
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe evolution of genome mining in microbes – a review
Nadine
Ziemert
*ab,
Mohammad
Alanjary
ab and
Tilmann
Weber
*c
aInterfaculty Institute for Microbiology and Infection Medicine Tübingen (IMIT), Microbiology and Biotechnology, University of Tuebingen, Germany. E-mail: nadine.ziemert@uni-tuebingen.de
bGerman Centre for Infection Research (DZIF), Partner Site Tübingen, Germany
cNovo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Hørsholm, Denmark. E-mail: tiwe@biosustain.dtu.dk
First published on 8th June 2016
Abstract
Covering: 2006 to 2016
The computational mining of genomes has become an important part in the discovery of novel natural products as drug leads. Thousands of bacterial genome sequences are publically available these days containing an even larger number and diversity of secondary metabolite gene clusters that await linkage to their encoded natural products. With the development of high-throughput sequencing methods and the wealth of DNA data available, a variety of genome mining methods and tools have been developed to guide discovery and characterisation of these compounds. This article reviews the development of these computational approaches during the last decade and shows how the revolution of next generation sequencing methods has led to an evolution of various genome mining approaches, techniques and tools. After a short introduction and brief overview of important milestones, this article will focus on the different approaches of mining genomes for secondary metabolites, from detecting biosynthetic genes to resistance based methods and “evo-mining” strategies including a short evaluation of the impact of the development of genome mining methods and tools on the field of natural products and microbial ecology.
 Nadine Ziemert | Nadine Ziemert received her Diploma and PhD degrees from the Humboldt University in Berlin, followed by a postdoc and project scientist position at the Scripps Institution of Oceanography in La Jolla, California. Since 2015, she is a Professor at the University of Tübingen, where she leads an interdisciplinary research group focusing on genome mining approaches and the evolution of secondary metabolites in bacteria and their diverse functions. |
 Mohammad Alanjary | Mohammad Alanjary obtained a B.S. in Biochemistry/Chemistry at the University of California San Diego (UCSD) before moving to an enterprising gene sequencing company, Ion Torrent; there he aided in the launch of the first commercial semi-conductor gene sequencing platform and developed several procedures for the optimization of sequencing performance. His interest in programming new analysis methods to deal with the overwhelming genomic tsunami of today led him to work on his PhD in bioinformatics at the University of Tübingen, Germany. |
 Tilmann Weber | Dr Tilmann Weber is Co-Principal Investigator of the New Bioactive Compound section at the Novo Nordisk Foundation Center for Biosustainability of the Technical University of Denmark. He is interested in integrating bioinformatics, genome mining, and systems biology approaches into Natural Products discovery and characterization and thus bridging the in silico and in vivo world. He obtained his PhD (supervisor Prof. Dr Wolfgang Wohlleben) and his habilitation at the Eberhard Karls University Tübingen, Germany. |
1. Introduction
The fast development of genome sequencing methods revolutionized almost every aspect of biology including natural product research. We have come a long way during the last three decades from the identification and manipulation of the first secondary metabolite genes to whole genome sequencing of thousands of bacterial genomes and metagenomes for a fast and automated discovery of promising new natural products and their role in the environment. With the wealth of genetic data available these days, there is no shortage of secondary metabolite gene clusters anymore; the challenge is now to effectively mine the data, connect whenever possible detected Biosynthetic Gene Clusters (BGC) to the vast amount of already known molecules and predict the ones that encode the most promising compounds. Plenty of tools are available to enable researchers to computationally mine genetic data and connect them to known secondary metabolites, and plenty of reviews are available that describe those tools and their applications. This review is focused on the development of genome mining methods over the last 10 years and the various strategies to detect and prioritize secondary metabolite gene clusters (Fig. 1). Rather than presenting extensive examples we focus on the rapid evolution of genome mining approaches and strategies and give some examples of when and how they were used for compound discovery, and which directions and challenges are remaining in the near future.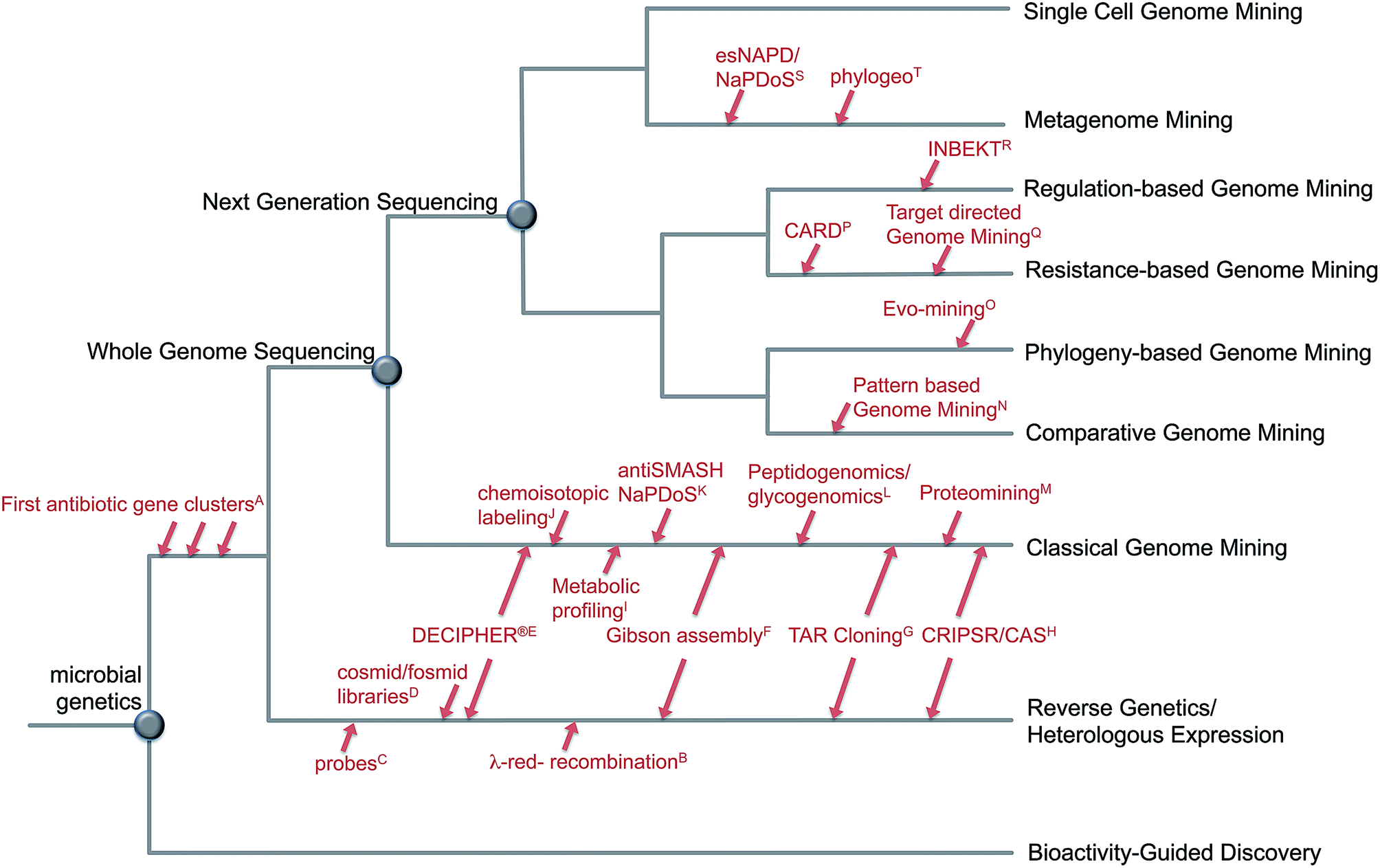 | ||
| Fig. 1 Evolution of genome mining. Overview about approaches and strategies to mine microbial genomes for novel secondary metabolites including selected developments in methods: A;4–8 B;15 C;9–12 D;16–18 E,19 F;20 G;21,22 H;23,24 I;25 J;26 K;27,28 L;29,30 M;31 N;32 O;33 P;34 Q;35 R;36 S;27,37 T.38 | ||
2. A short history of genome mining
Drug discovery efforts have traditionally been based on bioactivity screening efforts of natural sources such as plants, fungi or bacteria. Bioactive isolated chemical structures, so called natural products or derivatives of those have been used as drug leads for new antibiotics, anticancer agents or immunotherapeutics.1,2 Ignited by the discovery of penicillin and streptomycin, the golden age of antibiotics began and researchers discovered microbial secondary metabolites as an important source for new antibacterial compounds. Today, natural products remain the main source for new therapeutic agents. The genus Streptomyces has especially been chemically exploited for decades in search for new drugs.3With the establishment of Streptomyces genetics and the discovery of the first biosynthetic genes in the 70s and 80s, researchers started to understand the biosynthetic logic and genetic basis for the production of these compounds.4–8 Using classical genetics or reverse genetics approaches, it was possible to map and connect many biosynthetic gene clusters to known molecules.9–12
Beginning with the new millennium and the full genome sequences of two well studied natural product producing strains Streptomyces coelicolor13 and Streptomyces avermitilis14 scientist noticed the unexplored potential hidden in bacterial genomes. A Streptomyces genome contains on average about 30 secondary metabolite gene clusters and only two or three were known at the time.
At this time the classical idea of genome mining was born: predicting and isolating natural products based on genetic information without a structure at hand. Inspired by the observation that even well studied strains contain the genetic potential to synthesize many more compounds than detected analytically, the genome mining concept was expanded to other microbes where genome information became available, such as cyanobacteria,39,40 myxobacteria,41–43 and anaerobes.44,45 Nowadays, thousands of bacterial genomes have become available, whole culture collections are currently being sequenced, and new technologies like single cell genomics and metagenomics generate massive data to be analyzed.
The current largest collection of automatically mined gene clusters is the “Atlas of Biosynthetic gene Clusters”, a component of the “Integrated Microbial Genomes” Platform of the Joint Genome Institute (JGI IMG-ABC).46 As of February 2016, IMG-ABC contains entries for more than 960![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 putative gene clusters identified in JGI's huge genome and metagenome datasets and public databases. However, only a very small fraction of these predicted BGCs are characterized and their products described. In a recent community effort within the “Minimum Information for Biosynthetic Gene clusters” (MIBiG) standardization initiative a manual re-annotation of ∼1300 BGCs has been carried out now providing a highly curated reference dataset.47
000 putative gene clusters identified in JGI's huge genome and metagenome datasets and public databases. However, only a very small fraction of these predicted BGCs are characterized and their products described. In a recent community effort within the “Minimum Information for Biosynthetic Gene clusters” (MIBiG) standardization initiative a manual re-annotation of ∼1300 BGCs has been carried out now providing a highly curated reference dataset.47
3. Classical genome mining: search for enzymes involved in the biosynthesis of secondary metabolites
Mining for enzymes, or more precisely genes encoding enzymes putatively involved in secondary metabolite biosynthesis of interest is the most “classical” variant of genome mining. Although the diversity of secondary metabolites is huge, the biosynthetic principles and thus the biosynthetic machineries for many of these compounds are often strikingly conserved. This is reflected in the high amino acid sequence similarity of many of the core biosynthetic enzymes. Examples for classes of secondary metabolites using such conserved machineries are polyketides (PK), biosynthesized by polyketide synthases (PKS), non-ribosomally synthesized peptides (NRP), produced by non-ribosomal peptide synthetases (NRPS), ribosomally and post-translationally modified peptides (RiPPs), aminoglycosides, and many more.Even before large-scale genome sequence data were available, this sequence conservation was used in reverse genetics approaches to screen genetic libraries of producers for the presence of core biosynthetic genes.11 Conserved genes of characterized pathways (or fragments, e.g., PKS or NRPS domains) were labeled and used as probes in Southern hybridization experiments. Alternatively, primers were deduced from highly conserved motifs of these genes and used for PCR screening approaches. With improved quality and throughput of sequencing and the massive decrease of costs, many of these approaches are currently carried out in silico instead of involving tedious generation of libraries and experimental screening. But the general principle behind the in silico mining is the same for reverse genetics: one or multiple sequences encoding “reference” enzymes are used as seed sequences to identify homologues in the genome sequences of the organisms of interest. For this task, sequence based comparison software, like BLAST48 or DIAMOND49 or profile-based tools like HMMer50 are usually used. If the structure of the compound of interest is already known and a hypothetical biosynthetic route can be predicted, this approach often leads to an easy identification of the responsible biosynthetic gene cluster, as demonstrated for many pathways, for example: teixobactin,51 cypemycin,52 microbisporicin,53 ristomycin,54 microcyclamide,55 microviridin,56 poly(L-diaminopropionic acid),57 and many more. In the following sections, we are focusing on metabolites, in which the gene cluster was used as the starting point and the compound was not described before.
While mining for genes encoding conserved biosynthetic enzymes can, in principle, be easily done manually with BLAST or HMMer, integrated tools and databases were developed that greatly expedite this approach. To our best knowledge, the first reported tool for automated cluster mining was DECEIPHER®, a proprietary pipeline and database developed around 2001 by the former company Ecopia Biosciences Inc.19 In the following years additional tools were developed and became freely available including BAGEL,58 CLUSEAN59 and antiSMASH.28 To provide a comprehensive overview on such software and databases, the “The Secondary Metabolite Bioinformatics Portal” was recently launched at http://www.secondarymetabolites.org. The community-driven website provides a regularly updated and maintained catalogue of available secondary metabolism specialized software and databases and direct links to the respective tools and websites.60 As there have been multiple recent publications reviewing these tools,60–69 here we focus primarily on the application aspects. Several tools exist focusing on specific classes of secondary metabolite biosynthetic pathways, mostly PKS and/or NRPS70–74 or RiPPs.58,75,76 All these tools screen genomic data using profiles of known and highly conserved biosynthetic enzymes (e.g., PKS domains) and evaluate the results using pre-defined manually curated rules. The most comprehensive platform to perform such analyses currently is antiSMASH.28,77,78 In the current version 3.04, antiSMASH can identify 44 different gene cluster types based on hits against a library of enzymes/protein domains commonly observed in secondary metabolite biosynthetic pathways.
3.1 Mining for genes encoding core-biosynthetic enzymes
In the following section, several examples – from the beginning of genome mining, where most sequence analysis steps had to be carried out manually – until the present, where comprehensive bioinformatic software packages aid the scientists to identify novel compounds, are discussed for important families of bioactive secondary metabolites. A more extensive list of compounds, where genome mining has directly led to the identification of the metabolites, is included in Table 1.| Compound name | Gene cluster ID (MIBiG)/Genbank | Means of identification | Ref. |
|---|---|---|---|
| Type I polyketides | |||
| Asperfuranone | BGC0000022 | Search for PKS genes in A. nidulans whole genome sequence | 158 |
| Stambomycins | BGC0000151 | Search for modular PKS genes in S. ambofaciens | 83 |
| Salinilactam | BGC0000142 | Search for PKS/NRPS domain sequences (and other secondary metabolite biosynthesis related genes) in Salinispora tropica CNB-440 | 85 |
| ECO-02301 | BGC0000052 | Genome scanning for PKS genes25 | 80 |
|
|||
| trans-AT polyketides | |||
| Rhizopodin | BGC0001111 | Search for PKS/NRPS genes in S. aurantiaca Sg a15 using pipeline96 | 91 |
|
|||
| Type II polyketides | |||
| Hexaricins | Genbank: KT713752 | Search for type II gene cluster with antiSMASH 2 (ref. 77) | 93 |
|
|||
| Type III polyketides | |||
| Isogermicidin | Genbank: AL645882 | Analysis of genes encoding type III PKS of S. coelicolor A3(2) | 159 |
| Gene: SCO7221 | |||
|
|||
| NRPS | |||
| Aureusimines | BGC0000308 | Search for conserved NRPS genes in S. aureus and other Staphylococcus strains | 105 |
| Coelichelin | BGC0000325 | Search for NRPS genes in S. coelicolor M145 | 106 |
| Poeamide | BGC0001208 | Search for NRPS genes in Pseudomonas poae RE*1-1-14 | 160 |
| Orfamide | BGC0000399 | Search for NRPS genes in P. fluorescens Pf-5 | 26 |
| Viscosin-family lipopeptide | Genbank: AM181176 | Search for NRPS genes encoding cyclic lipopeptides | 161 |
| Locus: PFLU_2552, 2553, 4007 | |||
| S. peucetius siderophores | Not publicly available | Search for NRPS genes, analysis with NRPS–PKS162 | 163 |
| Thanapeptin | Genbank: CBLV010000330 | Search for NRPS with PKS/NRPS predictor,96 NP.searcher70 and antiSMASH28 | 164 |
| Locus: BN844_0667-0664 | |||
|
|||
| Hybrid PKS–NRPS | |||
| Isoflavipucine/dihydroisoflavipucine | BGC0001122 | Identification of the hybrid PKS/NRPS gene with SMURF165 | 166 |
| Aspyridones | BGC0000959 | Search for hybrid PKS/NRPS genes | 167 |
| Pyranonigirin E | BGC0001124 | Search for hybrid PKS/NRPS genes; comparison with pynA of A. niger CBS 513.88 | 168 |
| Haliamide | RefSeq: NC_013440 | Search for hybrid PKS/NRPS genes with antiSMASH28 | 169 |
| Locus: HOCH_RS34665-03960 | |||
| Carlosic acid, carlosic acid methyl ester | Genbank: ACJE01000021 | Search for hybrid PKS/NRPS genes | 170 |
| Locus: ASPNIDRAFT_176722 | |||
| Agglomerin F | Genbank: ACJE01000021 | Search for hybrid PKS/NRPS genes | 170 |
| Locus: ASPNIDRAFT_176722 | |||
| Mutanobactin | Genbank: AE014133 | Search for PKS and NRPS genes, analysis with NRPS–PKS162 | 171 and 172 |
| Locus: SMU_1334-1349 | |||
| Clarepoxcin A–E | BGC0001203 | Search for specific KS domain responsible for synthesizing the epoxyketone warhead173 with eSNAPD37 | 132 |
| Landepoxcin A/B | BGC0001202 | Search for specific KS domain responsible for synthesizing the epoxyketone warhead173 with eSNAPD37 | 132 |
|
|||
| RiPPs: lanthipeptides | |||
| Venezuelin | BGC0000563 | Search for genes encoding enzymes with N-terminal Ser/Thr kinase and C-terminal LanC-type domain | 109 |
| Streptocollin | BGC0001226 | Search for lanthipeptide gene clusters using antiSMASH 3 (ref. 78) | 114 |
| Informatipeptin | BGC0000518 | Combination of automated genome mining for RiPPs and mass spectrometric analysis using RiPPquest116 | 116 |
|
|||
| RiPPs: lasso peptides | |||
| Capistruin | BGC0000572 | Search for homologues of microcin J25 biosynthetic genes mcjBCD | 121 |
| Caulosegnins | BGC0000574 | Search for homologues of lasso peptide biosynthetic enzymes (B-/C-proteins) | 174 |
| Astexin-1 | BGC0000570 | Search for conserved patterns in lasso peptide precursor peptide using MEME175/MAST176 | 177 |
| Burhizin | BGC0000571 | Search for homologues of lasso peptide biosynthetic enzymes (B-/C-proteins) | 178 |
| Caulonodins | BGC0000573 | Search for homologues of lasso peptide biosynthetic enzymes (B-/C-proteins) | 178 |
| Rubrivinodin | BGC0000576 | Search for homologues of lasso peptide biosynthetic enzymes (B-/C-proteins) | 178 |
| Sphingonodins | BGC0000577 | Search for homologues of lasso peptide biosynthetic enzymes (B-/C-proteins) | 178 |
| Xanthomonins | BGC0000580 | Search for homologues of lasso peptide biosynthetic enzymes (B-/C-proteins) | 179 |
| Chaxapeptin | BGC0001307 | Search for homologues of B-protein LarB (lariatin biosynthesis) | 180 |
|
|||
| RiPPs: cyanobactins | |||
| Microcyclamide PCC7806A/B | BGC0000474 | Search for homologues of cyanobactin clusters in M. aeruginosa PCC7806 | 55 |
| Aeruginosamide | BGC0000483 | Search for cyanobactin gene clusters in cyanobacteria | 181 |
| Viridisamide | BGC0000471 | Search for cyanobactin gene clusters in cyanobacteria | 181 |
|
|||
| Terpenoids/isoprenoids | |||
| Cembrane | Genbank: AB738084, AB738085 | Blast search for homologs of CotB1, a geranyl-geranyl diphosphate synthase from cyclooctatin biosynthesis | 124 |
| Kolavelools | Genbank: ABX04785 | Blast search for homologs of Rv3377c (diterpene cyclase) and Rv3378c (diterpene synthase) | 182 |
| Locus: HAUR_2145, HAUR_2146 | |||
| Stellatic acid | Genbank: LC073704 | Search for homologs of AcOS, a sesterterpenoid synthase from ophiobolin F biosynthesis | 183 |
| Hydropyrene, hydropyrenol, and others | Genbank: CM000914 | HMM-based search for terpene synthases;127 heterologous expression | 126 |
| Locus: SCLAV_p0765 | |||
3.2 Polyketides
Even before whole genome sequencing became a routine endeavor, genomics guided approaches were used to identify novel biosynthetic pathways and new molecules. One of the first approaches was published in 2003, when researchers of the company Ecopia Biosciences Inc. reported the identification of 11 new enediyne BGCs.25 Enediynes are interesting drug candidates as their high cytotoxicity makes them promising tools for antibody–drug conjugates to treat cancer.79 As it was not affordable at that time to simply sequence whole genomes of the studied actinomycetes, Zazopoulos et al. generated plasmid libraries of the producers. By Sanger sequencing of 1000 plasmids per library, they were able to generate ∼700 bp long “sequence tags” covering the whole genome. The tags then were compared to sequences of genes involved in warhead formation of enediyne BGCs that were already known using Ecopia's DECIPHER® tool and database. Using this strategy, it was possible to identify 8 out of 50 strains that contain BGCs encoding enediyne biosynthesis pathways. Although no analytical proof of enediyne formation was provided in the original work, results of prophage induction assays indeed indicated that all of these strains have the potential to produce DNA damaging agents (as the enediynes are).25 A similar strategy was also applied to identify the antifungal agent ECO-02301, which is biosynthesized via a modular type I PKS complex.80With the easy and cheap availability of whole genome sequence data, many other gene clusters have been identified and associated with chemical products. One noteworthy example is the polyketide stambomycin. In the course of the analysis of the Streptomyces ambofaciens genome, which was known to code for the congocidine and spiramycin biosynthetic gene clusters,81,82 a 150 kb gene cluster was identified, which codes for 25 genes, among them 9 encode type I PKS, making this one of the largest PKS gene clusters known so far.83 The analysis with the software SEARCHPKS84 indicated that the PKS genes code for 112 enzymatic domains organized in 25 PKS modules, including a KSQ domain at the starter module and a TE domain at the last module. Based on the domain organization, specificity predictions for the AT domains and stereochemistry predictions of KR domains the authors were able to predict a planar structure of the PK product synthesized by the PKS – with only one ambiguity as the substrate for the AT of module 12 (corresponding to the PK-unit C-25–C-26) could not be predicted. A transcriptional analysis revealed that this gene cluster was not expressed under normal laboratory growth conditions. However Laureti et al. were able to activate the expression by constitutively expressing samR0484, a LuxR-type regulator. With this engineered strain it was now possible to isolate the biosynthesis product stambomycin and perform an NMR-based structure elucidation. The experimentally determined structure of stambomycin was in very good accordance with the theoretically predicted product of the PKS. However, several features were not predicted in silico: the NMR studies revealed that at position C-26 of the polyketide (corresponding to the ambiguous AT in module 12) extender units with diverse sidechains are used (leading to stambomycins A–D) and thus explains the lack of predictions for this module. In addition, the sites of macrolactonization, glycosylation and hydroxylations could not be predicted only based on the sequence analysis.83 However, this work nicely demonstrates the power of the genome mining approach and that genome sequence data can be used to predict structures of secondary metabolites.
A nice example where genetic data and structural data go hand in hand is the polyene macrolactam salinilactam. The salinilactam gene cluster is the biggest gene cluster that was detected by bioinformatic analysis in the Salinispora tropica CNB-440 genome.85 The high sequence identity and the repetitive nature of the various modules made correct assembly and closure of the genome problematic. However, based on characteristic UV chromophores the compound could be detected and initial structure elucidations suggested a 10-module PKS enzyme responsible for the biosynthesis of the compound, which facilitated assembly and therefore closure of the genome. The subsequent bioinformatic analysis of the AT domains on the other hand facilitated the final structure elucidation of the compound. This example aided the discovery of a whole family of macrolactams by implementing a “molecules-to-genes-to-molecules” approach by Schulze et al., who where able to detect the lobosamides and mirilactams by comparative genomic analysis.86
One important group of PKS enzymes are the trans-AT PKSs.87,88 These owe their name due to the lack of AT domains in each module; instead they encode one or more AT domains in trans, usually within each BGC. Genome mining efforts have shown that this group of enzymes is wide spread in bacteria, especially in chemically less studied genera.89trans-AT PKS systems have evolved independently from cis-AT PKSs90 and often include unusual biosynthetic enzymes leading to unique chemistry. Therefore, trans-AT PKS have been targeted by genome mining strategies in the past and led to the discovery of new compounds such as thailandamides,90 rhizopodin91 or tolytoxin.92 For an in depth review about these systems, we refer to Helfrich et al.88
Mining for PKS core enzymes is not restricted to cis-AT and trans-AT modular type I PKS; the technique can also be highly efficiently applied for other types of PK pathways: using the antiSMASH software, 20 gene clusters were predicted in the rare actinomycete Streptosporangium sp. CGMCC 4.7309, among them the hex-cluster coding for a type II PKS.93 Phylogenetic analyses indicated that the gene product of hex23, which encodes the KSβ of the type II PKS, can be assigned to a group of pathways producing pentangular polyphenols. Indeed, Tian and colleagues were able to detect and elucidate a new family of polyketides, named hexaricins and experimentally confirmed by gene knock-out that the gene cluster identified is responsible for the biosynthesis of the compound.
In the case of type II PKS pathways, the consideration of phylogenetic data can give more accurate predictions on the putative products on metagenomic datasets94 (for details, see Section 5).
3.3 Non-ribosomally synthesized peptides (NRPs)
The high degree of conservation of the core-enzymes involved in NRP biosynthesis, the good co-linearity of the modular domain organization of NRPS enzymes with their biosynthetic products, the possibility to predict substrate specificities, e.g. using software like NRPSpredictor,71,72 SEQL-NRPS95 or other A-domain specificity predictors96–98 have made this family of secondary metabolites interesting candidates for genome mining approaches.96,99 This is further supported by the availability of cheminformatic approaches like MS/MS networking29,100 or iSNAP101 that allow the automatic mapping of identified and in silico analyzed NRP (and RiPP) BGCs to mass spectrometric data.73,101–104Genome mining, for example, led to the identification of NRP secondary metabolites also from organisms, such as Staphylococcus aureus,105 which usually are not regarded as prolific secondary metabolite producers. In the case of the biosynthesis of the siderophore coelichelin from the model actinomycete Streptomyces coelicolor M145, genome mining revealed new biochemical insights about NRPS biochemistry. The coelichelin pathway, which was identified in the S. coelicolor genome by searching for NRPS-like genes, codes for a 3-module NRPS, but was experimentally confirmed to generate a tetrapeptide,106 indicating exceptions of the co-linearity rule.
In a global view, mining for genes encoding NRPS or NRPS-related enzymes has revealed a wide distribution of these pathways in Bacteria and Eukarya. In a comprehensive study by Wang et al.107 more than 3300 BGCs coding for more than 16500 NRPS(like) enzymes have been identified. Interestingly, a significant number of the in silico identified enzymes do not follow the classical modular organization of NRPSs. Only few of these pathways have currently been studied experimentally, for example the pathway for congocidin biosynthesis.81 This makes it very challenging to interpret these BGCs and develop bioinformatics algorithms to predict their products.
3.4 Ribosomally synthesized and post-translationally modified peptides (RiPPs)
Lanthipeptide/lantibiotics have also been subject to successful genome mining approaches. One of the first examples, where the identification of the biosynthetic gene cluster preceded the discovery of the compound is lichenicidin.108 Begley et al. used PSI-BLAST to mine publicly available microbial genome sequences for homologues of the LanM lanthipeptide dehydratase/cyclase, which catalyzes the dehydration and cyclization of the lanthipeptide prepeptide using the sequence of LtnM1 involved in lacticin 3147 biosynthesis as query. Using this strategy, they were able to identify 89 strains. 61 of these strains were not previously described as lanthipeptide producers. Among these hits was Bacillus licheniformis ATCC 14580, which inhibited growth of Gram-positive bacteria, including L. monocytogenes, methicillin resistant S. aureus and vancomycin resistant enterococci. Indeed a lanthipeptide named lichenicidin, which had the predicted molecular weight could be identified with mass spectrometry, isolated and re-tested demonstrating that it caused the antibacterial effect.Another interesting example is the lanthipeptide venezuelin.109 When analyzing the draft genome sequence of S. venezuelae, a novel type of lanthionine cyclase/dehydratase named VenL was identified, which was composed of an N-terminal serin/threonine kinase instead of the dehydratase present in LanM/type II lanthipeptide biosynthesis pathways, and a C-terminal LanC domain. Although it was not possible to isolate a lanthipeptide from S. venezuelae, the authors were able to express and purify VenL in E. coli and demonstrate its activity as lanthionine synthase in vitro using synthesized peptides.109,110 Using the datasets of Doroghazi et al.,111 only recently it was possible to identify further Streptomyces strains that possess BGCs that are similar to the venezuelin gene cluster.112 HPLC-MS analyses indicated, that some of these strains indeed produce venezuelin or closely related derivatives. The latest member of the venezuelin family of lanthipeptides is streptocollin, which was identified via antiSMASH-based genome mining of S. collinus Tü 365.113,114 This strain only produces traces of streptocollin under the tested laboratory conditions. However, by expressing the streptocollin biosynthesis genes under control of a constitutive promoter in S. collinus Tü 365 or by heterologously expressing the gene cluster in the optimized expression host S. coelicolor M1152,115 preparative amounts of streptocollin could be obtained. While no significant antibacterial or antiviral activity was detected for streptocollin, a moderate inhibitory activity towards protein-tyrosine-phosphatase 1B, a potential target to treat type 2 diabetes or obesity, was observed.113
Similar to NRPs, the combination of genome mining with computational mass spectrometry approaches provides valuable tools for the identification of novel compounds.103,116 One of the first peptides identified with this method was the type III lanthipeptide informatipeptin produced by Streptomyces viridochromogenes DSM 40736.116
Lasso peptides are another very interesting family of RiPPs where genome mining has contributed much knowledge. Like most RiPPs, biosynthesis of lasso peptides starts with the ribosomal synthesis of a precursor peptide (A-peptide). This precursor is post-translationally modified by an ATP-dependent cysteine protease (B-protein), which cleaves-off the leader peptide, and an ATP-dependent asparagine synthetase B-like protein (C-protein), which catalyzes the lactam formation between the N-terminal amino group and the side chain of an aspartate or glutamate residue in the peptide leading to a cyclized molecule. The latter reaction is catalyzed in a way such that the C-terminal peptide tail is placed inside the ring thus leading to the name-giving lasso structure. This structure is usually stabilized by bulky plug residues of the peptide tail that prevent the slip-out and disulfide bonds that stabilize the structure.117
The first gene cluster of a lasso peptide was microcin J25 of E. coli AY25,118–120 where it was demonstrated that the precursor peptide along with the genes mcjB and mcjC, which code for the B- and C-proteins respectively, are responsible for microcin J25 biosynthesis. In addition, the microcin J25 gene cluster contains mcjD, which codes for an ABC transporter that is involved in conferring immunity. In the first genome mining study that targeted novel lasso peptides, the sequences of McjB, McjC and McjD were used as in silico probes to mine genomic databases. Knappe et al.121 were able to detect genes coding for homologous enzymes in the genome sequence of Burkholderia thailandensis E264. In a manual reinvestigation of the DNA sequence upstream of the identified homologs the authors could identify an un-annotated open reading frame that was proposed to encode the precursor peptide. Indeed, the authors were successful in detecting traces of the predicted lasso peptide, named capistruin, in culture extracts of the strain. By optimizing the fermentation media and purification procedure, it was possible to obtain capistruin yields of 0.7 mg L−1, which provided sufficient amounts to confirm the structure of capistruin via NMR-based methods. Finally, it was possible to clone the capistruin BGC comprising of capABCD and express the pathway in E. coli with yields of 0.2 mg L−1, which is ∼30% of the B. thailandensis wild type yield.121 In the meantime, many additional lasso peptide biosynthetic gene clusters have been identified by genome mining (Table 1). Similar approaches have also been carried out for other classes of RiPPs, for example thiazole/oxazole modified microcins (TOMMs).112
3.5 Terpenoids/isoprenoids
With almost 400 distinct structural families comprising in total more than 55.000 described compounds, terpenoid/isoprenoid secondary metabolites are one of the largest class of bioactive metabolites, many produced by plants, fungi and also bacteria.122 The core units of terpenoids are assembled from a varying number of linked isoprene units (catalyzed by terpene synthases) that can undergo a multitude of intramolecular cyclizations, catalyzed by terpene cyclases, often followed by extensive tailoring steps. Although terpene synthases/cyclases are often not as highly conserved as, for example, NRPS or PKS it is possible to use individual characterized sequences as probes. Using these strategies, a variety of isoprenoids have been firstly identified and characterized by genome mining, e.g., the monoterpene cineolole,123 the diterpene cembrane,124 or the sesterterpene stellatic acid.125To cover a wide sequence space and to identify more distantly related enzymes, a profile-HMM-based approach, that uses HMM-profiles trained with 140 bacterial terpene synthases, was also successfully used to screen novel pathways and identify a diverse set of new terpenoids.126,127
3.6 Screening for tailoring enzymes
Currently, most genome mining approaches focus on identifying core biosynthetic enzymes in genomic or metagenomic data.128 However, also so-called tailoring enzymes, that are involved in modifying precursor molecules, can be valuable targets to identify new BGCs. Already in the pre-genome mining era, reverse genetics approaches have been carried out to identify, for example, pathways encoding halogenases.129With the availability of whole genome sequences, this strategy has expanded in silico. For example, sequences of bacterial desaturases/acetylenases were used as genome mining probes to identify biosynthetic gene clusters synthesizing compounds containing very rarely found alkyne groups.130,131 Other examples are the identification of BGCs encoding the biosynthesis of epoxyketone biosynthetic pathways in metagenomic datasets that led to the discovery of the clarepoxcins and landepoxcins, potent 20S proteasome inhibitors.132
4. Comparative genome mining
“Classical” genome mining, i.e. the specific search for genes encoding enzymes involved in the biosynthesis of secondary metabolites in (meta)genomic sequences, and sophisticated software aiding the scientists for this task, were one of the main innovations within the history of natural products research in the recent years.64,133 The method can be further refined by not only focusing on single genes, but on partial or complete gene clusters. This functionality is, for example, included in the software antiSMASH,78 which can compare the identified BGCs in user-submitted genomes with a huge collection of BGCs of other microorganisms and the curated MIBiG database,47 and thus indicate whether there are similar pathways in other organisms. The MultiGeneBlast algorithm, that provides such information, is also available as a stand-alone software,134 which can be used to identify similar gene clusters/operons for any given sequence.Another approach to assess novelty of identified BGCs is to use Genome Neighborhood Networks (GNNs),135 which are extended variants of sequence similarity networks136 (Fig. 2) that also take into account the neighborhood of the “target” gene. Such a study has recently been carried out to comprehensively study enediyne biosynthetic pathways and provide means for in silico prioritization.137
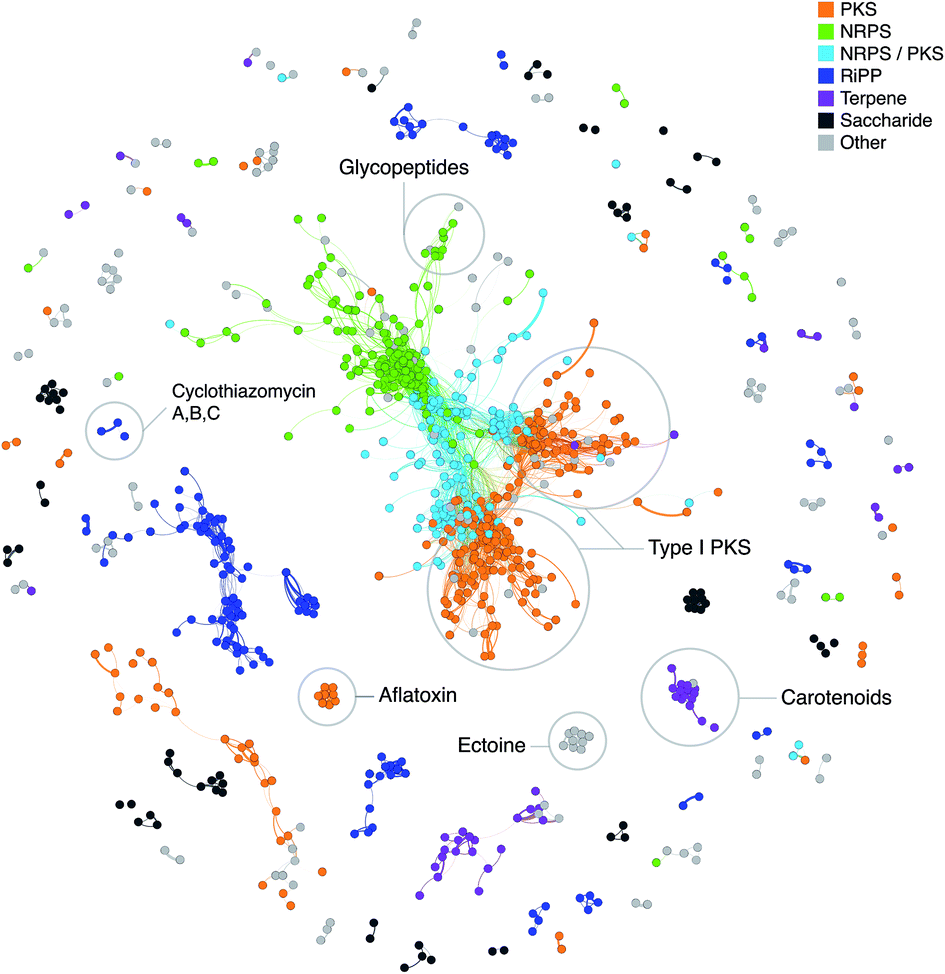 | ||
| Fig. 2 Network of known BGCs from the MIBiG database47 using Pfam composition similarity. Distances were calculated with an adapted Jaccard and domain duplication index149 with a threshold of 0.5 and clustered in Gephi156 using the Yifan Hu method.157 Colored regions depict gene cluster type with examples of known compounds circled based on MIBiG annotations. | ||
Although targeted genome mining approaches are very powerful and have led to the identification of new compounds and associations between biosynthetic pathways and molecules, they have one important limitation. As this approach requires sequences of known homologous enzymes or explicitly defined rules on what is considered a BGC, the results are limited to known biosynthetic types and families. Often, a difficult task is to connect new structural classes of compounds to their respective BGCs. Once the BGC and biochemistry is identified, homologous clusters and corresponding compounds can be detected in other organisms as well. A powerful example for this is the patellamide family of compounds.138 These highly modified cyclic peptides have been isolated from the marine ascidian Lissoclinum patella and were shown to be produced by cyanobacterial symbionts.139 Due to their highly modified amino acids, they were suspected to be produced by an NRPS dependent machinery for a long time. In contrast, Schmidt and colleagues were able to link the production to a class of ribosomal pathways140 later called cyanobactins that added to the great variety of RiPP pathways and led to the discovery of many more related molecules.141–143 Similarly, the microviridin BGCs have been discovered.56 These tricyclic peptides are biosynthesized by previously unknown classes of ATP grasp domain containing enzymes,56,144,145 which were shown to be wide spread within free-living cyanobacteria.146,147
One approach to find unknown types of biosynthetic gene clusters more systematically was recently reported by Takeda et al.148 The algorithm used to identify BGCs in filamentous fungi is based on identifying homologous and orthologous genes in related species, and evaluate syntenic and non-syntenic regions. Unfortunately, the implementation of the algorithm or a web server is not yet publicly available.
Another strategy was developed by Cimermancic et al.149 The ClusterFinder algorithm uses a two-state hidden Markov model trained on strings of contiguous Pfam domains identified in 677 validated gene clusters (BGC state) and strings of Pfam domains of random genome regions (non-BGC state). This probabilistic model then was used to screen 1154 prokaryotic genomes, leading to the identification of more than 33000 putative BGCs, 10700 of the hits with high confidence scores. The most abundant class of biosynthetic gene clusters identified in this study were saccharides, which comprised 40% of all hits. This was unexpected as this class of molecules is only represented with 13% of the validated test data set and thus indicates a huge potential for finding new molecules. A global similarity analysis of the high-confidence hits revealed several “cliques”/families of BGC that have not yet experimentally studied so far. Experimental studies on selected members of this family (comprising 811 BGCs) revealed that one family codes for the biosynthesis of aryl polyene lipids.149 ClusterFinder was also used in a study analyzing data from the Human Microbiome Consortium.150 In the human microbiome, saccharide and RiPP gene clusters were also observed frequently, whereas NRPS and PKS gene clusters were significantly depleted.151 Based on these data, Donia et al. were able to identify a novel thiopeptide (RiPP) lactocillin, associate it with the corresponding BGC, and demonstrate that the biosynthesis pathway is actually transcribed in the human body.150
In a large-scale genome mining approach for type I and II polyketides, NRPS, NRPS-independent siderophores, lanthipeptides and TOMMs in more than 800 actinobacterial genomes Doroghazi et al.152 were able to identify more than 11000 gene clusters which could be grouped into 4122 gene cluster families (GCFs). The GCF network was calculated based on the combination of three distance metrics on the number of shared homologous genes, the proportion of nucleotides involved in pairwise alignment and the amino acid sequence identity of domains of modular enzymes. 77 of these GCFs included at least one already known and characterized gene cluster. Based on these, the authors identified 1193 uncharacterized gene clusters in a subset of 344 genomes, which likely code for the biosynthesis of novel derivatives of known compounds. The GCF network then was correlated with large-scale high-resolution liquid chromatography/mass-spectrometry data of a subset of 178 strains allowing the link between metabolites and gene clusters.152 These datasets now are a valuable source for further studies, such as a global analysis of lanthipeptide biosynthetic pathways112 (see above).
These and other recent examples nicely demonstrate that especially combining genome mining with metabolomics/cheminformatics approaches, i.e. the automatic evaluation of mass spectrometric data using peptide- or glycogenomics,29,30 molecular networking,100 self-organizing metabolomic maps,153 or comprehensive MS/MS fragment databases, provide very powerful tools to identify novel secondary metabolites.32,102,103,116,154,155
5. Phylogeny based mining methods
The idea of comparing multiple bacterial genomes to detect and prioritize gene clusters is strongly connected to the idea of using phylogenetic methods to find promising secondary metabolite genes. As Dobzhansky stated: “Nothing in biology makes sense except in the light of evolution”.184 Understanding how nature creates the amazing structural diversity of chemicals observed and appreciated by natural product chemists is not just a basic research question, but gives important insights into bioprospecting efforts, ecological function of these molecules and synthetic biology approaches.The often modular structure of secondary metabolite gene clusters rendered the idea of a rapidly evolving defence system that develops new molecules by randomly shuffling and swapping domains and modules and develops analogous to an adaptive immune system.185 The first phylogenetic studies with selected PKS and NRPS systems clearly showed significant amounts of homologous recombination and gene duplication.186–189 Most of these studies, however, are limited case studies that include only a few related gene clusters that code for very similar molecules. The limited amount of data available at the time and the intensive computational demands on phylogenetic algorithms impeded more systematical analysis. With the diversity of tools for genome mining available now, more systematic approaches to infer the evolutionary history of nature's chemistry are available. One recent analysis of the evolution of about 10000 biosynthetic gene clusters revealed significantly higher rates of insertions, deletions and duplications in secondary metabolism compared to primary metabolism and demonstrated how successful nature mixes and matches sub-clusters to produce novel chemistry.190
Evolution and phylogenetic approaches have been used to guide genome mining efforts for a couple of years now. Two major approaches can be distinguished in natural product research.191,192 One uses species trees of natural product producing organisms based on conserved housekeeping genes or core genomes, and maps compound production subsequently on the tree. This way, talented chemistry producing lineages can be traced to develop more efficient sampling, isolation and genome mining techniques.193 The genome mining of 75 closely related Salinispora strains for example revealed major differences in the metabolic diversity of the three distinct species within the genus, proving S. pacifica as the most diverse compared to its sister taxa S. tropica and S. arenicola.194 In contrast, another study that compared secondary metabolite gene cluster diversity within the Streptomyces species S. pratensis revealed no differences in natural product BGCs, even when isolated from various distant geographic locations.195 An improved understanding of these phylogenetic patterns can guide bioprospecting efforts, especially in combination with the discovery of new families and clades within bacteria through directed cultivation efforts51 or metagenomic sequencing.196
However, species patterns and taxonomical correlations have to be interpreted carefully considering the tremendous amount of horizontal gene transfer, which proves to play a vital role in the evolution of secondary metabolites.189,190,194,197 A second approach uses gene trees of secondary metabolite genes directly. These gene trees infer the evolutionary history of biosynthetic genes and gene clusters, and can often be used to infer biosynthetic functions of the enzymes more precisely than simple sequence similarity approaches.198,199
For more in depth information about evolution and the use of phylogenetic methods of natural products, we refer to other reviews.191,192 Here we focus on examples of phylogeny based mining techniques. Ketosynthase (KS) domains in PKS gene clusters have been one of the first enzyme families where phylogenetic trees have been used to predict structures. Instead of sequencing full genomes, at a time when this was not even yet feasible, degenerated primers were constructed and amplified PCR products sequenced.186,200,201 Based on the phylogeny of these short sequence tags compound families and structures could be predicted. If characterized known KS domains claded closely with amplified PCR products, lineages without any known characterized KS domain indicated unknown or novel chemistry.
This concept was further developed by the Natural Product Domain Seeker (NaPDoS), a bioinformatic pipeline that allows the automated detection of KS and C domains from NRPS and PKS gene clusters and subsequently constructs a phylogenetic tree to infer novelty and potential of secondary metabolites from bacterial genetic data.27 NaPDoS works for PCR products but also draft and full bacterial genomes as well as assembled metagenomic data sets (http://napdos.ucsd.edu). The sequence tag approach is especially useful for screening complex environmental data such as soil and sediments, where complete gene cluster assemblies are still challenging using recent sequencing technologies, and can be used to infer diversity and genetic potential of natural products in specific environments.132,202–204 Brady and colleagues developed the webtool esNAPD37 and phylogeo, a specialized R package38 to facilitate richness and diversity analysis of NRPS and PKS sequence tags in soil and direct the discovery of bioactive natural products from metagenomes as shown for a novel pentangular polyphenol type II polyketide.94 This general concept of phylogeny of sequence tags as a guide for secondary metabolite genome mining was also expanded to other enzyme families such as the chromopyrrolic acid synthase responsible for the biosynthesis of rare tryptophan dimers205,206 and the epoxyketone family of compounds.132,173
A completely new aspect of using phylogeny and evolutionary distances for genome mining purposes has been recently developed by Barona-Gomez and colleagues.33 Their EvoMining approach is based on the concept that enzymes involved in secondary metabolism evolved by duplication and subsequent expansion of substrate specificity of primary enzymes and these expanded and repurposed enzyme families are detectable with phylogenetic methods. They developed a genome mining pipeline that detects homologues of certain classes of housekeeping genes and compared the average number and phylogenetic distance of each enzyme family.
As proof of principle two previously uncharacterized enzymes could be identified, an argininosuccinate lyase involved in the biosynthesis of leupeptin and an unusual AroA family enzyme involved in the biosynthesis of a putative novel arseno-organic compound. Both compounds in this case have been linked to known biosynthetic machineries such as NRPS and PKS based systems. However, this approach has not only the potential to detect novel highly unusual and previously uncharacterized enzyme families but could be useful to detect non-canonical secondary metabolic pathways, where no known machinery has been previously described.
6. Resistance/target based mining methods
Methods and tools to detect and extract secondary metabolite gene clusters are nowadays quite well established, and with all the microbial genomes available the biggest database at this point, the JGI IMG-ABC database, contains more than 960000 detected gene clusters,46 the majority are orphan. Analysing and characterizing each and every one of these orphan gene clusters in real wet-lab experiments is clearly not feasible. The challenge is now to prioritize and focus on the most promising gene clusters and gene cluster families. Which of the gene clusters is most interesting certainly depends on what exactly researchers are looking for and ranges from novel biosynthetic mechanisms, novel structures or derivatives of compounds to new bioactivities and modes of actions. However, predicting bioactivities and mechanisms of actions in silico remains one of the challenges of computational methods in drug discovery.
Resistance based and target based mining techniques are relatively recently developed genome mining approaches that aim to detect secondary metabolite gene clusters based on the self-resistant mechanisms of an antibiotic producing organism. The idea is based on the experience that BGCs not only contain the biochemical enzymes for compound production, but also encode additional important information such as regulatory elements, transporter proteins and resistance mechanisms. A bacterium that produces an antibiotic compound needs to develop self-resistant mechanisms in order to avoid suicide.207 The resistant mechanisms vary and include efflux pumps, degrading enzymes to remove toxic compounds, and modified target proteins to prevent binding of antibiotics to the active site of their targets.208 The gene clusters responsible for the biosynthesis of the antibiotics novobiocin,209 platensin210 and griselimycins211 are some examples were second copies of resistant housekeeping genes (gyrB, fabB/F and dnaN respectively) are directly encoded within the gene cluster.
The first proof of principle that resistance can be used as a discriminating criterion in antibiotic producing organisms has been published in 2013. Wright and co-authors were able to show that organisms resistant to glycopeptide and ansamycin-like antibiotics are more likely to produce similar chemical compounds,212 and an antibiotic resistance based discovery platform was developed for the isolation of scaffold-specific antibacterial producers.213 Moore and colleagues took this approach one step further and developed a target-directed genome mining approach.35 By screening 86 highly related strains of the marine actinomycete genus Salinispora for second copies of house keeping genes and relating those to their presence in biosynthetic gene clusters, they were able to identify a duplicated bacterial fatty acid synthase in the direct vicinity of an non-canonical hybrid PKS–NRPS gene cluster. Using direct cloning, heterologous expression and mutational analysis, the gene cluster was linked to the biosynthesis of thiolactomycin, a known fatty acid synthase inhibitor. Thus, correlating putative resistance genes with orphan secondary metabolite gene clusters can be one way to mine bacterial genomes specifically for antibacterial compounds, at least in those cases, where resistance is mediated by target modification.
With the rise of antibiotic resistant pathogens and the urgent need for new antibiotics with novel mode of actions, resistance-based genome mining techniques will be an important toolkit for the discovery of specifically antibacterial compounds and mechanism of action studies in the future. Well-curated databases of resistance genes such as CARD34 and ARDB214 will play an important part in these efforts. A recent example for an automated online tool that can connect genomic data, chemical structures and resistance genes is PRISM.215 Originally created to connect structural information and BGCs in a high throughput fashion, a resistance-determinant library of known antibiotic resistance has been added in order to enable the targeted search for compounds with uncommon modes of action.104 As proof of principle Johnston and colleagues investigated the telomycin family of natural products and identified a new mode of action for these compounds binding to the phospholipid cardiolipin.
7. Mining for regulators
Understanding the complex regulation of secondary metabolites in bacteria has been an important part of in the history of genetics of secondary metabolites.216 Especially after whole genome sequences unveiled the large amount of silent gene clusters not active under normal laboratory conditions, researchers are looking for effective ways to activate and optimize production of encoded compounds. Specifically bacteria of the genus Streptomyces have been extensively studied for their complex regulatory networks involved in secondary metabolism.216–218 Global and pathway specific regulators,216,218 precursors from primary metabolism,219 and small molecules220,221 including N-acetylglucosamine222 have been shown to play a role in regulation of natural products biosynthesis. Various tools and databases have been developed to predict regulatory elements in bacterial genomes and facilitate drug discovery and compound optimization.223,224One of the best-known systems of globally regulated secondary metabolites present in a wide range of bacteria are metal–chelating agents, including the well known iron binding siderophore compounds.225 Siderophores are produced when iron is limited to solubilize and facilitate the uptake of iron into the cells. Other metal chelators are able to bind zinc or copper, respectively.226 Involved in production regulation of these metal–chelating compounds are two large families of global regulators, the Fur and the IdeR family of transcriptional repressors.227,228 If enough of the respective trace metal is present in the cell, the repressor–metal complex binds upstream of the BGC and silences transcription and production of chelators. If metal concentration in the cytoplasm drops, the metal–regulator complex disintegrates and is no longer able to bind DNA, and production of compounds are activated. Using this knowledge Stegmann and colleagues recently developed a method called INBEKT (Identification of Natural compound Biosynthesis pathways by Exploiting Knowledge of Transcriptional regulation) to mine the genome of Amycolatopsis japonicum MG417-CF17 for the BGC for ethylenediamine-disuccinate (EDDS).36 EDDS is a biodegradable zinc–chelating alternative for the widely industrially applied ethylenediamine-tetraacetate (EDTA). No biosynthetic genes could be detected for EDDS using canonical genome mining tools. However, screening the genome of Amycolatopsis japonicum for zinc-binding regions led to the identification and characterization of the aes genes responsible for EDDS production. Considering that certain classes of regulators are more often connected to secondary metabolites than others, this approach could be further developed in the future to mine genomes for non-canonical secondary metabolite pathways.
Consideration of regulators is also an important technique to study fungal secondary metabolite biosynthetic gene clusters.
Many fungal gene clusters encode cluster-specific regulators, which result in activation of the gene cluster when artificially overexpressed.167 As the regulator binding sites are often highly conserved, they can be used to identify genes belonging to fungal gene clusters by computational methods, such as implemented in the software CASSIS.229 Especially in fungi, genes belonging to the same biosynthetic gene cluster are highly co-regulated. Thus integrating transcriptomics data into the genome mining workflows can provide additional means to identify BGCs and define gene cluster borders.230–232 While the first approaches to integrate such data have involved many manual steps, recently easy-to-use web-based implementations of this approach have become available.233
8. Culture independent mining: single cells and metagenomes
The amount and complexity of metagenomic data makes mining them for secondary metabolites a specifically challenging task. Especially large and highly repetitive gene clusters such as NRPS and PKS pathways are rarely fully assembled. However mining environmental DNA for natural products opens the possibility to explore the large microbial world present that is not yet cultured and study diversity and distribution of these compounds directly in their environment. Heterologous expression and synthetic biology approaches allow subsequent capturing of sequenced pathways and enable compound discovery.94,234 During the last decade assembly problems were bypassed by sequence tag approaches and phylogenetic classification to dereplicate and identify promising novel biosynthetic gene clusters. Brady and colleagues used sequence tag approaches to discover new compounds directly from soil DNA,94,132,206 a diverse environment full of yet uncultured bacterial diversity and hidden natural product potential.51,202,203How much potential the so-called microbial dark matter really contains in terms of chemistry and novel biosynthetic machineries, has been demonstrated by genome mining studies from obligate symbiotic bacteria of ascidians and sponges.140 Schmidt and colleagues were able to identify the biosynthetic machinery of the patellamides by screening fosmid libraries from enriched Prochloron DNA, the cyanobacterial symbiont of the ascidian Lissoclinum patella. Heterologous production in E. coli proved a highly unusual RiPP pathway was responsible for the biosynthesis of these cyclic peptides (chapter 4). Sequencing the microbiome of further ascidians and bryozoans allowed the assembly of genome sequences of symbiotic proteobacteria, led to the identification of the patellazole and bryostatin gene clusters, and showed the important role of secondary metabolites in these organisms.235-237
A combination of metagenomic sequencing and single cell genomics of a sponge microbiome recently revealed a whole new candidate phylum ‘Tectomicrobia’ widely distributed in sponges.196 Two different genomes of the candidate genus ‘Entotheonella’ were assembled with genomes larger than 9 megabases and multiple biosynthetic gene clusters including the highly unusual polytheonamide pathway.238
With sequencing cost constantly falling and improving read length from technologies such as PacBio239 and Nanopore,240 assembly and mining of single cells and complex environments will be easier in the future and help to uncover more of the microbial dark matter and the enormous amounts of cryptic gene clusters in unusual environments such as the human microbiome.150
9. Conclusions
Enabled by the fast development of genome sequencing technologies, genome mining techniques rapidly evolved during the last decade and are currently an important part of drug discovery efforts. Depending on focus and interest of researchers, different mining techniques and approaches have been developed to detect, dereplicate and prioritize secondary metabolite gene clusters. The sheer amount of data available is, and will be challenging in the future, and will drive the constant development of effective computational tools and algorithms to guide wet lab experiments. Sequencing of uncultured organisms and whole environments, hand in hand with the improvements in “omics” technologies, systems biology approaches and synthetic biology will drive automated high-throughput analysis connecting genomic, transcriptomic and metabolomic data to connect genes more efficiently to molecules and vice versa (Fig. 3). Furthermore, the possibilities of studying diversity and distribution of secondary metabolites in their direct environment provides the chance to learn more about the impact of these compounds in their natural habitat. Fascinating examples of complex symbiotic interactions of microbes and their environments regulated by small molecules and secondary metabolites prove their important impact on shaping environmental niches. A true understanding of natural products, their evolution and role in the environment will give important insights in regulation, distribution and mode of action studies and therefore facilitate and maintain their use as human therapeutics.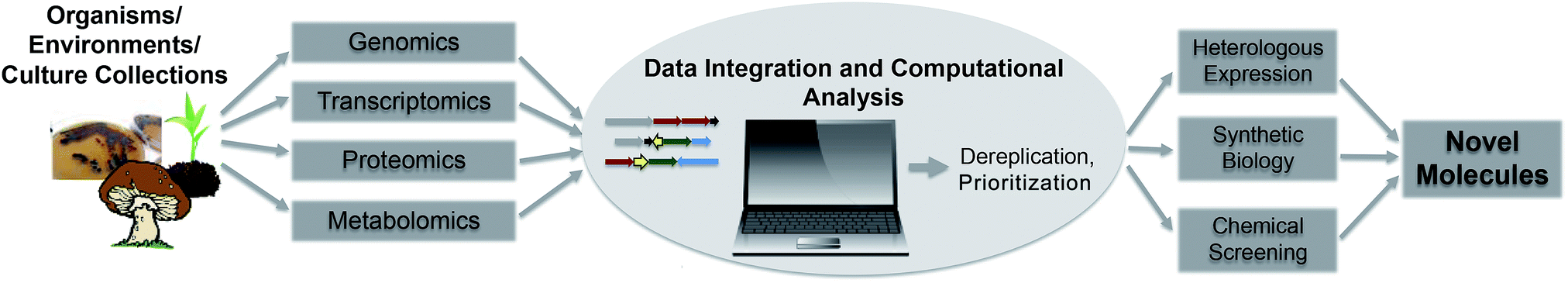 | ||
| Fig. 3 Flowchart of future automated genome mining approaches. | ||
10. Acknowledgements
The authors thank Harald Gross, Evi Stegmann, Yvonne Mast, Bradley Moore, and Michelle Schorn for helpful comments. NZ is funded by the German Center for Infection Biology (DZIF). The work of TW is funded by a grant of the Novo Nordisk Foundation.11. References
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed.
- G. L. Challis and D. A. Hopwood, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14555–14561 CrossRef CAS PubMed.
- L. F. Wright and D. A. Hopwood, J. Gen. Microbiol., 1976, 96, 289–297 CrossRef CAS PubMed.
- B. A. Rudd and D. A. Hopwood, J. Gen. Microbiol., 1979, 114, 35–43 CrossRef CAS PubMed.
- F. Malpartida and D. A. Hopwood, Nature, 1984, 309, 462–464 CrossRef CAS PubMed.
- D. A. Hopwood and H. M. Wright, J. Gen. Microbiol., 1983, 129, 3575–3579 CAS.
- H. Ikeda, H. Kotaki and S. Omura, J. Bacteriol., 1987, 169, 5615–5621 CAS.
- A. Bechthold, J. K. Sohng, T. M. Smith, X. Chu and H. G. Floss, Mol. Gen. Genet., 1995, 248, 610–620 CrossRef CAS PubMed.
- S. Pelzer, R. Süßmuth, D. Heckmann, J. Recktenwald, P. Huber, G. Jung and W. Wohlleben, Antimicrob. Agents Chemother., 1999, 43, 1565–1573 CAS.
- T. Weber, K. Welzel, S. Pelzer, A. Vente and W. Wohlleben, J. Biotechnol., 2003, 106, 221–232 CrossRef CAS PubMed.
- S. Donadio, M. J. Staver, J. B. McAlpine, S. J. Swanson and L. Katz, Science, 1991, 252, 675–679 CAS.
- S. Bentley, K. Chater, A.-M. Cerdeño-Tárraga, G. L. Challis, N. R. Thomson, K. D. James, D. E. Harris, M. A. Quail, H. Kieser, D. Harper, A. Bateman, S. Brown, G. Chandra, C. W. Chen, M. Collins, A. Cronin, A. Fraser, A. Goble, J. Hidalgo, T. Hornsby, S. Howarth, C.-H. Huang, T. Kieser, L. Larke, L. Murphy, K. Oliver, S. O'Neil, E. Rabbinowitsch, M.-A. Rajandream, K. Rutherford, S. Rutter, K. Seeger, D. Saunders, S. Sharp, R. Squares, S. Squares, K. Taylor, T. Warren, A. Wietzorrek, J. Woodward, B. G. Barrell, J. Parkhill and D. A. Hopwood, Nature, 2002, 417, 141–147 CrossRef PubMed.
- H. Ikeda, J. Ishikawa, A. Hanamoto, M. Shinose, H. Kikuchi, T. Shiba, Y. Sakaki, M. Hattori and S. Omura, Nat. Biotechnol., 2003, 21, 526–531 CrossRef PubMed.
- B. Gust, G. L. Challis, K. Fowler, T. Kieser and K. F. Chater, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1541–1546 CrossRef CAS PubMed.
- J. S. Tuan, J. M. Weber, M. J. Staver, J. O. Leung, S. Donadio and L. Katz, Gene, 1990, 90, 21–29 CrossRef CAS PubMed.
- U. J. Kim, H. Shizuya, P. J. de Jong, B. Birren and M. I. Simon, Nucleic Acids Res., 1992, 20, 1083–1085 CrossRef CAS PubMed.
- V. M. Chauthaiwale, A. Therwath and V. V. Deshpande, Microbiol. Rev., 1992, 56, 577–591 CAS.
- C. M. Farnet and E. Zazopoulos, Improving Drug Discovery From Microorganisms in Natural Products: Drug Discovery and Therapeutic Medicine, ed. L. Zhang and A. L. Demain, Humana Press, Totowa, NJ, 2005, pp. 95–106 Search PubMed.
- D. G. Gibson, L. Young, R. Chuang, J. C. Venter, C. a. Hutchison and H. O. Smith, Nat. Methods, 2009, 6, 343–345 CrossRef CAS PubMed.
- K. Yamanaka, K. A. Reynolds, R. D. Kersten, K. S. Ryan, D. J. Gonzalez, V. Nizet, P. C. Dorrestein and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1957–1962 CrossRef CAS PubMed.
- N. Kouprina and V. Larionov, Nat. Protoc., 2008, 3, 371–377 CrossRef CAS PubMed.
- S. H. Sternberg, S. Redding, M. Jinek, E. C. Greene and J. A. Doudna, Nature, 2014, 507, 62–67 CrossRef CAS PubMed.
- Y. Tong, P. Charusanti, L. Zhang, T. Weber and S. Y. Lee, ACS Synth. Biol., 2015, 4, 1020–1029 CrossRef CAS PubMed.
- E. Zazopoulos, K. Huang, A. Staffa, W. Liu, B. O. Bachmann, K. Nonaka, J. Ahlert, J. S. Thorson, B. Shen and C. M. Farnet, Nat. Biotechnol., 2003, 21, 187–190 CrossRef CAS PubMed.
- H. Gross, V. O. Stockwell, M. D. Henkels, B. Nowak-Thompson, J. E. Loper and W. H. Gerwick, Chem. Biol., 2007, 14, 53–63 CrossRef CAS PubMed.
- N. Ziemert, S. Podell, K. Penn, J. H. Badger, E. Allen and P. R. Jensen, PLoS One, 2012, 7, e34064 CAS.
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. a. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS PubMed.
- R. D. Kersten, Y.-L. Yang, Y. Xu, P. Cimermancic, S.-J. Nam, W. Fenical, M. A. Fischbach, B. S. Moore and P. C. Dorrestein, Nat. Chem. Biol., 2011, 7, 794–802 CrossRef CAS PubMed.
- R. D. Kersten, N. Ziemert, D. J. Gonzalez, B. M. Duggan, V. Nizet, P. C. Dorrestein and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E4407–E4416 CrossRef CAS PubMed.
- J. Gubbens, H. Zhu, G. Girard, L. Song, B. I. Florea, P. Aston, K. Ichinose, D. V. Filippov, Y. H. Choi, H. S. Overkleeft, G. L. Challis and G. P. Van Wezel, Chem. Biol., 2014, 21, 707–718 CrossRef CAS PubMed.
- K. R. Duncan, M. Crüsemann, A. Lechner, A. Sarkar, J. Li, N. Ziemert, M. Wang, N. Bandeira, B. S. Moore, P. C. Dorrestein and P. R. Jensen, Chem. Biol., 2015, 22, 460–471 CrossRef CAS PubMed.
- P. Cruz-Morales, C. E. Martínez-Guerrero, M. A. Morales-Escalante, L. A. Yáñez-Guerra, J. F. Kopp, J. Feldmann, H. E. Ramos-Aboites and F. Barona-Gomez, bioRxiv, 2015 DOI:10.1101/020503.
- A. G. McArthur, N. Waglechner, F. Nizam, A. Yan, M. A. Azad, A. J. Baylay, K. Bhullar, M. J. Canova, G. De Pascale, L. Ejim, L. Kalan, A. M. King, K. Koteva, M. Morar, M. R. Mulvey, J. S. O'Brien, A. C. Pawlowski, L. J. V. Piddock, P. Spanogiannopoulos, A. D. Sutherland, I. Tang, P. L. Taylor, M. Thaker, W. Wang, M. Yan, T. Yu and G. D. Wright, Antimicrob. Agents Chemother., 2013, 57, 3348–3357 CrossRef CAS PubMed.
- X. Tang, J. Li, N. Millán-Aguiñaga, J. J. Zhang, E. C. O'Neill, J. A. Ugalde, P. R. Jensen, S. M. Mantovani and B. S. Moore, ACS Chem. Biol., 2015, 10, 2841–2849 CrossRef CAS PubMed.
- M. Spohn, W. Wohlleben and E. Stegmann, Environ. Microbiol., 2016, 18(4), 1249–1263 CrossRef CAS PubMed.
- B. V. Reddy, A. Milshteyn, Z. Charlop-Powers and S. F. Brady, Chem. Biol., 2014, 21, 1023–1033 CrossRef CAS PubMed.
- Z. Charlop-Powers and S. F. Brady, Bioinformatics, 2015, 1–3 Search PubMed.
- A. Calteau, D. P. Fewer, A. Latifi, T. Coursin, T. Laurent, J. Jokela, C. a. Kerfeld, K. Sivonen, J. Piel and M. Gugger, BMC Genomics, 2014, 15, 1–14 CrossRef PubMed.
- M. Welker, E. Dittmann and H. Von Döhren, Methods Enzymol., 2012, 517, 23–46 CAS.
- S. C. Wenzel and R. Müller, Comprehensive Natural Products II, 2010 Search PubMed.
- K. Gerth, S. Pradella, O. Perlova, S. Beyer and R. Müller, J. Biotechnol., 2003, 106, 233–253 CrossRef CAS PubMed.
- S. C. Wenzel and R. Müller, Mol. BioSyst., 2009, 5, 567–574 RSC.
- S. Behnken and C. Hertweck, Appl. Microbiol. Biotechnol., 2012, 96, 61–67 CrossRef CAS PubMed.
- A.-C. Letzel, S. J. Pidot and C. Hertweck, Nat. Prod. Rep., 2013, 30, 392–428 RSC.
- M. Hadjithomas, I.-M. A. Chen, K. Chu, A. Ratner, K. Palaniappan, E. Szeto, J. Huang, T. B. K. Reddy, P. Cimermancic, M. A. Fischbach, N. N. Ivanova, V. M. Markowitz, N. C. Kyrpides and A. Pati, MBio, 2015, 6, 1–10 CrossRef PubMed.
- M. H. Medema, R. Kottmann, P. Yilmaz, M. Cummings, J. B. Biggins, K. Blin, I. de Bruijn, Y. H. Chooi, J. Claesen, R. C. Coates, P. Cruz-Morales, S. Duddela, S. Dusterhus, D. J. Edwards, D. P. Fewer, N. Garg, C. Geiger, J. P. Gomez-Escribano, A. Greule, M. Hadjithomas, A. S. Haines, E. J. Helfrich, M. L. Hillwig, K. Ishida, A. C. Jones, C. S. Jones, K. Jungmann, C. Kegler, H. U. Kim, P. Kotter, D. Krug, J. Masschelein, A. V. Melnik, S. M. Mantovani, E. A. Monroe, M. Moore, N. Moss, H. W. Nutzmann, G. Pan, A. Pati, D. Petras, F. J. Reen, F. Rosconi, Z. Rui, Z. Tian, N. J. Tobias, Y. Tsunematsu, P. Wiemann, E. Wyckoff, X. Yan, G. Yim, F. Yu, Y. Xie, B. Aigle, A. K. Apel, C. J. Balibar, E. P. Balskus, F. Barona-Gomez, A. Bechthold, H. B. Bode, R. Borriss, S. F. Brady, A. A. Brakhage, P. Caffrey, Y. Q. Cheng, J. Clardy, R. J. Cox, R. De Mot, S. Donadio, M. S. Donia, W. A. van der Donk, P. C. Dorrestein, S. Doyle, A. J. Driessen, M. Ehling-Schulz, K. D. Entian, M. A. Fischbach, L. Gerwick, W. H. Gerwick, H. Gross, B. Gust, C. Hertweck, M. Hofte, S. E. Jensen, J. Ju, L. Katz, L. Kaysser, J. L. Klassen, N. P. Keller, J. Kormanec, O. P. Kuipers, T. Kuzuyama, N. C. Kyrpides, H. J. Kwon, S. Lautru, R. Lavigne, C. Y. Lee, B. Linquan, X. Liu, W. Liu, A. Luzhetskyy, T. Mahmud, Y. Mast, C. Mendez, M. Metsa-Ketela, J. Micklefield, D. A. Mitchell, B. S. Moore, L. M. Moreira, R. Muller, B. A. Neilan, M. Nett, J. Nielsen, F. O'Gara, H. Oikawa, A. Osbourn, M. S. Osburne, B. Ostash, S. M. Payne, J. L. Pernodet, M. Petricek, J. Piel, O. Ploux, J. M. Raaijmakers, J. A. Salas, E. K. Schmitt, B. Scott, R. F. Seipke, B. Shen, D. H. Sherman, K. Sivonen, M. J. Smanski, M. Sosio, E. Stegmann, R. D. Süssmuth, K. Tahlan, C. M. Thomas, Y. Tang, A. W. Truman, M. Viaud, J. D. Walton, C. T. Walsh, T. Weber, G. P. van Wezel, B. Wilkinson, J. M. Willey, W. Wohlleben, G. D. Wright, N. Ziemert, C. Zhang, S. B. Zotchev, R. Breitling, E. Takano and F. O. Glöckner, Nat. Chem. Biol., 2015, 11, 625–631 CrossRef CAS PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS PubMed.
- B. Buchfink, C. Xie and D. H. Huson, Nat. Methods, 2015, 12, 59–60 CrossRef CAS PubMed.
- R. D. Finn, J. Clements and S. R. Eddy, Nucleic Acids Res., 2011, 39 DOI:10.1093/nar/gkr367.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schaberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen, K. Lewis, T. F. Schaberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- J. Claesen and M. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16297–16302 CrossRef CAS PubMed.
- L. C. Foulston and M. J. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13461–13466 CrossRef CAS PubMed.
- M. Spohn, N. Kirchner, A. Kulik, A. Jochim, F. Wolf, P. Muenzer, O. Borst, H. Gross, W. Wohlleben and E. Stegmann, Antimicrob. Agents Chemother., 2014, 58, 6185–6196 CrossRef PubMed.
- N. Ziemert, K. Ishida, P. Quillardet, C. Bouchier, C. Hertweck, N. T. de Marsac and E. Dittmann, Appl. 0, 2008, 74, 1791–1797 CAS.
- N. Ziemert, K. Ishida, A. Liaimer, C. Hertweck and E. Dittmann, Angew. Chem., Int. Ed. Engl., 2008, 47, 7756–7759 CrossRef CAS PubMed.
- Z. Xu, Z. Sun, S. Li, Z. Xu, C. Cao, Z. Xu, X. Feng and H. Xu, Sci. Rep., 2015, 5, 17400 CrossRef CAS PubMed.
- A. de Jong, S. a F. T. van Hijum, J. J. E. Bijlsma, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2006, 34, 273–279 CrossRef PubMed.
- T. Weber, C. Rausch, P. Lopez, I. Hoof, V. Gaykova, D. H. Huson and W. Wohlleben, J. Biotechnol., 2009, 140, 13–17 CrossRef CAS PubMed.
- T. Weber and H. U. Kim, Synthetic and Systems Biotechnology, 2016, 1, 69–79, DOI:10.1016/j.synbio.2015.12.002.
- B. O. Bachmann, S. G. Van Lanen and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2014, 41, 175–184 CrossRef CAS PubMed.
- C. N. Boddy, J. Ind. Microbiol. Biotechnol., 2014, 41, 443–450 CrossRef CAS PubMed.
- E. J. N. Helfrich, S. Reiter and J. Piel, Curr. Opin. Biotechnol., 2014, 29, 107–115 CrossRef CAS PubMed.
- M. H. Medema and M. A. Fischbach, Nat. Chem. Biol., 2015, 11, 639–648 CrossRef CAS PubMed.
- M. Nett, Prog. Chem. Org. Nat. Prod., 2014, 99, 199–245 CAS.
- R. J. Scheffler, S. Colmer, H. Tynan, A. L. Demain and V. P. Gullo, Appl. Microbiol. Biotechnol., 2013, 97, 969–978 CrossRef CAS PubMed.
- J. I. Tietz and D. A. Mitchell, Curr. Top. Med. Chem., 2015, 16, 1645–1694 CrossRef.
- T. Weber, Int. J. Med. Microbiol., 2014, 304, 230–235 CrossRef CAS PubMed.
- J. Yaegashi, B. R. Oakley and C. C. Wang, J. Ind. Microbiol. Biotechnol., 2014, 41, 433–442 CrossRef CAS PubMed.
- M. H. Li, P. M. Ung, J. Zajkowski, S. Garneau-Tsodikova and D. H. Sherman, BMC Bioinf., 2009, 10, 185 CrossRef PubMed.
- C. Rausch, T. Weber, O. Kohlbacher, W. Wohlleben and D. H. Huson, Nucleic Acids Res., 2005, 33, 5799–5808 CrossRef CAS PubMed.
- M. Röttig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef PubMed.
- M. A. Skinnider, C. W. Johnston, R. Zvanych and N. A. Magarvey, ChemBioChem, 2015, 16, 223–227 CrossRef CAS PubMed.
- A. Starcevic, J. Zucko, J. Simunkovic, P. F. Long, J. Cullum and D. Hranueli, Nucleic Acids Res., 2008, 36, 6882–6892 CrossRef CAS PubMed.
- A. de Jong, A. J. van Heel, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2010, 38, W647–W651 CrossRef CAS PubMed.
- A. J. van Heel, A. de Jong, M. Montalban-Lopez, J. Kok, O. P. Kuipers, M. Montalbán-López, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2013, 41, W448–W453 CrossRef PubMed.
- K. Blin, M. H. Medema, D. Kazempour, M. A. Fischbach, R. Breitling, E. Takano and T. Weber, Nucleic Acids Res., 2013, 41, W204–W212 CrossRef PubMed.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. a. Fischbach, R. Müller, W. Wohlleben, R. Breitling, E. Takano and M. H. Medema, Nucleic Acids Res., 2015, 43, W237–W243 CrossRef PubMed.
- B. Shen, Hindra, X. Yan, T. Huang, H. Ge, D. Yang, Q. Teng, J. D. Rudolf and J. R. Lohman, Bioorg. Med. Chem. Lett., 2015, 25, 9–15 CrossRef CAS PubMed.
- J. B. McAlpine, B. O. Bachmann, M. Piraee, S. Tremblay, A. M. Alarco, E. Zazopoulos and C. M. Farnet, J. Nat. Prod., 2005, 68, 493–496 CrossRef CAS PubMed.
- M. Juguet, S. Lautru, F. X. Francou, S. Nezbedova, P. Leblond, M. Gondry and J. L. Pernodet, Chem. Biol., 2009, 16, 421–431 CrossRef CAS PubMed.
- F. Karray, E. Darbon, N. Oestreicher, H. Dominguez, K. Tuphile, J. Gagnat, M. H. Blondelet-Rouault, C. Gerbaud and J. L. Pernodet, Microbiology, 2007, 153, 4111–4122 CrossRef CAS PubMed.
- L. Laureti, L. Song, S. Huang, C. Corre, P. Leblond, G. L. Challis and B. Aigle, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6258–6263 CrossRef CAS PubMed.
- G. Yadav, R. S. Gokhale and D. Mohanty, Nucleic Acids Res., 2003, 31, 3654–3658 CrossRef CAS PubMed.
- D. W. Udwary, L. Zeigler, R. N. Asolkar, V. Singan, A. Lapidus, W. Fenical, P. R. Jensen and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10376–10381 CrossRef CAS PubMed.
- C. J. Schulze, M. S. Donia, J. L. Siqueira-Neto, D. Ray, J. A. Raskatov, R. E. Green, J. H. McKerrow, M. A. Fischbach and R. G. Linington, ACS Chem. Biol., 2015, 10, 2373–2381 CrossRef CAS PubMed.
- J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14002–14007 CrossRef CAS PubMed.
- E. J. N. Helfrich and J. Piel, Nat. Prod. Rep., 2016, 33, 231–316 RSC.
- R. V. O'Brien, R. W. Davis, C. Khosla and M. E. Hillenmeyer, J. Antibiot., 2014, 67, 89–97 CrossRef PubMed.
- T. Nguyen, K. Ishida, H. Jenke-Kodama, E. Dittmann, C. Gurgui, T. Hochmuth, S. Taudien, M. Platzer, C. Hertweck and J. Piel, Nat. Biotechnol., 2008, 26, 225–233 CrossRef CAS PubMed.
- D. Pistorius and R. Müller, ChemBioChem, 2012, 13, 416–426 CrossRef CAS PubMed.
- R. Ueoka, A. R. Uria, S. Reiter, T. Mori, P. Karbaum, E. E. Peters, E. J. N. Helfrich, B. I. Morinaka, M. Gugger, H. Takeyama, S. Matsunaga and J. Piel, Nat. Chem. Biol., 2015, 11, 705–712 CrossRef CAS PubMed.
- J. Tian, H. Chen, Z. Guo, N. Liu, J. Li, Y. Huang, W. Xiang and Y. Chen, Appl. Microbiol. Biotechnol., 2016, 100, 4189–4199 CrossRef CAS PubMed.
- H. Kang and S. F. Brady, J. Am. Chem. Soc., 2014, 136, 18111–18119 CrossRef CAS PubMed.
- M. Knudsen, D. Sondergaard, C. Tofting-Olesen, F. T. Hansen, D. E. Brodersen and C. N. Pedersen, Bioinformatics, 2016, 32, 325–329 CrossRef PubMed.
- B. O. Bachmann and J. Ravel, Methods Enzymol., 2009, 458, 181–217 CAS.
- D. Baranašić, J. Zucko, J. Diminic, R. Gacesa, P. F. Long, J. Cullum, D. Hranueli and A. Starcevic, J. Ind. Microbiol. Biotechnol., 2014, 41, 461–467 CrossRef PubMed.
- C. Prieto, C. Garcia-Estrada, D. Lorenzana and J. F. Martin, Bioinformatics, 2012, 28, 426–427 CrossRef CAS PubMed.
- B. S. E. Editor and J. M. Walker, 1401.
- D. D. Nguyen, C.-H. H. Wu, W. J. Moree, A. Lamsa, M. H. Medema, X. Zhao, R. G. Gavilan, M. Aparicio, L. Atencio, C. Jackson, J. Ballesteros, J. Sanchez, J. D. Watrous, V. V. Phelan, C. van de Wiel, R. D. Kersten, S. Mehnaz, R. De Mot, E. a. Shank, P. Charusanti, H. Nagarajan, B. M. Duggan, B. S. Moore, N. Bandeira, B. Ø. O. Palsson, K. Pogliano, M. Gutiérrez and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2611–E2620 CrossRef CAS PubMed.
- A. Ibrahim, L. Yang, C. Johnston, X. Liu, B. Ma and N. A. Magarvey, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19196–19201 CrossRef CAS PubMed.
- H. Mohimani, W. T. Liu, R. D. Kersten, B. S. Moore, P. C. Dorrestein and P. A. Pevzner, J. Nat. Prod., 2014, 77, 1902–1909 CrossRef CAS PubMed.
- M. H. Medema, Y. Paalvast, D. D. Nguyen, A. Melnik, P. C. Dorrestein, E. Takano and R. Breitling, PLoS Comput. Biol., 2014, 10, e1003822 Search PubMed.
- C. W. Johnston, M. A. Skinnider, C. A. Dejong, P. N. Rees, G. M. Chen, C. G. Walker, S. French, E. D. Brown, J. Bérdy, D. Y. Liu and N. A. Magarvey, Nat. Chem. Biol., 2016 DOI:10.1038/nchembio.2018.
- M. A. Wyatt, W. Wang, C. M. Roux, F. C. Beasley, D. E. Heinrichs, P. M. Dunman and N. A. Magarvey, Science, 2010, 329(80), 294–296 CrossRef CAS PubMed.
- S. Lautru, R. J. Deeth, L. M. Bailey and G. L. Challis, Nat. Chem. Biol., 2005, 1, 265–269 CrossRef CAS PubMed.
- H. Wang, D. P. Fewer, L. Holm, L. Rouhiainen and K. Sivonen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9259–9264 CrossRef CAS PubMed.
- M. Begley, P. D. Cotter, C. Hill and R. P. Ross, Appl. Environ. Microbiol., 2009, 75, 5451–5460 CrossRef CAS PubMed.
- Y. Goto, B. Li, J. Claesen, Y. Shi, M. J. Bibb and W. A. van der Donk, PLoS Biol., 2010, 8, e1000339 Search PubMed.
- L. S. Family, Y. Goto, A. Okesli and W. A. van der Donk, Biochemistry, 2011, 50, 891–898 CrossRef PubMed.
- J. R. Doroghazi and W. W. Metcalf, BMC Genomics, 2013, 14, 611–623 CrossRef CAS PubMed.
- C. L. Cox, J. R. Doroghazi and D. A. Mitchell, BMC Genomics, 2015, 16, 778 CrossRef PubMed.
- D. Iftime, A. Kulik, T. Härtner, S. Rohrer, T. H. Niedermeyer, E. Stegmann, T. Weber and W. Wohlleben, J. Ind. Microbiol. Biotechnol., 2015, 43, 277–291 CrossRef PubMed.
- D. Iftime, M. Jasyk, A. Kulik, J. F. Imhoff, E. Stegmann, W. Wohlleben, R. D. Süssmuth and T. Weber, ChemBioChem, 2015, 16, 2615–2623 CrossRef CAS PubMed.
- J. P. Gomez-Escribano and M. J. Bibb, Methods Enzymol., 2012, 517, 279–300 CAS.
- H. Mohimani, R. D. Kersten, W. T. Liu, M. Wang, S. O. Purvine, S. Wu, H. M. Brewer, L. Pasa-Tolic, N. Bandeira, B. S. Moore, P. A. Pevzner and P. C. Dorrestein, ACS Chem. Biol., 2014, 9, 1545–1551 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS PubMed.
- S. Duquesne, D. Destoumieux-Garzon, S. Zirah, C. Goulard, J. Peduzzi and S. Rebuffat, Chem. Biol., 2007, 14, 793–803 CrossRef CAS PubMed.
- J. O. Solbiati, M. Ciaccio, R. N. Farias, J. E. Gonzalez-Pastor, F. Moreno and R. A. Salomon, J. Bacteriol., 1999, 181, 2659–2662 CAS.
- J. O. Solbiati, M. Ciaccio, R. N. Farias and R. A. Salomon, J. Bacteriol., 1996, 178, 3661–3663 CAS.
- T. A. Knappe, U. Linne, S. Zirah, S. Rebuffat, X. Xie and M. A. Marahiel, J Am. Chem. Soc., 2008, 130, 11446–11454 CrossRef CAS PubMed.
- D. W. Christianson, Curr. Opin. Chem. Biol., 2008, 12, 141–150 CrossRef CAS PubMed.
- C. Nakano, H. K. Kim and Y. Ohnishi, ChemBioChem, 2011, 12, 1988–1991 CrossRef CAS PubMed.
- A. Meguro, T. Tomita, M. Nishiyama and T. Kuzuyama, ChemBioChem, 2013, 14, 316–321 CrossRef CAS PubMed.
- Y. Matsuda, T. Mitsuhashi, Z. Quan and I. Abe, Org. Lett., 2015, 17, 4644–4647 CrossRef CAS PubMed.
- Y. Yamada, S. Arima, T. Nagamitsu, K. Johmoto, H. Uekusa, T. Eguchi, K. Shin-ya, D. E. Cane and H. Ikeda, J. Antibiot., 2015, 68, 385–394 CrossRef CAS PubMed.
- Y. Yamada, T. Kuzuyama, M. Komatsu, K. Shin-Ya, S. Omura, D. E. Cane and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 857–862 CrossRef CAS PubMed.
- I. Takeda, M. Umemura, H. Koike, K. Asai and M. Machida, DNA Res., 2014, 1–11 Search PubMed.
- A. Hornung, M. Bertazzo, A. Dziarnowski, K. Schneider, K. Welzel, S. E. Wohlert, M. Holzenkampfer, G. J. Nicholson, A. Bechthold, R. D. Süßmuth, A. Vente and S. Pelzer, ChemBioChem, 2007, 8, 757–766 CrossRef CAS PubMed.
- C. Ross, K. Scherlach, F. Kloss and C. Hertweck, Angew. Chem., Int. Ed. Engl., 2014, 53, 7794–7798 CrossRef CAS PubMed.
- X. Zhu, M. Su, K. Manickam and W. Zhang, ACS Chem. Biol., 2015, 10, 2785–2793 CrossRef CAS PubMed.
- J. G. Owen, Z. Charlop-Powers, A. G. Smith, M. a. Ternei, P. Y. Calle, B. V. B. Reddy, D. Montiel and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 201501124 CrossRef PubMed.
- T. Weber, P. Charusanti, E. M. Musiol-Kroll, X. Jiang, Y. Tong, H. U. Kim and S. Y. Lee, Trends Biotechnol., 2015, 33, 15–26 CrossRef CAS PubMed.
- M. H. Medema, E. Takano and R. Breitling, Mol. Biol. Evol., 2013, 30, 1218–1223 CrossRef CAS PubMed.
- S. Zhao, A. Sakai, X. Zhang, M. W. Vetting, R. Kumar, B. Hillerich, B. San Francisco, J. Solbiati, A. Steves, S. Brown, E. Akiva, A. Barber, R. D. Seidel, P. C. Babbitt, S. C. Almo, J. A. Gerlt and M. P. Jacobson, eLife, 2014, 3:e03275 Search PubMed.
- H. J. Atkinson, J. H. Morris, T. E. Ferrin and P. C. Babbitt, PLoS One, 2009, 4, e4345 Search PubMed.
- J. D. Rudolf, X. Yan and B. Shen, J. Ind. Microbiol. Biotechnol., 2016, 43, 261–276 CrossRef CAS PubMed.
- Y. In, M. Doi, M. Inoue, T. Ishida, Y. Hamada and T. Shioiri, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 432–434 CrossRef.
- E. W. Schmidt, S. Sudek and M. G. Haygood, J. Nat. Prod., 2004, 67, 1341–1345 CrossRef CAS PubMed.
- E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315–7320 CrossRef CAS PubMed.
- M. S. Donia and E. W. Schmidt, Chem. Biol., 2011, 18, 508–519 CrossRef CAS PubMed.
- N. Leikoski, D. P. Fewer and K. Sivonen, Appl. Environ. Microbiol., 2009, 75, 853–857 CrossRef CAS PubMed.
- M. S. Donia, B. J. Hathaway, S. Sudek, M. G. Haygood, M. J. Rosovitz, J. Ravel and E. W. Schmidt, Nat. Chem. Biol., 2006, 2, 729–735 CrossRef CAS PubMed.
- B. Philmus, G. Christiansen, W. Y. Yoshida and T. K. Hemscheidt, ChemBioChem, 2008, 9, 3066–3073 CrossRef CAS PubMed.
- A. R. Weiz, K. Ishida, K. Makower, N. Ziemert, C. Hertweck and E. Dittmann, Chem. Biol., 2011, 18, 1413–1421 CrossRef CAS PubMed.
- N. Ziemert, K. Ishida, A. Weiz, C. Hertweck and E. Dittmann, Appl. Environ. Microbiol., 2010, 76, 3568–3574 CrossRef CAS PubMed.
- M. L. Micallef, P. M. D'Agostino, D. Sharma, R. Viswanathan and M. C. Moffitt, BMC Genomics, 2015, 16, 669 CrossRef PubMed.
- I. Takeda, M. Umemura, H. Koike, K. Asai and M. Machida, DNA Res., 2014, 1–11 Search PubMed.
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 412–421 CrossRef CAS PubMed.
- M. S. Donia, P. Cimermancic, C. J. Schulze, L. C. Wieland Brown, J. Martin, M. Mitreva, J. Clardy, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 1402–1414 CrossRef CAS PubMed.
- M. S. Donia and M. A. Fischbach, Science, 2015, 349(80), 1254766 CrossRef PubMed.
- J. R. Doroghazi, J. C. Albright, A. W. Goering, K.-S. S. Ju, R. R. Haines, K. a. Tchalukov, D. P. Labeda, N. L. Kelleher and W. W. Metcalf, Nat. Chem. Biol., 2014, 10, 963–968 CrossRef CAS PubMed.
- C. R. Goodwin, B. C. Covington, D. K. Derewacz, C. R. McNees, J. P. Wikswo, J. A. McLean and B. O. Bachmann, Chem. Biol., 2015, 22, 661–670 CrossRef CAS PubMed.
- M. Maansson, N. G. Vynne, A. Klitgaard, J. L. Nybo, J. Melchiorsen, D. D. Nguyen, L. M. Sanchez, N. Ziemert, P. C. Dorrestein, M. R. Andersen and L. Gram, mSystems, 2016, 1, e00028-15 CrossRef.
- K. Kleigrewe, J. Almaliti, I. Y. Tian, R. B. Kinnel, A. Korobeynikov, E. A. Monroe, B. M. Duggan, V. Di Marzo, D. H. Sherman, P. C. Dorrestein, L. Gerwick and W. H. Gerwick, J. Nat. Prod., 2015, 78, 1671–1682 CrossRef CAS PubMed.
- M. Bastian, S. Heymann and M. Jacomy, Third Int. AAAI Conf. Weblogs Soc. Media, 2009, pp. 361–362 Search PubMed.
- Y. Hu, Math. J., 2005, 10, 37–71 Search PubMed.
- Y. M. Chiang, E. Szewczyk, A. D. Davidson, N. Keller, B. R. Oakley and C. C. Wang, J. Am. Chem. Soc., 2009, 131, 2965–2970 CrossRef CAS PubMed.
- L. Song, F. Barona-Gomez, C. Corre, L. Xiang, D. W. Udwary, M. B. Austin, J. P. Noel, B. S. Moore and G. L. Challis, J. Am. Chem. Soc., 2006, 128, 14754–14755 CrossRef CAS PubMed.
- C. Zachow, G. Jahanshah, I. de Bruijn, C. Song, F. Ianni, Z. Pataj, H. Gerhardt, I. Pianet, M. Lammerhofer, G. Berg, H. Gross and J. M. Raaijmakers, Mol. Plant-Microbe Interact., 2015, 28, 800–810 CrossRef CAS PubMed.
- I. de Bruijn, M. J. de Kock, M. Yang, P. de Waard, T. A. van Beek and J. M. Raaijmakers, Mol. Microbiol., 2007, 63, 417–428 CrossRef CAS PubMed.
- M. Z. Ansari, G. Yadav, R. S. Gokhale and D. Mohanty, Nucleic Acids Res., 2004, 32, 405–413 CrossRef PubMed.
- H. M. Park, B. G. Kim, D. Chang, S. Malla, H. S. Joo, E. J. Kim, S. J. Park, J. K. Sohng and P. I. Kim, Appl. Microbiol. Biotechnol., 2013, 97, 1213–1222 CrossRef CAS PubMed.
- M. van der Voort, H. J. Meijer, Y. Schmidt, J. Watrous, E. Dekkers, R. Mendes, P. C. Dorrestein, H. Gross and J. M. Raaijmakers, Front. Microbiol., 2015, 6, 693 Search PubMed.
- N. Khaldi, F. T. Seifuddin, G. Turner, D. Haft, W. C. Nierman, K. H. Wolfe and N. D. Fedorova, Fungal Genet. Biol., 2010, 47, 736–741 CrossRef CAS PubMed.
- M. Gressler, C. Zaehle, K. Scherlach, C. Hertweck and M. Brock, Chem. Biol., 2011, 18, 198–209 CrossRef CAS PubMed.
- S. Bergmann, J. Schumann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef CAS PubMed.
- T. Awakawa, X. L. Yang, T. Wakimoto and I. Abe, ChemBioChem, 2013, 14, 2095–2099 CrossRef CAS PubMed.
- Y. Sun, T. Tomura, J. Sato, T. Iizuka, R. Fudou and M. Ojika, Molecules, 2016, 21, 59 CrossRef PubMed.
- X. L. Yang, T. Awakawa, T. Wakimoto and I. Abe, ChemBioChem, 2014, 15, 1578–1583 CrossRef CAS PubMed.
- C. Wu, R. Cichewicz, Y. Li, J. Liu, B. Roe, J. Ferretti, J. Merritt and F. Qi, Appl. Environ. Microbiol., 2010, 76, 5815–5826 CrossRef CAS PubMed.
- P. M. Joyner, J. Liu, Z. Zhang, J. Merritt, F. Qi and R. H. Cichewicz, Org. Biomol. Chem., 2010, 8, 5486–5489 CAS.
- M. Schorn, J. Zettler, J. P. Noel, P. C. Dorrestein, B. S. Moore and L. Kaysser, ACS Chem. Biol., 2014, 9, 301–309 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, J. Am. Chem. Soc., 2013, 135, 210–222 CrossRef CAS PubMed.
- T. L. Bailey and C. Elkan, Proc. Int. Conf. Intell. Syst. Mol. Biol., 5th, 1995, 3, 21–29 CAS.
- T. L. Bailey and M. Gribskov, J. Comput. Biol., 1998, 5, 211–221 CrossRef CAS PubMed.
- M. O. Maksimov, I. Pelczer and A. J. Link, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15223–15228 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, S. Zhu, D. Klug and M. A. Marahiel, Biopolymers, 2013, 100, 527–542 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, S. Zhu, H. Steuber, K. Harms, X. Xie and M. A. Marahiel, Angew. Chem., Int. Ed. Engl., 2014, 53, 2230–2234 CrossRef CAS PubMed.
- S. S. Elsayed, F. Trusch, H. Deng, A. Raab, I. Prokes, K. Busarakam, J. A. Asenjo, B. A. Andrews, P. van West, A. T. Bull, M. Goodfellow, Y. Yi, R. Ebel, M. Jaspars and M. E. Rateb, J. Org. Chem., 2015, 80, 10252–10260 CrossRef CAS PubMed.
- N. Leikoski, L. Liu, J. Jokela, M. Wahlsten, M. Gugger, A. Calteau, P. Permi, C. A. Kerfeld, K. Sivonen and D. P. Fewer, Chem. Biol., 2013, 20, 1033–1043 CrossRef CAS PubMed.
- C. Nakano, M. Oshima, N. Kurashima and T. Hoshino, ChemBioChem, 2015, 16, 772–781 CrossRef CAS PubMed.
- Y. Matsuda, T. Mitsuhashi, Z. Quan and I. Abe, Org. Lett., 2015, 17, 4644–4647 CrossRef CAS PubMed.
- F. J. Ayala, J. Hered., 1975, 68, 3–10 Search PubMed.
- M. A. Fischbach, C. T. Walsh and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4601–4608 CrossRef CAS PubMed.
- M. C. Moffitt and B. a. Neilan, J. Mol. Evol., 2003, 56, 446–457 CrossRef CAS PubMed.
- H. Jenke-Kodama, T. Börner and E. Dittmann, PLoS Comput. Biol., 2006, 2, 1210–1218 CAS.
- H. Jenke-Kodama and E. Dittmann, Phytochemistry, 2009, 70, 1858–1866 CrossRef CAS PubMed.
- J. Zucko, P. F. Long, D. Hranueli and J. Cullum, J. Ind. Microbiol. Biotechnol., 2012, 39, 1541–1547 CrossRef CAS PubMed.
- M. H. Medema, P. Cimermancic, A. Sali, E. Takano and M. A. Fischbach, PLoS Comput. Biol., 2014, 10, e1004016 Search PubMed.
- I. Schmitt and F. K. Barker, Nat. Prod. Rep., 2009, 26, 1585–1602 RSC.
- N. Ziemert and P. R. Jensen, in Methods in Enzymology, Elsevier Inc., 2012, vol. 517, pp. 161–182 Search PubMed.
- U. R. Abdelmohsen, C. Yang, H. Horn, D. Hajjar, T. Ravasi and U. Hentschel, Mar. Drugs, 2014, 12, 2771–2789 CrossRef PubMed.
- N. Ziemert, A. Lechner, M. Wietz, N. Millán-Aguiñaga, K. L. Chavarria and P. R. Jensen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1130–E1139 CrossRef CAS PubMed.
- J. R. Doroghazi and D. H. Buckley, BMC Genomics, 2014, 15, 970 CrossRef PubMed.
- M. C. Wilson, T. Mori, C. Rückert, A. R. Uria, M. J. Helf, K. Takada, C. Gernert, U. a E. Steffens, N. Heycke, S. Schmitt, C. Rinke, E. J. N. Helfrich, A. O. Brachmann, C. Gurgui, T. Wakimoto, M. Kracht, M. Crüsemann, U. Hentschel, I. Abe, S. Matsunaga, J. Kalinowski, H. Takeyama and J. Piel, Nature, 2014, 506, 58–62 CrossRef CAS PubMed.
- P. Liras, A. Rodríguez-García and J. F. Martín, Int. Microbiol., 1998, 1, 271–278 CAS.
- J. A. Eisen and M. Wu, Theor. Popul. Biol., 2002, 61, 481–487 CrossRef PubMed.
- J. A. Eisen and C. M. Fraser, Science, 2003, 300(80), 1706–1707 CrossRef CAS PubMed.
- M. Metsä-Ketelä, L. Halo, E. Munukka, J. Hakala, P. Mäntsäla and K. Ylihonko, Appl. Environ. Microbiol., 2002, 68, 4472–4479 CrossRef.
- E. a. Gontang, S. P. Gaudêncio, W. Fenical and P. R. Jensen, Appl. Environ. Microbiol., 2010, 76, 2487–2499 CrossRef CAS PubMed.
- H. Morlon, T. K. O'Connor, J. a. Bryant, L. K. Charkoudian, K. M. Docherty, E. Jones, S. W. Kembel, J. L. Green and B. J. M. Bohannan, PLoS One, 2015, 10, e0130659 Search PubMed.
- Z. Charlop-Powers, J. G. Owen, B. V. B. Reddy, M. a. Ternei and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3757–3762 CrossRef CAS PubMed.
- J. N. Woodhouse, L. Fan, M. V. Brown, T. Thomas and B. A. Neilan, ISME J., 2013, 7, 1842–1851 CrossRef CAS PubMed.
- F. Y. Chang, M. A. Ternei, P. Y. Calle and S. F. Brady, J. Am. Chem. Soc., 2013, 135, 17906–17912 CrossRef CAS PubMed.
- F.-Y. Chang, M. A. Ternei, P. Y. Calle and S. F. Brady, J. Am. Chem. Soc., 2015, 137, 6044–6052 CrossRef CAS PubMed.
- V. M. D'Costa, C. E. King, L. Kalan, M. Morar, W. W. L. Sung, C. Schwarz, D. Froese, G. Zazula, F. Calmels, R. Debruyne, G. B. Golding, H. N. Poinar and G. D. Wright, Nature, 2011, 477, 457–461 CrossRef PubMed.
- G. Cox and G. D. Wright, Int. J. Med. Microbiol., 2013, 303, 287–292 CrossRef CAS PubMed.
- M. Steffensky, A. Mühlenweg, Z. X. Wang, S. M. Li and L. Heide, Antimicrob. Agents Chemother., 2000, 44, 1214–1222 CrossRef CAS PubMed.
- R. M. Peterson, T. Huang, J. D. Rudolf, M. J. Smanski and B. Shen, Chem. Biol., 2014, 21, 389–397 CrossRef CAS PubMed.
- A. Kling, P. Lukat, D. V. Almeida, A. Bauer, E. Fontaine, S. Sordello, N. Zaburannyi, J. Herrmann, S. C. Wenzel, C. Konig, N. C. Ammerman, M. B. Barrio, K. Borchers, F. Bordon-Pallier, M. Bronstrup, G. Courtemanche, M. Gerlitz, M. Geslin, P. Hammann, D. W. Heinz, H. Hoffmann, S. Klieber, M. Kohlmann, M. Kurz, C. Lair, H. Matter, E. Nuermberger, S. Tyagi, L. Fraisse, J. H. Grosset, S. Lagrange and R. Muller, Science, 2015, 348(80), 1106–1112 CrossRef CAS PubMed.
- M. N. Thaker, W. Wang, P. Spanogiannopoulos, N. Waglechner, A. M. King, R. Medina and G. D. Wright, Nat. Biotechnol., 2013, 31, 922–927 CrossRef CAS PubMed.
- M. N. Thaker, N. Waglechner and G. D. Wright, Nat. Protoc., 2014, 9, 1469–1479 CrossRef CAS PubMed.
- B. Liu and M. Pop, Nucleic Acids Res., 2009, 37, D443–D447 CrossRef CAS PubMed.
- M. A. Skinnider, C. A. Dejong, P. N. Rees, C. W. Johnston, H. Li, A. L. H. L. H. Webster, M. A. Wyatt and N. A. Magarvey, Nucleic Acids Res., 2015, 43, 9645–9662 CAS.
- G. P. van Wezel and K. J. McDowall, Nat. Prod. Rep., 2011, 28, 1311–1333 RSC.
- M. J. Bibb, Curr. Opin. Microbiol., 2005, 8, 208–215 CrossRef CAS PubMed.
- G. Liu, K. F. Chater, G. Chandra, G. Niu and H. Tan, Microbiol. Mol. Biol. Rev., 2013, 77, 112–143 CrossRef CAS PubMed.
- J. Thykaer, J. Nielsen, W. Wohlleben, T. Weber, M. Gutknecht, A. E. Lantz and E. Stegmann, Metab. Eng., 2010, 12, 455–461 CrossRef CAS PubMed.
- C. Corre, L. Song, S. O'Rourke, K. F. Chater and G. L. Challis, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17510–17515 CrossRef CAS PubMed.
- J. H. Shin, A. K. Singh, D. J. Cheon and J. H. Roe, J. Bacteriol., 2011, 193, 75–81 CrossRef CAS PubMed.
- M. Craig, S. Lambert, S. Jourdan, E. Tenconi, S. Colson, M. Maciejewska, M. Ongena, J. F. Martin, G. van Wezel and S. Rigali, Environ. Microbiol. Rep., 2012, 4, 512–521 CrossRef CAS PubMed.
- E. Michta, K. Schad, K. Blin, R. Ort-Winklbauer, M. Röttig, O. Kohlbacher, W. Wohlleben, E. Schinko and Y. Mast, Environ. Microbiol., 2012, 14, 3203–3219 CrossRef CAS PubMed.
- S. Hiard, R. Marée, S. Colson, P. A. Hoskisson, F. Titgemeyer, G. P. van Wezel, B. Joris, L. Wehenkel and S. Rigali, Biochem. Biophys. Res. Commun., 2007, 357, 861–864 CrossRef CAS PubMed.
- R. Saha, N. Saha, R. S. Donofrio and L. L. Bestervelt, J. Basic Microbiol., 2013, 53, 303–317 CrossRef PubMed.
- E. Ahmed and S. J. M. Holmström, Microb. Biotechnol., 2014, 7, 196–208 CrossRef CAS PubMed.
- M. F. Fillat, Arch. Biochem. Biophys., 2014, 546, 41–52 CrossRef CAS PubMed.
- S. Ranjan, S. Yellaboina and A. Ranjan, Crit. Rev. Microbiol., 2006, 32, 69–75 CrossRef CAS PubMed.
- T. Wolf, V. Shelest, N. Nath and E. Shelest, Bioinformatics, 2015, 32, 1138–1143 CrossRef PubMed.
- M. R. Andersen, J. B. Nielsen, A. Klitgaard, L. M. Petersen, M. Zachariasen, T. J. Hansen, L. H. Blicher, C. H. Gotfredsen, T. O. Larsen, K. F. Nielsen and U. H. Mortensen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E99–E107 CrossRef CAS PubMed.
- M. Umemura, H. Koike, N. Nagano, T. Ishii, J. Kawano, N. Yamane, I. Kozone, K. Horimoto, K. Shin-ya, K. Asai, J. Yu, J. W. Bennett and M. Machida, PLoS One, 2013, 8, e84028 Search PubMed.
- M. Umemura, H. Koike and M. Machida, Front. Microbiol., 6, 371, DOI:10.3389/fmicb.2015.00371.
- T. C. Vesth, J. Brandl and M. R. Andersen, Synthetic and Systems Biotechnology, 2016, 1–8, DOI:10.1016/j.synbio.2016.01.002.
- M. Katz, B. M. Hover and S. F. Brady, J. Ind. Microbiol. Biotechnol., 2015, 43, 1–13 Search PubMed.
- E. W. Schmidt, M. S. Donia, J. A. McIntosh, W. F. Fricke and J. Ravel, J. Nat. Prod., 2012, 75, 295–304 CrossRef CAS PubMed.
- J. C. Kwan, M. S. Donia, A. W. Han, E. Hirose, M. G. Haygood and E. W. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20655–20660 CrossRef CAS PubMed.
- S. Sudek, N. B. Lopanik, L. E. Waggoner, M. Hildebrand, C. Anderson, H. Liu, A. Patel, D. H. Sherman and M. G. Haygood, J. Nat. Prod., 2007, 70, 67–74 CrossRef CAS PubMed.
- M. F. Freeman, C. Gurgui, M. J. Helf, B. I. Morinaka, A. R. Uria, N. J. Oldham, H.-G. Sahl, S. Matsunaga and J. Piel, Science, 2012, 338(80), 387–390 CrossRef CAS PubMed.
- A. C. English, S. Richards, Y. Han, M. Wang, V. Vee, J. Qu, X. Qin, D. M. Muzny, J. G. Reid, K. C. Worley and R. A. Gibbs, PLoS One, 2012, 7, e47768 CAS.
- E. Karlsson, A. Lärkeryd, A. Sjödin, M. Forsman and P. Stenberg, Sci. Rep., 2015, 5, 11996 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |