chatterjee, M. K. Gnanasammandhan and Y. Zhang, Small, 2010, 6, 2781–2795 CrossRef CAS PubMed.
- F. Wang, D. Banerjee, Y. Liu, X. Chen and X. Liu, Analyst, 2010, 135, 1839–1854 RSC.
- D. Vennerberg and Z. Lin, Sci. Adv. Mater., 2011, 3, 26–40 CrossRef.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- Z. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765–4769 CrossRef CAS.
- G. Chen, T. Y. Ohulchanskyy, S. Liu, W.-C. Law, F. Wu, M. T. Swihart, H. Hans Ågren and P. N. Prasad, ACS Nano, 2012, 6, 2969–2977 CrossRef CAS PubMed.
- Q. Liu, Y. Sun, T. Yang, W. Feng, C. Li and F. Li, J. Am. Chem. Soc., 2011, 133, 17122–17125 CrossRef CAS PubMed.
- J. Liu, Y. Liu, W. Bu, J. Bu, Y. Sun, J. Du and J. Shi, J. Am. Chem. Soc., 2014, 136, 9701–9709 CrossRef CAS PubMed.
- Y. Sun, X. Zhu, J. Peng and F. Li, ACS Nano, 2013, 7, 11290–11300 CrossRef CAS PubMed.
- Y.-F. Wang, G.-Y. Liu, L.-D. Sun, J.-W. Xiao, J.-C. Zhou and C.-H. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef CAS PubMed.
- R. Naccache, P. Chevallier, J. Lagueux, Y. Gossuin, S. Laurent, L. Vander Elst, C. Chilian, J. A. Capobianco and M.-A. Fortin, Adv. Healthcare Mater., 2013, 2, 1478–1488 CrossRef CAS PubMed.
- R. Qiao, C. Liu, M. Liu, H. Hu, C. Liu, Y. Hou, K. Wu, Y. Lin, J. Liang and M. Gao, ACS Nano, 2015, 9, 2120–2129 CrossRef CAS PubMed.
- Y. Deng, H. Wang, W. Gu, S. Li, N. Xiao, C. Shao, Q. Xu and L. Ye, J. Mater. Chem. B, 2014, 2, 1521–1529 RSC.
- H. Groult, J. Ruiz-Cabello, J. Pellico, A. V. Lechuga-Vieco, R. Bhavesh, M. Zamai, E. Almarza, I. Martin-Padura, E. Cantelar, M. P. Martinez-Alcazar and F. Herranz, Bioconjugate Chem., 2015, 26, 153–160 CrossRef CAS PubMed.
- C. Liu, W. Ma, Z. Gao, J. Huang, Y. Hou, C. Xu, W. Yang and M. Gao, J. Mater. Chem. C, 2014, 2, 9637–9642 RSC.
- R. Wang, X. Li, L. Zhou and F. Zhang, Angew. Chem., Int. Ed., 2014, 126, 12282–12286 CrossRef.
- H. Xing, S. Zhang, W. Bu, X. Zheng, L. Wang, Q. Xiao, D. Ni, J. Zhang, L. Zhou, W. Peng, K. Zhao, Y. Hua and J. Shi, Adv. Mater., 2014, 26, 3867–3872 CrossRef CAS PubMed.
- Q. Zhan, J. Qian, H. Liang, G. Somesfalean, D. Wang, S. He, Z. Zhang and S. Andersson-Engels, ACS Nano, 2011, 5, 3744–3757 CrossRef CAS PubMed.
- D. Yang, P. Ma, Z. Hou, Z. Cheng, C. Li and J. Lin, Chem. Soc. Rev., 2015, 44, 1416–1448 RSC.
- Y. Dai, H. Xiao, J. Liu, Q. Yuan, P. Ma, D. Yang, C. Li, Z. Cheng, Z. Hou, P. Yang and J. Lin, J. Am. Chem. Soc., 2013, 135, 18920–18929 CrossRef CAS PubMed.
- Y. I. Park, H. M. Kim, J. H. Kim, K. C. Moon, B. Yoo, K. T. Lee, N. Lee, Y. Choi, W. Park, D. Ling, K. Na, W. K. Moon, S. H. Choi, H. S. Park, S.-Y. Yoon, Y. D. Suh, S. H. Lee and T. Hyeon, Adv. Mater., 2012, 24, 5755–5761 CrossRef CAS PubMed.
- G. Tian, Z. Gu, L. Zhou, W. Yin, X. Liu, L. Yan, S. Jin, W. Ren, G. Xing, S. Li and Y. Zhao, Adv. Mater., 2012, 24, 1226–1231 CrossRef CAS PubMed.
- D. Yang, X. Kang, P. Ma, Y. Dai, Z. Hou, Z. Cheng, C. Li and J. Lin, Biomaterials, 2013, 34, 1601–1612 CrossRef CAS PubMed.
- L. Wang, J. Liu, Y. Dai, Q. Yang, Y. Zhang, P. Yang, Z. Cheng, H. Lian, C. Li, Z. Hou, P. Ma and J. Lin, Langmuir, 2014, 30, 13042–13051 CrossRef CAS PubMed.
- R. Naccache, F. Vetrone and J. A. Capobianco, ChemSusChem, 2013, 6, 1308–1311 CrossRef CAS PubMed.
- H. Lian, Z. Hou, M. Shang, D. Geng, Y. Zhang and J. Lin, Energy, 2013, 57, 270–283 CrossRef CAS.
- J. Shen, Z. Li, R. Cheng, Q. Luo, Y. Luo, Y. Chen, X. Chen, Z. Sun and S. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 17454–17462 CAS.
- G.-B. Shan, H. Assaaoudi and G. P. Demopoulos, ACS Appl. Mater. Interfaces, 2011, 3, 3239–3243 CAS.
- H. Zhu, X. Chen, L. M. Jin, Q. J. Wang, F. Wang and S. F. Yu, ACS Nano, 2013, 7, 11420–11426 CrossRef CAS PubMed.
- Y. Zhou, S.-T. Han, X. Chen, F. Wang, Y.-B. Tang and V. Roy, Nat. Commun., 2014, 5, 1–8 CrossRef.
- D. Peng, Q. Ju, X. Chen, R. Ma, B. Chen, G. Bai, J. Hao, X. Qiao, X. Fan and F. Wang, Chem. Mater., 2015, 27, 3115–3120 CrossRef.
- W. J. Kim, M. Nyk and P. N. Prasad, Nanotechnology, 2009, 20, 185301 CrossRef PubMed.
- Y. Lu, J. Zhao, R. Zhang, Y. Liu, D. Liu, E. M. Goldys, X. Yang, P. Xi, A. Sunna, J. Lu, Y. Shi, R. C. Leif, Y. Huo, J. Shen, J. A. Piper, J. P. Robinson and D. Jin, Nat. Photonics, 2014, 8, 32–36 CrossRef CAS.
- J. M. Meruga, A. Baride, W. Cross, J. J. Kellar and P. S. May, J. Mater. Chem. C, 2014, 2, 2221–2227 RSC.
- G.-S. Yi and G.-M. Chow, Chem. Mater., 2007, 19, 341–343 CrossRef CAS.
- Y.-F. Wang, L.-D. Sun, J.-W. Xiao, W. Feng, J.-C. Zhou, J. Shen and C.-H. Yan, Chemistry, 2012, 18, 5558–5564 CrossRef CAS PubMed.
- D. Chen and P. Huang, Dalton Trans., 2014, 43, 11299–11304 RSC.
- G. Chen, H. Agren, T. Y. Ohulchanskyy and P. N. Prasad, Chem. Soc. Rev., 2015, 44, 1680–1713 RSC.
- M.-K. Tsang, G. Bai and J. Hao, Chem. Soc. Rev., 2015, 44, 1585–1607 RSC.
- F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924–2929 CrossRef CAS.
- Y. Liu, D. Tu, H. Zhu, R. Li, W. Luo and X. Chen, Adv. Mater., 2010, 22, 3266–3271 CrossRef CAS PubMed.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- M. Quintanilla, F. Ren, D. Ma and F. Vetrone, ACS Photonics, 2014, 1, 662–669 CrossRef CAS.
- C. Dong, A. Korinek, B. Blasiak, B. Tomanek and F. C. J. M. van Veggel, Chem. Mater., 2012, 24, 1297–1305 CrossRef CAS.
- Q. Su, S. Han, X. Xie, H. Zhu, H. Chen, C.-K. Chen, R.-S. Liu, X. Chen, F. Wang and X. Liu, J. Am. Chem. Soc., 2012, 134, 20849–20857 CrossRef CAS PubMed.
- H. Wen, H. Zhu, X. Chen, T. F. Hung, B. Wang, G. Zhu, S. F. Yu and F. Wang, Angew. Chem., Int. Ed., 2013, 125, 13661–13665 CrossRef.
- X. Li, R. Wang, F. Zhang and D. Zhao, Nano Lett., 2014, 14, 3634–3639 CrossRef CAS PubMed.
- M. Bottrill, L. Kwok and N. J. Long, Chem. Soc. Rev., 2006, 35, 557–571 RSC.
- E.-J. Cho and S.-J. Oh, Phys. Rev. B: Condens. Matter, 1999, 59, R15613–R15616 CrossRef CAS.
- W. Burian, J. Szade, T. O'Keevan and Z. Celinski, Phys. Status Solidi B, 2004, 241, R15–R18 CrossRef CAS.
- V. Kachkanov, M. J. Wallace, G. van der Laan, S. S. Dhesi, S. A. Cavill, Y. Fujiwara and K. P. O'Donnell, Sci. Rep., 2012, 2, 1–5 Search PubMed.
- G. H. Dieke, Spectra and energy levels of rare-earth ions in crystals, Interscience Publishers, New York, 1968 Search PubMed.
- P. Ptacek, H. Schaefer, K. Koempe and M. Haase, Adv. Funct. Mater., 2007, 17, 3843–3848 CrossRef CAS.
- C. Li, Z. Quan, J. Yang, P. Yang and J. Lin, Inorg. Chem., 2007, 46, 6329–6337 CrossRef CAS PubMed.
- C. M. Brodbeck and L. E. Iton, J. Chem. Phys., 1985, 83, 4285–4299 CrossRef CAS.
- R. Komban, J. P. Klare, B. Voss, J. Nordmann, H.-J. Steinhoff and M. Haase, Angew. Chem., Int. Ed., 2012, 51, 6506–6510 CrossRef CAS PubMed.
- J. Szade and M. Neumann, J. Phys.: Condens. Matter, 2001, 13, 2717 CrossRef CAS.
- F. Li, C. Li, X. Liu, Y. Chen, T. Bai, L. Wang, Z. Shi and S. Feng, Chem. – Eur. J., 2012, 18, 11641–11646 CrossRef CAS PubMed.
- L. Margheriti, D. Chiappe, M. Mannini, P.-E. Car, P. Sainctavit, M.-A. Arrio, F. B. de Mongeot, J. C. Cezar, F. M. Piras, A. Magnani, E. Otero, A. Caneschi and R. Sessoli, Adv. Mater., 2010, 22, 5488–5493 CrossRef CAS PubMed.
- G. Ziółkowski and A. Chrobak, Acta Phys. Pol., A, 2015, 127, 597–598 CrossRef.
- G. Champion, N. Lalioti, V. Tangoulis, M.-A. Arrio, P. Sainctavit, F. Villain, A. Caneschi, D. Gatteschi, C. Giorgetti, F. Baudelet, M. Verdaguer and C. Cartier dit Moulin, J. Am. Chem. Soc., 2003, 125, 8371–8376 CrossRef CAS PubMed.
- B. J. Ruck, H. J. Trodahl, J. H. Richter, J. C. Cezar, F. Wilhelm, A. Rogalev, V. N. Antonov, B. D. Le and C. Meyer, Phys. Rev. B: Condens. Matter, 2011, 83, 174404 CrossRef.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- C. Liu, H. Wang, X. Zhang and D. Chen, J. Mater. Chem., 2009, 19, 489–496 RSC.
- B. Voss and M. Haase, ACS Nano, 2013, 7, 11242–11254 CrossRef CAS PubMed.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Hausermann, High Pressure Res., 1996, 14, 235–248 CrossRef.
- P. Juhas, T. Davis, C. Farrow and S. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef CAS.
- M. Zangrando, M. Finazzi, G. Paolucci, G. Comelli, B. Diviacco, R. P. Walker, D. Cocco and F. Parmigiani, Rev. Sci. Instrum., 2001, 72, 1313–1319 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C5NR06915G |
| This journal is © The Royal Society of Chemistry 2016 |