
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Applicability of avidin protein coated mesoporous silica nanoparticles as drug carriers in the lung†
S. H.
van Rijt‡
*a,
D. A.
Bölükbas‡
a,
C.
Argyo
b,
K.
Wipplinger‡
a,
M.
Naureen‡
a,
S.
Datz
b,
O.
Eickelberg‡
a,
S.
Meiners‡
a,
T.
Bein
*b,
O.
Schmid‡
a and
T.
Stoeger‡
a
aComprehensive Pneumology Center (CPC), University Hospital, Ludwig-Maximilians University and Helmholtz Zentrum München, Munich, Germany. E-mail: sabine.vanrijt@helmholtz-muenchen.de
bDepartment of Chemistry and Center for NanoScience (CeNS), University of Munich (LMU), Butenandtstrasse 5-13(E), 81377 Munich, Germany. E-mail: bein@lmu.de
First published on 10th March 2016
Abstract
Mesoporous silica nanoparticles (MSNs) exhibit unique drug delivery properties and are thus considered as promising candidates for next generation nano-medicines. In particular, inhalation into the lungs represents a direct, non-invasive delivery route for treating lung disease. To assess MSN biocompatibility in the lung, we investigated the bioresponse of avidin-coated MSNs (MSN-AVI), as well as aminated (uncoated) MSNs, after direct application into the lungs of mice. We quantified MSN distribution, clearance rate, cell-specific uptake, and inflammatory responses to MSNs within one week after instillation. We show that amine-functionalized (MSN-NH2) particles are not taken up by lung epithelial cells, but induced a prolonged inflammatory response in the lung and macrophage cell death. In contrast, MSN-AVI co-localized with alveolar epithelial type 1 and type 2 cells in the lung in the absence of sustained inflammatory responses or cell death, and showed preferential epithelial cell uptake in in vitro co-cultures. Further, MSN-AVI particles demonstrated uniform particle distribution in mouse lungs and slow clearance rates. Thus, we provide evidence that avidin functionalized MSNs (MSN-AVI) have the potential to serve as versatile biocompatible drug carriers for lung-specific drug delivery.
Introduction
In the past decades, the use of nanoparticles as inert carriers for therapeutic agents has revolutionized the field of drug delivery research. Several nanoscale drug delivery systems, especially non-functionalized particle formulations, have been approved by the FDA and European Medicines Agency for treatment of cancer.1 Nanocarriers offer major improvements such as increased bio-availability of the incorporated agent, site specificity, and the ability to overcome multi-drug resistance.2,3 Particularly, mesoporous silica nanoparticles (MSNs) are novel drug carriers with unique properties, such as high loading capacity with tunable pore sizes and volumes for transport of a wide variety of cargo molecules.4 Importantly, these particles can be selectively functionalized at specific sites within their structure. For example, the particle core offers a site for covalent attachment of fluorescent dye molecules for particle tracking in biological studies.5 In addition, the surface of the MSNs can be selectively modified to introduce controlled drug release functions for optimized drug delivery.4 For example, in our recent work we developed mesoporous silica nanoparticles with protease-responsive avidin caps for controllable drug release in lung tumour areas.6 Due to the tight sealing of the mesopores by the avidin caps and the selective cleavage of these caps at high protease concentrations found in lung tumours, these drug carriers were able to efficiently release a combination of chemotherapeutic drugs in vitro and ex vivo (mouse and human) with high tumour-selectivity. Other interesting examples of functionalized MSNs that have shown in vivo effectiveness include MSNs functionalized with PEG chains,7 folic acid,8 or transferrin.9 Due to their unique properties, MSNs have been considered as promising candidates for next generation nano-medicines.10 However, before these carriers can be used in the clinic, their biocompatibility needs to be proven. Although MSNs are generally considered to be biocompatible, several reports suggest that their bioresponse is strongly affected by their size,11 shape,12 porosity,13 and surface chemistry.11,14 In addition, the administration route has been found to play an important role for their biodistribution and bioresponse.15 Encouragingly, in vivo toxicity studies on different types of MSNs administered using several application routes, indicate that the use of these particles is safe for drug delivery purposes,16–20 however, not many of these studies deal with the applicability of MSNs directly in the lung.Direct application of nanoparticles into the lung (i.e. inhalation therapy) would be beneficial for treatment of (chronic) lung diseases such as idiopathic pulmonary fibrosis (IPF), chronic obstructive pulmonary disease (COPD) and asthma, as drugs are directly administered in the target organ. This is advantageous because high local doses at the site of disease (lung) can be accomplished allowing for high efficacy combined with low prevalence of side effects. Moreover, in contrast to oral administered delivery, inhaled drugs bypass the gastrointestinal tract and the liver, avoiding problems associated with drug degradation in these organs and/or stability in blood circulation. Moreover, pulmonary application of drugs presents a non-invasive route for systemic delivery of drugs, since the huge surface area of the alveolar lung epithelium (ca. 100 m2) presents an effective portal of entry into the blood stream. Many drug delivery materials such as polymers are commonly accepted as biocompatible for systemic applications, but some have shown to develop adverse cytotoxic and pro-inflammatory properties in the lungs and seem therefore not suited for direct application into the lung.21 Moreover, the inflammatory potential of nanoparticles is especially relevant for the treatment of inflammatory lung diseases such as COPD and asthma, as the additional inflammation caused by drug delivery particles could lead to worsening of the symptoms.
Here, we investigated the suitability of direct application of avidin-capped MSNs in the lung, namely their distribution, clearance rate, cell specific uptake, and the inflammatory response induced over the course of one-week time. For that purpose, single high doses (20 or 100 μg per mouse) of avidin-capped MSNs (MSN-AVI) were instilled in adult BALB/c mice, and their bioresponse after 1, 3, and 7 days was studied. To analyse the impact of the avidin protein coating, we also included non-protein coated MSNs containing propyl amines on the outer shell (MSN-NH2) in the study. Labelling of the particle core with a fluorescent dye (ATTO 633) allowed for whole lung dosimetry as well as cell specific particle tracking in lung cryo-sections and cytospins of bronchoalveolar lavage fluid (BALF) recovered cells. Moreover, supporting studies using in vitro cell cultures were performed. This provided evidence that MSNs capped with avidin are significantly more biocompatible than amino functionalized MSNs as proven by analyses of their inflammation and toxicity profiles, biodistribution, and cell specific internalization rates, and thus hold potential for use in future lung disease therapy.
Materials and methods
Materials
Core–shell functionalized MSNs containing thiol groups in the core and propyl amines on the shell (MSN-SHinNH2,out), avidin coated MSNs (MSN-AVI) and outer shell non-functionalised MSN particles (nonMSN) were synthesized as previously reported.5,6 Characterisation of the MSN-SHinNH2,out, and MSN-AVI can be found in Fig. S1.† Details on the synthesis and characterization of non-functionalised MSNs can be found in Fig. S2.†The MSNs suspended in bi-distilled water feature BET surface areas of 1150 m2 g−1 (MSN-NH2) and 90 m2 g−1 (MSN-AVI), zeta-potentials of −30 mV (MSN-NH2) and +30 mV (MSN-AVI) at pH 7.4, and diameter of 106 ± 9 nm (MSN-NH2) and 164 ± 15 nm (MSN-AVI). The cores of the MSNs were covalently labelled with ATTO 633 dye and the outer surface contained either NH2 groups (MSN-NH2) or avidin protein (MSN-AVI) covalently attached to the MSNs through a peptide linker. Further details on the synthesis and characterization of the MSNs can be found in the ESI.†
Cleaved caspase-3 antibody (Asp175) (Cell signaling, 9661), T1α antibody (R&D, AF3244), Pro-SPC antibody (Millipore, AB3786), MAC3 antibody (BD biosciences, BD 550292), were used as received. Bi-distilled water was obtained from a Millipore system (Milli-Q Academic A10). The mouse cell lines, MH-S (murine alveolar macrophages), MLE-12 (murine lung epithelial cells), and CCL-206 (murine lung fibroblasts) were obtained from ATCC (American Type Culture Collection, Manassas, USA). The MLE-12 cell line was maintained in RPMI 1640 medium (Gibco, Life Technologies); the MH-S cell line was maintained in RPMI 1640 medium supplemented with 1 mM Na-pyruvate, 10 mM HEPES, and 50 μM 2-ME (all AppliChem). The CCL-206 cell line was maintained in DMEM-F12 medium (Gibco, Life Technologies). All media were supplemented with 10% FBS (Biochrom) and 1% penicillin/streptomycin (Life Technologies). All cells were grown at 37 °C in a sterile humidified atmosphere containing 5% CO2.
Study design
The nanoparticles were instilled into Balb/c mice (20 or 100 μg) and after 1, 3, or 7 days the lungs were excised (n = 10 per group). Characterisation of the MSN dispersion over time was performed to assess the agglomeration of the nanoparticles in cell culture medium (RPMI supplemented with FCS). MSN-NH2 particles of 100 nm primary size agglomerated after 1 hour to form microparticles of about 1 μm, while MSN-AVI particles agglomerated to a much lesser extent (Fig. S3A†). It is important to note that the particles were instilled into the mouse lungs as a homogeneous mixture by vortexing before application (i.e. minimal agglomeration had taken place). Four lungs were directly prepared for cryo-slicing. An additional six mice were used for bronchoalveolar lavage (BAL); BAL fluid (BALF) was collected and separated into cells and supernatant by centrifugation. In addition, BAL recovered cells on cytospins were prepared for all mice for differential cell count. Furthermore, the lavaged lungs and the BAL were used for dosimetry analyses (see Fig. S3B† for a scheme of the experimental set-up).Methods
The dosimetric method presented here was validated using reference mice with a known pulmonary MSN dose as previously described.26 In brief, these reference mice received 100 μL of the MSN-AVI suspension via intratracheal instillation and were sacrificed immediately after the procedure to avoid dose bias due to partial clearance of the applied MSN-AVI particles from the lung. The actually instilled volume of the MSN-AVI suspension was determined for each mouse by gravimetric analysis of the instillation wear prior and after instillation.26 The spectrophotometrically measured amount of MSN-AVI agreed with the applied amount of MSN-AVI within the expected experimental uncertainty of 15%. Finally, 7 d-incubation of MSN-AVI particles in the supernatant of the BAL and subsequent centrifugation revealed that no detectable amount of the fluorescent tracer (ATTO 633) was leaking out of the MSN-AVI. This is a prerequisite for reliable pulmonary dosimetry based on fluorescence analysis.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and HBBS medium was removed, the pellet was resuspended in 500 μL mouse BAL. 500 μL of each MSN particle type in HBBS 1 mg mL−1 was used as control. The samples were incubated at 37 °C for 16 hours while gently shaking. After the incubation period, the samples were vortexed briefly followed by centrifugation at 18000 rpm for 40 min at 15 °C. The supernatant was removed and the remaining pellets were washed with PBS (centrifugation at 18000 rpm for 40 min at 15 °C after each wash). Finally, the pellet was dissolved in 50 μL of PBS and run on a 10% native gel for 90 minutes at 100 V. The native gel was stained with page blue™ and the whole gel was imaged using a ChemiDoc imaging system (Bio-rad).
000 rpm and HBBS medium was removed, the pellet was resuspended in 500 μL mouse BAL. 500 μL of each MSN particle type in HBBS 1 mg mL−1 was used as control. The samples were incubated at 37 °C for 16 hours while gently shaking. After the incubation period, the samples were vortexed briefly followed by centrifugation at 18000 rpm for 40 min at 15 °C. The supernatant was removed and the remaining pellets were washed with PBS (centrifugation at 18000 rpm for 40 min at 15 °C after each wash). Finally, the pellet was dissolved in 50 μL of PBS and run on a 10% native gel for 90 minutes at 100 V. The native gel was stained with page blue™ and the whole gel was imaged using a ChemiDoc imaging system (Bio-rad).
Results and discussion
Pulmonary inflammation analyses
In vivo inflammatory cell recruitment into the airspace was first assessed by BAL differential cell count. Total BAL cell numbers were not significantly different from sham control (PBS) at day 1, but increased at day 3 for 100 μg of MSN-AVI and at day 7 for 100 μg of MSN-NH2 particles (Fig. 1A). These changes are mainly related to the increases in macrophage numbers, which in general represent the majority of BAL leukocytes (Fig. 1C). A particular severe inflammatory response was detected by the acute increase of polymorphonuclear neutrophils (PMNs): after 1 day of treatment with the high dose (100 μg) of MSN-NH2 particles giving rise to 60% of total BAL cells (Fig. 1B and Table 1). The same treatment with MSN-AVI particles also resulted in a distinct PMN influx into the airspace; however, comparatively less pronounced to what was observed for the MSN-NH2 particles (33% of total BAL cells, Fig. 1B). This almost 2 fold higher PMN influx into the alveolar airspace at day 1 after instillation for MSN-NH2 compared to MSN-AVI particles indicates an improved biocompatibility of the MSN surface due to avidin capping. In addition, for both particle types, a time-dependent resolution of the neutrophilic inflammation was noted, but only for the MSN-AVI particles inflammation had completely resolved after 7 days (Fig. 1B), further indicating improved biocompatibility for MSN-AVI.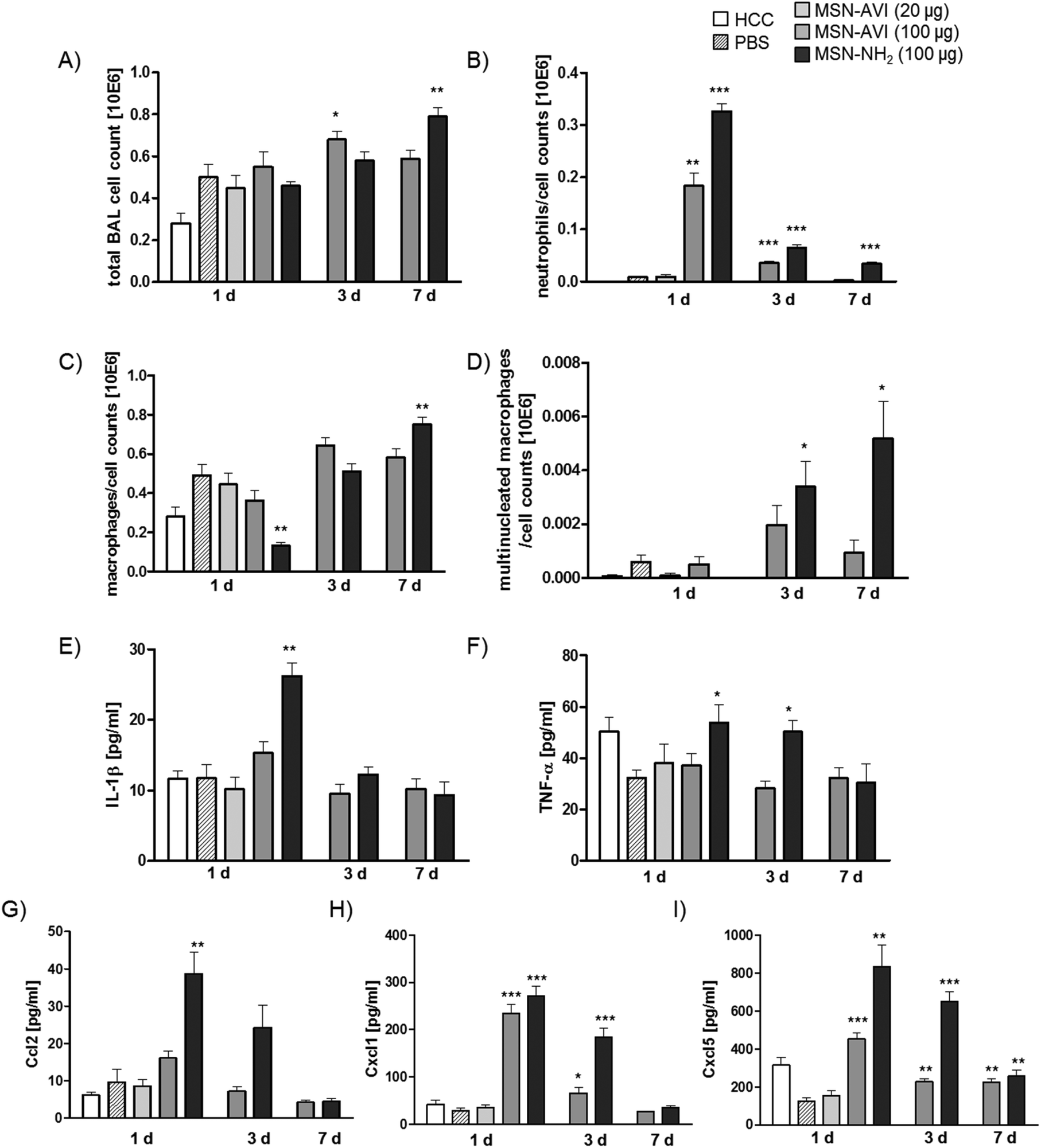 | ||
| Fig. 1 BAL cell analysis and cytokine release in BALF at three time points after MSN instillation into Balb/c mice. Bronchoalveolar lavage (BAL) cells were counted and dead cells were discriminated by Tryphan blue staining; differentiation was analyzed by May-Grünwald staining. Total BAL cell count is shown in A, total neutrophil numbers in B, macrophage numbers in C, and multinucleated macrophages in D. Release of cytokines was measured in bronchoalveolar lavage fluid (BALF) for each animal in each group, interleukin-1 beta (IL-1β) is shown in E, TNF-α is shown in F, monocyte chemotactic protein 1 (CCL2) is shown in G, Cxcl1 is shown in H, and Cxcl5 is shown in I. Values are indicated as mean ± SD, n = 6, asterisks represent significance compared to PBS control groups (100 μL PBS instilled animals) with ***p < 0.001, **p < 0.01, *p < 0.05. HCC = home cage control (non-treated animals). | ||
| Instilled amount (μg) | PMN cell count × 103 (%) | Macrophages cell count × 103 (%) | Multinucleated macrophages cell count × 103 (%) | Detected cytokines | ||
|---|---|---|---|---|---|---|
| a HCC = home cage control animals (non-treated mice). b PBS = 100 μL PBS instilled mice (vehicle/sham control). | ||||||
| 1 d | HCCa | — | 0.2 (0) | 280 (100) | 0.1 (0) | — |
| PBSb | — | 7.9 (1.6) | 491 (98) | 0.6 (0.1) | — | |
| MSN-AVI | 20 | 9.2 (2) | 444 (98) | 0.1 (0) | — | |
| MSN-AVI | 100 | 183 (33) | 363 (66) | 0.5 (0.1) | CXCL5, CXCL1 | |
| MSN-NH2 | 100 | 326 (60) | 131 (29) | 0 (0) | CXCL5, CXCL1, CCL2, IL-1β, TNF-α | |
| 3 d | MSN-AVI | 100 | 35 (5) | 644 (95) | 1.9 (0.3) | CXCL5, CXCL1 |
| MSN-NH2 | 100 | 65 (11) | 511 (88) | 3.4 (0.6) | CXCL5, CXCL1, TNF-α | |
| 7 d | MSN-AVI | 100 | 2 (0.4) | 581 (99) | 0.9 (0.2) | CXCL5 |
| MSN-NH2 | 100 | 34 (4.3) | 751 (95) | 5.2 (0.7) | CXCL5 | |
In line with that, only MSN-NH2 instillation resulted in macrophage accumulation in the airspace from day 3 to day 7 (Fig. 1C and Table 1) as well as the formation of giant cells (multinucleated macrophages, Fig. 1D and Table 1), both of which are signs for chronic inflammation and foreign body response.27 Lymphocyte numbers remained below 2000 (<0.5% of total BAL cells) for all conditions investigated (Fig. S3C†).
Inflammation caused by MSN-NH2 was further assessed on the molecular level by BALF cytokine profiling, assessing IL-1β, and TNF-α as the major pro-inflammatory master cytokines, CCL2 as classical monocyte/macrophage and CXCL1 and CXCL5 as the most relevant neutrophil chemoattractants. Treatment with MSN-NH2 caused the highest pro-inflammatory cytokine response; 1 day after treatment yielded high levels of all cytokines investigated (Fig. 1E–I). BALF levels of the two master cytokines for inflammation, IL-1β and TNFα, were high during the acute phase after MSN-NH2, but not after MSN-AVI treatment and returned to baseline levels by day 7 (Fig. 1E and F). Levels of chemokine CCL2, a monocyte attractant, correlated rather with PMN than macrophage numbers and were highest 1 day after MSN-NH2 treatment (Fig. 1G), where the lowest macrophage numbers were detected (Fig. 1C). This argues for significant macrophage cell death during the acute phase after MSN-NH2 instillation, causing a depletion of the alveolar macrophage pool uncompensated by the high levels of the CCL2 monocyte/macrophage attractant. The concentrations of the functional murine IL-8 homologues, CXCL1 and -5 (Fig. 1H and I) correlated well with the number of BAL PMNs (Fig. 1B) and showed accordingly higher levels for MSN-NH2 than MSN-AVI treated lungs. Elevated levels of the epithelial derived CXCL5 at day 7 could be interpreted as a sign for prolonged epithelial pro-inflammatory response to MSN particles, however similar levels are observed for untreated controls at day 1, thus questioning a physiological relevance. No evidence for inflammation was detected at the lower dose of 20 μg MSN-AVI per mouse, suggesting 1 mg kg−1 as a safe dose for pulmonary applications for this particle type.
The impact of the particle characteristics, agglomeration state, zeta-potential and specific surface area are of particular interest for the development of safe nanocarrier systems for pulmonary drug delivery. Recently, an indirect correlation between the agglomerate size of instilled particles and the acute inflammatory response was described for different dispersions of nickel-oxide nanoparticles with size distributions ranging from 100 nm to 4 μm.28 Based on their findings the authors argued that the reduced biologically accessible surface area of poorly dispersed suspensions might be limiting the bioactivity and toxicity of the instillation delivered nanoparticles. High zeta-potential of nanoparticle dispersions are generally appreciated for their enhanced stability, but have also been associated with increased toxicity and inflammation.29,30 As opposed to this broad rule, we show an improved biocompatibility for MSN-AVI preparations characterized by low agglomerate size and even higher zeta-potential as compared to the MSN-NH2 material. The particle surface of crystalline silica (i.e. quartz), is well known to induce lung inflammation (silicosis) upon inhalation. Toxicity has also been shown for some non-crystalline (amorphous) silica particles when applied directly in the lung, although highly dependent on size, surface and preparation route of the silica particles.31 The release of IL-1β from pulmonary macrophages is described as a central mechanism triggering their toxicity in the lung.32 In agreement with the latter, the highest levels of IL-1β release, and inflammatory cell accumulation were detected for MSN-NH2 particles, where the silicate particle surface area is not coated and hidden by the basically charged glycoprotein, avidin. The observation of an increased inflammatory response for MSN-NH2 compared to MSN-AVI particles suggests that the partially exposed silica surface of the MSN-NH2 particles, but not the avidin protein covered surface, induces inflammation. The MSN-NH2 particles have a large BET surface area of 1150 m2 g−1 and a zeta potential of −30 mV, while MSN-AVI particles have a BET surface area of only 90 m2 g−1 and a zeta potential of 30 mV (measured at pH = 7).6 This dramatic difference in accessible surface area and charge indicates that the avidin protein covers the surface of the MSN, thus significantly changing the surface characteristics of the particles. Further analysis of the MSN's incubated with isolated mouse lung alveolar lining fluid (BALF) revealed that MSN-NH2 particles form a significantly larger protein corona compared to MSN-AVI particles (Fig. S3D†). This may be explained by the partially exposed silica surface of the MSN-NH2 particles, compared to the already protein-coated MSN-AVI particles. This result highlights that MSN's coated with a protein corona do not behave similarly compared to MSN particles that have a protein coat that is covalently attached to the surface of the particle. Interestingly, when instilling 100 μL of MSN-NH2 or non-functionalised MSNs (with hydroxyl groups on the surface; nonMSN) into mice, we observed that nonMSN cause significantly less acute inflammation (pulmonary PMNs influx) compared to MSN-NH2 after 24 h (Table S1†). Both particles have a large BET surface area arguing against surface area as major driver of toxicity in this case. It is possible that the amine groups are responsible for the increased inflammatory effect. Similarly, polystyrene particles with outer NH2 groups have been reported to be more toxic then their hydroxyl counterparts.33–35 In addition to their differential functionalities, our observations may also be explained by the fact that MSN-NH2 particles form larger agglomerates (about 1000 nm) compared to MSN-AVI and nonMSN particles (about 200 nm) after only a few hours in suspension (Fig. S3A†). Large agglomerates (bigger than 400 nm) are more effectively phagocytized by macrophages than endocytosed by epithelial cells.36 In addition, the formation of a larger protein corona on MSN-NH2 particles compared to the MSN-AVI (Fig. S3†) may also contribute to enhanced bio-activity and even to the observed increased macrophage particle uptake for the MSN-NH2. Size-dependent nanoparticle uptake has been observed previously in lung macrophage and epithelial cells.37 All in all, our data suggests that avidin protein modified MSNs are safe to use for lung application at 1 mg per kg dose and induce a minor inflammatory response at 5 mg per kg dose that completely resolved after 7 days.
Cytotoxicity analysis
The toxic effects of the two particle types in the mouse model were investigated with cytospins of bronchoalveolar lavage (BAL) cells and on lung cryo-sections (from non-lavaged mice) using immunofluorescence staining. For this approach, cells and tissues were stained with an apoptotic cell marker (cleaved caspase-3, green signal in Fig. 2A). The MSNs, covalently labelled with ATTO 633 dye, could easily be recognised in both the cytospins and the cryo-sections (red signal in Fig. 2A). Exposure to high doses of MSN-AVI particles did not cause any significant apoptotic toxicity in BAL cells, compared to the vehicle/sham control (PBS instilled mice) or the low dose of MSN-AVI (Fig. 2A upper panel, controls are shown in Fig. S4A†). Significant apoptosis, however, could be observed for BAL cells exposed to MSN-NH2 particles, which did not subside 7 days after exposure (Fig. 2A lower panel). Neither particle type induced any distinguishable toxicity in lung cryo-sections of the lavaged lungs (Fig. 2A, right panel). Furthermore, particle uptake and burden of the macrophages correlated with increased apoptosis marker staining (Fig. 2B). Macrophage MSN particle internalization was confirmed with phalloidin staining in confocal microscopy, showing actin (green) encapsulated MSN agglomerates (Fig. 2C). In addition, in in vitro exposure experiments of the three major cell types of the lung supported our in vivo observation; only MSN-NH2 showed significant cytotoxicity towards MH-S murine alveolar macrophage cells, whereas no effect was observed in murine lung epithelial (MLE-12) or fibroblast (CCL-206) cell lines at high doses for either material (Fig. 2D). In line with that, also no structural changes of the alveolar microarchitecture could be observed in paraffin lung sections due to particle exposure (Fig. S4B†). In accord with our pulmonary inflammation analysis, the MSN-NH2 particles appear to cause selective macrophage toxicity, but no toxicity in lung parenchyma or in epithelial and fibroblast cell lines. It seems therefore likely that a phagocytic uptake of the large MSN-NH2 agglomerates is required to generate the cytotoxic response while non-professional phagocytes might be spared from the harmful effects of non-avidin coated MSNs. In summary, the nanoparticle coating appears to be crucial for their inflammatory and toxic response in the lung, where protein coating enhances biocompatibility.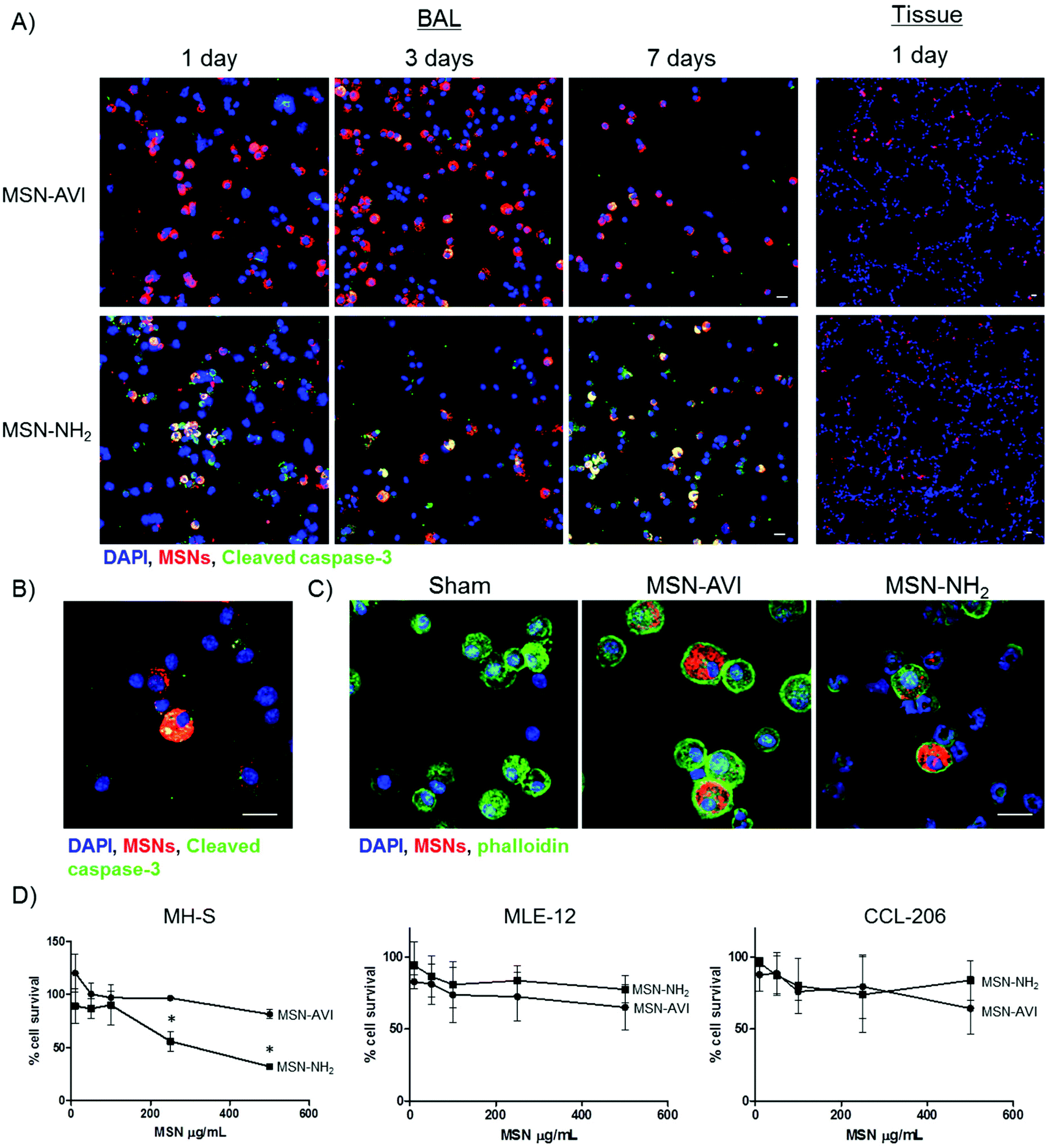 | ||
| Fig. 2 Cytotoxicity assessment of MSN-AVI and MSN-NH2 particles. (A) Toxicity of MSN-AVI or MSN-NH2 particles in Balb/c mice after instillation with 100 μg for 1, 3, or 7 days, analyzed from cytospins of BAL cells (left) and lung cryo-slices (right). Cell nuclei are shown in blue (DAPI), ATTO 633 labelled MSNs are shown in red, and the apoptotic marker (cleaved caspase-3) is shown in green. (B) Toxicity of MSN-NH2 in Balb/c mice exposed to 100 μg for 7 days in cytospins of BAL recovered cells using a 63× objective; high particle loading correlates with toxicity. Yellow staining (in A and B) is due to overlap of MSNs (red) and apoptotic cells (green). (C) MSN uptake in macrophages after 1 day exposure to, from left to right, 100 μL PBS, 100 μg MSN-AVI or MSN-NH2 (cytoskeleton labelled with phalloidin, green), (D) Cytotoxicity of MSN-AVI and MSN-NH2 particles as determined by MTT assay in MH-S, MLE-12, and CCL-206 cells after 16 h of exposure. Values are indicated as mean ± SD, n = 3, asterisks represent significance with *p < 0.05. Scale bar is 20 μm. | ||
Pulmonary dose and clearance of MSNs
The pulmonary dose of MSN-AVI particles at the three time points was determined by quantifying the fluorescent signal of the particles in three pulmonary compartments, namely homogenized lung tissue (after BAL), BAL fluid (BALF) and BAL cells. Using gravimetric analysis of the instillation wear prior and after instillation, we determined that 87.8 ± 3.5 μg and 17.6 ± 0.6 μg of the nominally applied 100 μg and 20 μg MSN dose was delivered to the lungs, respectively. It is important to note that while the absolute pulmonary dose (in μg) determines the toxicological (and pharmacological) response, the pulmonary clearance (removal) and biodistribution of particles is typically expressed in terms of relative dose, i.e. pulmonary dose normalized to applied dose (here: 87.8 or 17.6 μg). For the following, we refer to the pulmonary dose as the relative dose. As seen in Fig. 3A, the pulmonary dose slowly decreased from 80.1% of the applied dose at day 1 to 55.5% at day 7, indicating relatively slow clearance kinetics (for both 20 and 100 μg MSN doses), which can be attributed to macrophage clearance from the alveolar surface.36 By fitting the measured total lung dose to an exponential function (relative dose = 0.851e−0.061t, where t represents time in units of days), the alveolar clearance half time was determined as 8.7 days. During this period, the MSN dose decreased by 50% corresponding to an average clearance rate of 5.7% per day. Consequently, we found a relatively high retention of MSN-AVI particles in pulmonary tissue at day 7 after instillation (56% of the applied dose). When extrapolating the exponential fit curve to t = 0 (time point of MSN application), one obtains a relative dose of 0.851, i.e. macrophage clearance can only account for 85.1% of the actually applied dose. The residual 14.9% can be attributed to a faster clearance mechanism known as mucociliary clearance from the bronchial region, which is typically completed within 1 d after application.36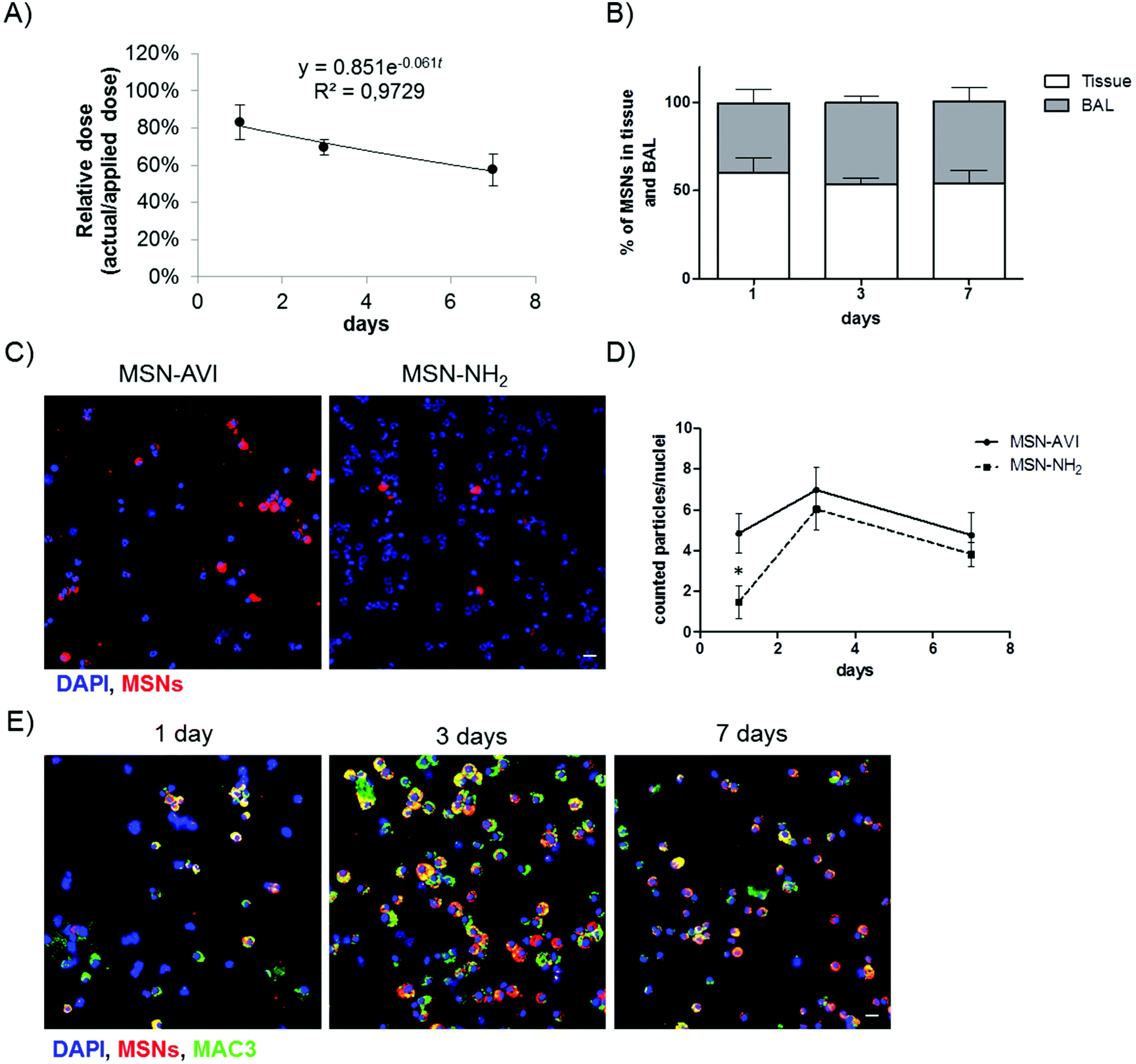 | ||
| Fig. 3 Pulmonary dose and clearance of MSNs in the lungs of Balb/c mice up to seven days after particle instillation. (A) Temporal decrease of pulmonary dose (normalized to applied dose) and derived exponential clearance kinetics of MSN-AVI particles up to day 7 (n = 3). (B) MSN-AVI particle distribution in lung tissue and BAL over time (n = 3). (C) MSN-AVI and MSN-NH2 particles (red) distribution in BAL cells (nuclei DAPI staining, blue) after 1 day. (D) Quantification of particle clearance in BAL cells depicted as the number of MSN agglomerates (larger than 1 μm) per counted nuclei (n = 3 animals, 2 images per animal). (E) MSN-AVI particles (red) in BAL recovered cells with macrophage marker co-staining (MAC3, green). Cell nuclei are shown in blue (DAPI), ATTO 633 labelled MSNs are shown in red. Scale bar is 20 μm. | ||
The phagocytic clearance of MSNs was also studied on cytospins of BAL cells by counting the amount of MSN agglomerates larger than 1 μm (the smallest detectable cross sectional area) per nuclei on cytospins (i.e. the amount of >1 μm MSN agglomerates over the total amount of counted nuclei per image). After 1 day, more agglomerates of MSN-AVI could be observed in the BAL compared to MSN-NH2 (Fig. 3C). The ratio of estimated MSN agglomerates per nuclei showed a transient increase at day 3 which declined again by day 7 after treatment (Fig. 3D). The observed difference in clearance rate may be explained by the fact that at day 1 the number of macrophages in MSN-NH2 treated lungs had declined to about one third of MSN-AVI treated mice due to the pronounced phagocyte toxicity of MSN-NH2. However, the macrophage number recovered to similar levels of MSN-AVI treated mice at the later time points (Fig. 1C).
Particle uptake specifically by alveolar macrophages was confirmed, by counterstaining with a macrophage marker (MAC3, green channel). All detected MSN-AVI were found in macrophage marker positive cells with round nuclei, but not in polymorphonuclear neutrophils, thus significant uptake by other BAL cells could be excluded (Fig. 2E). The type A scavenger receptor MARCO (macrophage receptor with collagenous structure) has been described to function as an important receptor of alveolar macrophages mediating the interaction with unopsonized particles such as silica or bacteria.38 Since the expression of MARCO is restricted to subpopulations of tissue macrophages and particularly high in spleen marginal zone, resident peritoneal, and alveolar macrophages but is low in monocyte derived macrophages,39 the choice of cells studied is very important. Surface modification, such as by avidin might reduce the interaction of MSN particles with these scavenger receptors and thus effect a rapid phagocytosis and related clearance by alveolar macrophages. In summary, MSN-AVI particles showed the bimodal lung clearance kinetics (i.e. slow alveolar macrophage clearance, and fast mucociliary clearance from the bronchial region) which is typical for particle removal from the lungs.36 The MSN-AVI had a slow clearance kinetics which is particularly promising for pulmonary therapy as it increases the residence time and hence the bioavailability of the encapsulated drugs.
Particle distribution in the lung
The distribution of MSNs in the lungs was assessed by preparing cryo-sections of (non-lavaged) lungs, which were analysed by immunofluorescence. ATTO 633 labelled MSNs could easily be detected by confocal microscopy on 14 μm thick lung cryo-sections (red channel, Fig. 4A). The MSNs were distributed evenly over the tissue, also reaching the alveolar region of the lung. Furthermore, the nanoparticles showed widespread and significant accumulation in the lungs for at least 7 days, confirming our previous dosimetry findings (Fig. 4A and S5†). | ||
| Fig. 4 MSN distribution in mouse lungs up to 7 days after particle instillation. (A) Lung cryo-slices of Balb/c mice exposed to 100 μg MSN-AVI for 1, 3, or 7 days with phalloidin co-staining (upper panel) and the same images showing only MSNs (lower panel, MSNs are shown in white to increase the contrast). Lung cryo-slices of Balb/c mice exposed to (B) 100 μg MSN-AVI for 1 day (left) and 7 days (right)(arrowheads represent locations of MSN-AVI) and (C) 100 μg MSN-NH2 for 1 day, (D) 100 μg MSN-AVI with epithelial cell type 1 co-staining (T1α, green) for 1 day (left) and 7 days (right), and (E) 100 μg MSN-AVI with epithelial cell type 2 co-staining (pro-SPC, green) for 1 day (left) and 7 days (right). Cell nuclei are shown in blue (DAPI), ATTO 633 labelled MSNs are shown in red. Images are representative images for n = 4 animals. Scale bar is 20 μm. | ||
After 1 day, many particle agglomerates could be observed, while after 3 and 7 days, fewer but bigger MSN-AVI agglomerates remained (Fig. 4A lower panel). High magnification images indicated that a fraction of the MSN-AVI particles remained on the epithelium, evidence of which was still present after 7 days (Fig. 4B and S5†). In contrast, only limited evidence was obtained that MSN-NH2 particles were taken up by epithelial cells. In fact, when investigating cryo-sections at higher magnifications, we mainly observed macrophage uptake of MSN-NH2 particles (Fig. 4C). Furthermore, z-stack images of non-phagocytized MSN-NH2 particles revealed that these nanoparticles appear to associate with the tissue rather than being internalized into the cells (Fig. 4C, right panel). Counterstaining the cryo-sections with epithelial type I and II cell markers, podoplanin (T1α) and pro-surfactant associated protein C (Pro-SPC), respectively, revealed that MSN-AVI particles are internalized by epithelial lung cells (Fig. 4D, E and S4†) as confirmed by confocal z-stack imaging (Fig. 5A). To investigate the cell specific uptake further, we prepared co-cultures of a murine alveolar macrophage cell line (MH-S) and an alveolar epithelial cell line (MLE-12) to study the fate of the particles in an in vitro model. Clearly distinguishable cell populations could be observed by fluorescence-activated cell sorting (FACS) analysis excluding leakage of the dye to the other cell type at the studied time-points (Fig. S7†). Co-cultures indeed showed enhanced MSN-AVI particle uptake in epithelial cells compared to macrophage cell uptake (Fig. 5B, left panel). In contrast, MSN-NH2 particles showed almost exclusively uptake in macrophages in the same co-culture set-up (Fig. 5B, right panel).
 | ||
| Fig. 5 Cellular uptake comparison of MSN-AVI and MSN-NH2. (A) Z-stack images (63× objective) of cryo-slices of Balb/c mice exposed to MSN-AVI for 1 day after instillation, co-stained with alveolar epithelial cell type 1 (ATI) marker (T1α, left image) and alveolar epithelial cell type 2 (ATII) marker (pro-SPC, right image) in green. DAPI staining in blue, scale bar is 20 μm. (B) Representative (of n = 3) FACS histograms of co-cultures of MH-S and MLE-12 cells, showing differential cell uptake of MSN-AVI and MSN-NH2 particles. MSN particles (labelled with Alexafluor 488) uptake could be quantified by gating MLE-12 DiD labelled cell population (630 nm) and non-labelled MH-S cell population. | ||
In summary, cell type specific immunostainings revealed that MSN-NH2 particles are preferentially internalized by macrophages in vivo and in vitro and that this leads to macrophage cytotoxicity. In contrast, MSN-AVI particles were – in addition to being phagocytically cleared by macrophages – also efficiently internalized by alveolar epithelial type 1 and type 2 cells, as investigated in lung tissue cryo-sections. Moreover, MSN-AVI also showed higher uptake in epithelial cells compared to macrophage cell uptake in an in vitro co-culture model. Previous studies have shown that cellular uptake of MSNs is highly dependent on surface charge, surface modifications, and is cell type-specific.40,41 Furthermore, several in vitro studies have shown that the cell uptake of MSNs is cell-type-, dosage- and time-dependent.42,43 Interestingly, it has been reported that amination of MSN particles prevents particle endocytosis in T-lymphocyte cells (Jurkat) and in a human neuroblast cell line.36,41 These findings highlight the importance of outer surface modifications and their interactions with different cell types, but also that avidin coating may be a good strategy to overcome issues related to macrophage uptake and particle toxicity.
Conclusions
In this study, we investigated the relevance of avidin-capped MSNs (MSN-AVI) for pulmonary therapy by looking at their pulmonary distribution, clearance rate, cell specific uptake, and induction of inflammatory response after direct (intratracheal) instillation in the lungs of mice. In a recent publication we showed that these particles are promising carriers for lung cancer therapy as they could release a combination of drugs efficiently and tumor-selectively in in vitro and in human and mouse ex vivo lung tissue.6 Since MSNs allow for multiple functionalizations, which have been found to be important for their bioresponse, we also included non-capped (only amino-functionalized; MSN-NH2) particles in this study. We show that MSN avidin surface modification had an effect not only on toxicity, but also on cell specific uptake and tissue distribution in the lungs. In particular, non-capped (MSN-NH2) particles were found to be cytotoxic to macrophages, caused an enhanced inflammatory response, and were hardly taken up by epithelial cells. In contrast, MSN-AVI particles co-localized with alveolar epithelial type 1 and type 2 cells in the lung tissue and showed preferential epithelial cell uptake in in vitro co-cultures. These findings, in combination with the low surface specific toxicity, wide distribution of the particles in the mouse lungs and slow clearance rate is promising for the treatment of chronic lung diseases such as COPD, IPF, and lung cancer, where (alveolar) epithelial cells play an important role in the pathogenesis. Moreover, the inflammatory potential of drug delivery particles is most critical in inflammatory lung diseases such as asthma and COPD, due to additive effects leading to worsening of the symptoms. In this context, the observation that the dose of 1 mg kg−1 of MSN-AVI did not cause any detectable inflammatory response is particularly promising for treatment of these devastating lung diseases. Thus, we believe that avidin-coated MSNs offer potential for inhalative application as therapeutic drug carriers in chronic lung diseases. In addition, the finding that surface modifications greatly affect toxicity and cell type specific uptake highlights the importance of these types of studies for future development of nanomedicines.Acknowledgements
We thank the Alexander von Humboldt foundation for providing funding for S. H. van Rijt. Furthermore, financial support from the Nanosystems Initiative Munich (NIM) and the DFG (SFB 749) are gratefully acknowledged.References
- C. M. Dawidczyk, C. Kim, J. H. Park, L. M. Russell, K. H. Lee, M. G. Pomper and P. C. Searson, J. Controlled Release, 2014, 187, 133–144 CrossRef CAS PubMed.
- C. A. Schutz, L. Juillerat-Jeanneret, H. Mueller, I. Lynch, M. Riediker and N. Consortium, Nanomedicine, 2013, 8, 449–467 CrossRef PubMed.
- S. H. van Rijt, T. Bein and S. Meiners, Eur. Respir. J., 2014, 44, 765–774 CrossRef CAS PubMed.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26(1), 435–451 CrossRef CAS.
- V. Cauda, A. Schlossbauer, J. Kecht, A. Zurner and T. Bein, J. Am. Chem. Soc., 2009, 131, 11361–11370 CrossRef CAS PubMed.
- S. H. van Rijt, D. A. Bölükbas, C. Argyo, S. Datz, M. Lindner, O. Eickelberg, M. Koenigshoff, T. Bein and S. Meiners, ACS Nano, 2015, 9(3), 2377–2389 CrossRef CAS PubMed.
- H. K. Na, M. H. Kim, K. Park, S. R. Ryoo, K. E. Lee, H. Jeon, R. Ryoo, C. Hyeon and D. H. Min, Small, 2012, 8, 1752–1761 CrossRef CAS PubMed.
- J. Lu, Z. Li, J. I. Zink and F. Tamanoi, Nanomedicine, 2012, 8, 212–220 CAS.
- H. Liu, T. Liu, X. Wu, L. Li, L. Tan, D. Chen and F. Tang, Adv. Mater., 2012, 24, 755–761 CrossRef CAS PubMed.
- Y. Chen, H. Chen and J. Shi, Adv. Mater., 2013, 25, 3144–3176 CrossRef CAS PubMed.
- Q. He, Z. Zhang, F. Gao, Y. Li and J. Shi, Small, 2011, 7, 271–280 CrossRef CAS PubMed.
- X. Huang, L. Li, T. Liu, N. Hao, H. Liu, D. Chen and F. Tang, ACS Nano, 2011, 5, 5390–5399 CrossRef CAS PubMed.
- Y. S. Lin and C. L. Haynes, J. Am. Chem. Soc., 2010, 132, 4834–4842 CrossRef CAS PubMed.
- Y. Zhao, X. Sun, G. Zhang, B. G. Trewyn, I. I. Slowing and V. S. Lin, ACS Nano, 2011, 5, 1366–1375 CrossRef CAS PubMed.
- O. Taratula, O. B. Garbuzenko, A. M. Chen and T. Minko, J. Drug Targeting, 2011, 19, 900–914 CrossRef CAS PubMed.
- J. Lu, M. Liong, Z. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794–1805 CrossRef CAS PubMed.
- N. Kupferschmidt, X. Xia, R. H. Labrador, R. Atluri, L. Ballell and A. E. Garcia-Bennett, Nanomedicine, 2013, 8, 57–64 CrossRef CAS PubMed.
- H. Vallhov, N. Kupferschmidt, S. Gabrielsson, S. Paulie, M. Stromme, A. E. Garcia-Bennett and A. Scheynius, Small, 2012, 8, 2116–2124 CrossRef CAS PubMed.
- T. Yu, K. Greish, L. D. McGill, A. Ray and H. Ghandehari, ACS Nano, 2012, 6, 2289–2301 CrossRef CAS PubMed.
- S. P. Hudson, R. F. Padera, R. Langer and D. S. Kohane, Biomaterials, 2008, 29, 4045–4055 CrossRef CAS PubMed.
- A. Beyerle, A. Braun, A. Banerjee, N. Ercal, O. Eickelberg, T. H. Kissel and T. Stoeger, Biomaterials, 2011, 32, 8694–8701 CrossRef CAS PubMed.
- E. F. Burguera, M. Bitar and A. Bruinink, Eur. Cells Mater., 2010, 19, 166–179 CAS.
- T. Stoeger, C. Reinhard, S. Takenaka, A. Schroeppel, E. Karg, B. Ritter, J. Heyder and H. Schulz, Environ. Health Perspect., 2006, 114, 328–333 CrossRef PubMed.
- O. M. Merkel, A. Beyerle, D. Librizzi, A. Pfestroff, T. M. Behr, B. Sproat, P. J. Barth and T. Kissel, Mol. Pharm., 2009, 6, 1246–1260 CrossRef CAS PubMed.
- A. A. Gotz, A. Vidal-Puig, H. G. Rodel, M. H. de Angelis and T. Stoeger, Part. Fibre Toxicol., 2011, 8, 28 CrossRef PubMed.
- N. Barapatre, P. Symvoulidis, W. Moller, F. Prade, N. C. Deliolanis, S. Hertel, G. Winter, A. O. Yildirim, T. Stoeger, O. Eickelberg, V. Ntziachristos and O. Schmid, J. Pharm. Biomed. Anal., 2015, 102, 129–136 CrossRef CAS PubMed.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS PubMed.
- T. Sager, M. Wolfarth, M. Keane, D. Porter, V. Castranova and A. Holian, Nanotoxicology, 2015, 1–11, DOI:10.3109/17435390.2015.1025883.
- W. S. Cho, R. Duffin, F. Thielbeer, M. Bradley, I. L. Megson, W. Macnee, C. A. Poland, C. L. Tran and K. Donaldson, Toxicol. Sci., 2012, 126, 469–477 CrossRef CAS PubMed.
- M. Simko, D. Nosske and W. G. Kreyling, Int. J. Environ. Res. Public Health, 2014, 11, 4026–4048 CrossRef PubMed.
- L. M. Costantini, R. M. Gilberti and D. A. Knecht, PLoS One, 2011, 6, e14647 CAS.
- W. J. Sandberg, M. Lag, J. A. Holme, B. Friede, M. Gualtieri, M. Kruszewski, P. E. Schwarze, T. Skuland and M. Refsnes, Part. Fibre Toxicol., 2012, 9, 32 CrossRef CAS PubMed.
- S. Bhattacharjee, D. Ershov, K. Fytianos, J. van der Gucht, G. M. Alink, I. M. Rietjens, A. T. Marcelis and H. Zuilhof, Part. Fibre Toxicol., 2012, 9, 11 CrossRef CAS PubMed.
- H. W. Chiu, T. Xia, Y. H. Lee, C. W. Chen, J. C. Tsai and Y. J. Wang, Nanoscale, 2015, 7, 736–746 RSC.
- P. Ruenraroengsak and T. D. Tetley, Part. Fibre Toxicol., 2015, 12, 19 CrossRef PubMed.
- W. G. Kreyling, M. Semmler-Behnke, S. Takenaka and W. Moller, Acc. Chem. Res., 2013, 46, 714–722 CrossRef CAS PubMed.
- D. A. Kuhn, D. Vanhecke, B. Michen, F. Blank, P. Gehr, A. Petri-Fink and B. Rothen-Rutishauser, Beilstein J. Nanotechnol., 2014, 5, 1625–1636 CrossRef CAS PubMed.
- M. S. Arredouani, A. Palecanda, H. Koziel, Y. C. Huang, A. Imrich, T. H. Sulahian, Y. Y. Ning, Z. Yang, T. Pikkarainen, M. Sankala, S. O. Vargas, M. Takeya, K. Tryggvason and L. Kobzik, J. Immunol., 2005, 175, 6058–6064 CrossRef CAS.
- S. Mukhopadhyay, Y. Chen, M. Sankala, L. Peiser, T. Pikkarainen, G. Kraal, K. Tryggvason and S. Gordon, Eur. J. Immunol., 2006, 36, 940–949 CrossRef CAS PubMed.
- T. H. Chung, S. H. Wu, M. Yao, C. W. Lu, Y. S. Lin, Y. Hung, C. Y. Mou, Y. C. Chen and D. M. Huang, Biomaterials, 2007, 28, 2959–2966 CrossRef CAS PubMed.
- Z. Tao, B. B. Toms, J. Goodisman and T. Asefa, Chem. Res. Toxicol., 2009, 22, 1869–1880 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Lin, Small, 2010, 6, 1952–1967 CrossRef CAS PubMed.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of MSN particles. Characterisation of MSN particles (Fig. S1 and S2), DLS measurements of MSNs over time, lymphocyte and PMN cell count after MSN exposure (Fig. S3). Toxicity in BAL cytospins controls, phalloidin staining on BAL cytospins of MSN-NH2 exposed mice (Fig. S4), nanoparticle distribution in lung cryo-slices of Balb/c mice exposed to 100 μg MSNs (Fig. S5). Balb/c mice cryo-slices exposed to MSN-AVI for 1 or 7 days, co-stained with alveolar epithelial cell type 1 marker or with alveolar epithelial cell type 2 marker (Fig. S6), DiD selective labeling in a co-culture set-up (Fig. S7). See DOI: 10.1039/c5nr04119h |
| ‡ Member of the German Center for Lung Research (DZL), Germany. |
| This journal is © The Royal Society of Chemistry 2016 |