
Bacterial terpene cyclases
Jeroen S.
Dickschat
University of Bonn, Kekulé-Institute of Organic Chemistry and Biochemistry, Gerhard-Domagk-Straße 1, 53121 Bonn, Germany. E-mail: dickschat@uni-bonn.de
First published on 13th November 2015
Abstract
Covering: up to 2015
This review summarises the accumulated knowledge about characterised bacterial terpene cyclases. The structures of identified products and of crystallised enzymes are included, and the obtained insights into enzyme mechanisms are discussed. After a summary of mono-, sesqui- and diterpene cyclases the special cases of the geosmin and 2-methylisoborneol synthases that are both particularly widespread in bacteria will be presented. A total number of 63 enzymes that have been characterised so far is presented, with 132 cited references.
 Jeroen S. Dickschat | Jeroen S. Dickschat studied chemistry at TU Braunschweig, followed by a PhD with Stefan Schulz on bacterial volatiles in 2005. He subsequently moved for postdoctoral stays with Rolf Müller to Saarland University and with Peter F. Leadlay to the University of Cambridge, UK. In 2008 he started his independent career at TU Braunschweig and since 2014 he is a professor of organic chemistry at the University of Bonn. His main research interests include the biosynthesis of natural products with a special focus on bacterial and fungal terpenoids. |
1 Introduction
With more than 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 known compounds terpenes make up the largest class of natural products. As was already noticed by Otto Wallach, the pioneer of terpene chemistry, all compounds from this class are generally made up from isoprene units.1 The discovery of the mevalonate and deoxyxylulose phosphate pathways towards isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), the two activated forms of isoprene, today give a satisfying explanation for Wallach's observations. The monomers IPP and DMAPP are fused by oligoprenyl diphosphate synthases to geranyl (GPP), farnesyl (FPP) and geranylgeranyl diphosphate (GGPP), that are the linear precursors for the biosynthesis of mono-, sesqui- and diterpenes. The conversions of the linear precursors into (poly)cyclic terpene hydrocarbons or alcohols by terpene cyclases certainly belong to the most interesting and complex enzymatic reactions in Nature. While nearly all of the early work on terpene cyclases was performed on plant enzymes, only since about two decades more and more knowledge about bacterial enzymes accumulates. The chemistry of the enzyme products and the mechanisms of bacterial terpene cyclases will be comprehensively presented in this review article for the first time. Special types of cyclisation reactions, e.g. those catalysed by vanadium-dependent haloperoxidases as in the biosynthesis of the meroterpenoids napyradiomycin and merochlorin,2,3 will not be covered in this article. Previous important reviews that were published during the past ten years have addressed such unusual cyclisation reactions in the biosynthesis of terpenes,4 the structural biology of terpene cyclases,5,6 the application of quantum chemical calculations for studying terpene biosynthesis,7 and stereochemical aspects of terpene biosynthesis.8
000 known compounds terpenes make up the largest class of natural products. As was already noticed by Otto Wallach, the pioneer of terpene chemistry, all compounds from this class are generally made up from isoprene units.1 The discovery of the mevalonate and deoxyxylulose phosphate pathways towards isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), the two activated forms of isoprene, today give a satisfying explanation for Wallach's observations. The monomers IPP and DMAPP are fused by oligoprenyl diphosphate synthases to geranyl (GPP), farnesyl (FPP) and geranylgeranyl diphosphate (GGPP), that are the linear precursors for the biosynthesis of mono-, sesqui- and diterpenes. The conversions of the linear precursors into (poly)cyclic terpene hydrocarbons or alcohols by terpene cyclases certainly belong to the most interesting and complex enzymatic reactions in Nature. While nearly all of the early work on terpene cyclases was performed on plant enzymes, only since about two decades more and more knowledge about bacterial enzymes accumulates. The chemistry of the enzyme products and the mechanisms of bacterial terpene cyclases will be comprehensively presented in this review article for the first time. Special types of cyclisation reactions, e.g. those catalysed by vanadium-dependent haloperoxidases as in the biosynthesis of the meroterpenoids napyradiomycin and merochlorin,2,3 will not be covered in this article. Previous important reviews that were published during the past ten years have addressed such unusual cyclisation reactions in the biosynthesis of terpenes,4 the structural biology of terpene cyclases,5,6 the application of quantum chemical calculations for studying terpene biosynthesis,7 and stereochemical aspects of terpene biosynthesis.8
2 Monoterpene synthases
Bacterial monoterpene synthases are comparably rare, and in consequence only a few enzymes of this class have been discovered and thoroughly characterised. Monoterpene synthases sensu stricto catalyse the conversion of GPP into a monoterpene hydrocarbon or alcohol. The substrate of the 2-methylisoborneol synthase, an enzyme that is present in many bacteria from different taxonomic groups, is its methylated derivative (E)-2-methyl-GPP. The amino acid sequences of 2-methylisoborneol synthases exhibit significant differences to regular bacterial monoterpene synthases, and this enzyme will be discussed in a separate section of this article (Chapter 5).The cyclisation of GPP into a monoterpene not only requires substrate ionisation, but also an initial isomerisation step to linalyl diphosphate (LPP) by reattack of diphosphate to the geranyl cation (A) at C-3 (Scheme 1). This mechanism allows the subsequent cyclisation of LPP to the α-terpinyl cation (B), while its direct formation from GPP is hindered by the presence of the E-configured double bond between C-2 and C-3. As has been elucidated in great detail by extensive studies with plant enzymes,9,10 formation of the (4R)-α-terpinyl cation proceeds via (3R)-LPP, whereas the (4S)-α-terpinyl cation arises via (3S)-LPP. Cation B is a common intermediate towards all cyclic monoterpene hydrocarbons and alcohols, while acyclic products may arise from cation A or LPP.
 | ||
| Scheme 1 Cyclisation of GPP to the α-terpinyl cation (A). | ||
2.1 1,8-Cineole synthase
The first bacterial monoterpene synthase was identified from Streptomyces clavuligerus ATCC 27064 and was described by Ohnishi and coworkers.11 Its product 1,8-cineole (2) is one of the very few known achiral terpenes that is, however, made via the chiral intermediate α-terpineol (1) of unknown absolute configuration (Scheme 2). The purified recombinant enzyme was characterised by incubation with GPP, and the enzyme product was identified by GC/MS and comparison to an authentic standard. While the original report mentioned that 2 could not be detected in liquid culture extracts of S. clavuligerus, the compound was subsequently found in headspace extracts obtained from an agar plate culture by use of a closed-loop stripping apparatus (CLSA).12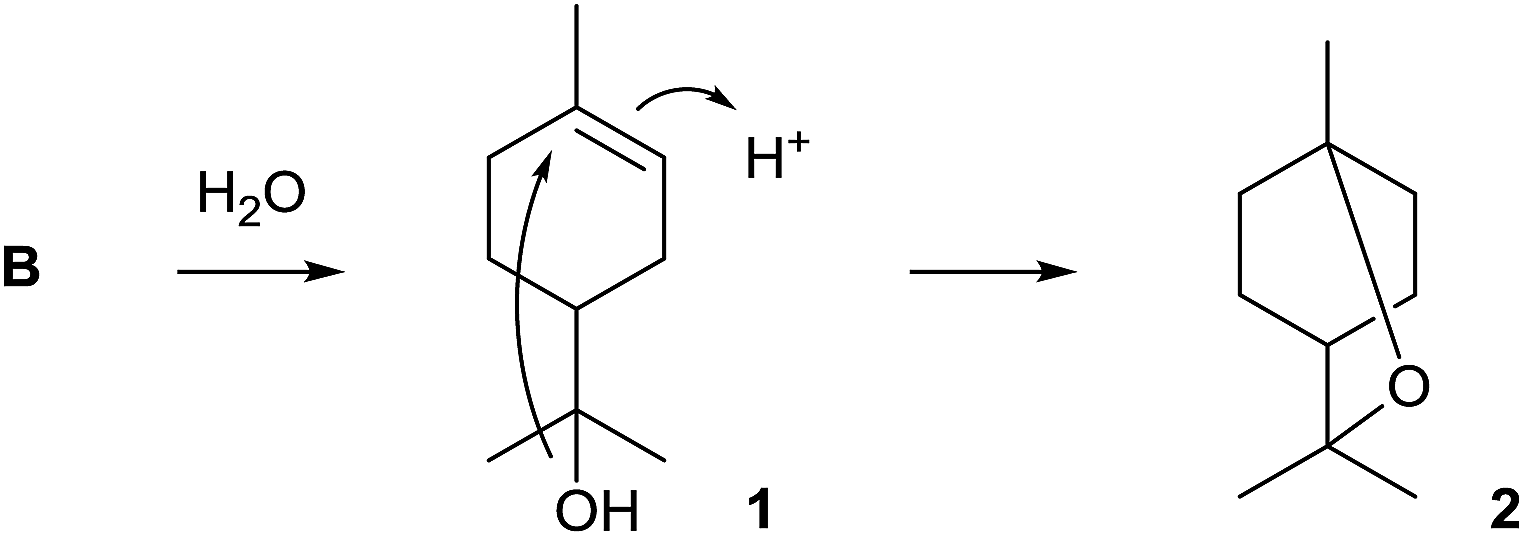 | ||
| Scheme 2 Biosynthesis of 1,8-cineole (2). | ||
The 1,8-cineole synthase is composed of 330 amino acids and contains the typical aspartate-rich motif (81DDHFD) and the NSE triad (220NDVLSLEKE, Table 1). No closely related orthologs of this enzyme have been found in other bacteria (Fig. 1). Several distantly related 1,8-cineol synthases have been characterised from plants13–17 and fungi,18 and isotopic labelling experiments revealed that the enzyme from Salvia officinalis converts GPP via (4R)-1 into 2.19 Detailed mechanistic studies with the bacterial 1,8-cineole synthase have so far not been performed. The function of the 1,8-cineol synthase has recently also been confirmed by heterologous expression in S. avermitilis.20
| (Main) product | Via | Strain | Accession no.b | PDBc | DDXXDd | Rd | NSE triadd | RYd | Ref.e |
|---|---|---|---|---|---|---|---|---|---|
| a Intermediates in mono- and sesquiterpene biosynthesis as shown in Schemes 1 and 4 (n.d. = not discussed for diterpenes). b NCBI accession numbers of characterised bacterial terpene cyclases. c Protein data bank identification code of representative structures of crystallised bacterial terpene cyclases. d Highly conserved motifs that are important for binding of the substrate and the Mg2+ cofactor. e Author's selection of the most important literature references for each bacterial terpene cyclase. | |||||||||
| Monoterpene synthases | |||||||||
| 1,8-Cineole (2) | A, B | Streptomyces clavuligerus ATCC 27064 | WP_003952918 | — | 81DDHFD | 174R | 220NDVLSLEKE | 314RY | 11 |
| (3R)-Linalool (3) | A | Streptomyces clavuligerus ATCC 27064 | WP_003957954 | — | 83DDQFD | 176R | 222NELHSFEKD | 312RY | 21 |
|
|||||||||
| Sesquiterpene synthases | |||||||||
| Pentalenene (8) | D | Streptomyces exfoliatus UC5319 | Q55012 | 1PS1 | 80DDLFD | 173R | 219NDIASLEKE | 314RY | 29,30 and 35 |
| epi-Isozizaene (11) | (R)-NPP, (R)-F | Streptomyces coelicolor A3(2) | NP_629369 | 3KB9 | 99DDRHD | 194R | 240NDLCSLPKE | 338RY | 43,46 and 47 |
| Germacrene A (26) | C | Nostoc sp. PCC7120 | WP_010998816 | — | 82DDQCD | 176R | 222NDIFSAPRE | 310RY | 55 |
| Germacrene A (26) | C | Anabaena variabilis ATCC 29413 | WP_011318775 | — | 82DDQCD | 176R | 222NDIFSAPRE | 310RY | 20 |
| 8-epi-α-Selinene (28) | C | Nostoc punctiforme PCC73102 | WP_012410187 | — | 82DDQCD | 176R | 222NDIFSASRE | 310RY | 55 |
| Avermitilol (34) | (R)-C | Streptomyces avermitilis MA-4680 | WP_010981512 | — | 80DDQFD | 173R | 219NDVYSLEKE | 313RY | 58 |
| (−)-δ-Cadinene (43) | NPP, (S)-H | Streptomyces clavuligerus ATCC 27074 | WP_003954606 | — | 85DDRID | 176R | 222NDLMTVDKE | 316RY | 60 |
| (+)-T-Muurolol (44) | NPP, (S)-H | Streptomyces clavuligerus ATCC 27074 | WP_003956090 | — | 83DDEYCD | 179R | 225NDILSHHKE | 312RY | 60 |
| (−)-Germacradien-4-ol (51) | (R)-C | Streptomyces citricolor NBRC 13005 | AB621338 | — | 80DDQFD | 172R | 218NDVRSFAQE | 312RY | 66 |
| (−)-Germacradien-4-ol (51) | (R)-C | Streptomyces lactacystinaeus OM-6519 | BAP82213 | — | 79DDQFD | 171R | 217NDVHSYPLE | 311RY | 21 |
| (−)-epi-α-Bisabolol (52) | NPP, (S)-F | Streptomyces citricolor NBRC 13005 | AB621339 | — | 84DDRFD | 179R | 225NDLCSLQRE | 310RY | 66 |
| (+)-Caryolan-1-ol (55) | D | Streptomyces griseus NBRC 13350 | WP_012378966 | — | 83DDEFD | 174R | 220NDICSFEKE | 314RY | 69 |
| (+)-epi-Cubenol (57) | (R)-NPP, (S)-H | Streptomyces griseus NBRC 13350 | WP_012381690 | — | 81DDQLD | 180R | 226NDVYSFEKE | 314RY | 71 and 74 |
| (2Z,6E)-Hedycaryol (58) | (R)-NPP, (S)-H | Kitasatospora setae KM-6054 | WP_014133196 | 4MC3 | 82DDQID | 175R | 221NDVFSVERE | 315RY | 78 |
| Selina-4(15),7(11)-diene (60) | C | Streptomyces pristinaespiralis ATCC 25486 | WP_005317515 | 4OKZ | 82DDGHCE | 178R | 224NDIFSYHKE | 310RY | 53 and 79 |
| Selina-3,7(11)-diene (61) | C | Streptomyces tsukubaensis NRRL 18488 | WP_006348376 | — | 78DDGYCE | 174R | 220NDIFSHHKE | 306RY | 24 |
| Selina-3,7(11)-diene (61) | C | Streptomyces sp. SK 1894 | BAP82223 | — | 82DDGYCE | 178R | 224NDIFSHHKE | 310RY | 20 |
| Corvol ether B (69) | NPP, H | Kitasatospora setae KM-6054 | WP_014134444 | — | 78DDLFVD | 171R | 217NDVQSLKME | 313RY | 80 |
| (+)-Eremophilene (72) | (S)-C | Sorangium cellulosum so ce56 | WP_012241161 | — | 91DDAYD | 185R | 231NDYFSLGKE | 317RY | 20 and 81 |
| Isoafricanol (73) | D | Streptomyces violaceusniger Tü 4113 | WP_043239846 | — | 95DDQFD | 199R | 245NDIASLDKE | 339RY | 83 |
| Germacrene A (26) | C | Chitinophaga pinensis DSM 2588 | WP_012789469 | — | 81DDIFD | 202R | 248NDLYSLGKE | 337RY | 79 |
| γ-Cadinene (74) | NPP, H | Chitinophaga pinensis DSM 2588 | WP_012792334 | — | 82DDQCD | 174R | 220NDIFSCAKE | 309RY | 79 |
| α-Amorphene (75) | NPP, H | Streptomyces viridochromogenes DSM 40736 | WP_039931950 | — | 102DDRAE | 193R | 239PDLFSAVKE | 324RY | 79 |
| 7-epi-α-Eudesmol (76) | C | Streptomyces viridochromogenes DSM 40736 | WP_003994861 | — | 80DDQFD | 177R | 223NDIHSFERE | 317RY | 79 |
| (+)-T-Muurolol (44) | NPP, H | Roseiflexus castenholzii DSM 13941 | WP_012119179 | — | 81DDQCD | 175R | 221NDVLSYPKE | 309RY | 20 and 79 |
| (+)-T-Muurolol (44) | NPP, H | Roseiflexus sp. RS-1 | WP_011958209 | — | 81DDQCD | 175R | 221NDMLSYPKE | 309RY | 20 |
| (E)-β-Caryophyllene (54) | D | Saccharothrix espanaensis DSM 44229 | WP_041318180 | — | 85DDQFD | 179R | 225NDVASTIKE | 320RY | 25 |
| Hedycaryol (77) | C | Saccharopolyspora spinosa NRRL 18395 | WP_010314578 | — | 87DDSLD | 181R | 227NDLVSARNE | 321RY | 25 |
| epi-Cubebol (78) | NPP, H | Streptosporangium roseum DSM 43021 | WP_012893303 | — | 91DDAFCE | 184R | 230NDLISYAKE | 316RY | 25 |
| African-2-ene (79) | D | Streptomyces clavuligerus ATCC 27074 | WP_003963391 | — | 73DEQFD | 166R | 212NDIVSLPKE | 306RY | 20 |
| Intermedeol (80) | C | Streptomyces clavuligerus ATCC 27074 | WP_003955204 | — | 91DDLAL | 197R | 242NDIVSYERE | 338RY | 20 |
| α-Selinene (81) | C | Herpetosiphon aurantiacus DSM 785 | WP_012190525 | — | 93DDQCD | 187R | 233NDLVSVKKE | 317RY | 20 |
| (+)-Dauca-8,11-diene (82) | NPP, G | Streptomyces venezuelae ATCC 10712 | CCA53839 | — | 92DDYFA | 190R | 236NDVASYERE | 332RY | 20 |
| (+)-Allohedycaryol (83) | C | Mycobacterium marinum M | WP_012394883 | — | 82DDVFD | 179R | 225NDLYSAPKE | 318RY | 20 |
| Bicyclosesquiphellandrene (84) | NPP, H | Sorangium cellulosum so ce56 | WP_012238985 | — | 79DDVCE | 174R | 220NDIYSLRKE | 308RY | 20 |
| (−)-Isohirsut-1-ene (85) | D | Streptomyces clavuligerus ATCC 27074 | EFG04889 | — | 98DDQFD | 193R | 239NDLCSAEKE | 332RY | 20,87 and 88 |
| (−)-Isohirsut-4-ene (86) | D | Streptomyces lactacystinaeus OM-6519 | BAP82216 | — | 91DDALD | 181R | 227NDIVSLEKD | 322RY | 20 and 87 |
| (+)-(1(10)E,4E,6S,7R)-Germacradien-6-ol (87) | (S)-C | Streptomyces pratensis ATCC 33331 | ADW03055 | — | 86DDEYCD | 181R | 227NDLVSYHKE | 314RY | 89 |
|
|||||||||
| Diterpene synthases (type I) | |||||||||
| Cyclooctat-9-en-7-ol (88) | n.d. | Streptomyces melanosporofaciens MI614-43F2 | BAI44338 | 4OMG | 110DDMD | 175R | 220NDFYSYDRE | 294RY | 90,92 and 93 |
| Hydropyrene (93) | n.d. | Streptomyces clavuligerus ATCC 27074 | WP_003963279 | — | 82DDRAID | 179R | 225NDLHSFARE | 313RY | 20 and 87 |
| Clavulatriene A (96) | n.d. | Streptomyces clavuligerus ATCC 27074 | EFG04655 | — | 155DDMLE | 286R | 332NDLFSYRKE | 431RY | 20 and 87 |
| Tsukubadiene (99) | n.d. | Streptomyces tsukubaensis NRRL 18488 | EIF90392 | — | 75DDHLD | 165R | 212SDLHSFQLE | 298RY | 20 and 87 |
| Odyverdiene A (100) | n.d. | Streptomyces sp. ND90 | BAP82229 | — | 79EDEDC | 173R | 221NDTHSLERE | 316RY | 20 and 87 |
| Cyclooctat-7(8),10(14)-diene (102) | n.d. | Streptomyces lactacystinaeus OM-6519 | BAP82203 | — | 83DDDLD | 178R | 224NDLISIHRE | 310RY | 20 and 87 |
| Cembrene C (103) | n.d. | Rubrobacter xylanophilus DSM 9941 | WP_041328593 | — | 77DDLAD | 172R | 218NDIISLAKE | 306RY | 12 and 20 |
| Obscuronatin (104) | n.d. | Herpetosiphon aurantiacus DSM 785 | WP_012190524 | — | 94DDQLD | 188R | 234NDIISLRKE | 323RY | 20 |
|
|||||||||
| Geosmin and 2-MIB synthases | |||||||||
| Geosmin (120), N-term. Domain | (R)-C | Streptomyces coelicolor A3(2) | WP_011030632 | — | 86DDHFLE | 184R | 229NDLFSYQRE | 325RY | 124 |
| Geosmin (120), C-term. Domain | — | Streptomyces coelicolor A3(2) | WP_011030632 | — | 455DDYYP | 552R | 598NDVFSYQKE | 694RY | 124 |
| Geosmin (120), N-term. Domain | (R)-C | Nostoc punctiforme PCC73102 | YP_001866236 | — | 90DDHFLE | 188R | 234NDLFSYQRE | 330RY | 55 and 125 |
| Geosmin (120), C-term. Domain | — | Nostoc punctiforme PCC73102 | YP_001866236 | — | 470DDYFP | 568R | 614NDIFSYQKE | 710RY | 55 and 125 |
| 2-Methylisoborneol (121) | — | Streptomyces coelicolor A3(2) | WP_011031839 | 3V1V | 197DDCYCE | 300R | 345NDLYSYTKE | 433RY | 127 and 131 |
| 2-Methylisoborneol (121) | — | Pseudoanabaena limnetica Castaic Lake | HQ630883 | — | 145DDYYAD | 250R | 295NDLLSVAKD | 381RY | 129 |
| 2-Methylenebornane (127) | — | Pseudomonas fluorescens PfO-1 | WP_011333305 | — | 95DDHYCD | 200R | 245NDLYSAYKE | 332RY | 130 |
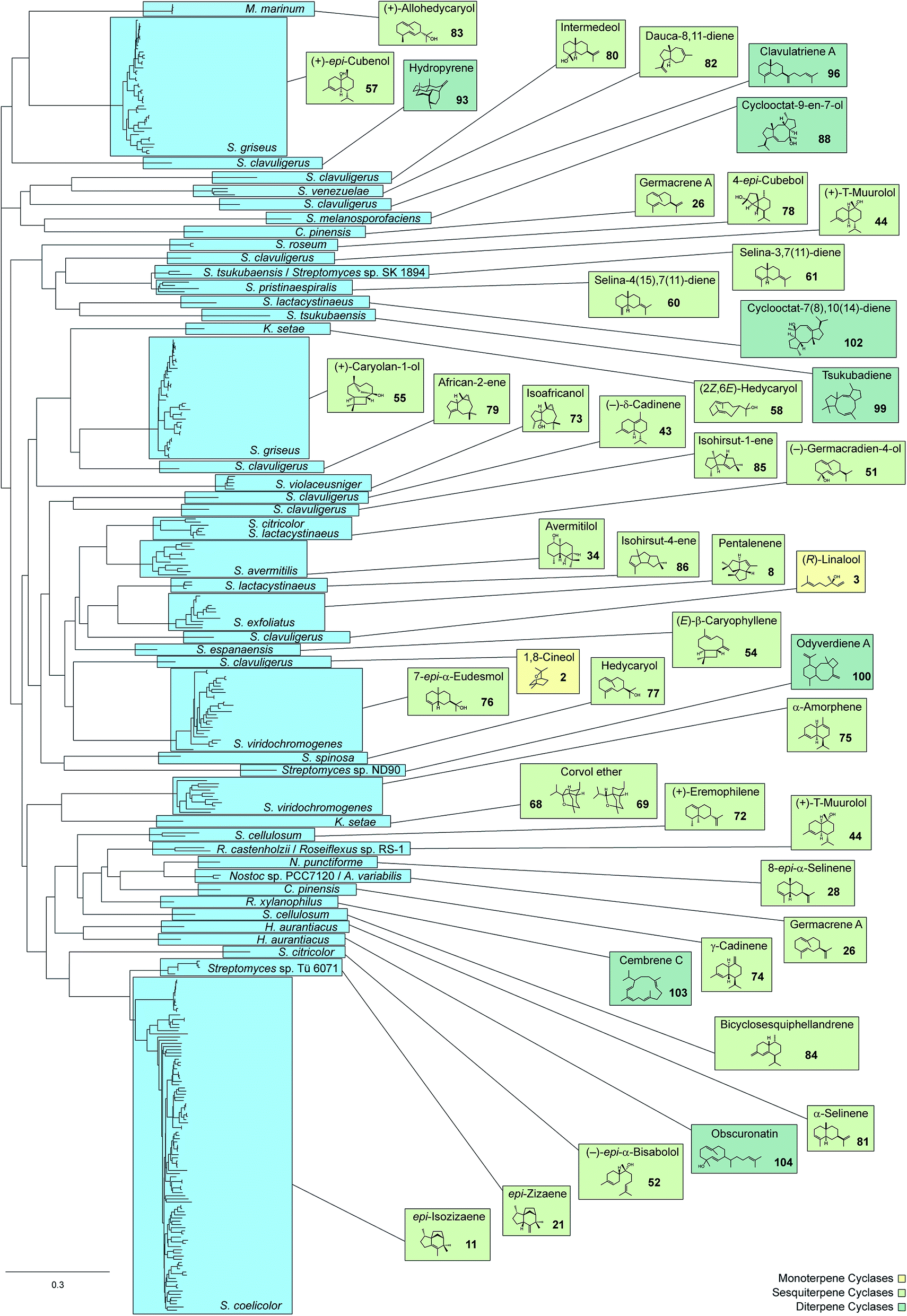 | ||
| Fig. 1 Phylogenetic tree of characterised bacterial terpene cyclases and closely related enzymes (313 sequences included) and their main products. Species names in blue boxes refer to species from which the terpene cyclases were characterised. | ||
2.2 Linalool/nerolidol synthase
The second identified monoterpene synthase was the linalool/nerolidol synthase from S. clavuligerus.21 This enzyme is the only known bacterial terpene synthase producing acyclic compounds (apart from albaflavenone synthase, a bacterial cytochrome P450 monooxygenase that contains a second domain with activity as farnesene synthase, Chapter 3). The products linalool (3) and nerolidol (4) were identified by incubation of the purified enzyme with GPP and FPP, followed by GC/MS analysis, while their absolute configurations (3R)-3 and (3R)-4 were established by GC analysis on a chiral stationary phase and comparison to authentic samples (Scheme 3). The enzyme converts FPP with higher efficiency than GPP, but GGPP is not accepted as substrate. Both compounds 3 and 4 could not be detected in liquid culture extracts from S. clavuligerus, but heterologous expression of the linalool/nerolidol synthase gene in Streptomyces lividans and Streptomyces avermitilis resulted in the production of 3, but not 4,20,21 suggesting that the enzyme may naturally function only as monoterpene synthase. Both terpene alcohols were also detected in CLSA headspace extracts from S. clavuligerus agar plate cultures.12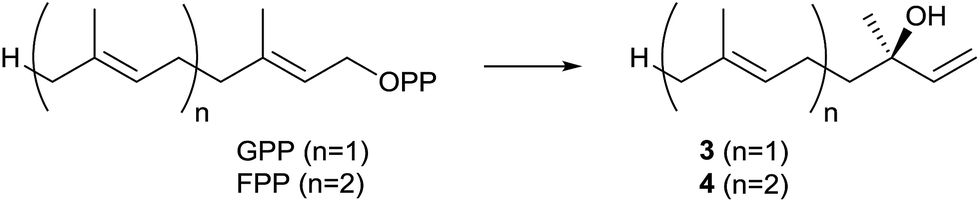 | ||
| Scheme 3 Biosynthesis of (3R)-linalool (3) and (3R)-nerolidol (4). | ||
The linalool/nerolidol synthase from S. clavuligerus is a typical bacterial terpene synthase made up from 333 amino acids, exhibiting the aspartate-rich motif (83DDQFD) and a slightly modified NSE triad with a terminal aspartate instead of the usual glutamate (222NELHSFEKD, Table 1).21 No closely related homologs are found to be encoded within the genomes of other sequenced bacteria (Fig. 1), while a large number of linalool and nerolidol synthases yielding either of both enantiomers, in some cases as a mixture, are known from plants.22 The S. clavuligerus linalool/nerolidol synthase has not been investigated for its mechanistic details.
Linalool production was also reported for the marine Streptomyces sp. GWS-BW-H5 (ref. 23) and several other streptomycetes,12,24 suggesting that these organisms may also encode a linalool synthase, albeit with low sequence homology to the S. clavuligerus enzyme. However, heterologous expression of the epi-cubebol synthase from Streptosporangium roseum in Escherichia coli resulted in the production of linalool and a few other monoterpenes, demonstrating that some sesquiterpene synthases may accept GPP as substrate.25
3 Sesquiterpene synthases
The majority of characterised bacterial terpene synthases are sesquiterpene synthases. Their substrate FPP is qualified to react in diverse cyclisation cascades, thereby resulting in a large structural variety of (poly)cyclic sesquiterpene skeletons. As outlined above, all cyclic monoterpenes are generated via the α-terpinyl cation (A), while six different initial cyclisation reactions of FPP are possible (Scheme 4). This includes the direct 1,10- or 1,11-cyclisation to the (E,E)-germacrenyl (C) or (E,E)-humulyl cation (D), respectively. A 1,6- or 1,7-cyclisation requires isomerisation of the 2E double bond of FPP via the mechanism discussed above for GPP: ionisation of FPP to the farnesyl cation (E) is followed by reattack of diphosphate at C-3 to yield nerolidyl diphosphate (NPP). This enables a 1,6-cyclisation to the bisabolyl cation (F) or a 1,7-cyclisation to the cycloheptenyl cation (G). In addition, a 1,10-cyclisation to the (E,Z)-germacrenyl cation (helmintogermacrenyl cation, H) or a 1,11-cyclisation to the (E,Z)-humulyl cation (I) are possible. In case of NPP and the chiral cations C, F, G and H both enantiomers may potentially be formed. | ||
| Scheme 4 Possible cyclisation reactions of FPP. | ||
3.1 Pentalenene synthase
The pentalenene synthase from Streptomyces exfoliatus was the first characterised bacterial terpene synthase and is thus the prototype of this enzyme class. It is also the best investigated bacterial terpene cyclase, and gives a good example of the problems of gene annotations in the post genomic era. Many automatic annotations of putative bacterial terpene cyclase genes refer to the pentalenene synthase that was for a long time the only characterised enzyme, although the functions of most of the enzymes encoded by these genes is different or unknown.Pentalenene (8, Scheme 5) is the parent hydrocarbon of the antibiotic pentalenolactone (10) that was isolated from Streptomyces roseogriseus in 1957.26 The pentalenene synthase is encoded by a terpene cyclase gene within the pentalenolactone biosynthetic gene cluster (Fig. 2A)27 and was partially purified from a cell lysate of Streptomyces UC5319 (later assigned as S. exfoliatus),28 followed by gene cloning and expression in E. coli.29 The crystallisation of pentalenene synthase yielded the first X-ray structure of a bacterial terpene cyclase in 1997 in its open conformation.30 Although the amino acid sequence is very different from the sequences of avian farnesyl diphosphate synthase and tobacco 5-epi-aristolochene synthase whose structures were also known at that time,31,32 all three enzymes were found to share a common α-helical topology (Fig. 2B). The bottom of the active site cavity of pentalenene synthase is composed of aromatic and aliphatic residues that contour a template for FPP binding in a suitable conformation for its cyclisation to 8. The top region contains the aspartate residues of the DDXXD motif (80DDLFD) and residues of the NSE triad (219NDIASLEKE) involved in Mg2+ binding (Table 1). Although the crystal structure of pentalenene synthase was not obtained in complex with the Mg2+ cofactor, comparison to the structure of tobacco 5-epi-aristolochene synthase supported this interpretation of the apo-structure. The critical role of several of these residues including D80, D81 and N219 for enzyme functionality was later shown by site-directed mutagenesis, while mutation of D84 proved to be less effective.33 The suggested function of the active site residue H309 as a proton shuttle in the terpene cyclase reaction30 could not be confirmed experimentally.33,34
 | ||
| Scheme 5 Mechanism for the cyclisation of cation D to pentalenene (dyR = dyotropic rearrangement). | ||
 | ||
| Fig. 2 Pentalenene synthase. (A) Biosynthetic gene cluster for pentalenolactone from Streptomyces avermitilis (dark blue: pentalenene synthase, light blue: oxidising enzymes, grey: other enzymes), (B) structure of pentalenene synthase. | ||
A generally accepted mechanism for the cyclisation of FPP to 8 that is based on quantum chemical calculations is shown in Scheme 5.35 The initial cyclisation of FPP to the (E,E)-humulyl cation (D, Scheme 4) is followed by a 1,2-hydride shift, either directly or via a deprotonation–protonation sequence, to yield 5. Its cyclisation results in the protoilludyl cation 6 that reacts in a dyotropic rearrangement to 7 from which 8 is formed by deprotonation. The dyotropic rearrangement is an unusual elementary step in this terpene cyclisation, especially because the cationic centre seems to be not involved and rather plays a role as a spectator, but certainly the neighbouring migrating C–C-bond of the cyclobutane is substantially weakened and thus more reactive due to the presence of the cation. Experimental proof for this mechanism that is a modification of an initially suggested reaction cascade34 was obtained by an isotopic labelling experiment.36 Incubation of (6-2H)FPP (Ha = 2H) with the H309A mutant of pentalenene synthase, a mutant that produces substantial amounts of protoilludene (9),34 gave a decreased formation of this side product in comparison to the conversion of non-labelled FPP. This kinetic isotope effect is in agreement with cation 6 as a branching point towards 8 and 9. Several stereochemical details of the reaction catalysed by pentalenene synthase have also been investigated37,38 and have been discussed in a previous review.8
As the available genome sequences reveal, a few other streptomycetes also encode terpene cyclases that are closely related to the pentalenene synthase from S. exfoliatus (Fig. 1), and among these sequenced strains 8 has been detected in headspace extracts from Streptomyces avermitilis, Streptomyces arenae and Streptomyces collinus.12,24
3.2 epi-Isozizaene synthase
The epi-isozizaene synthase (EIZS) is, with the exception of the geosmin synthase that is due to its unique biosynthesis discussed in a separate Chapter 5, the most widespread sesquiterpene synthase in all bacteria whose genomes have been sequenced so far (Fig. 1). In fact, the coding gene appears in >100 bacterial strains, invariably from the genus Streptomyces, and production of epi-isozizaene (11, Scheme 6) was reported from at least 20 of the sequenced streptomycetes.12,24,39 The enzyme's main product 11 is the parent hydrocarbon of a series of oxidised sesquiterpenes including the albaflavenols 12 and 13, the antibiotic albaflavenone (14),40 and 4β,5β-epoxy-2-epi-zizaan-6β-ol (15).41 Interestingly, the albaflavenone synthase that catalyses the formation of these oxidation products from 11 contains a second sesquiterpene synthase domain that produces farnesenes from FPP.42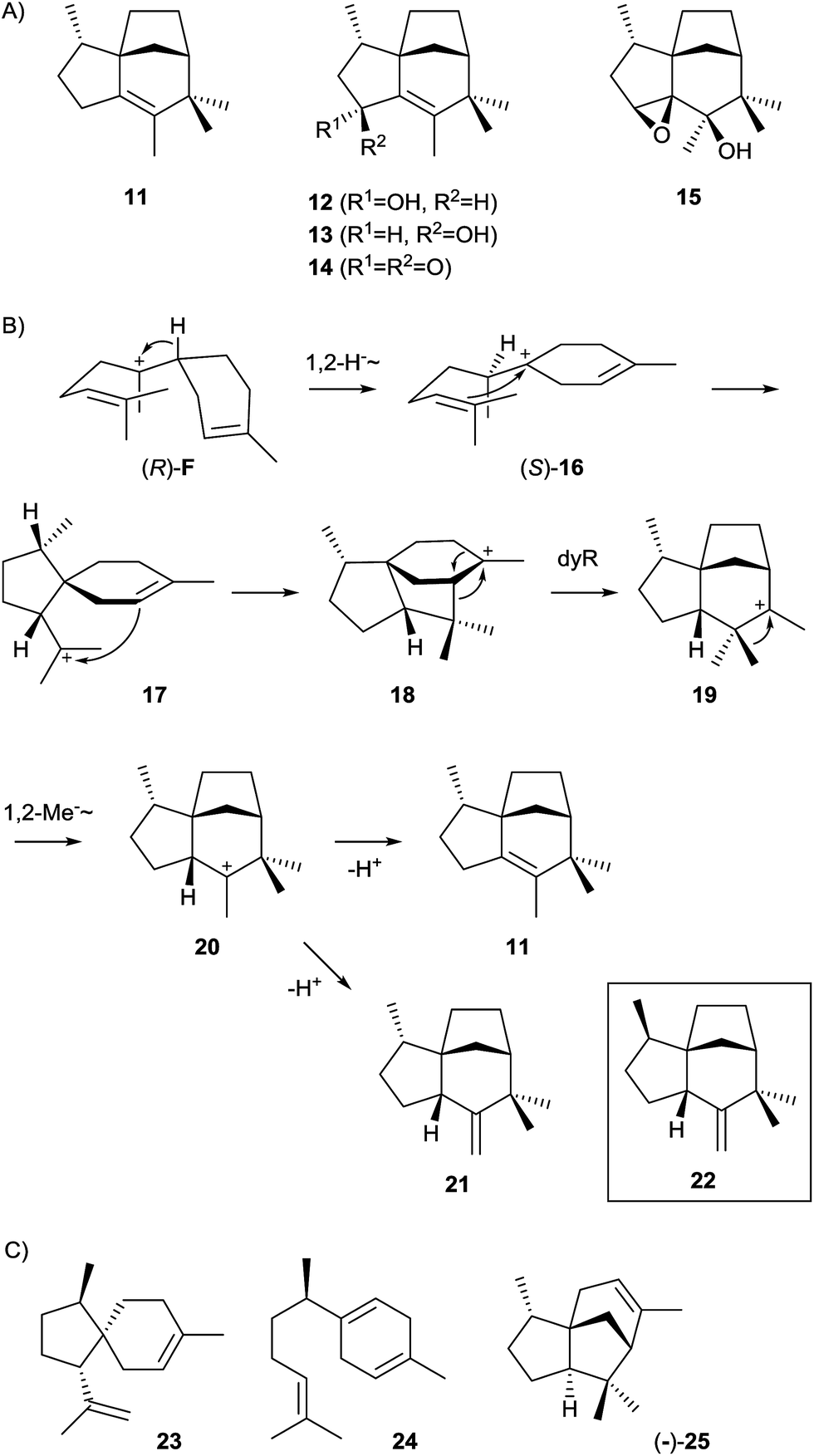 | ||
| Scheme 6 epi-Isozizaene synthase. (A) Structures of epi-isozizaene (11) and its oxidation products, (B) mechanism for the cyclisation of cation F to 11 (dyR = dyotropic rearrangement), (C) structures of products obtained from mutants of epi-isozizaene synthase. | ||
The EIZS from Streptomyces coelicolor A3(2) is a protein composed of 361 amino acid residues including the aspartate-rich motif (99DDRHD) and the NSE triad (240NDLCSLPKE, Table 1).43 Mechanistically, the cyclisation of FPP to 11 proceeds via (R)-NPP and the (R)-bisabolyl cation (F, Scheme 4) from which the (S)-homobisabolyl cation (16) is formed in a 1,2-hydride shift (Scheme 6).43,44 A subsequent cyclisation generates the acorenyl cation (17) that reacts to the cedryl cation (18) and then by dyotropic rearrangement to 19. This mechanism is supported by quantum chemical calculations45,46 and avoids a previously suggested secondary cation intermediate. A final 1,2-methyl migration to 20 and deprotonation yield 11. The quantum chemical models for this pathway were recently refined by docking of relevant conformers of (R)-F into EIZS, revealing that some downstream dynamic trajectories can directly reach the cedryl cation 18, which explains why deprotonation products of the early pathway intermediates 16 and 17 are not observed.46 The stereochemical implications of the pathway were delineated from enzymatic conversions of (1R)- and (1S)-[1-2H]FPP and NMR-based stereochemical analysis of the site of incorporation of the labelling into 11, allowing for the conclusion that the key intermediate (R)-F is formed via (R)-NPP.43 This was subsequently confirmed by competitive incubation experiments with mixtures of deuterated and non-labelled (S)- and (rac)-NPP.44 Furthermore, incubation of [12,12,12-2H3]FPP and of [13,13,13-2H3]FPP revealed that specifically the original C-13 methyl group of FPP migrates in the methyl shift from 19 to 20.44
A small group of streptomycetes encodes a sesquiterpene synthase that separates slightly, but also very clearly from the large group of epi-isozizaene synthases in a phylogenetic analysis of bacterial terpene cyclases (Fig. 1). One of these streptomycetes, Streptomyces sp. Tü 6071, was shown to produce a compound with a mass spectrum that is very similar to the mass spectrum of zizaene (22), but also shows some subtile differences that together with biosynthetic considerations resulted in the tentative identification of the compound as epi-zizaene (21).12
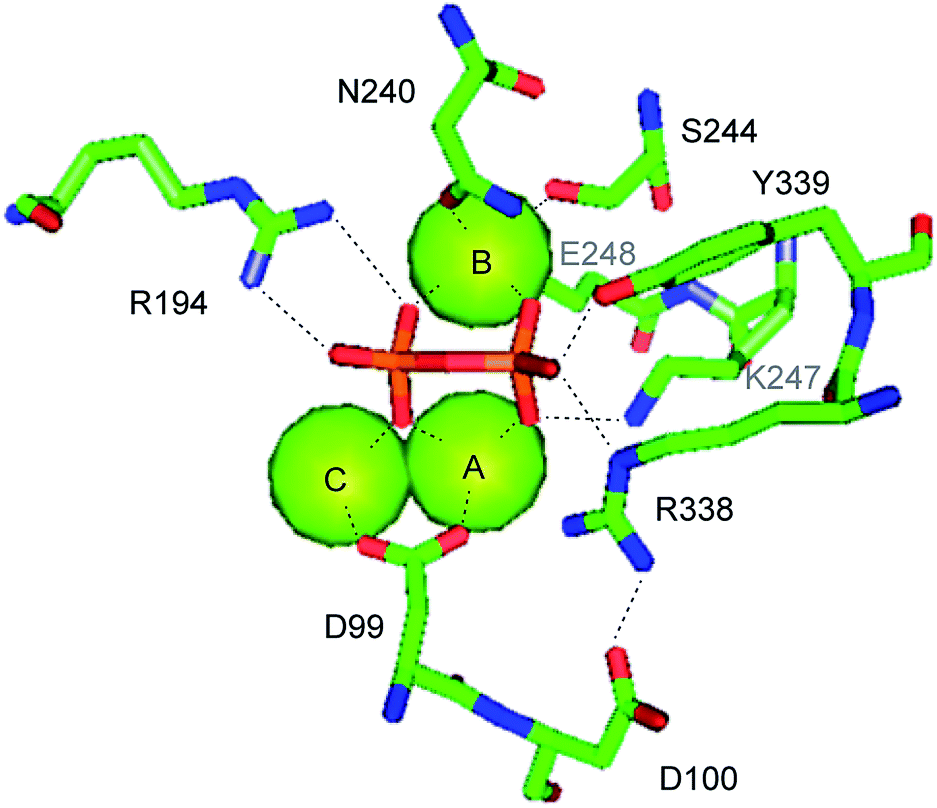
The crystal structure of EIZS was obtained in a closed Mg2+ and pyrophosphate (PPi) bound state and gave important insights into general structural aspects of bacterial terpene cyclases.47 The active site contains a (Mg2+)3 cluster in which each of the individual Mg2+ cations is octahedrally coordinated by specific amino acid residues, oxygen atoms of PPi, or water (Fig. 3). While in plant terpene cyclases the first and the third aspartate of the aspartate-rich motif coordinate to Mg2+A and Mg2+C,48–50 in EIZS only the first aspartate (D99) is bound to these cations, similar to the situation known from fungal enzymes.51,52 Accordingly, the D99N mutant is inactive, while the D100N mutant shows a strongly decreased activity (<5% of wildtype enzyme).44 The latter finding is explained by a disrupted hydrogen bonding network that is observed between D100, R338 and PPi (Fig. 3). The third D103 points away from the active centre which is in agreement with the only slightly reduced catalytic activity of a corresponding pentalenene synthase mutant.33 Within the NSE triad the residues N240, S244 and E248 are involved in cation binding, and mutation of each of these residues resulted in a strongly decreased enzyme functionality.44 K247 may have a critical function due to a hydrogen bridge to PPi, but its role was not tested by mutation. Furthermore, the crystallographic results seem to suggest that also R194 and the dimer R338/Y339 may be important for enzymatic activity, which is further supported by the fact that these residues are highly conserved in all functionally characterised terpene cyclases (Table 1). These three residues were also not interrogated for EIZS, but their critical role was demonstrated later for the selina-4(15),7(11)-diene synthase (vide infra).53
 | ||
| Fig. 3 Mechanism for the cyclisation of cation F to epi-isozizaene (11). | ||
EIZS generates besides the main product 11 a few side products from FPP.47 This moderate promiscuity prompted investigations of whether it is possible to alter the product profile of the enzyme by mutation of aromatic or aliphatic active site residues.54 The most dramatic changes in the product profile were observed upon mutation of aromatic residues that are presumably involved in the stabilisation of cationic intermediates by cation-π-interactions. The major product obtained from a F95M mutant was identified by GC/MS as β-acoradiene (23, Scheme 6, 68% of total product mixture), while the F95H substitution yielded mainly β-curcumene (24, 50%). Particularly interesting is the outcome of the F198L mutation that resulted in a 4:1 mixture of (−)- and (+)-β-cedrene (25, sum of enantiomers: 61%), demonstrating that subtile changes in the active site can indeed redirect the strict conversion of FPP via (S)-16 as observed for the wildtype to a relaxed stereochemical course that proceeds via both enantiomers of 16. Quantum chemical calculations by the Tantillo group demonstrated impressively that the conformational arrangement of the bisabolyl cation in the enzyme's active site determines the downstream stereochemical course and thus the structures of the observed products.45
3.3 Germacrene A and 8-epi-α-selinene synthases from cyanobacteria
Two genes putatively encoding sesquiterpene synthase homologs, one from the cyanobacterium Nostoc sp. PCC7120 and one from Nostoc punctiforme PCC73102, were cloned into an expression vector and heterologously expressed in Escherichia coli, resulting in the detection of a sesquiterpene hydrocarbon in both cases.55 The product of the first enzyme from Nostoc sp. PCC7120 was readily identified as germacrene A (26) by GC/MS, partially detected as its Cope rearrangement product β-elemene (27) due to a thermal reaction in the GC injection port (Scheme 7A). The product of the second terpene cyclase from N. punctiforme PCC73102 was isolated in large scale for structure elucidation by NMR spectroscopy and identified as 8-epi-α-selinene (28). The absolute configurations of both terpene cyclase products remain to be determined. | ||
| Scheme 7 Germacrene A and 8-epi-α-selinene synthases. (A) Structures of enzyme and Cope products, (B) cyclisation mechanism via neutral intermediate 26, (C) alternative mechanism via proton sandwiches 30 and 32. | ||
Interestingly, a phylogenetic analysis of bacterial terpene cyclases shows that the two characterised enzymes from Nostoc are related (Fig. 1),12 which coincides with the fact that 26 is a neutral intermediate along the cyclisation cascade from FPP via cation C towards 28 (Schemes 4 and 7B).55 This suggests that the 8-epi-α-selinene synthase from N. punctiforme PCC73102 may have its evolutionary origin in the germacrene A synthase from Nostoc sp. PCC7120. In fact, the extended function of the terpene cyclase for 28 in comparison to the enzyme making 26 may result from only subtile changes within the amino acid sequences such as the presence or absence of a precisely located acidic amino acid residue for reprotonation of the neutral intermediate 26 for its further conversion into 28. An alternative suggestion for the cyclisation from C to 28 involved proton sandwiches (Scheme 7C),55 non-classical carbocations with tetracoordinate protons sandwiched between two olefinic π-bonds.56 However, calculations demonstrated that 30 is not at an energy minimum, while 32 indeed is.57
The two Nostoc enzymes exhibit almost exactly the same highly conserved motifs which reflects again their close relationship (Table 1). Furthermore, the genetic environment of their coding genes is similar: both genes for the germacrene A and 8-epi-α-selinene synthases in Nostoc are flanked by genes encoding cytochrome P450 monooxygenases. Heterologous coexpression of the terpene cyclase and P450 gene from Nostoc sp. PCC 7120 in E. coli resulted in the detection of three unidentified oxidised sesquiterpenes in culture extracts by GC/MS, while a similar coexpression of the genes from N. punctiforme PCC73102 gave no oxidation products.55
3.4 Avermitilol synthase
Streptomyces avermitilis MA-4860 encodes four terpene cyclases, three of which were highly homologous to the synthases for pentalenene, epi-isozizaene, and geosmin. The fourth gene was shown to encode an enzyme that converts FPP into avermitilol (34), a previously unknown sesquiterpene alcohol, besides small amounts of the germacrenes A (26) and B (35), and viridiflorol (36, Scheme 8A). The structure of 34 was rigorously established by NMR-spectroscopy, while its absolute configuration was determined by the Mosher ester method.58 In the avermitilol synthase all highly conserved motifs for enzyme function are found (Table 1).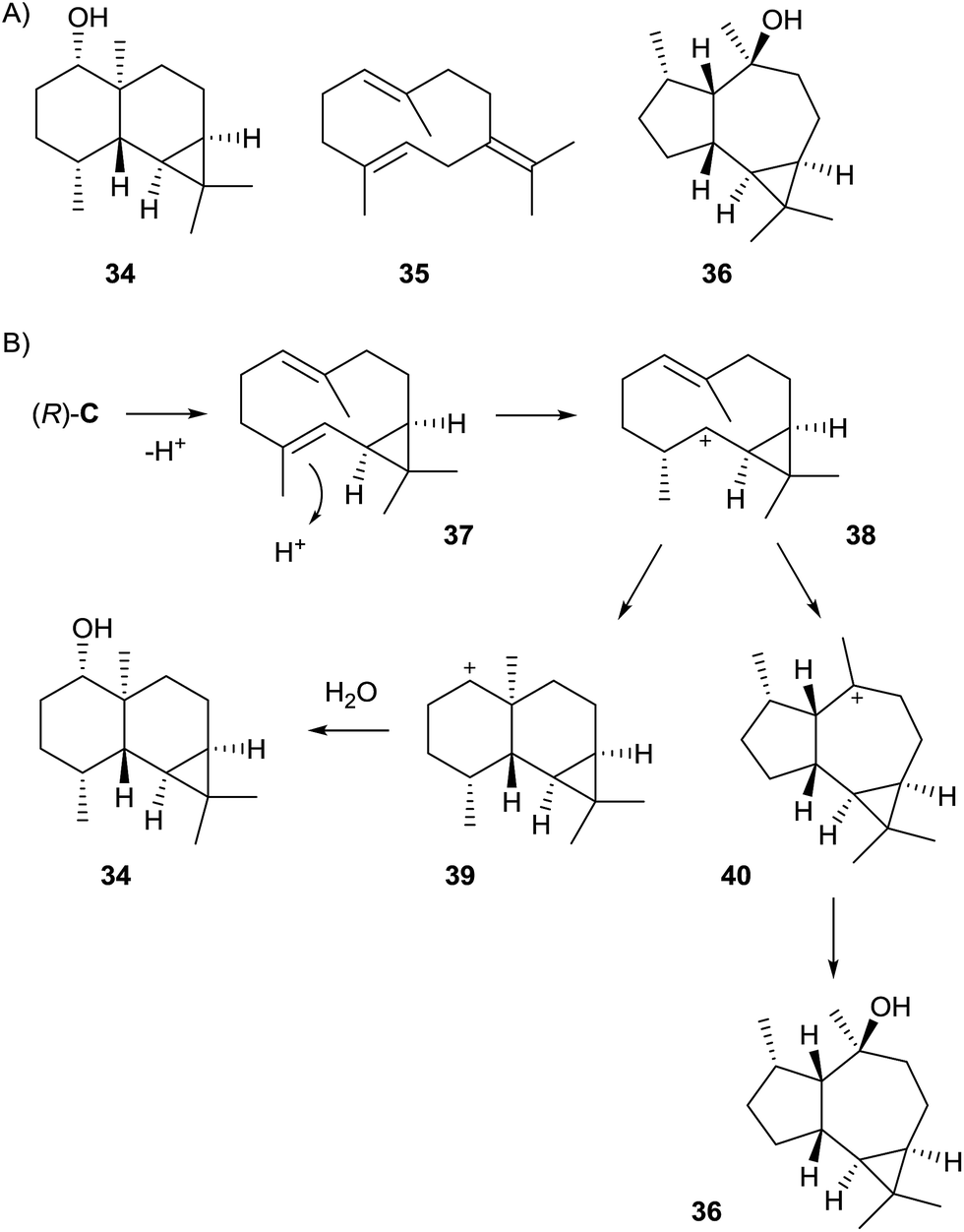 | ||
| Scheme 8 Avermitilol synthase. (A) Structures of main and side products, (B) cyclisation mechanism from cation (R)-Cvia the cyclopropylcarbinyl cation 38 to the main product 34 and alternative pathway to 36. | ||
The biosynthesis of 34 was suggested to proceed from FPP via cation (R)-C (Scheme 4) and the neutral intermediate bicyclogermacrene (37) that is formed upon deprotonation (Scheme 8B). Alternative modes of deprotonation of (R)-C give rise to the side products 26 and 35. Reprotonation of 37 yields the cyclopropylcarbinyl cation 38 that cyclises to the secondary cation 39, and a final attack of water results in 34. Similarly, the alternative cyclisation of 38 to the tertiary cation 40 and attack of water explain the formation of 36. The stereochemical course of the cascade includes a specific loss of the 1-pro-R proton of FPP, as established in isotopic labelling experiments with both enantiomers of (1-2H)FPP.58
Quantum chemical calculations demonstrated that the secondary cation 38 is at a minimum of the potential energy surface, but reacts almost barrierless to the only slightly more stable 40 and is thus best described as a metastable intermediate.59 The direct conversion of 38 into 39 seemed not possible as 39 is not located at a minimum. Instead, the reactions from 38 to the main product 34 must be described as a concerted, albeit asynchronous process.
3.5 (−)-δ-Cadinene and (+)-T-muurolol synthases from Streptomyces clavuligerus
Genome sequencing of S. clavuligerus ATCC 27064 revealed the presence of an exceptionally large number of about 20 putative terpene cyclase genes, two of which have been described above as 1,8-cineol synthase and linalool/nerolidol synthase (Chapter 2). The first characterised enzymes from this strain were the synthases for (−)-δ-cadinene (43) and (+)-T-muurolol (44, Scheme 9).60 Their structures were elucidated by GC/MS and NMR spectroscopy, while their absolute configurations were established by GC on a chiral stationary phase or from the optical rotary power, and comparison to authentic samples. Both enzymes exhibit all highly conserved motifs required for enzyme functionality, only for the (+)-T-muurolol synthase the DDXXD motif is slightly modified to a 83DDEYCD sequence (Table 1).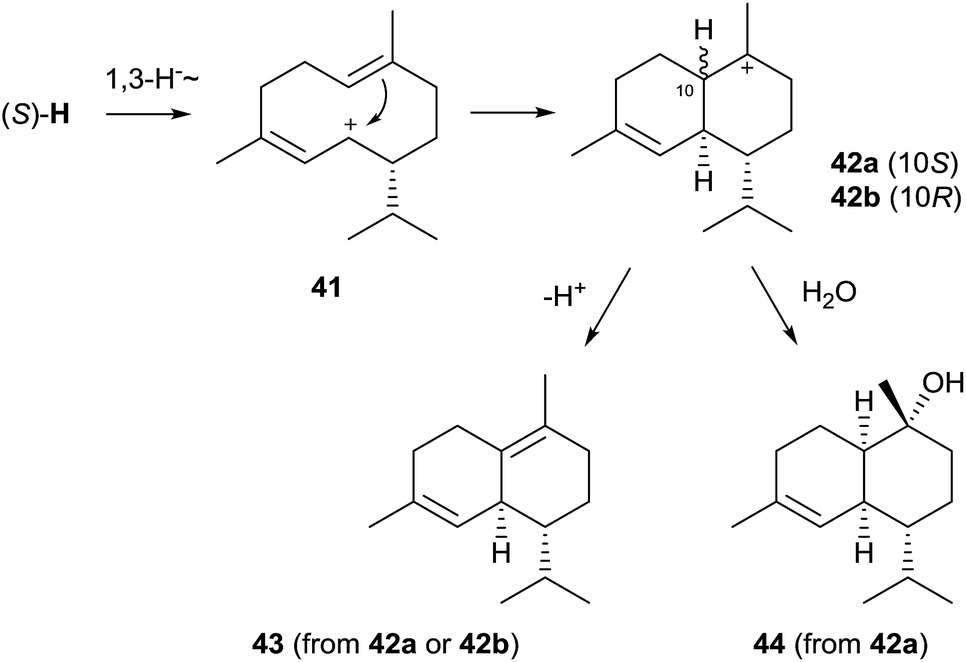 | ||
| Scheme 9 Biosynthesis of (−)-δ-cadinene (43) and (+)-T-muurolol (44). | ||
Although (−)-δ-cadinene and (+)-T-muurolol synthases from S. clavuligerus show only a low sequence identity and appear at distant locations in the phylogenetic tree of bacterial terpene cyclases (Fig. 1), their suggested biosynthesis mechanisms are very similar.60 Both cyclisation reactions proceed via (R)-NPP and the (S)-H cation (Scheme 4) that reacts to 41 upon a 1,3-hydride migration. The downstream steps of the biosynthesis of 44 must proceed via cyclisation to the muurolyl cation (42a) and attack of water, while the stereochemistry of the intermediate 42 at C-10 in the formation of 43 is unclear – it could be represented by either the muurolyl (42a) or the cadinyl cation (42b) – due to the loss of information in the final deprotonation step. This pathway to 43 and 44 is in agreement with isotopic labelling experiments using stereospecifically deuterated (R)- and (S)-(2H)FPP, but it is pointed out in the original publication that the formation of the cation (S)-Hvia (S)-NPP is also possible.60
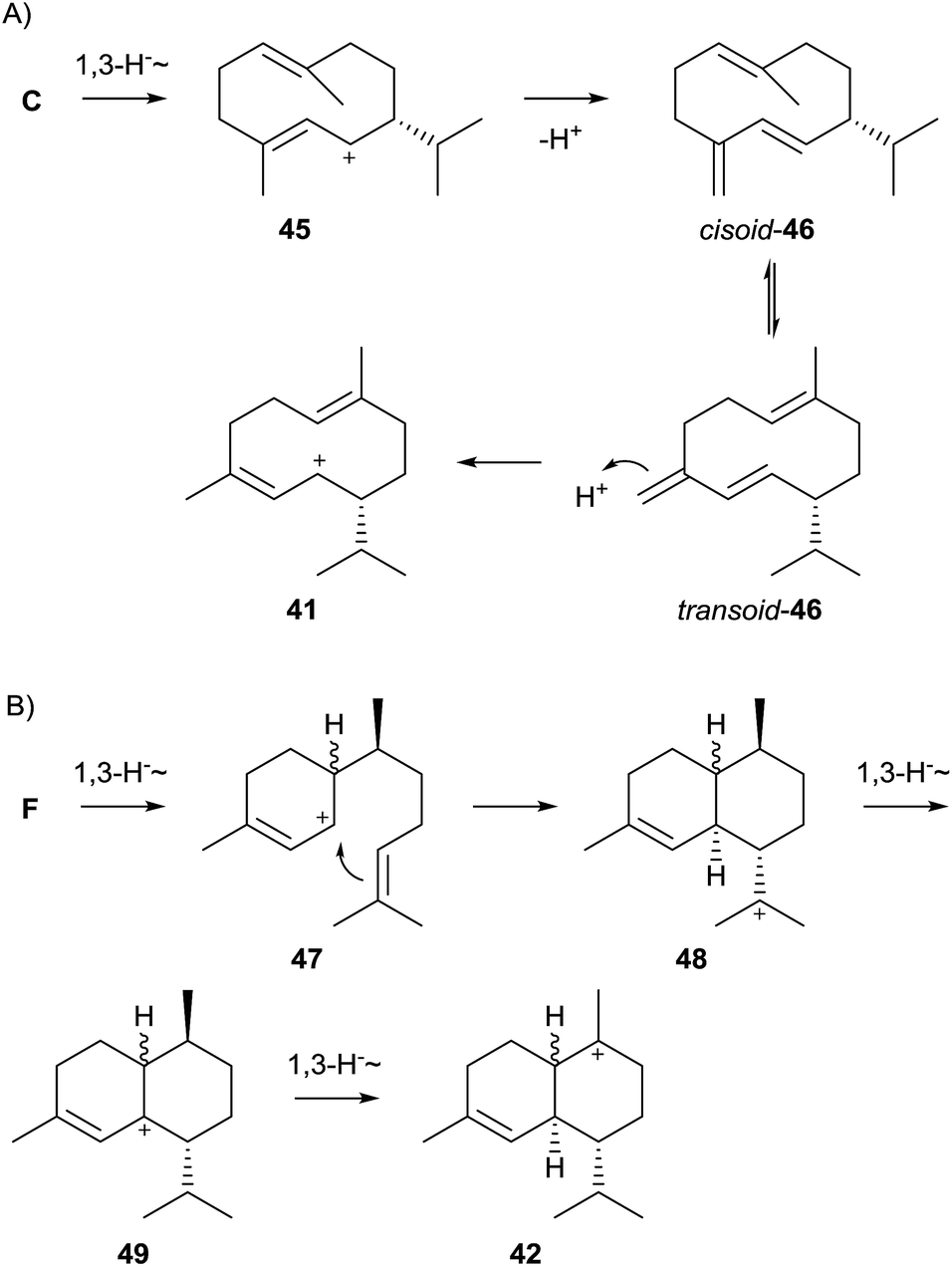
While the pathway to 43 shown in Scheme 9 is plausible, some alternatives have also been suggested. Arigoni pointed out that installation of the (3Z) double bond in 43 does not necessarily require NPP as pathway intermediate, but may also be explained by a 1,10-cyclisation of FPP to the (E,E)-germacrenyl cation (C, Scheme 4), followed a 1,3-hydride shift to 45 and its deprotonation to germacrene D (46, Scheme 10A). Its conformational rearrangement from the cisoid to the transoid conformation and reprotonation may also give rise to 41.61 The reported incorporation of deuterium from D2O into a series of cadinanes by a multi-product terpene cyclase from Medicago truncatula gave experimental proof for this pathway. Further support was obtained by investigation of a mutated enzyme in which a tyrosine residue believed to be involved in the reprotonation of 46 was exchanged by phenylalanine, resulting in the nearly exclusive formation of germacranes.62
 | ||
| Scheme 10 Alternative biosynthetic pathways towards 43 (A) via germacrene D, and (B) via the bisabolyl cation. | ||
A third alternative proceeds by 1,6-cyclisation of FPP via NPP to the bisabolyl cation (F, Scheme 4), followed by a 1,3-hydride shift to 47 and cyclisation to 48. Two subsequent 1,3-hydride shifts give access to 42via49. Since the 1,3-hydride migration from 49 to 42 must proceed suprafacially, the stereochemistry at the methyl group in 49 and all preceeding intermediates can be inferred from the stereochemical requirements of 42.63
For both (−)-δ-cadinene and (+)-T-muurolol synthase from S. clavuligerus no closely related homologs are found among genome sequenced bacteria (Fig. 1), but another distant T-muurolol synthase with only 30% sequence identity is known from Roseiflexus castenholzii (vide infra). The production of 43 and 44 was also shown for S. clavuligerus, but appeared to be highly dependent on the culture medium,12 while 43 also occurs in the North Sea isolate Streptomyces sp. GWS-BW-H5.23 Very interesting is the finding that the bacterial (−)-δ-cadinene synthase makes the optical antipode in comparison to well-known plant enzymes from cotton.64,65
The assigned functions of both terpene cyclase genes from S. clavuligerus have also recently been confirmed by heterologous expression in S. avermitilis. The genes for both enzymes are accompanied by a gene coding for a cytochrome P450 monooxygenase in their native host. Surprisingly, coexpression of the (+)-T-muurolol synthase gene together with its cytochrome P450 partner in S. avermitilis results in the formation of (−)-drimenol (50, Scheme 11).20
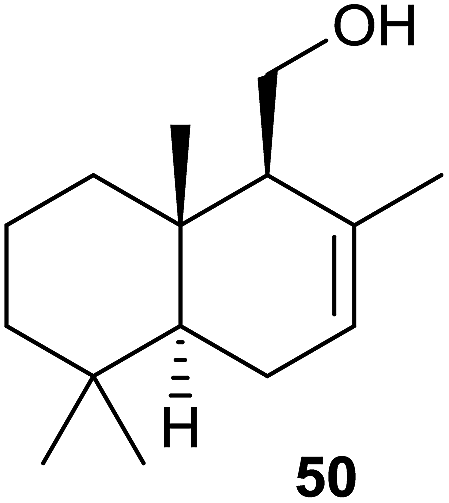 | ||
| Scheme 11 Structure of (−)-drimenol (50). | ||
3.6 (−)-Germacradien-4-ol and (−)-epi-α-bisabolol synthases from Streptomyces citricolor
In a genome mining approach two sesquiterpene cyclase homologue genes from Streptomyces citricolor NBRC 13005 were heterologously expressed in Streptomyces lividans. The GC/MS analysis of culture extracts from the heterologous host resulted in the detection of one sesquiterpene alcohol in each case. Both compounds were purified from large scale cultures and their structures were elucidated by NMR spectroscopy and from their optical rotary powers, resulting in the identification of (−)-germacradien-4-ol (51) and (−)-epi-α-bisabolol (52, Scheme 12),66 two compounds that are identical in all aspects to previously reported plant constituents.67,68 The production of 51, but not of 52, was also observed in S. citricolor.66 | ||
| Scheme 12 Biosynthesis of (−)-germacradien-4-ol (51) and (−)-epi-α-bisabolol (52). | ||
The biosynthesis of both sesquiterpene alcohols is a comparably simple process. Starting from cation (R)-C (Scheme 4), 51 is accessible through a 1,3-hydride shift and capture with water, while 52 arises by attack of water to cation (S)-F. The highly conserved motifs for substrate and Mg2+ cofactor binding are fully established in the two enzymes from S. citricolor (Table 1). While no close homolog of the (−)-epi-α-bisabolol synthase is observed in any sequenced bacterium, a few other (−)-germacradien-4-ol synthases are encoded in Streptomyces spp. and the recently sequenced Saccharothrix sp. ST-888 (Fig. 1). One of these related enzymes from Streptomyces lactacystinaeus OM-659 was recently characterised by heterologous expression in S. avermitilis and shown to produce also 51.20
3.7 (+)-Caryolan-1-ol synthase from Streptomyces griseus
Transcriptome analyses revealed that the expression of a terpene cyclase gene in Streptomyces griseus NBRC 13350 is under control of the A-factor (2-isocapryloyl-3R-hydroxymethyl-γ-butyrolactone), a mibcrobial hormone that is involved in many regulatory processes in streptomycetes.69 Heterologous expression of the gene in S. lividans resulted in the production of a sesquiterpene alcohol that was isolated and identified as (+)-caryolan-1-ol (55) by NMR spectroscopy and determination of the optical rotary power. Incubation experiments with the recombinant enzyme yielded 55 and the minor compound (+)-(E)-β-caryophyllene (54) that was identified by GC on a chiral capillary and is a biosynthetic intermediate towards 55 (Scheme 13). Both compounds 54 and 55 are the enantiomers of known plant compounds.70 | ||
| Scheme 13 Biosynthesis of (+)-caryolan-1-ol (55). | ||
All highly conserved motifs of bacterial terpene cyclases are present in the (+)-caryolan-1-ol synthase (Table 1). Close homologs to the S. griseus (+)-caryolan-1-ol synthase occur in various other strains of the genus Streptomyces. Interestingly, the (+)-caryolan-1-ol synthase co-occurs in most cases with the (+)-epi-cubenol synthase that is discussed below. The production of 55 under laboratory culturing conditions was shown for S. griseus,69Streptomyces filamentosus strains NRRL 15998 and NRRL 11379, Streptomyces anulatus NRRL B-2873, Streptomyces californicus NRRL B-3320, Streptomyces cyaneofuscatus NRRL B-2570, and Streptomyces mediolani NRRL WC3934.12,24,39
The mechanism of (+)-caryolan-1-ol synthase was investigated by enzymatic conversion of FPP in D2O, resulting in the stereospecific incorporation of labelling into 9α-H, confirming the reprotonation step of the neutral intermediate 54. Unexpectedly, the deuterium labelling was also incorporated into both hydrogens at C-12. This finding was explained by the repeated reprotonation of 54 to 53, leading to a complete H,D-exchange at C-12 (Scheme 13).69
3.8 (+)-epi-Cubenol synthase from Streptomyces griseus
(+)-epi-Cubenol (57) synthase is known from streptomycetes since the 1990s and its stereochemical course and mechanism have been investigated using the partially purified enzyme from cell lysates and (stereo-)specifically labelled FPP isotopomers (Scheme 14).71–73 Shortly after identification of the (+)-caryolan-1-ol synthase gene from S. griseus NBRC 13350, a second gene for a sesquiterpene cyclase homolog from this organism was cloned and identified as (+)-epi-cubenol synthase by heterologous expression in S. lividans and incubation of the recombinant enzyme with FPP.74 The enzyme's product was isolated from a large scale culture of S. lividans and structure elucidation was performed by NMR spectroscopy. The determined optical rotary power pointed to the same enantiomer as in the liverwort Heteroscyphus planus,75 thus establishing the structure of the enzyme product as (+)-epi-cubenol, in agreement with a report on the isolation of this compound from streptomycetes.76 The compound is the opposite enantiomer as found in cubeb oil77 and was also reported from several other streptomycetes.12,24,39 | ||
| Scheme 14 Biosynthesis of (+)-epi-cubenol (57). | ||
The active site's highly conserved motifs are all fully established in the S. griseus (+)-epi-cubenol synthase (Table 1). The biosynthesis of 57 proceeds via the helmintogermacrenyl cation (S)-H (Scheme 4) that reacts to 41 by a 1,3-hydride shift.71 A subsequent cyclisation yields the cationic intermediate 42a for which the stereochemistry can be inferred from the suprafacial 1,2-hydride shift to 56.72 A final attack of water results in 57. The formation of (S)-Hvia (R)-NPP was directly proven by enzymatic conversion of (1Z,3R)-[1-3H]NPP, while the corresponding (3S)-enantiomer failed to react.73
3.9 (2Z,6E)-Hedycaryol synthase from Kitasatospora setae
The actinomycete Kitasatospora setae KM-6054 encodes five terpene synthase homologs, two of which are readily identified from their sequences as synthases for geosmin and 2-methylisoborneol. A third uncharacterised enzyme was recently characterised by gene cloning and heterologous expression in E. coli. Incubation of FPP with the purified protein yielded mainly (2Z,6E)-hedycaryol (58), while GPP and GGPP were not accepted as substrates.78 All active site residues for catalytic functionality are present (Table 1), while no closely related enzymes are found among the sequenced bacteria (the closest relative is an uncharacterised terpene cyclase from Streptomyces viridochromogenes NRRL 3414 with 59% identical residues). The biosynthesis of 58 proceeds via (R)-NPP and the helmintogermacrenyl cation (S)-H (Scheme 4) that is directly trapped with water to yield 58 (Scheme 15). | ||
| Scheme 15 Biosynthesis of (2Z,6E)-hedycaryol (58). | ||
The crystal structure of the hedycaryol synthase (HcS) from K. setae was obtained in complex with a substrate surrogate, (R)-nerolidol, closely resembling the structure of the intermediate (R)-NPP.78 Its folding in the active centre gave interesting insights into the cyclisation reaction of HcS and also allowed for an assignment of the stereochemistry of the enzyme's product. Assuming a comparable conformational fold of (R)-NPP as observed for (R)-nerolidol, the anti-SN′ attack of C-10 from the Re side (front view) to the Re face of C-1 (downside, anti to OH/OPP) that is only 2.6 Å away will yield (2Z,6E)-hedycaryol (Fig. 4A). The intermediately formed helmintogermacrenyl cation H can be stabilised by cation-π-interaction with F149 that is in close proximity to C-11. Accordingly, a F149L substitution resulted in a strongly decreased production of 58, while the F149W mutation had no effect.
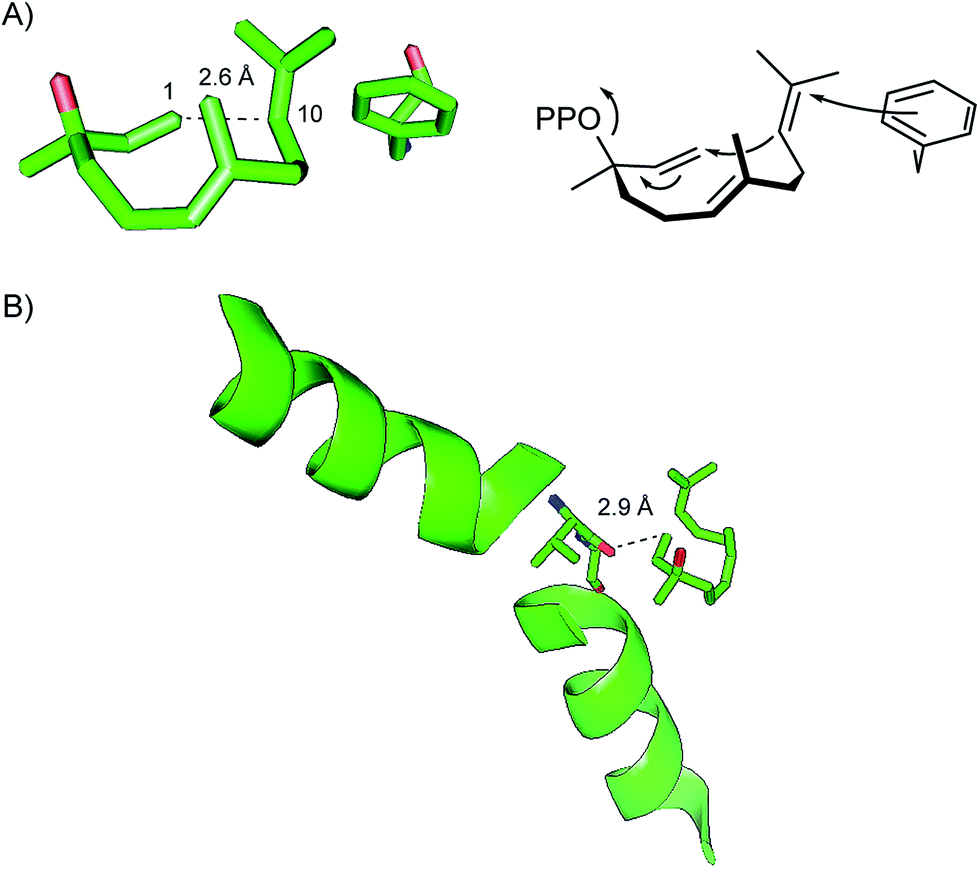 | ||
| Fig. 4 Structural features of HcS. (A) Folding of the substrate surrogate (R)-nerolidol in the active site of HcS and cyclisation of (R)-NPP that adopts the same conformational fold. (B) Helix break motif of helix G and function of V179 at the helix break. | ||
The reported HcS:(R)-nerolidol structure lacks the trinuclear Mg2+ cluster that was observed for EIZS:PPi complex,47 which underlines the bidirectional stabilisation of the metal ion cluster and PPi. Another interesting structural aspect that was observed for the HcS:(R)-nerolidol complex is a helix break motif of helix G. The carbonyl oxygen of V179 that is located directly at this helix break is perfectly aligned with the G1-helix dipole, both pointing directly at C-1 of (R)-nerolidol. The short distance of only 2.9 Å between the carbonyl oxygen of V179 and C-1 of (R)-nerolidol suggests an important role for reionisation of the (R)-NPP intermediate.78
3.10 Selina-4(15),7(11)-diene synthase from Streptomyces pristinaespiralis and related selinadiene synthases
In a screening approach of bacterial terpene cyclases that is discussed in more detail below one enzyme from Streptomyces pristinaespiralis ATCC 25486 was identified as selina-4(15),7(11)-diene synthase (SdS).79 All highly conserved motifs are fully established, only the DDXXD motif is altered to 82DDGHCE, which is similar to the situation in the (+)-T-muurolol synthase from S. clavuligerus (Table 1). Closely related homologs are found in a couple of species (Fig. 1), including Streptomyces rimosus ATCC 10970 with 70% identical sites for which the production of selina-4(15),7(11)-diene (60, Scheme 16) was also shown.24Streptomyces tsukubaensis NRRL 18488, a species encoding a slightly more distant enzyme (67% identity), was reported to produce the related compound selina-3,7(11)-diene (61),24 a product that was also observed during heterologous expression of its neighbour in the phylogenetic tree from Streptomyces sp. SK 1894 in S. avermitilis.20 | ||
| Scheme 16 Biosynthesis of selina-4(15),7(11)-diene (60) and selina-3,7(11)-diene (61). | ||
The biosynthesis of 60 proceeds via one of the two enantiomers of the germacrenyl cation (C, Scheme 4) that is deprotonated to the achiral neutral intermediate germacrene B (35, Scheme 16). Its reprotonation initiates a second cyclisation to the cation 59 that yields 60 by loss of a proton. An alternative deprotonation, possibly caused by only a subtile change in the amino acid sequence of the responsible terpene cyclase, may result in 61, the observed product in S. tsukubaensis.
The crystal structure of SdS was obtained in its open (apo) and closed form, in complex with the substrate surrogate 2,3-dihydrofarnesyl diphosphate (DHFPP).53 Following the milestone of pentalenene synthase obtained in the open (apo) form,30 EIZS in complex with PPi gave a refined picture of a bacterial sesquiterpene cyclase in the closed form, i.e. with a fully established trinuclear Mg2+ cluster that requires PPi binding for its stabilisation.47 The HcS:(R)-nerolidol complex gave a detailed view of the conformational folding of a substrate surrogate, only the functionally relevant Mg2+ cluster was not observed due to the missing PPi.78 The recently obtained open and fully closed structures of SdS allowed for a direct comparison of the conformational rearrangement of a bacterial sesquiterpene cyclase upon substrate binding and concomitant installation of the metal ion cluster.53 The dynamic enzyme processes upon substrate binding involve most dramatic movements of R178, D181 and G182 that are directly located at the helix G kink (Fig. 5A). Upon these conformational changes the “pyrophosphate sensor” R178, a strictly conserved residue in bacterial terpene cyclases located in nearly all cases exactly 46 amino acid residues upstream of the NSE triad (Table 1), forms together with D181 a network of hydrogen bonds to the substrate's diphosphate, while G182 (the “effector”) points with its carbonyl oxygen towards C-3 of the substrate analog and likely assists in substrate ionisation by stabilising the positive charge of the formed farnesyl cation. This is similar to the situation observed for V179 in HcS (Fig. 4B).78 Site-directed mutagenesis of SdS R178 established the critical role of the pyrophosphate sensor.53
 | ||
| Fig. 5 Structural features of SdS. (A) Superposition of helix G1/2 of open (apo, grey) and closed (DHFPP and Mg2+ bound, green) SdS. Red arrows indicate movements and black dotted lines distances for the closed conformation (both in Å). (B) Binding network in the active site of SdS (closed conformation) showing important hydrogen and coordinate bonds (black dotted lines). Spheres represent Mg2+ cations. | ||
A network of important active site residues is shown in Fig. 5B. The overall binding situation is similar to that observed for EIZS (Fig. 3).47 Specific differences include the presence of two hydrogen bonds between D83 and R310 of SdS, while only one hydrogen bond is observed between the corresponding D100 and R338 of EIZS, and of a coordinate bond from E87 to Mg2+ in SdS that is missing in EIZS. As noted above, E87 represents part of a slight modification of the DDXXD motif, and this subtile sequence deviation may account for the observed different binding modes in the active centres of SdS and EIZS.
In both structures of SdS and EIZS hydrogen bonds are observed between R310/R338 and the substrate's diphosphate that are extended by another hydrogen bond from the diphosphate to Y311/Y339. In fact, the same RY dimer is found in all other characterised bacterial terpene cyclases (Table 1) about 80–90 amino acid residues downstream of the NSE triad, suggesting an important role for catalytic activity, that was confirmed by site directed mutagenesis of Y311 of SdS.53
3.11 Corvol ether synthase from Kitasatospora setae
Besides HcS a second sesquiterpene cyclase from K. setae KM-6054 was recently investigated.80 The purified His-tagged enzyme converted FPP into two sesquiterpene ethers with new carbon skeletons in a 1:2 ratio, named corvol ethers A and B (68 and 69, Scheme 17), while GPP and GGPP were not accepted as substrates. Corvol ether synthase exhibits all functionally relevant motifs (Table 1). No homologs of corvol ether synthase are found in bacterial genomes (Fig. 1); the closest relative is an uncharacterised sesquiterpene cyclase from Granulicella mallensis MP5ACTX8 (58% identical sites) that likely produces different compounds.
 | ||
| Scheme 17 Biosynthesis of corvol ethers A (68) and B (69). | ||
The biosynthesis of corvol ethers proceeds via NPP and cation H (Scheme 4) that is converted by a 1,3-hydride migration to 42, followed by attack of water to the neutral intermediate 51. Its protonation-initiated reaction either to the bicyclic cation 62 and a subsequent 1,3-hydride shift or to the stereochemically different bicyclic cation 63 followed by two sequential 1,2-hydride shifts results in 65. This key intermediate is a branching point towards the two enzyme products and can either react by another 1,2-hydride shift and intramolecular attack of the hydroxy function to corvol ether A (68), or by Wagner–Meerwein rearrangement and intramolecular attack of the hydroxy group to corvol ether B (69). The intermediate secondary cation 67 along the path towards 69 may be omitted by a concerted rather than a stepwise reaction from 65, a question that may be of future interest for theoretical chemists.
The correctness of this suggested biosynthetic pathway was proven by enzymatic conversion of (1-13C)FPP and (2-13C)FPP, followed by 13C-NMR analysis of the obtained product mixture. In particular, the experiment with (2-13C)FPP was performed via incubation in a H2O/D2O mixture (1:1), resulting in strongly enhanced triplets for the carbons of corvol ethers corresponding to C-2 of FPP. The observed triplets are due to 13C-2H-couplings and gave proof for the reprotonation of intermediate 51 at the original C-2 of FPP.80
3.12 (+)-Eremophilene synthase from Sorangium cellulosum
Nearly all so far characterised bacterial terpene cyclases are from actinomycetes, reflecting that most terpene cyclase sequences are found within this taxon. A few exceptions are the above discussed germacrene A and 8-epi-α-selinene synthases from cyanobacteria, and a recently characterised (+)-eremophilene synthase from the myxobacterium Sorangium cellulosum So ce56.81 The enzymatic product was isolated in large quantities (ca. 19 mg L−1) from an E. coli overproduction strain that not only harboured the (+)-eremophilene synthase, but also mevalonate pathway genes on an expression vector.82 Structure elucidation was performed by NMR spectroscopy, X-ray diffraction and determination of the specific rotation.81 The (+)-eremophilene synthase shows all highly conserved motifs (Table 1) and within bacteria only one closely related enzyme with 71% identity can be found in Sorangium cellulosum So0157-2 (Fig. 1). The eremophilene synthase, albeit without assignment of the absolute configuration of its product, was independently identified by heterologous expression in S. avermitilis.20The biosynthesis of (+)-eremophilene (72) proceeds via cation (S)-C (Scheme 4) that looses a proton to yield (−)-germacrene A (26, Scheme 18). Reprotonation and cyclisation to 70 is followed by a sequence of a 1,2-hydride migration, a 1,2-methyl shift, and deprotonation to 72. This sesquiterpene hydrocarbon was shown to be a substrate for a cytochrome P450 monooxygenase whose gene is located adjacent to the eremophilene synthase gene, but the structure of the observed oxidation product remains to be determined.81
 | ||
| Scheme 18 Biosynthesis of (+)-eremophilene (72). | ||
3.13 Isoafricanol synthases from Streptomyces violaceusniger and Streptomyces rapamycinicus
Headspace extracts of Streptomyces violaceusniger TÜ 4113 were reported to contain the volatile sesquiterpene alcohol isoafricanol (73, Scheme 19) as a main constituent. Since the producing organism encoded, besides the readily identified geosmin and 2-methylisoborneol synthases, only one uncharacterised terpene cyclase, this finding was used to assign its function as isoafricanol synthase.83 This was subsequently confirmed by the production of 73 also by Streptomyces rapamycinicus NRRL 5491 that encodes a highly homologous enzyme (91% identity).39 The stereochemical course of the biosynthesis of 73 in S. violaceusniger was investigated by isotopic labelling experiments.83 A biochemical characterisation of a bacterial isoafricanol synthase has not yet been performed. | ||
| Scheme 19 Structures of bacterial sesquiterpene cyclase products identified from heterologous expression experiments. The structure shown for γ-cadinene in the original publication79 was confused with that of α-cadinene and must be revised. Asterisks indicate compounds for which the absolute configurations were reported. | ||
3.14 Rapid screening approaches for the characterisation of bacterial terpene cyclases
As described above, initial work on the characterisation of bacterial terpene cyclases was performed in the 1990s and during the first years of the new millennium. The very early work was performed with enzymes that were (partially) purified from cell lysates, while progress in the development of genome sequencing techniques later allowed for the molecular cloning of terpene cyclase genes. Consequently, among the first identified terpene cyclases are those from the first genome sequenced strains S. coelicolor A3(2) and S. avermitilis MA-4860.84,85 Only one decade ago genome sequencing, but not the molecular cloning and characterisation of genes, was the bottleneck of research on bacterial terpene cyclases. The fast developments of sequencing techniques during the last couple of years completely changed this situation, and today several genome sequences containing genes for bacterial terpene cyclases are released every week. Therefore, the permanent screening of all this information and homology-based identification of bacterial terpene cyclases is today required to keep track with these developments, while the functional characterisation of bacterial terpene cyclases of unknown functions requires rapid screening approaches.A recent study meeting these requirements was performed by cloning of terpene cyclase genes into the pET28c expression plasmid.79 Heterologous expression of the cloned genes in E. coli BL21 resulted in the production of terpenes by this fast growing and easy-to-handle bacterium. Product identification was performed by capturing the volatiles from the expression strains on charcoal traps, followed by extraction and GC/MS analysis.86 Using this approach six bacterial terpene cyclases were chemically characterised as germacrene A (26) and γ-cadinene (74) synthases from Chitinophaga pinensis DSM 2588, α-amorphene (75) and 7-epi-α-eudesmol (76) synthases from Streptomyces viridochromogenes DSM 40736, the above discussed selina-4(15),7(11)-diene (60) synthase from Streptomyces pristinaespiralis ATCC 25486, and T-muurolol (44) synthase from Roseiflexus castenholzii DSM 13941 (structures of newly mentioned products in this articles are shown in Scheme 19, for structures of other compounds vide supra). Notably, the γ-cadinene and T-muurolol synthases identified in this study79 are not related to the δ-cadinene and T-muurolol synthases from S. clavuligerus (Fig. 1).60 Large clusters of closely related enzymes are found for the two enzymes from S. viridochromogenes, i.e. the α-amorphene and 7-epi-α-eudesmol synthases, while no (germacrene A and γ-cadinene synthase) or only one (T-muurolol synthase) relative is seen for the other identified enzymes in bacteria. In particular, the germacrene A synthase from C. pinensis is distant from the cyanobacterial enzyme (Fig. 1). All highly conserved motifs are found in the bacterial sesquiterpene cyclases, only the distance between the aspartate-rich motif and the pyrophosphate sensor in the C. pinensis germacrene A synthase is with 116 amino acid residues exceptionally long, and instead of the usual asparagine a proline residue is present in the NSE triad of S. viridochromogenes (Table 1).
In a follow-up study three terpene cyclase genes were cloned into the pET28c-derived expression vector pYE-Express that is replicable in yeast and E. coli.25 Cloning was performed by homologous recombination in Saccharomyces cerevisiae. Gene expression in E. coli followed by capturing the volatiles allowed for characterisation of their terpene products. The three terpene cyclase were from Saccharothrix espanaensis DSM 44229 yielding (E)-β-caryophyllene (54), from Saccharopolyspora spinosa NRRL 18395 making hedycaryol (77), and from Streptosporangium roseum DSM 43021 that produces epi-cubebol (78, Scheme 19). The highly conserved motifs are for all three enzymes found as expected, only for the epi-cubebol synthase the aspartate-rich motif is modified to 91DDAFCE (Table 1). The phylogenetic analysis of bacterial terpene cyclases reveals that no homologs of the (E)-β-caryophyllene and hedycaryol synthases have been found in bacteria (Fig. 1), while another epi-cubebol synthase with 97% identity is found in S. roseum NRRL B-2638. The production of 54 was also shown for the native host S. espanaensis.24
Recently, a variety of bacterial terpene cyclases was investigated by gene expression in an engineered Streptomyces avermitilis host from which the native genes for secondary metabolite biosynthesis were deleted.20 Using this approach the previously assigned functions of the 8-epi-a-selinene synthase from Nostoc punctiforme PCC 73102, of the germacrene D synthase from Nostoc PCC7120, of the (+)-caryolan-1-ol and (+)-epi-cubenol synthases from S. griseus NBRC13350, and of the T-muurolol synthase from R. castenholzii DSM 13941 were confirmed. The absolute configuration of T-muurolol was determined as (+)-44, and a closely related enzyme from Roseiflexus sp. RS-1 was shown to make the same product, while an enzyme from Anabaena variabilis ATCC 29413 with high homology to the germacrene A synthase from Nostoc sp. PCC7120 (93% sequence identity) was also assigned as germacrene A synthase. Besides several newly identified diterpene cyclases (Chapter 4) also a series of sesquiterpene cyclases was newly characterised. This includes synthases for the known compounds african-2-ene (79) and intermedeol (80) from S. clavuligerus ATCC 27064, for α-selinene (81) from Herpetosiphon aurantiacus DSM 785, for (+)-dauca-8,11-diene (82) from Streptomyces venezuelae ATCC 10712, for (+)-allohedycaryol (83) from Mycobacterium marinum M, and for bicyclosesquiphellandrene (84) from Sorangium cellulosum So ce56, and for the new compounds isohirsut-1-ene (85) from S. clavuligerus ATCC 27064 and isohirsut-4-ene (86) from S. lactacystinaeus OM-6519 for which the planar structures were determined by NMR spectroscopy.87
3.15 Recent developments in computational chemistry
Poulter and coworkers recently reported about a very interesting approach to predict the product of a bacterial terpene cyclase by computational chemistry methods.88 The strategy is to model the substrate of a terpene cyclase into the active site of its crystal structure or homology model, followed by computing carbocationic reactions inside the enzyme pocket. The automated computational workflow predicted the structure of a linear triquinane scaffold for the isohirsut-1-ene synthase from S. clavuligerus ATCC 27064 (the computational work was performed independently of the above described identification of isohirsut-1-ene synthase). Biochemical characterisation resulted in the full stereochemical assignment as (5S,7S,10R,11S)-85, named cucumene in Poulter's paper. Since terpene cyclases show only weak sequence homology, while their overall structures are highly conserved, an amino acid sequence-based product prediction for terpene cyclases is not possible. In this light the presented computational work shows how this problem can be tackled. However, the method needs further improvement in the future, because the product's stereochemistry could still only be solved by classical chemical methods.3.16 (+)-(1(10)E,4E,6S,7R)-Germacradien-6-ol synthase from Streptomyces pratensis
Streptomyces pratensis ATCC 33331 encodes five terpene cyclases, four of which can be readily identified by their homology to the characterised synthases for epi-cubenol (57) and 7-epi-α-eudesmol (76, vide supra), geosmin and 2-methylisoborneol (Chapter 5). The fifth gene was recently characterised to encode a (+)-(1(10)E,4E,6S,7R)-germacradien-6-ol synthase, a previously unknown enzyme.89 As is typical for germacrene sesquiterpenes, the compound (1(10)E,4E,6S,7R)-germacradien-6-ol (87) exists as two interconverting conformers (87a and 87b, Scheme 20) for which distinct sets of peaks are observable in the NMR spectra recorded at low temperature. Due to multiple signal overlappings a full assignment of the NMR data was not possible. This problem was solved by synthesis of all fifteen isotopomers of (13C1)FPP and of (13C15)FPP and their enzymatic conversion. Extraction of the isotopically labelled enzyme products and NMR analysis allowed for a full assignment of NMR data. The obtained products were also used to investigate the EI-MS-fragmentation mechanism of 87. | ||
| Scheme 20 Conformations of (1(10)E,4E,6S,7R)-germacradien-6-ol (87). | ||
The (+)-(1(10)E,4E,6S,7R)-germacradien-6-ol synthase is a typical bacterial sesquiterpene cyclase that exhibits the highly conserved motifs 86DDEYCD (aspartate-rich motif), the pyrophosphate sensor 181R, the NSE triad 227NDLVSYHKE, and the 314RY dimer (Table 1). Only one closely related enzyme occurs in Streptomyces sp. PAMC26508 that has 99.4% identical sites and likely catalyses the same reaction.
4 Diterpene synthases
Bacterial diterpene biosynthesis either requires a single type I terpene cyclase that can be recognised from its highly conserved motifs, i.e. the aspartate-rich motif, the pyrophosphate sensor, the NSE triad and the RY dimer. In these enzymes substrate ionisation proceeds via diphosphate abstraction by the enzyme mechanisms as discussed above. Such bacterial diterpene cyclases cannot be distinguished from mono- or sesquiterpene synthases from their amino acid sequences alone. On the other hand, bacterial diterpene biosynthesis is in other cases a two-step process catalysed by two distinct enzymes. The first acting enzyme is a type II cyclase usually with a conserved DXDD motif for substrate ionisation by protonation. After cyclisation the diphosphate moiety remains in the product, thus allowing for another type I conversion. These two classes will be discussed separately.4.1 Bacterial type I diterpene cyclases
The first characterised bacterial type I diterpene cyclase was the cyclooctat-9-en-7-ol (88) synthase (CotB2) from Streptomyces melanosporofaciens MI614-43F2 (Scheme 21).90 Its gene is part of a gene cluster that also encodes a GGPP synthase and two cytochrome P450 monooxygenases responsible for the biosynthesis of the lysophospholipase inhibitor cyclooctatin (89). Site-specific CotB2 mutants were constructed that catalyse the conversion of GGPP into altered products, e.g. the active site mutation F149L results in cyclooctat-7-en-3-ol (90).91 Recently, the crystal structure of CotB2 was obtained in the open state that resembles the first structure of a bacterial diterpene cyclase and shows the classical α-helical fold.92 Isotopic labelling experiments revealed an unexpected carbon backbone rearrangement along the cyclisation cascade from GGPP to 88 (Scheme 21B).93 | ||
| Scheme 21 Cyclooctat-9-en-7-ol synthase. (A) Structures of 88, 89 and 90, and (B) cyclisation mechanism to 88. | ||
Two diterpene cyclases have been identified from Streptomyces sp. SANK 60404. The first one (DtcycA) produces the main compound (R)-nephthenol (91), while the second (DtcycB) produces, besides (R)-cembrene A, the main compounds 91 and the related diterpene alcohol 92 in a 1:1 ratio (Scheme 22).94 In both diterpene cyclases the aspartate-rich motifs are deviating from the regular DDXXD sequence: DtcycA shows a 91DDLRFE motif and DtcycB exhibits a hardly recognisable 86QEIRD motif (Table 1).
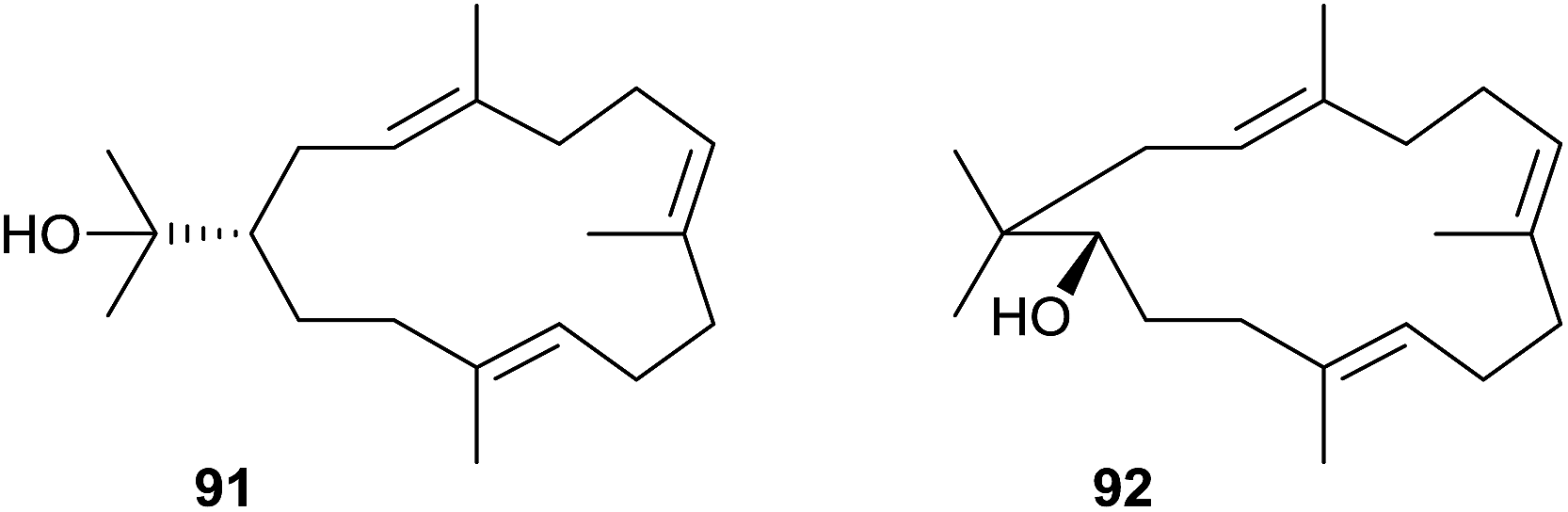 | ||
| Scheme 22 Structures of (R)-nephthenol (91) and the related diterpene alcohol 92. | ||
Using the approach of heterologously expressing bacterial terpene cyclases in S. avermitilis Ikeda and coworkers have recently identified various bacterial type I diterpene cyclases with new products (Scheme 23).20,87 The hydropyrene synthase from S. clavuligerus ATCC 27064 makes hydropyrene (93) and the side products hydropyrenol (94) and isoelisabethatriene A (known from sea plumes Pseudopterogorgia elisabethae)95 and B (new, 95). Structure elucidation of compounds 93 and 94 with a new 6-6-6-6 tetracyclic skeleton was performed by NMR spectroscopy and X-ray diffraction of the corresponding chemically obtained epoxides. The absolute configurations are unknown. The clavulatriene synthase from the same organism produces seven diterpenes including the main compounds clavulatrienes A (96) and B (97),20,87 and known prenylgermacrene B (98)96 that is likely a neutral intermediate towards 97. For the corresponding precursor of 96 the Cope rearrangement product was isolated. The clavulatrienes are new compounds with so far unknown absolute configuration. A diterpene cyclase from Streptomyces tsukubaensis NRRL 18488 generates one main compound, tsukubadiene (99).20,87 The planar structure was determined by NMR spectroscopy and resembles a new tricyclic 5–9–5 skeleton. The odyverdiene synthase from Streptomyces sp. ND90 produces the odyverdienes A (100) and B (101) in a 1:1 ratio.20,87 Their planar structures were determined by NMR and exhibit a novel 6–8–4 ring system. An enzyme from Streptomyces lactacystinaeus OM-6519 that is surprisingly not related to the cyclooctat-9-en-7-ol synthase described above produces the compound cyclooctat-7(8),10(14)-diene (102).
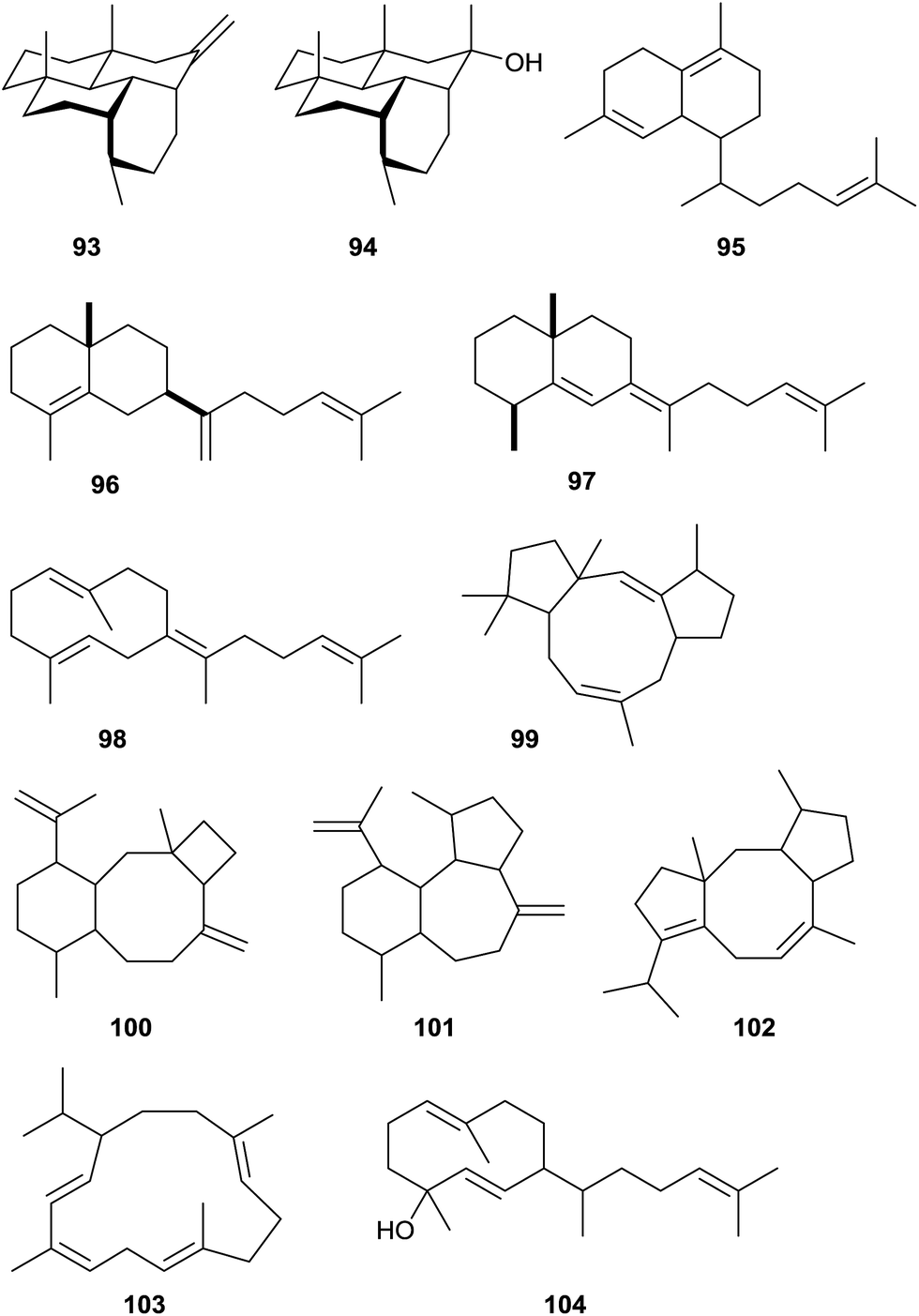 | ||
| Scheme 23 Products of bacterial type I terpene cyclases investigated by heterologous expression in S. avermitilis. | ||
Furthermore, two diterpene cyclases making known compounds were identified: the cembrene C (103) synthase from Rubrobacter xylanophilus DSM 9941 and the obscuronatin (104) synthase from Herpetosiphon aurantiacus DSM 785.20 The function of the cembrene C synthase was previously suggested based on a correlation of metabolome data with genetic information.12 The stereostructures of 103 and 104 remain to be determined.
4.2 Bacterial type II diterpene cyclases
Only a handful of bacterial type II diterpene cyclases is known. This class of enzymes is not in the main focus of this article, and therefore, these enzymes are only briefly mentioned, but not discussed in detail here. For more detailed information the reader is referred to a recent review by Shen and coworkers.97 A review about labdane-related diterpenoids has been published by Peters in 2010.98The first reported bacterial type II diterpene cyclase was the terpentedienyl diphosphate synthase from Streptomyces griseolosporus MF730-N6 that makes terpentedienyl diphosphate (105), the substrate for a type I enzyme catalysing the further conversion into terpentetriene (106) that is itself the precursor of the antibiotic terpentecin (Scheme 24).99,100 Four bacterial ent-copalyl diphosphate (ent-107) synthases from Streptomyces sp. KO-3988,101Bradyrhizobium japonicum USDA 110,102 and two strains of Streptomyces platensis (MA7327 and MA7339)103 are known. In Streptomyces sp. KO-3988 ent-107 is further converted by a type I enzyme into (−)-pimara-9(11),15-diene (108),104 while in B. japonicum ent-107 is converted into ent-kaurene (109), the parent hydrocarbon for gibberellin biosynthesis (gibberellic acid GA3, 111).102 In Streptomyces platensis ent-107 is either transformed into 109 or ent-atiserene (110) that serve as precursors for the mammalian fatty acid synthase inhibitors platensimycin (112) and platencin.103 The crystal structure of the ent-kaurene synthase from B. japonicum was recently reported.105
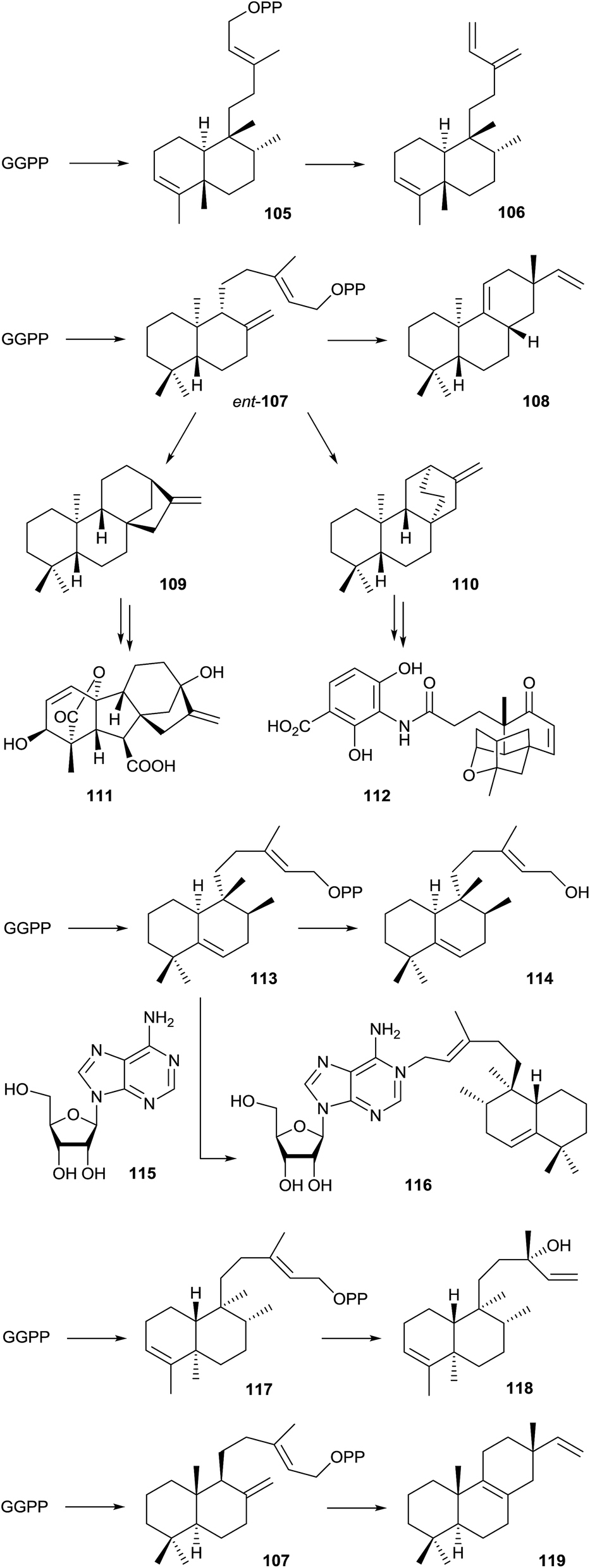 | ||
| Scheme 24 Products of bacterial type II terpene cyclases and further conversion by type I enzymes. | ||
A type II diterpene cyclase from Mycobacterium tuberculosis, encoded by the Rv3377c gene, converts GGPP into tuberculosinyl diphosphate (113).106,107 The neighbouring gene Rv3378c encodes a type I enzyme that converts 113 into tuberculosinol (114) and both stereoisomers of isotuberculosinol in in vitro incubation experiments,108 while the previously reported structure of edaxadiene was revised.109 However, the natural function of the type I enzyme is the production of 1-tuberculosinyladenosine (116) by ionisation of 113 and nucleophilic attack of the cosubstrate adenosine (115). Both genes are only present in Mycobacterium species that cause tuberculosis and the terpene nucleoside 116 may play an important role in the pathogenicity of these bacteria.110 Isotopically labelled GGPP was used in incubation experiments to investigate the stereochemical course of the terpene cyclisation by tuberculosinyl diphosphate synthase.107,111 Recently, a biosynthetic gene cluster containing genes for a type II and a type I diterpene cyclase was identified from Herpetosiphon aurantiacus. The type II enzyme was shown to convert GGPP into kolavenyl diphosphate (117) that is further transformed into (+)-kolavelool (118) by the type I enzyme.112 A type II diterpene synthase from the marine actinomycete Salinispora arenicola catalyses the conversion of GGPP into copalyl diphosphate (107), the opposite enantiomer as observed from the B. japonicum enzyme, that is subsequently transformed into isopimara-8,15-diene (119) by a type I terpene cyclase.113
5 Geosmin and 2-methylisoborneol synthases
The earthy and musty odorants geosmin (120) and 2-methylisoborneol (121) belong to the first identified terpenoids from streptomycetes (Scheme 25).114,115 Both compounds are important drinking water contaminants and are particularly widespread in bacteria that occur not only in actinomycetes, but also in myxobacteria and cyanobacteria.116 In 1981 Bentley and Meganathan reported the incorporation of radioactivity from [1-14C]- and [2-14C]acetate into both metabolites, while radioactivity from L-[methyl-14C]methionine was only incorporated into 121.117 They concluded that 120 is a degraded sesquiterpene, while 121 was suggested to be a methylated monoterpene.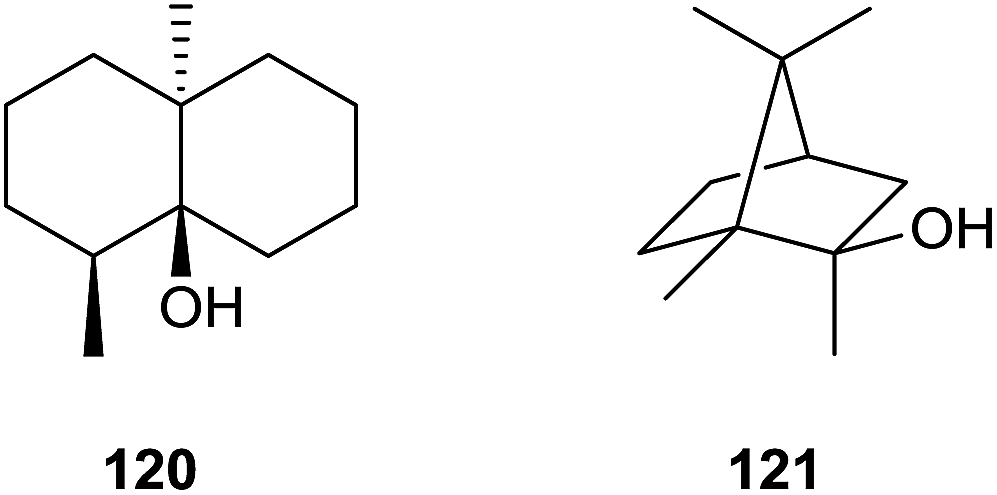 | ||
| Scheme 25 Structures of geosmin (120) and 2-methylisoborneol (121). | ||
5.1 Geosmin synthase
A mechanistic model for the biosynthesis of 120 that is based on feeding experiments with deuterated mevalonolactone isotopomers was developed more than two decades after the reported incorporation of radioactive acetate.118 The mechanism starts with FPP cyclisation to (R)-C that is attacked by water to yield hedycaryol (77, pathway a in Scheme 26). Its isomerisation by a protonation–deprotonation sequence gives rise to (1(10)E,5E)-germacradien-11-ol (123), a sesquiterpene alcohol that always co-occurs with 120. A protonation-initiated second cyclisation followed by retro-Prins fragmentation results in the trans-octalin 124 and acetone.119,120 A final sequence of protonation, 1,2-hydride shift and attack of water yields 120. Alternatively, the transformation of (R)-C into 123 may proceed via deprotonation to isolepidozene (122), followed by its protonation with opening of the cyclopropyl ring and attack of water (pathway b).121 While it is difficult to distinguish experimentally between the options of pathways a and b, e.g. by isotopic labelling experiments, the detection of trace amounts of 122 in headspace extracts from some actinomycetes is in favour of pathway b.12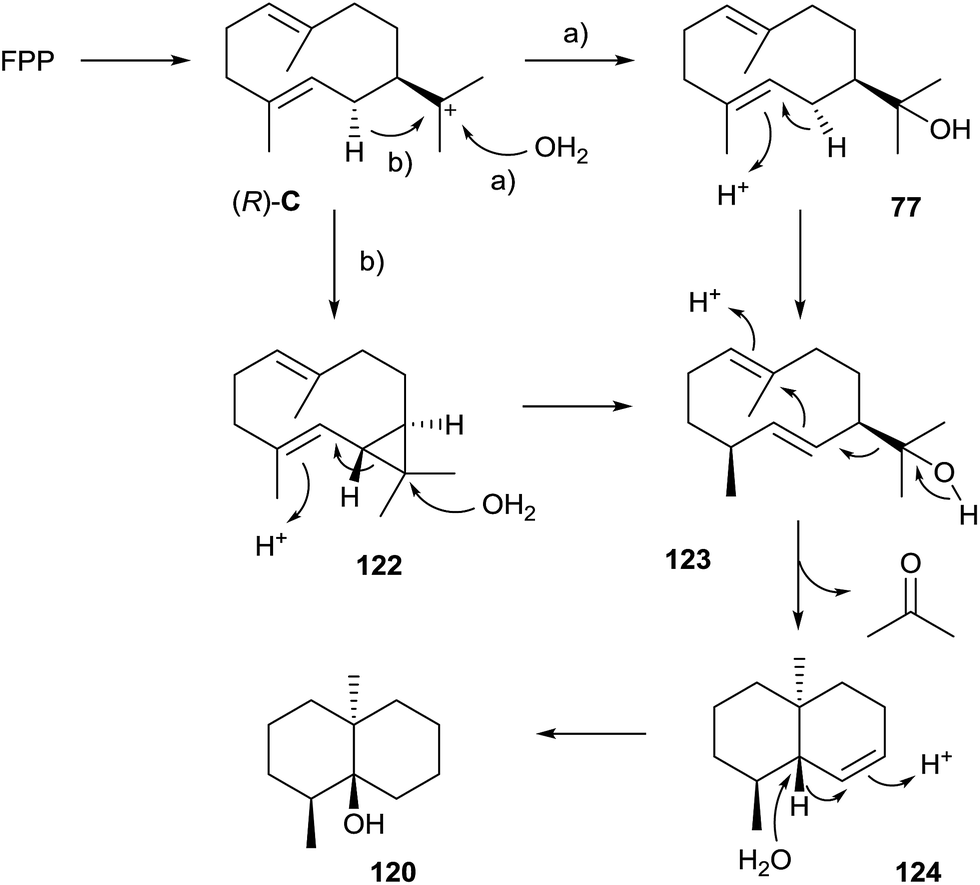 | ||
| Scheme 26 Biosynthesis of geosmin (120). | ||
The coding gene for the geosmin synthase was identified by a gene replacement, demonstrating that the encoded enzyme is composed of two domains.122 First attempts to characterise the enzyme from Streptomyces coelicolor A3(2) in vitro only revealed a terpene cyclase activity for the N-terminal domain for FPP conversion to 123, while the function of the C-terminal domain remained unclear.123 A later study resulted in a deep mechanistic understanding, demonstrating that the N-terminal domain has the previously reported activity, while the C-terminal domain catalyses the transformation of 123 into 120.124
A BLAST search using the amino acid sequence of the geosmin synthase from S. coelicolor as a probe demonstrates that more than 375 geosmin synthase homologs are encoded in bacteria. The majority of these enzymes (>250 sequences) is found in the genus Streptomyces. Both the N- and the C-terminal domains contain the highly conserved motifs of type I enzymes (Table 1). These are found in the N-terminal domain of the S. coelicolor enzyme at 86DDHFLE (aspartate-rich motif), 184R (pyrophosphate sensor), 229NDLFSYQRE (NSE triad) and 325RY (RY dimer). Interestingly, a second NSE triad is present at 267NDVLTSRLHQFE, but this is unusually long and less conserved among all geosmin synthases than the first NSE triad. Accordingly, site-directed mutagenesis of S233A resulted in a drastically decreased enzyme activity, while the mutations T271A and S272A only exhibited small effects,124 suggesting that the first, but not the second NSE triad of the N-terminal domain is important for substrate and Mg2+ cofactor binding. The C-terminal domain of the S. coelicolor enzyme exhibits the highly conserved motifs at 455DDYYP (modified aspartate-rich motif), 552R (pyrophosphate sensor), 598NDVFSYQKE (NSE triad) and 694RY (RY dimer). In subsequent work the geosmin synthases from the cyanobacterium N. punctiforme PCC73102 was also functionally characterised.55,125
5.2 2-Methylisoborneol synthase
A detailed biosynthetic model for the biosynthesis of 2-methylisoborneol (121) was obtained by feeding of deuterated isotopomers of mevalonolacatone and [methyl-13C]methionine to Nannocystis exedens in combination with GC/MS analysis of headspace extracts from this myxobacterium.126 The delineated model starts with the S-adenoyl-L-methionine (SAM) dependent methylation of GPP at C-2, followed by terpene cyclisation of the resulting (E)-2-methyl-GPP (125) to 121 (Scheme 27). Similar to the cyclisation of GPP to monoterpenes, this step requires the isomerisation of 125 to 2-methyllinalyl diphosphate (126) by abstraction and reattack of diphosphate. Subsequently, a gene cluster composed of three genes encoding a C-methyltransferase, a terpene cyclase and a regulator were identified in Streptomyces coelicolor A3(2).127 The C-methyltransferase and the terpene cyclase genes were cloned and expressed to yield the His-tagged proteins that were purified and used in in vitro incubation experiments. The C-methyltransferase catalysed the SAM-dependent methylation of GPP to 125, while the terpene cyclase converted 125 into 121. | ||
| Scheme 27 Biosynthesis of 2-methylisoborneol (121). | ||
As for the geosmin synthase the biosynthetic genes for 121 are particularly widespread in actinomycetes, myxobacteria and cyanobacteria, in agreement with the production of the compound by many bacteria from these taxa.12,24,39,116 A BLAST search using the sequence of the 2-methylisoborneol synthase from S. coelicolor as probe yields at least 120 homologous enzymes from bacteria. The 2-methylisoborneol synthase is compared to other type I terpene cyclases from bacteria with ca. 400–500 amino acid residues unusually long. The highly conserved motifs of the enzyme from S. coelicolor are found at 197DDCYCE (modified aspartate-rich motif), 300R (pyrophosphate sensor), 345NDLYSYTKE (NSE triad) and 433RY (RY dimer, Table 1). The enzymes for the biosynthesis of 121 were also studied from Saccharopolyspora erythraea NRRL 2338 and a few other streptomycetes by heterologous expression in S. avermitilis,128 and from the cyanobacterium Pseudoanabaena limnetica Castaic Lake.129Pseudomonas fluorescens PfO-1 encodes a very similar gene cluster, but the encoded terpene cyclase was shown to produce 2-methylenebornane (127) and not 121, due to a final deprotonation step instead of the attack of water in the terpene cyclisation (Scheme 27).130
The crystal structure of 2-methylisoborneol synthase from S. coelicolor has been obtained in complex with the substrate analogs geranyl S-thiolodiphosphate (GSPP) and 2-fluorogeranyl diphosphate (2FGPP).131 The structure reveals a disordered N-terminal proline-rich domain of unknown function, while the C-terminal domain shows the typical class I terpene synthase fold. Two Mg2+ cations are found in the active site, one of which is coordinated by N345, S349 and E353 of the NSE triad, while the second one is coordinated by D197 (in the enzyme complex with GSPP) of the aspartate-rich motif that is itself disordered. In conclusion, the active site was described as “incompletely closed”.
Recently, several side products of the 2-methylisoborneol synthase were identified by GC/MS and synthesis of reference compounds.132 This includes the methylated monoterpene hydrocarbons and alcohols 2-methylmyrcene (128), 2-methyllimonene (129), 2-methyllinalool (130), 2-methyl-α-terpineol (131) and 2-methyl-β-fenchol (132, Scheme 28). Their formation was explained by different side reactions of cations along the cyclisation cascade from 125 to 121. Furthermore, isotopic labelling studies were performed that gave insights into the stereochemical course of the terpene cyclisation.132
 | ||
| Scheme 28 Side products of 2-methylisoborneol biosynthesis. | ||
6 Conclusions and outlook
As demonstrated in this article terpene cyclases are widespread in bacteria. The recent developments in sequencing techniques have yielded a large number of presumptive terpene cyclase genes, including more than 600 genes for regular type I terpene cyclases, nearly 400 genes for geosmin synthases and >120 genes for 2-methylisoborneol synthases. More than 50 of the encoded enzymes have already been characterised for their products, either by heterologous expression in E. coli or S. avermitilis, or by incubation of purified enzymes with oligoprenyl diphosphates (Table 1). The results of this work also allow for an assignment of the functions of various closely related terpene cyclases by sequence homology (summarised in Fig. 1), but the functions of more than 300 presumptive bacterial terpene cyclases are still unknown, and due to further genome sequencing this number will grow.Our mechanistic understanding has profited much from the structural work, starting with the crystal structures of pentalenene synthase and followed by the epi-isozizaene synthase, 2-methylisoborneol synthase, and recently by the (2Z,6E)-hedycaryol synthase and selina-4(15),7(11)-diene synthase. These structures gave important insights into the active site architecture and allowed for the development of mechanistic models that were subsequently interrogated by site-directed mutagenesis. Today it is clear that besides the well-known aspartate-rich motif DDXX(X)(D,E) and the NSE triad NDXXSXX(R,K)(E,D) also the pyrophosphate sensor (R) and the RY dimer are of critical importance for substrate and Mg2+ cofactor binding, and consequently for enzyme function. As a sequence analysis of all characterised bacterial terpene cyclases reveals, these motifs are found in all cases, and only very few and usually small deviations from these generalised patterns are observed (Table 1).
Certainly, for our deep mechanistic understanding of bacterial terpene biosynthesis also isotopic labelling experiments and computational chemistry approaches were of utmost importance. While the classical labelling technique continues to be important, because it gives direct experimental evidence, computational chemistry is still a young field that will further develop in the future. In just a few years it may become possible to calculate the structure of an enzyme from its amino acid sequence, and the structure of its product including the absolute stereochemistry by modeling the substrate and its cyclisation into a terpene within the active site. Will this make experimental work superfluous? Surely not, because chemistry will always aim at making molecules with interesting functions, and for this purpose we can use terpene cyclases, natural enzymes or specifically designed mutants, as tools. This very interesting class of sophisticated enzymes catalyses reactions that will outcompete pure chemical methods in most cases.
7 References
- O. Wallach, Justus Liebigs Ann. Chem., 1885, 227, 277 CrossRef.
- J. M. Winter, M. C. Moffitt, E. Zazopoulos, J. B. McAlpine, P. C. Dorrestein and B. S. Moore, J. Biol. Chem., 2007, 282, 16362 CrossRef CAS PubMed.
- R. Teufel, R. Kaysser, M. T. Villaume, S. Diethelm, M. K. Carbullido, P. S. Baran and B. S. Moore, Angew. Chem., Int. Ed., 2014, 53, 11019 CrossRef CAS PubMed.
- M. Baunach, J. Franke and C. Hertweck, Angew. Chem., Int. Ed., 2015, 54, 2604 CrossRef CAS PubMed.
- D. W. Christianson, Chem. Rev., 2006, 106, 3412 CrossRef CAS PubMed.
- E. Oldfield and F.-Y. Lin, Angew. Chem., Int. Ed., 2012, 51, 1124 CrossRef CAS PubMed.
- D. J. Tantillo, Nat. Prod. Rep., 2011, 28, 1035 RSC.
- J. S. Dickschat, Nat. Prod. Rep., 2011, 28, 1917 RSC.
- M. L. Wise and R. Croteau, Monoterpene Biosynthesis, in Comprehensive Natural Products Chemistry, ed. D. E. Cane, 1999, vol. 2, p. 97 Search PubMed.
- E. M. Davis and R. Croteau, Cyclisation Enzymes in the Biosynthesis of Monoterpenes, Sesquiterpenes, and Diterpenes, in Topics in Current Chemistry, ed. F. J. Leeper and J. C. Vederas, 2000, vol. 209, p. 54 Search PubMed.
- C. Nakano, H.-K. Kim and Y. Ohnishi, ChemBioChem, 2011, 12, 1988 CrossRef CAS.
- C. A. Citron, J. Gleitzmann, G. Laurenzano, R. Pukall and J. S. Dickschat, ChemBioChem, 2012, 13, 202 CrossRef CAS PubMed.
- M. L. Wise, T. J. Savage, E. Katahira and R. Croteau, J. Biol. Chem., 1998, 273, 14891 CrossRef CAS PubMed.
- F. Chen, D.-K. Ro, J. Petri, J. Gershenzon, J. Bohlmann, E. Pichersky and D. Tholl, Plant Physiol., 2004, 135, 1956 CrossRef CAS PubMed.
- T. Shimada, T. Endo, H. Fujii, M. Hara and M. Omura, Plant Sci., 2005, 168, 987 CrossRef CAS.
- S. Roeder, A.-M. Hartmann, U. Effmert and B. Piechulla, Plant Mol. Biol., 2007, 65, 107 CrossRef CAS PubMed.
- S. C. Kampranis, D. Ioannidis, A. Purvis, W. Mahrez, E. Ninga, N. A. Katerelos, S. Anssour, J. M. Dunwell, J. Degenhardt, A. M. Makris, P. W. Goodenough and C. B. Johnson, Plant Cell, 2007, 19, 1994 CrossRef CAS PubMed.
- J. J. Shaw, T. Berbasova, T. Sasaki, K. Jefferson-George, D. J. Spakowicz, B. F. Dunican, C. E. Portero, A. Narvaez-Trujillo and S. A. Strobel, J. Biol. Chem., 2015, 290, 8511 CrossRef CAS PubMed.
- R. Croteau, W. R. Alonso, A. E. Koepp and M. A. Johnson, Arch. Biochem. Biophys., 1994, 309, 184 CrossRef CAS PubMed.
- Y. Yamada, T. Kuzuyama, M. Komatsu, K. Shin-ya, S. Omura, D. E. Cane and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 857–862 CrossRef CAS PubMed.
- C. Nakano, H.-K. Kim and Y. Ohnishi, ChemBioChem, 2011, 12, 2403 CrossRef CAS PubMed.
- J. Degenhardt, T. G. Köllner and J. Gershenzon, Phytochemistry, 2009, 70, 1621 CrossRef CAS PubMed.
- J. S. Dickschat, T. Martens, T. Brinkhoff, M. Simon and S. Schulz, Chem. Biodiversity, 2005, 2, 837 CAS.
- P. Rabe, C. A. Citron and J. S. Dickschat, ChemBioChem, 2013, 14, 2345 CrossRef CAS PubMed.
- J. S. Dickschat, K. A. K. Pahirulzaman, P. Rabe and T. A. Klapschinski, ChemBioChem, 2014, 15, 810 CrossRef CAS PubMed.
- B. K. Koe, B. A. Sobin and W. D. Celmer, Antibiot. Annu., 1957, 672 Search PubMed.
- C. N. Tetzlaff, Z. You, D. E. Cane, S. Takamatsu, S. Omura and H. Ikeda, Biochemistry, 2006, 45, 6179 CrossRef CAS PubMed.
- D. E. Cane and C. Pargellis, Arch. Biochem. Biophys., 1987, 254, 421 CrossRef CAS PubMed.
- D. E. Cane, J. K. Sohng, C. R. Lamberson, S. M. Rudnicki, Z. Wu, M. D. Lloyd, J. S. Oliver and B. R. Hubbard, Biochemistry, 1994, 33, 5846 CrossRef CAS PubMed.
- C. A. Lesburg, G. Zhai, D. E. Cane and D. W. Christianson, Science, 1997, 277, 1820 CrossRef CAS PubMed.
- L. C. Tarshis, M. Yan, C. D. Poulter and J. C. Sacchettini, Biochemistry, 1994, 33, 10871 CrossRef CAS PubMed.
- C. M. Starks, K. Back, J. Chappell and J. P. Noel, Science, 1997, 277, 1815 CrossRef CAS PubMed.
- M. Seemann, G. Zhai, J.-W. de Kraker, C. M. Paschall, D. W. Christianson and D. E. Cane, J. Am. Chem. Soc., 2002, 124, 7681 CrossRef CAS PubMed.
- M. Seemann, G. Zhai, K. Umezawa and D. E. Cane, J. Am. Chem. Soc., 1999, 121, 591 CrossRef CAS.
- P. Gutta and D. J. Tantillo, J. Am. Chem. Soc., 2006, 128, 6172 CrossRef CAS PubMed.
- L. Zu, M. Xu, M. W. Lodewyk, D. E. Cane, R. J. Peters and D. J. Tantillo, J. Am. Chem. Soc., 2012, 134, 11369 CrossRef CAS PubMed.
- D. E. Cane, J. S. Oliver, P. H. M. Harrison, C. Abell, B. R. Hubbard, C. T. Kane and R. Lattman, J. Am. Chem. Soc., 1990, 112, 4513 CrossRef CAS.
- D. E. Cane, C. Abell, P. H. M. Harrison, B. R. Hubbard, C. T. Kane, R. Lattman, J. S. Oliver and S. W. Weiner, Philos. Trans. R. Soc. London, Ser. B, 1991, 332, 123 CrossRef CAS PubMed.
- C. A. Citron, L. Barra, J. Wink and J. S. Dickschat, Org. Biomol. Chem., 2015, 13, 2673 CAS.
- B. Zhao, X. Lin, D. C. Lamb, S. L. Kelly, M. R. Waterman and D. E. Cane, J. Biol. Chem., 2008, 283, 8183 CrossRef CAS PubMed.
- S. Takamatsu, X. Lin, A. Nara, M. Komatsu, D. E. Cane and H. Ikeda, Microb. Biotechnol., 2011, 4, 184 CrossRef CAS PubMed.
- B. Zhao, L. Lei, D. G. Vassylyev, X. Lin, D. E. Cane, S. L. Kelly, H. Yuan, D. C. Lamb and M. R. Waterman, J. Biol. Chem., 2009, 284, 36711 CrossRef CAS PubMed.
- X. Lin, R. Hopson and D. E. Cane, J. Am. Chem. Soc., 2006, 128, 6022 CrossRef CAS PubMed.
- X. Lin and D. E. Cane, J. Am. Chem. Soc., 2009, 131, 6332 CrossRef CAS PubMed.
- Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2009, 131, 7999 CrossRef CAS PubMed.
- R. P. Pemberton, K. C. Ho and D. J. Tantillo, Chem. Sci., 2015, 6, 2347 RSC.
- J. A. Aaron, X. Lin, D. E. Cane and D. W. Christianson, Biochemistry, 2010, 49, 1787 CrossRef CAS PubMed.
- D. A. Whittington, M. L. Wise, M. Urbansky, R. M. Coates, R. B. Croteau and D. W. Christianson, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15375 CrossRef CAS PubMed.
- D. C. Hyatt, B. Youn, Y. Zhao, B. Santhamma, R. M. Coates, R. B. Croteau and C. Kang, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5360 CrossRef CAS PubMed.
- H. A. Gennadios, V. Gonzalez, L. Di Costanzo, A. Li, F. Yu, D. J. Miller, R. K. Allemann and D. W. Christianson, Biochemistry, 2009, 48, 6175 CrossRef CAS PubMed.
- M. J. Rynkiewicz, D. E. Cane and D. W. Christianson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13543 CrossRef CAS PubMed.
- E. Y. Shishova, L. Di Costanzo, D. E. Cane and D. W. Christianson, Biochemistry, 2007, 46, 1941 CrossRef CAS PubMed.
- P. Baer, P. Rabe, K. Fischer, C. A. Citron, T. A. Klapschinski, M. Groll and J. S. Dickschat, Angew. Chem., Int. Ed., 2014, 53, 7652 CrossRef CAS PubMed.
- R. Li, W. K. W. Chou, J. A. Himmelberger, K. M. Litwin, G. G. Harris, D. E. Cane and D. W. Christianson, Biochemistry, 2014, 53, 1155 CrossRef CAS PubMed.
- S. A. Agger, F. Lopez-Gallego, T. R. Hoye and C. Schmidt-Dannert, J. Bacteriol., 2008, 190, 6084 CrossRef CAS PubMed.
- P. Gutta and D. J. Tantillo, Angew. Chem., Int. Ed., 2005, 44, 2719 CrossRef CAS PubMed.
- D. J. Tantillo, Chem. Soc. Rev., 2010, 39, 2847 RSC.
- W. K. W. Chou, I. Fanizza, T. Uchiyama, M. Komatsu, H. Ikeda and D. E. Cane, J. Am. Chem. Soc., 2010, 132, 8850 CrossRef CAS PubMed.
- Y. J. Hong and D. J. Tantillo, Org. Lett., 2011, 13, 1294 CrossRef CAS PubMed.
- Y. Hu, W. K. W. Chou, R. Hopson and D. E. Cane, Chem. Biol., 2011, 18, 32 CrossRef CAS PubMed.
- D. Arigoni, Pure Appl. Chem., 1975, 41, 219 CrossRef CAS.
- S. Garms, T. G. Köllner and W. Boland, J. Org. Chem., 2010, 75, 5590 CrossRef CAS PubMed.
- I. Alchanati, J. A. Acreman Patel, J. Liu, C. R. Benedikt, R. D. Stipanovic, A. A. Bell, Y. Cui and C. W. Magill, Phytochemistry, 1998, 47, 961 CrossRef CAS.
- E. M. Davis, J. Tsuji, G. D. Davis, M. L. Pierce and M. Essenberg, Phytochemistry, 1996, 41, 1047 CrossRef CAS PubMed.
- X.-Y. Chen, M. Wang, Y. Chen, V. J. Davisson and P. Heinstein, J. Nat. Prod., 1996, 59, 944 CrossRef CAS PubMed.
- C. Nakano, F. Kudo, T. Eguchi and Y. Ohnishi, ChemBioChem, 2011, 12, 2271 CrossRef CAS PubMed.
- F. Bohlmann, C. Zdero, R. M. King and H. Robinson, Phytochemistry, 1984, 23, 1798 CrossRef CAS.
- G. Ohloff, W. Giersch, R. Näf and F. Delay, Helv. Chim. Acta, 1986, 69, 698 CrossRef CAS.
- C. Nakano, S. Horinouchi and Y. Ohnishi, J. Biol. Chem., 2011, 286, 27980 CrossRef CAS PubMed.
- R. Misra, R. C. Pandey and S. Dev, Tetrahedron, 1979, 35, 2301 CrossRef CAS.
- D. E. Cane, M. Tandon and P. C. Prabhakaran, J. Am. Chem. Soc., 1993, 115, 8103 CrossRef CAS.
- D. E. Cane and M. Tandon, Tetrahedron Lett., 1994, 35, 5355 CrossRef CAS.
- D. E. Cane and M. Tandon, J. Am. Chem. Soc., 1995, 117, 5602 CrossRef CAS.
- C. Nakano, T. Tezuka, S. Horinouchi and Y. Ohnishi, J. Antibiot., 2012, 65, 551 CrossRef CAS PubMed.
- K. Nabeta, K. Kigure, M. Fujita, T. Nagoya, T. Ishikawa, H. Okuyama and T. Takasawa, J. Chem. Soc., Perkin Trans. 1, 1995, 1935 RSC.
- N. N. Gerber, Phytochemistry, 1971, 10, 185 CrossRef CAS.
- Y. Ohta and Y. Hirose, Tetrahedron Lett., 1967, 8, 2073 CrossRef.
- P. Baer, P. Rabe, C. A. Citron, C. C. de Oliveira Mann, N. Kaufmann, M. Groll and J. S. Dickschat, ChemBioChem, 2014, 15, 213 CrossRef CAS PubMed.
- P. Rabe and J. S. Dickschat, Angew. Chem., Int. Ed., 2013, 52, 1810 CrossRef CAS PubMed.
- P. Rabe, K. A. K. Pahirulzaman and J. S. Dickschat, Angew. Chem., Int. Ed., 2015, 54, 6041 CrossRef CAS PubMed.
- A. Schifrin, T. T. B. Ly, N. Günnewich, J. Zapp, V. Thiel, S. Schulz, F. Hannemann, Y. Khatri and R. Bernhardt, ChemBioChem, 2015, 16, 337 CrossRef CAS PubMed.
- H. Harada, K. Shindo, K. Iki, A. Teraoka, S. Okamoto, F. Yu, J. Hattan, R. Utsumi and N. Misawa, Appl. Microbiol. Biotechnol., 2011, 90, 467 CrossRef CAS PubMed.
- R. Riclea, C. A. Citron, J. Rinkel and J. S. Dickschat, Chem. Commun., 2014, 50, 4228 RSC.
- S. D. Bentley, K. F. Chater, A.-M. Cerdeno-Tarraga, G. L. Challis, N. R. Thomson, K. D. James, D. E. Harris, M. A. Quail, H. Kieser, D. Harper, A. Bateman, S. Brown, G. Chandra, C. W. Chen, M. Collins, A. Cronin, A. Fraser, A. Goble, J. Hidalgo, T. Hornsby, S. Howarth, C.-H. Huang, T. Kieser, L. Larke, L. Murphy, K. Oliver, S. O'Neil, E. Rabbinowitsch, M.-A. Rajandream, K. Rutherford, S. Rutter, K. Seeger, D. Saunders, S. Sharp, R. Squares, S. Squares, K. Taylor, T. Warren, A. Wietzorrek, J. Woodward, B. G. Barrell, J. Parkhill and D. A. Hopwood, Nature, 2002, 417, 141 CrossRef PubMed.
- H. Ikeda, J. Ishikawa, A. Hanamoto, M. Shinose, H. Kikuchi, T. Shiba, Y. Sakaki, M. Hattori and S. Omura, Nat. Biotechnol., 2003, 21, 526 CrossRef PubMed.
- J. S. Dickschat, Nat. Prod. Rep., 2014, 31, 838 RSC.
- Y. Yamada, S. Arima, T. Nagamitsu, K. Johmoto, H. Uekusa, T. Eguchi, K. Shin-ya, D. E. Cane and H. Ikeda, J. Antibiot., 2015, 68, 385 CrossRef CAS PubMed.
- J.-Y. Chow, B.-X. Tian, G. Ramamoorthy, B. S. Hillerich, R. D. Seidel, S. C. Almo, M. P. Jacobson and C. D. Poulter, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5661 CrossRef CAS PubMed.
- P. Rabe, L. Barra, J. Rinkel, R. Riclea, C. A. Citron, T. A. Klapschinski, A. Janusko and J. S. Dickschat, Angew. Chem., Int. Ed., 2015, 54, 13448–13451 CrossRef CAS PubMed.
- S.-Y. Kim, P. Zhao, M. Igarashi, R. Sawa, T. Tomita, M. Nishiyama and T. Kuzuyama, Chem. Biol., 2009, 16, 736 CrossRef CAS PubMed.
- C. Görner, I. Häuslein, P. Schrepfer, W. Eisenreich and T. Brück, ChemCatChem, 2013, 5, 3289 CrossRef.
- R. Janke, C. Görner, M. Hirte, T. Brück and B. Loll, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1528 CAS.
- A. Meguro, Y. Motoyoshi, K. Teramoto, S. Ueda, Y. Totsuka, Y. Ando, T. Tomita, S.-Y. Kim, T. Kimura, M. Igarashi, R. Sawa, T. Shinada, M. Nishiyama and T. Kuzuyama, Angew. Chem., Int. Ed., 2015, 54, 4353 CrossRef CAS PubMed.
- A. Meguro, T. Tomita, M. Nishiyama and T. Kuzuyama, ChemBioChem, 2013, 14, 316 CrossRef CAS PubMed.
- A. C. Kohl and R. G. Kerr, Mar. Drugs, 2003, 1, 54 CrossRef CAS.
- J. Jakupovic, C. Zdero, M. Grenz, F. Tsichritzis, L. Lehmann, S. M. Hashemi-Nejad and F. Bohlmann, Phytochemistry, 1989, 28, 1119 CrossRef CAS.
- M. J. Smanski, R. M. Peterson, S.-X. Huang and B. Shen, Curr. Opin. Chem. Biol., 2012, 16, 132 CrossRef CAS PubMed.
- R. J. Peters, Nat. Prod. Rep., 2010, 27, 1521 RSC.
- T. Dairi, Y. Hamano, T. Kuzuyama, N. Itoh, K. Furihata and H. Seto, J. Bacteriol., 2001, 183, 6085 CrossRef CAS PubMed.
- Y. Hamano, T. Kuzuyama, N. Itoh, K. Furihata, H. Seto and T. Dairi, J. Biol. Chem., 2002, 277, 37098 CrossRef CAS PubMed.
- T. Kawasaki, T. Kuzuyama, Y. Kuwamori, N. Matsuura, N. Itoh, K. Furihata, H. Seto and T. Dairi, J. Antibiot., 2004, 57, 739 CrossRef CAS PubMed.
- D. Morrone, J. Chambers, L. Lowry, G. Kim, A. Anterola, K. Bender and R. J. Peters, FEBS Lett., 2009, 583, 475 CrossRef CAS PubMed.
- M. J. Smanski, Z. Yu, J. Casper, S. Lin, R. M. Peterson, Y. Chen, E. Wendt-Pienkowski, S. R. Rajski and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13498 CrossRef CAS PubMed.
- T. Kawasaki, Y. Hayashi, T. Kuzuyama, K. Furihata, N. Itoh, H. Seto and T. Dairi, J. Bacteriol., 2006, 188, 1236 CrossRef CAS PubMed.
- W. Liu, X. Feng, Y. Zheng, C.-H. Huang, C. Nakano, T. Hoshino, S. Bogue, T.-P. Ko, C.-C. Chen, Y. Cui, J. Li, I. Wang, S.-T. Danny Hsu, E. Oldfield and R. T. Guo, Sci. Rep., 2014, 4, 6214 CrossRef CAS PubMed.
- C. Nakano, T. Okamura, T. Sato, T. Dairi and T. Hoshino, Chem. Commun., 2005, 1016 RSC.
- C. Nakano and T. Hoshino, ChemBioChem, 2009, 10, 2060 CrossRef CAS PubMed.
- C. Nakano, T. Ootsuka, K. Takayama, T. Mitsui, T. Sato and T. Hoshino, Biosci., Biotechnol., Biochem., 2011, 75, 75 CrossRef CAS PubMed.
- F. M. Mann, M. Xu, X. Chen, D. B. Fulton, D. G. Russell and R. J. Peters, J. Am. Chem. Soc., 2009, 131, 17526 CrossRef CAS PubMed.
- E. Layre, H. J. Lee, D. C. Young, A. J. Martinot, J. Buter, A. J. Minnaard, J. W. Annand, S. M. Fortune, B. B. Snider, I. Matsunaga, E. J. Rubin, T. Alber and D. B. Moody, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2978 CrossRef CAS PubMed.
- C. A. Citron, P. Rabe, L. Barra, C. Nakano, T. Hoshino and J. S. Dickschat, Eur. J. Org. Chem., 2014, 7684 CrossRef CAS.
- C. Nakano, M. Oshima, N. Kurashima and T. Hoshino, ChemBioChem, 2015, 16, 772 CrossRef CAS PubMed.
- M. Xu, M. L. Hillwig, A. L. Lane, M. S. Tiernan, B. S. Moore and R. J. Peters, J. Nat. Prod., 2014, 77, 2144 CrossRef CAS PubMed.
- N. N. Gerber and H. A. Lechevalier, Appl. Microbiol., 1965, 13, 935 CAS.
- L. L. Medsker, D. Jenkins, J. F. Thomas and C. Koch, Environ. Sci. Technol., 1969, 3, 476 CrossRef CAS.
- S. Schulz and J. S. Dickschat, Nat. Prod. Rep., 2007, 24, 814 RSC.
- R. Bentley and R. Meganathan, FEBS Lett., 1981, 125, 220 CrossRef CAS.
- J. S. Dickschat, H. B. Bode, T. Mahmut, R. Müller and S. Schulz, J. Org. Chem., 2005, 70, 5174 CrossRef CAS PubMed.
- T. Nawrath, J. S. Dickschat, R. Müller, J. Jiang, D. E. Cane and S. Schulz, J. Am. Chem. Soc., 2008, 130, 430 CrossRef CAS PubMed.
- J. Jiang and D. E. Cane, J. Am. Chem. Soc., 2008, 130, 428 CrossRef CAS PubMed.
- D. E. Cane, X. He, S. Kobayashi, S. Omura and H. Ikeda, J. Antibiot., 2006, 59, 471 CrossRef CAS PubMed.
- B. Gust, G. L. Challis, K. Fowler, T. Kieser and K. F. Chater, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1541 CrossRef CAS PubMed.
- D. E. Cane and R. M. Watt, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1547 CrossRef CAS.
- J. Jiang, X. He and D. E. Cane, Nat. Chem. Biol., 2007, 3, 711 CrossRef CAS PubMed.
- S. Giglio, J. Jiang, C. P. Saint, D. E. Cane and P. T. Monis, Environ. Sci. Technol., 2008, 42, 8027 CrossRef CAS PubMed.
- J. S. Dickschat, T. Nawrath, V. Thiel, B. Kunze, R. Müller and S. Schulz, Angew. Chem., Int. Ed., 2007, 46, 8287 CrossRef CAS PubMed.
- C.-M. Wang and D. E. Cane, J. Am. Chem. Soc., 2008, 130, 8908 CrossRef CAS PubMed.
- M. Komatsu, M. Tsuda, S. Omura, H. Oikawa and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7422 CrossRef CAS PubMed.
- S. Giglio, W. K. W. Chou, H. Ikeda, D. E. Cane and P. T. Monis, Environ. Sci. Technol., 2011, 45, 992 CrossRef CAS PubMed.
- W. K. W. Chou, H. Ikeda and D. E. Cane, Tetrahedron, 2011, 67, 6627 CrossRef CAS PubMed.
- M. Köksal, W. K. W. Chou, D. E. Cane and D. W. Christianson, Biochemistry, 2012, 51, 3011 CrossRef PubMed.
- N. L. Brock, S. R. Ravella, S. Schulz and J. S. Dickschat, Angew. Chem., Int. Ed., 2013, 52, 2100 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |