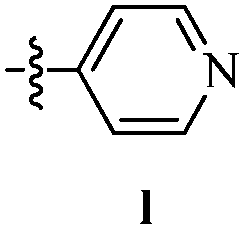
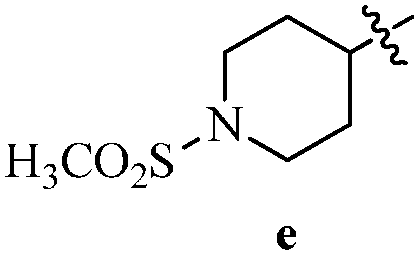
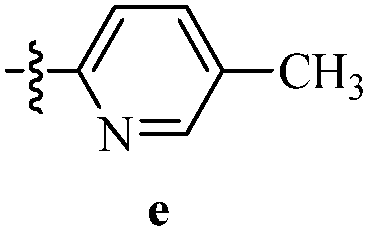
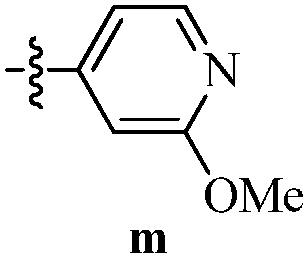
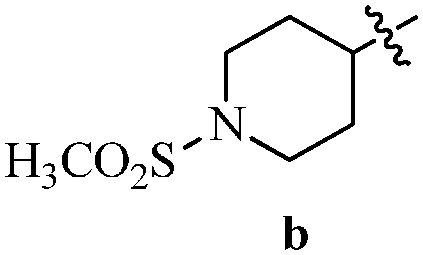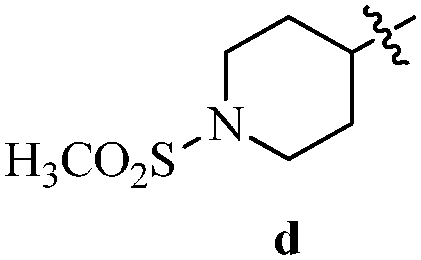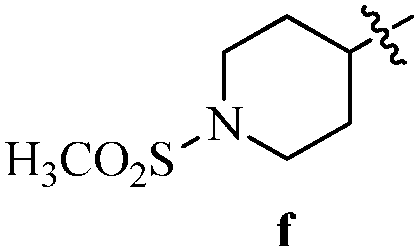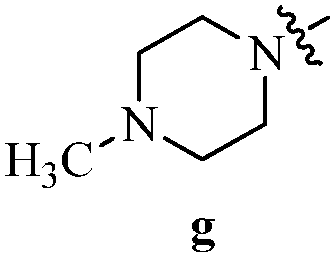DOI:
10.1039/C6NJ03181A
(Perspective)
New J. Chem., 2017,
41, 16-41
Review: biologically active pyrazole derivatives
Received
(in Montpellier, France)
10th October 2016
, Accepted 16th November 2016
First published on 17th November 2016
Abstract
Nitrogen-containing heterocyclic compounds and their derivatives have historically been invaluable as a source of therapeutic agents. Pyrazole, which has two nitrogen atoms and aromatic character, provides diverse functionality and stereochemical complexity in a five-membered ring structure. In the past decade, studies have reported a growing body of data on different pyrazole derivatives and their innumerable physiological and pharmacological activities. In part, such studies attempted to reveal the wide range of drug-like properties of pyrazole derivatives along with their structure–activity relationships in order to create opportunities to harness the full potentials of these compounds. Here, we summarize strategies to synthesize pyrazole derivatives and demonstrate that this class of compounds can be targeted for the discovery of new drugs and can be readily prepared owing to recent advances in synthetic medicinal chemistry.

Anam Ansari
| Anam Ansari was born in Uttar Pradesh (India) in 1992. She received her BSc in Chemistry in 2012 and MSc with a specialization in Organic Chemistry in 2014 from Aligarh Muslim University, Aligarh. At present, she is pursuing a PhD as a Junior Research Fellow under the supervision of Prof. Shamsuzzaman in the Department of Chemistry, AMU, Aligarh. Her research interest focuses on steroidal derivatives incorporated within heterocyclic systems. |

Abad Ali
| Abad Ali received his BSc (Hons.) degree in Chemistry from Aligarh Muslim University in 2009. He obtained his MSc degree in Organic Chemistry from the same institution in 2011. He has already submitted a PhD thesis under the supervision of Prof. Shamsuzzaman entitled “Synthesis, Characterization and Biological Evaluation of Steroidal Heterocyclic Compounds.” |

Mohd Asif
| Mohd Asif was born in 1986 in Aligarh (U.P.). He obtained his BSc degree in chemistry from Aligarh Muslim University and received his MSc from the same university. Presently, he is working as a research scholar under the supervision of Prof. Shamsuzzaman at AMU, Aligarh. |

Shamsuzzaman
| Prof. Shamsuzzaman obtained a PhD in 1983 in synthetic organic chemistry from AMU. In 1985, he joined Queen's University, Belfast (UK) as a Leverhulme Commonwealth/USA Visiting Fellow. In Jan. 1989, he joined JMI, New Delhi as Assistant Professor. Later, in 1998, he joined AMU, Aligarh as Associate Professor. Presently, he is working as Professor of Chemistry in the Department of Chemistry, Aligarh Muslim University, Aligarh. He has published about 100 research papers in reputed international journals. His research interests are mainly steroids and their synthetic and biological evaluation. |
Introduction
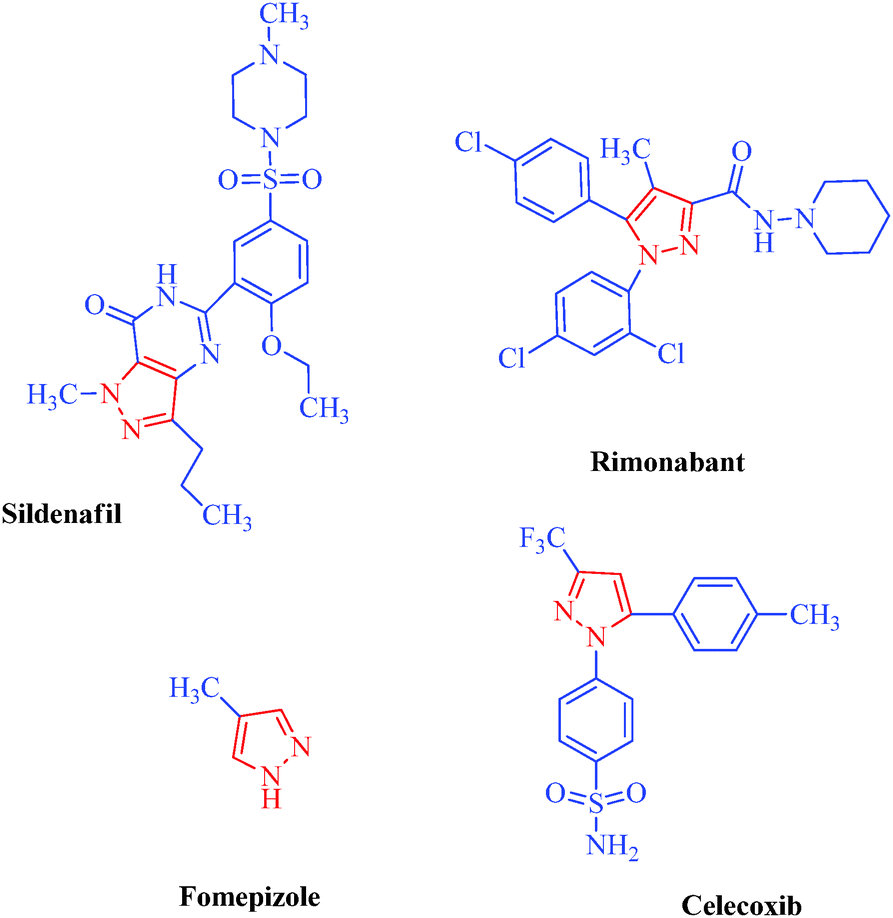
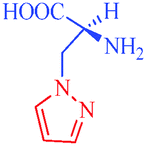
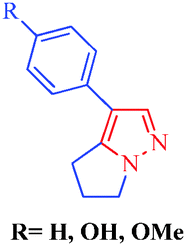
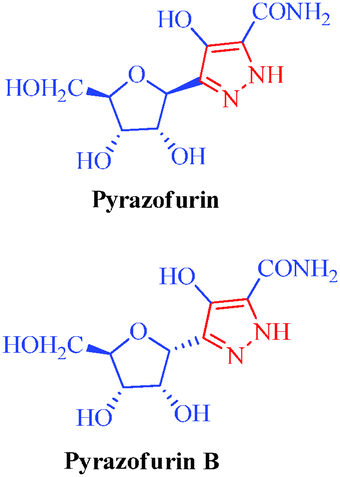
Heterocycles are an extraordinarily important and unique class of compounds; they make up more than half of all known organic compounds and have a wide range of physical, chemical and biological properties spanning a broad spectrum of reactivity and stability.1 Heterocycles are widely distributed in nature and play a vital role in metabolism because their structural subunits exist in many natural products, including vitamins, hormones, antibiotics, and alkaloids as well as pharmaceuticals, agrochemicals, dyes, and many others.2 In addition to naturally occurring compounds, a large number of synthetic heterocyclic compounds with important physiological and pharmacological properties are also known.3 These compounds provide scaffolds on which pharmacophores can arrange to yield potent and selective drugs.4 Moreover, compounds having heterocyclic moieties display enhanced solubility and salt-formation properties that enable their oral absorption and bioavalability.5 Among heterocyclic compounds, nitrogen-containing heterocycles are the core structures of numerous biologically active compounds and exhibit numerous application in chemistry, biology and other sciences.6 They are the building blocks of life due to their wide occurrence in nature and central roles in the chemical reactions that occur in all organisms.7 Furthermore, nitrogen-containing heterocycles play an important role in coordination chemistry.8 Pyrazoles are well-known examples of aromatic heterocycles containing two nitrogen atoms in their five-membered rings.9 They constitute an important heterocyclic family covering a broad range of synthetic as well as natural products that display innumerable chemical, biological, agrochemical and pharmacological properties.10 Pyrazole derivatives represent one of the most active classes of compounds and possess a wide spectrum of biological activities.11–22 In recent years, several drugs have been developed from pyrazole derivatives. For example, celecoxib demonstrates anti-inflammatory effects and inhibits COX-2; rimonabant functions as a cannabinoid receptor and is utilized to treat obesity; fomepizole inhibits alcohol dehydrogenase; and sildenafil inhibits phosphodiesterase23 (Fig. 1). Some of these compounds have important applications in material chemistry24 and as brightening agents,25 and some exhibit significant solvatochromic26 and electroluminescence27 properties. Moreover, they act as semiconductors,28 liquid crystals,29 and organic light-emitting diodes.30 Heterocycles having pyrazole rings are also of considerable interest because of their synthetic utility as synthetic reagents in multicomponent reactions,31 chiral auxillaries32 and guanylating agents.33 It has also been found that various substituted pyrazoles are used as chelation and extraction reagents for many metal ions.34 In addition, there are well-known natural products containing pyrazole moieties that have various physiological, pharmacological and toxic properties35 (Table 1). In light of the significance of pyrazoles in countless areas, particularly medicinal chemistry, the present review focuses on the synthesis of diverse pyrazole derivatives and their biological activities.
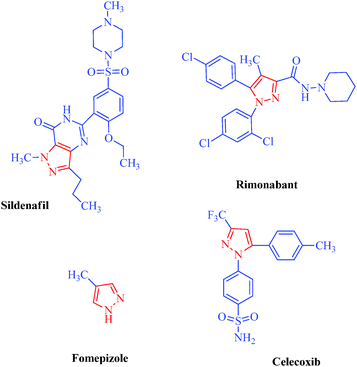 |
| | Fig. 1 Drug molecules containing pyrazole scaffolds. | |
Table 1 Natural products containing pyrazole moieties
| Name |
Isolated from |
Structure |
Applications |
|
L-α-Amino-β (pyrazolyl-N)-propanoic acid
– First natural product containing pyrazole
|
Citrullus vulgaris
|
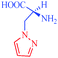
|
– Antidiabetic |
|
|
|
Withasomnine
4′-Hydroxywithasomnine
4′-Methoxywithasomnine
|
Withania somnifera Dun
|
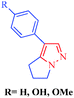
|
– Analgesic
– Anti-inflammatory
Depressant to
– CNS (Central Nervous System)
– Circulatory system
|
|
|
|
Pyrazofurin
Pyrazofurin B
|
Streptomyces candidus
|
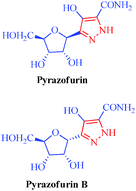
|
– Antitumor
– Antiviral
|
|
|
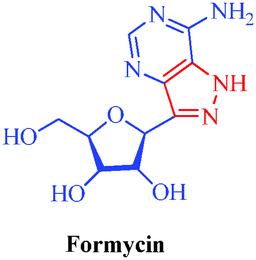
| Formycin |
Streptomyces candidus, Streptomyces lavendulae and Nocardia interforma |
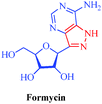
|
– Antiviral
– Antitumor
|
|
|
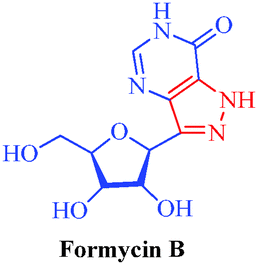
| Formycin B |
Streptomyces lavendulae and Nocardia interforma |
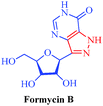
|
– Antiviral
– Antitumor
|
|
|
|
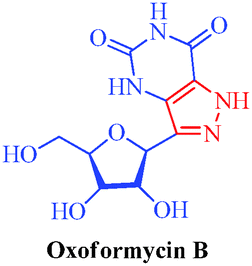
Oxoformycin B
– A metabolite of Formycin and Formycin B
|
Streptomyces lavendulae and Nocardia interforma |
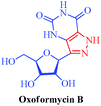
|
– Antiviral
– Antitumor
|
|
|
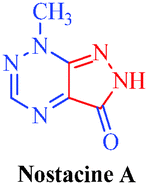
| Nostacine A |
Nostoc spongiaeforme
|
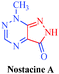
|
– Cytotoxic |
|
|
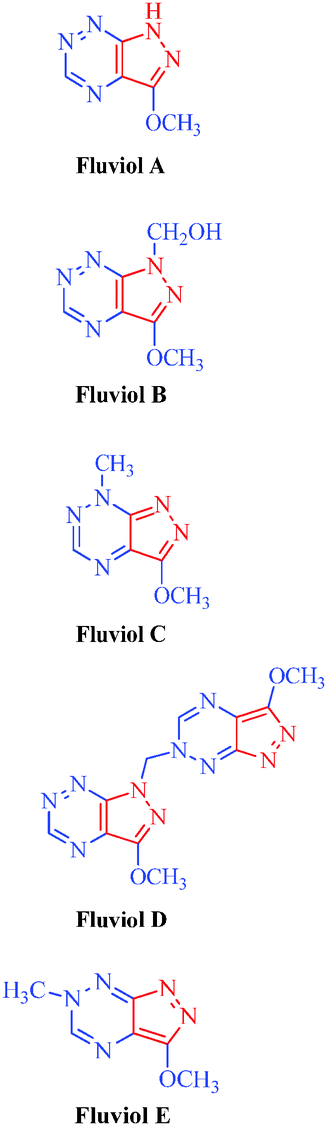
| Fluviols (A–E) |
Pseudomonas fluoresences
|
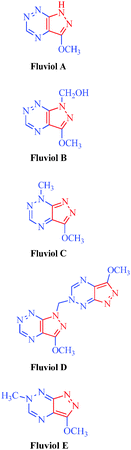
|
– Antimicrobial |
|
|
Chemistry
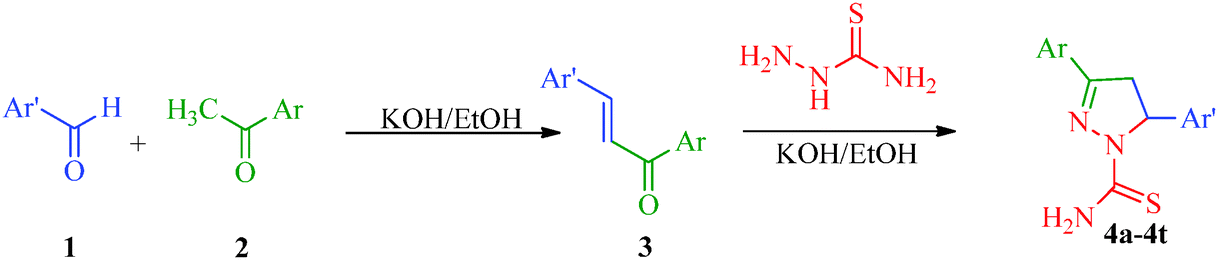
The pyrazole ring is a prominent structural motif found in numerous pharmaceutically active compounds. This is mainly due to its ease of preparation and pharmacological activity. It has also been found that the selective functionalization of pyrazole with diverse substituents improves their range of action in various fields. As a part of the continuing interest in pyrazole derivatives, numerous compounds with pyrazole rings have been reported. Chimenti et al.36 put forward a synthetic route for a series of N1-thiocarbamoyl-3,5-di(hetero)aryl-4,5-dihydro-(1H)-pyrazole derivatives that act as monoamine oxidase inhibitors (Scheme 1). In this protocol, the starting 1,3-di(hetero)aryl-2-propen-1-ones (3) have been synthesized by Claisen-Schmidt condensation under reflux between substituted aryl or heteroaryl aldehyde (1) and an appropriate substituted aryl or heteroaryl ketone (2).37 The intermediates were treated with thiosemicarbazide in KOH/EtOH to afford the desired products in good yields without further purification (Table 2).
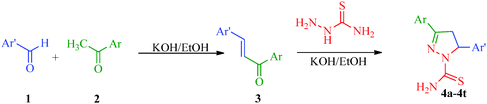 |
| | Scheme 1 Synthesis of N1-thiocarbamoyl-3,5-di(hetero)aryl-4,5-dihydro-(1H)-pyrazole derivatives (4a–4t) as monoamine oxidase inhibitors. | |
Table 2
N1-Thiocarbamoyl-3,5-di(hetero)aryl-4,5-dihydro-(1H)-pyrazole derivatives (4a–4t)
| Entry |
Compound |
Ar |
Ar′ |
| 1 |
|
4a
|
Ph |
Ph |
| 2 |
4b
|
Ph |
4′-CH3-Ph |
| 3 |
4c
|
Ph |
4′-Cl-Ph |
| 4 |
4d
|
Ph |
Fur-2′-yl |
| 5 |
4e
|
4′-CH3-Ph |
4′-F-Ph |
| 6 |
4f
|
4′-CH3-Ph |
Thiophen-2′-yl |
| 7 |
4g
|
4′-F-Ph |
Thiophen-2′-yl |
| 8 |
4h
|
4′-Cl-Ph |
4′-CH3-Ph |
| 9 |
4i
|
4′-Cl-Ph |
4′-F-Ph |
| 10 |
4j
|
4′-Cl-Ph |
4′-Cl-Ph |
| 11 |
4k
|
4′-Cl-Ph |
Pyrrol-2′-yl |
| 12 |
4l
|
Fur-2′-yl |
4′-CH3-Ph |
| 13 |
4m
|
Fur-2′-yl |
4′-F-Ph |
| 14 |
4n
|
Fur-2′-yl |
Thiophen-2′-yl |
| 15 |
4o
|
Thiophen-2′-yl |
Pyrrol-2′-yl |
| 16 |
4p
|
Pyrrol-2′-yl |
Ph |
| 17 |
4q
|
Pyrrol-2′-yl |
4′-F-Ph |
| 18 |
4r
|
Pyrrol-2′-yl |
4′-Cl-Ph |
| 19 |
4s
|
Pyrrol-2′-yl |
Fur-2′-yl |
| 20 |
4t
|
Pyrrol-2′-yl |
Thiophen-2′-yl |
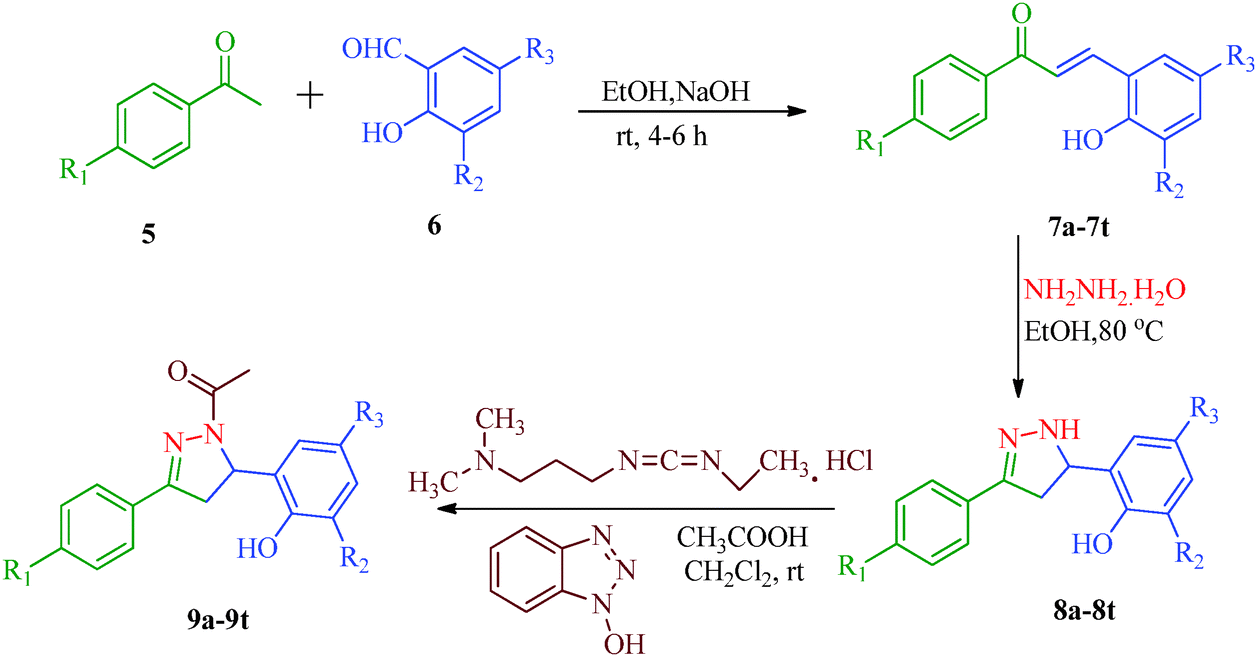
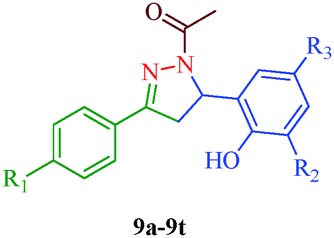
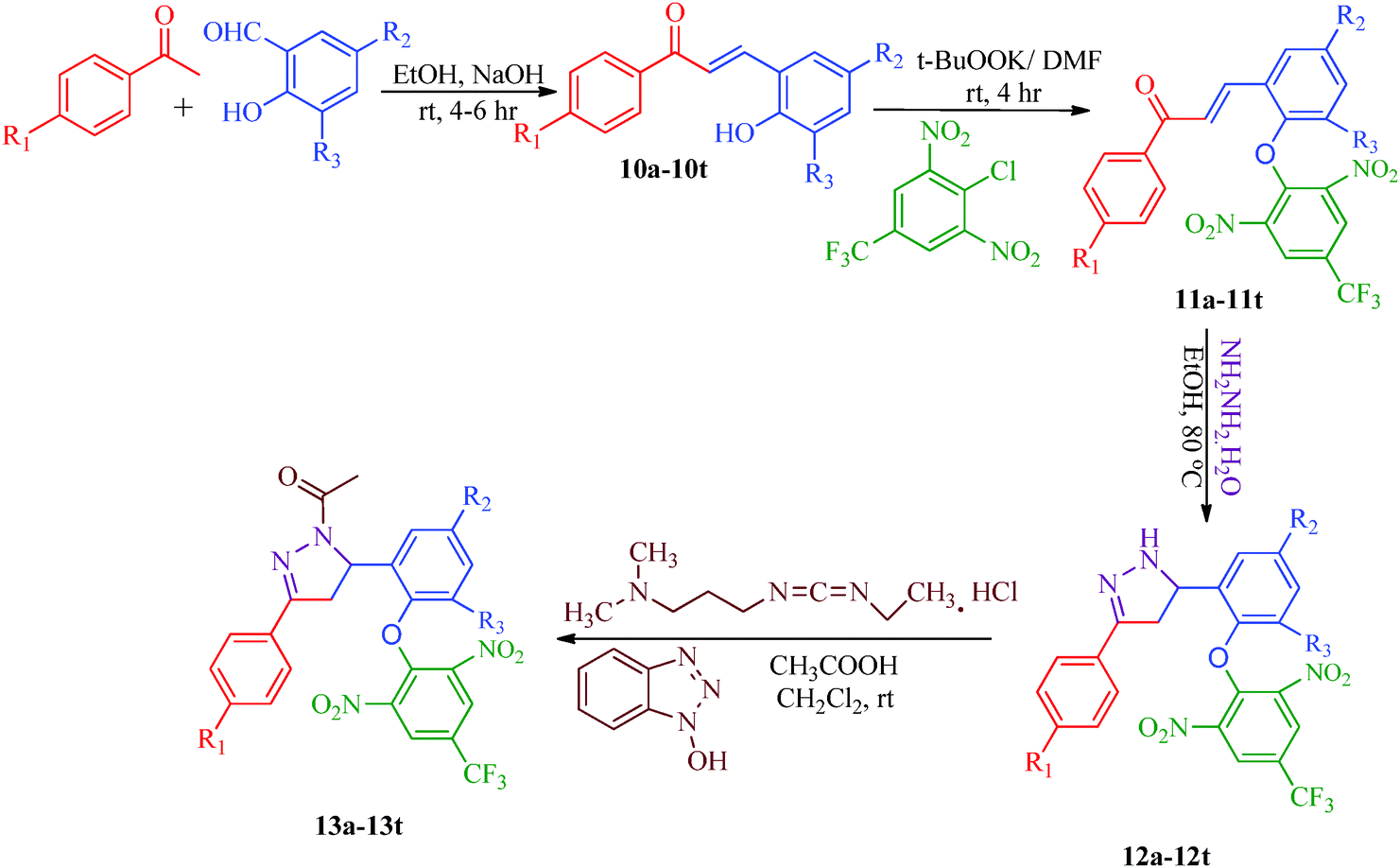
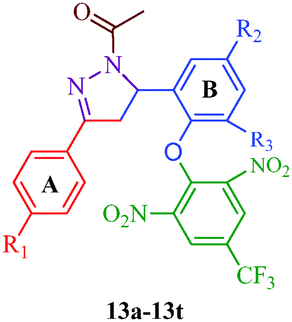
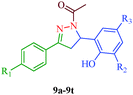
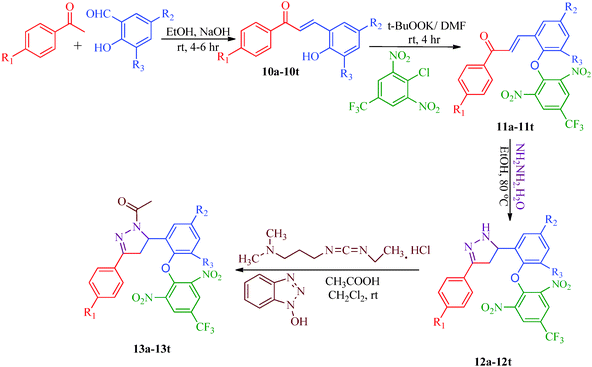
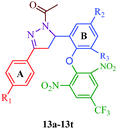
Liu et al.38 reported the synthesis of 4,5-dihydro-2H-pyrazole-2-hydroxyphenyl derivatives as BRAF inhibitors with hydrazine hydrate and chalcones (Scheme 2). The chalcones (7a–7t) were obtained from substituted salicylaldehydes (6) and substituted acetophenones (5) using 40% KOH as catalyst. The N-acetyl group in the final products (9a–9t) was introduced by treating a solution of compounds 8a–8t with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and hydroxybenzotriazole (HOBT) (Table 3). They further reported a slightly modified synthetic route to 4,5-dihydro-1H-pyrazole derivatives (13a–13t) as DNA gyrase inhibitors39 (Scheme 3). The chalcones (10a–10t) were first allowed to react with 1-chloro-2,6-dinitro-4-trifluoromethylbenzene in the presence of potassium tert-butoxide to furnish 11a–11t, which were then reacted with hydrazine hydrate in refluxing ethanol for 8 h. Subsequently, the desired products (13a–13t) were obtained by the treatment of solutions of compounds 12a–12t with EDC (I) and HOBT (II) (Table 4).
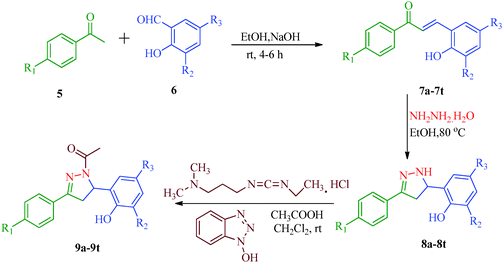 |
| | Scheme 2 Synthesis of 4,5-dihydro-2H-pyrazole-2-hydroxyphenyl derivatives (9a–9t) as BRAF inhibitors. | |
Table 3 4,5-Dihydro-2H-pyrazole-2-hydroxyphenyl derivatives (9a–9t)
| Entry |
Compound |
R1 |
R2 |
R3 |
| 1 |
|
9a
|
CH3 |
Br |
Br |
| 2 |
9b
|
CH3 |
Cl |
Cl |
| 3 |
9c
|
CH3 |
H |
Br |
| 4 |
9d
|
CH3 |
H |
Cl |
| 5 |
9e
|
OCH3 |
Br |
Br |
| 6 |
9f
|
OCH3 |
Cl |
Cl |
| 7 |
9g
|
OCH3 |
H |
Br |
| 8 |
9h
|
OCH3 |
H |
Cl |
| 9 |
9i
|
F |
Br |
Br |
| 10 |
9j
|
F |
Cl |
Cl |
| 11 |
9k
|
F |
H |
Br |
| 12 |
9l
|
F |
H |
Cl |
| 13 |
9m
|
Cl |
Br |
Br |
| 14 |
9n
|
Cl |
Cl |
Cl |
| 15 |
9o
|
Cl |
H |
Br |
| 16 |
9p
|
Cl |
H |
Cl |
| 17 |
9q
|
Br |
Br |
Br |
| 18 |
9r
|
Br |
Cl |
Cl |
| 19 |
9s
|
Br |
H |
Br |
| 20 |
9t
|
Br |
H |
Cl |
 |
| | Scheme 3 Synthesis of 4,5-dihydro-1H-pyrazole derivatives (13a–13t) as DNA gyrase inhibitors. | |
Table 4 4,5-Dihydro-1H-pyrazole derivatives (13a–13t)
| Entry |
Compound |
R1 |
R2 |
R3 |
| 1 |
|
13a
|
CH3 |
Br |
Br |
| 2 |
13b
|
CH3 |
Cl |
Cl |
| 3 |
13c
|
CH3 |
Br |
H |
| 4 |
13d
|
CH3 |
Cl |
H |
| 5 |
13e
|
OCH3 |
Br |
Br |
| 6 |
13f
|
OCH3 |
Cl |
Cl |
| 7 |
13g
|
OCH3 |
Br |
H |
| 8 |
13h
|
OCH3 |
Cl |
H |
| 9 |
13i
|
F |
Br |
Br |
| 10 |
13j
|
F |
Cl |
Cl |
| 11 |
13k
|
F |
Br |
H |
| 12 |
13l
|
F |
Cl |
H |
| 13 |
13m
|
Cl |
Br |
Br |
| 14 |
13n
|
Cl |
Cl |
Cl |
| 15 |
13o
|
Cl |
Br |
H |
| 16 |
13p
|
Cl |
Cl |
H |
| 17 |
13q
|
Br |
Br |
Br |
| 18 |
13r
|
Br |
Cl |
Cl |
| 19 |
13s
|
Br |
Br |
H |
| 20 |
13t
|
Br |
Cl |
H |
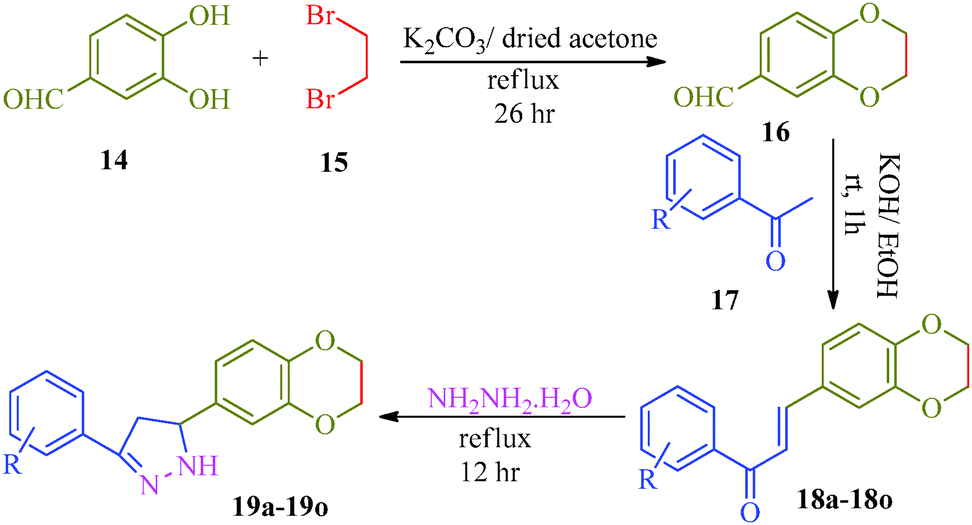
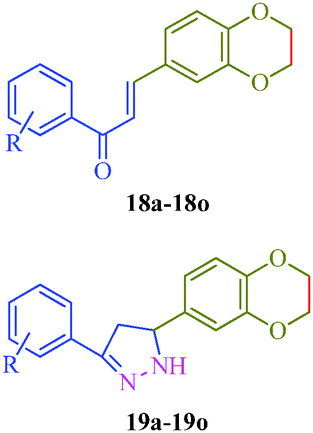
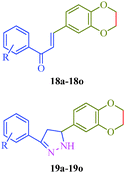
Khalilullah et al.40 put forward a slightly altered route to synthesize a series of pyrazole derivatives (19a–19o) as antihepatotoxic agents from chalcones and hydrazine hydrate (Scheme 4). The starting material 2,3-dihydro-1,4-benzodioxane-6-carbaldehyde (16) was prepared by the reaction of 3,4-dihydroxybenzaldehyde (14) and ethylenedibromide (15) in the presence of anhydrous K2CO3. On condensation with substituted acetophenone (17), 15 afforded corresponding chalcones (18a–18o), which was treated with hydrazine to obtain pyrazole derivatives (19a–19o) (Table 5).
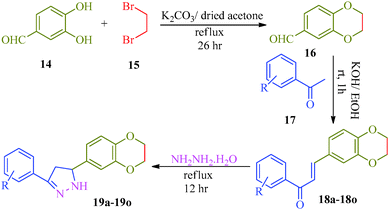 |
| | Scheme 4 Synthetic route for the preparation of 5-(2,3-dihydro-1,4-(benzodioxane-6-yl)-3-phenyl)-4,5-dihydro-1H-pyrazole derivatives (19a–19o). | |
Table 5 5-(2,3-Dihydro-1,4-(benzodioxane-6-yl)-3-phenyl)-4,5-dihydro-1H-pyrazole derivatives (19a–19o) as antihepatotoxic agents
| Entry |
Compound |
R |
| 1 |
|
18a, 19a |
H |
| 2 |
18b, 19b |
2-OH |
| 3 |
18c, 19c |
4-OH |
| 4 |
18d, 19d |
2,4-Dihydroxy |
| 5 |
18e, 19e |
2-Cl |
| 6 |
18f, 19f |
4-Cl |
| 7 |
18g, 19g |
4-F |
| 8 |
18h, 19h |
4-Br |
| 9 |
18i, 19i |
4-OMe |
| 10 |
18j, 19j |
3,4-Dimethoxy |
| 11 |
18k, 19k |
2-CH3 |
| 12 |
18l, 19l |
3-CH3 |
| 13 |
18m, 19m |
4-CH3 |
| 14 |
18n, 19n |
4-NO2 |
| 15 |
18o, 19o |
N(CH3)2 |
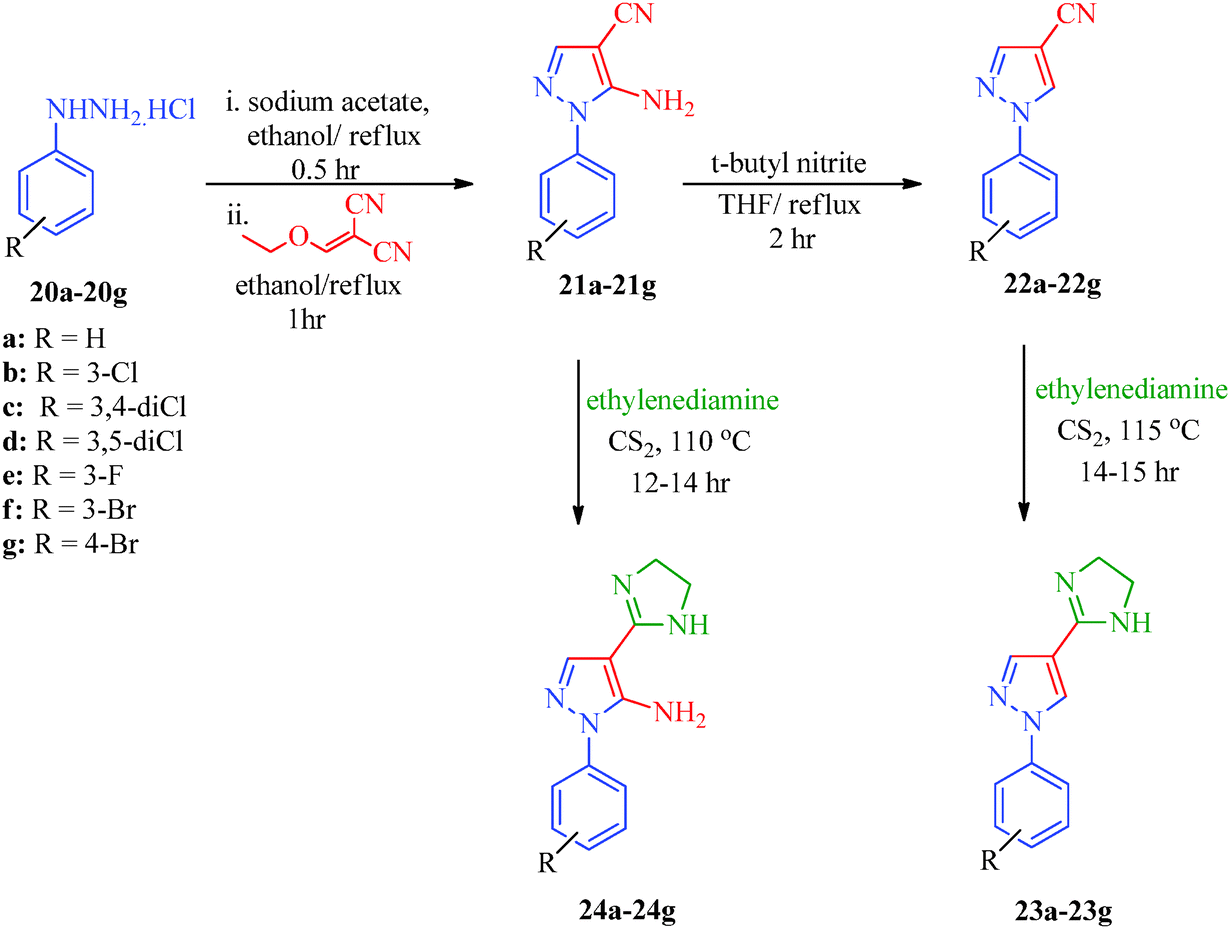
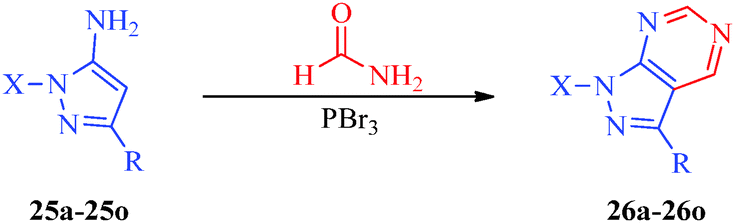
An efficient synthetic route for 1-aryl-4-(4,5-dihydro-1H-imidazol-2-yl)-1H-pyrazoles (23a–23g) and 5-amino-1-aryl-4-(4,5-dihydro-1H-imidazol-2-yl)-1H-pyrazoles (24a–24g) as antileishmanial agents (Scheme 5) was reported by Santos et al.41 The intermediates (21a–21g) were obtained from arylhydrazine hydrochlorides (20a–20g) and ethoxymethylene malononitrile42 and were converted to pyrazole carbonitriles (22a–22g) by aprotic deamination using t-butyl nitrite. Finally, the targets were obtained using carbon disulphide and ethylenediamine. Huang et al.43 reported an expeditious, straight-forward, one-pot methodology for the synthesis of pyrazolo[3,4-d]pyrimidines (26a–26o) showing antiproliferative activity (Scheme 6). The procedure involved the treatment of 5-aminopyrazoles (25a–25o) with PBr3 in formamide (Table 6).
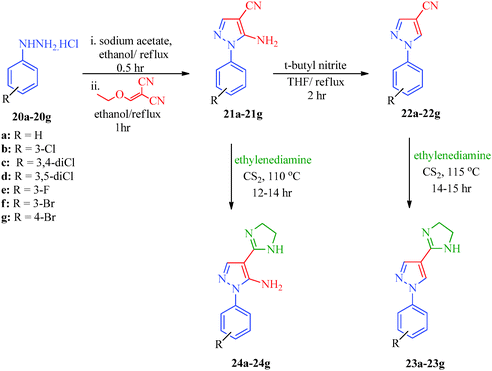 |
| | Scheme 5 Synthetic path for the preparation of 1-aryl-4-(4,5-dihydro-1H-imidazol-2-yl)-1H-pyrazoles (23a–23g) and 5-amino-1-aryl-4-(4,5-dihydro-1H-imidazol-2-yl)-1H-pyrazoles (24a–24g). | |
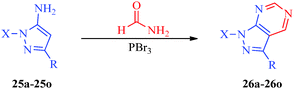 |
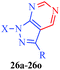
| | Scheme 6 One-pot synthesis of pyrazolo[3,4-d]pyrimidines derivatives (26a–26o). | |
Table 6 Cytotoxic pyrazolo[3,4-d]pyrimidines derivatives (26a–26o)43
| Entry |
Compounds |
X(N-1) |
R(C-3) |
| 1 |
|
a
|
Ph |
Ph |
| 2 |
b
|
o-Cl-Ph |
Ph |
| 3 |
c
|
p-Cl-Ph |
Ph |
| 4 |
d
|
p-Br-Ph |
Ph |
| 5 |
e
|
p-OMe-Ph |
Ph |
| 6 |
f
|
Ph |
Me |
| 7 |
g
|
Ph |
t-Bu |
| 8 |
h
|
Ph |
p-Cl-Ph |
| 9 |
i
|
p-Cl-Ph |
p-Cl-Ph |
| 10 |
j
|
2-Pyridinyl |
Me |
| 11 |
k
|
2-Pyridinyl |
Ph |
| 12 |
l
|
2-Quinolinyl |
Me |
| 13 |
m
|
2-Quinolinyl |
t-Bu |
| 14 |
n
|
2-Quinolinyl |
p-Me-Ph |
| 15 |
o
|
2-Quinolinyl |
p-OMe-Ph |
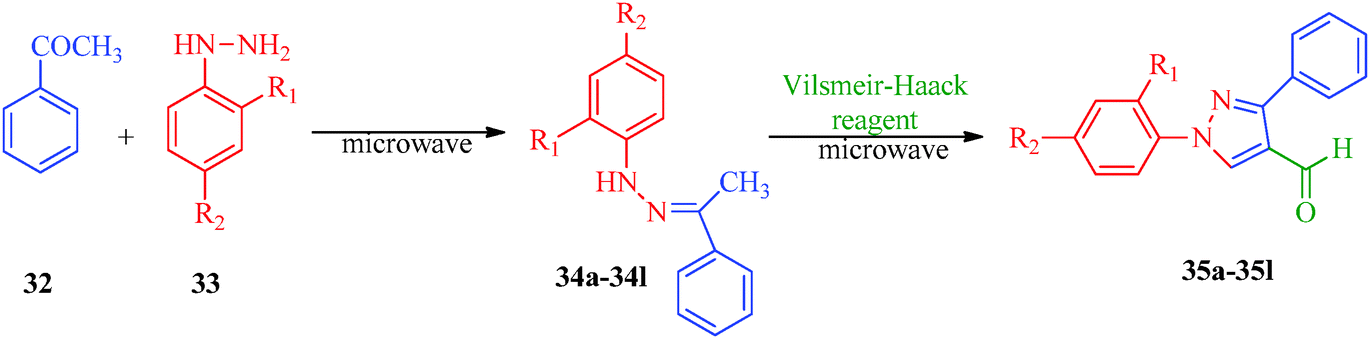
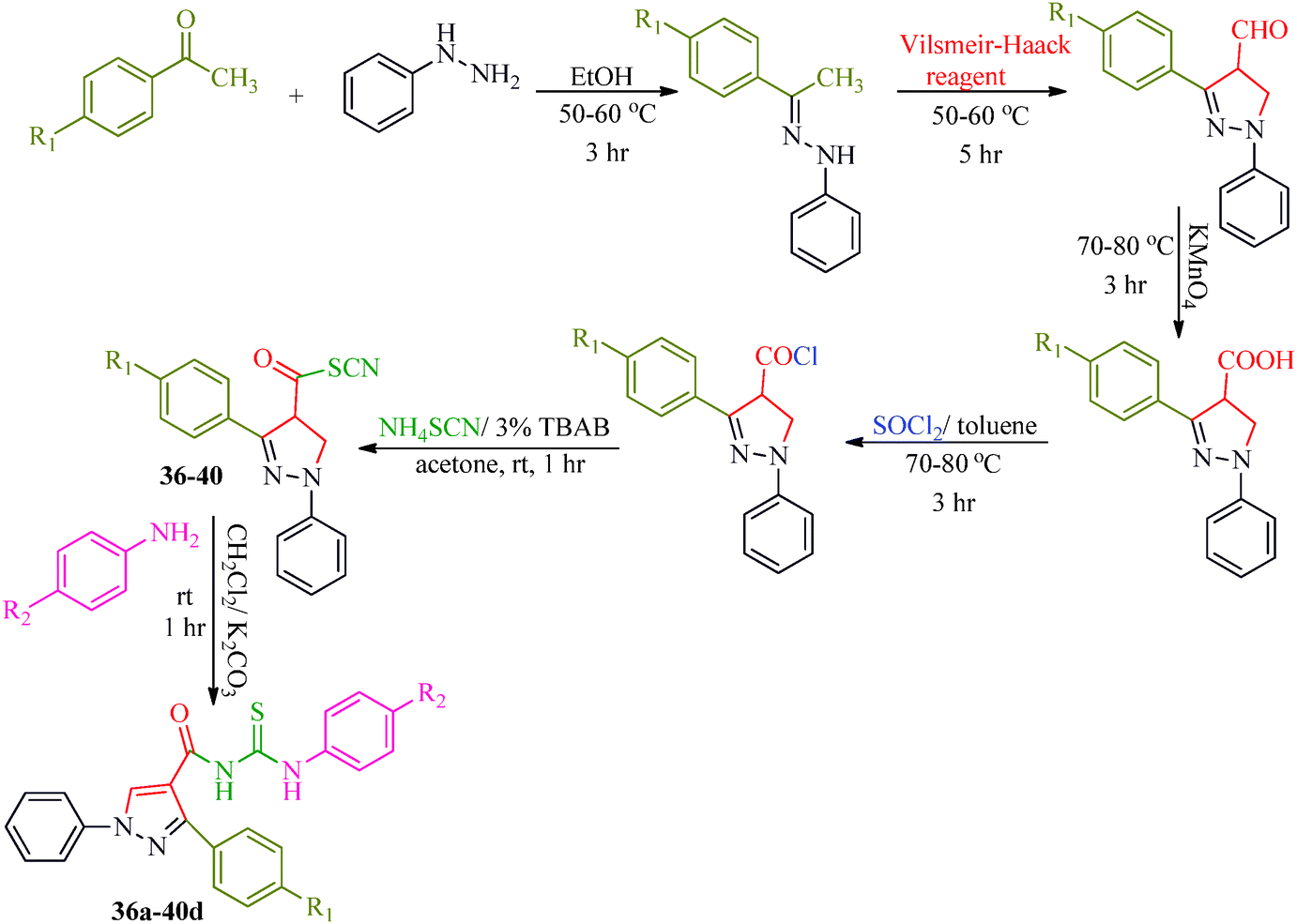
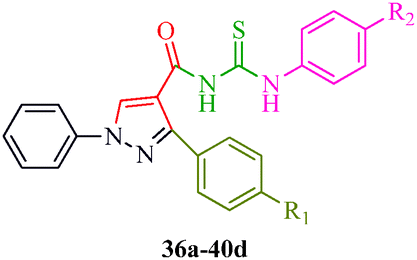
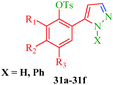
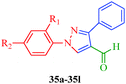
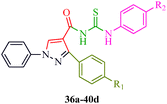
Some new 1H-pyrazole derivatives containing aryl sulfonate moieties acting as COX-2 inhibitors were synthesized by Kendre et al.44 (Scheme 7). In this synthetic route, the intermediate (29a–29e) was prepared by the reaction of 2-hydroxy acetophenone45,46 (27a–27e) with N,N-dimethyl formamide dimethyl acetal (DMF-DMA) (28) under microwave irradiation, which furnished 30a–30e when reacted with p-toluene sulfonyl chloride in the presence of anhydrous K2CO3 as catalyst. The desired product (31a–31f) was obtained by the action of hydrazine hydrate (Table 7). Another microwave-assisted synthetic route to synthesize pyrazole-4-carbaldehyde with analgesic and anti-inflammatory activity was reported by Selvam et al.47 (Scheme 8). In this approach, acetophenone (32) and substituted aryl hydrazine (33) were exposed to microwave to give 1-substituted phenyl-2-(1-phenylethylidene) hydrazine (34a–34l), which undergo Vilsmeier–Haack reactions to furnish the target compounds (35a–35l) (Table 8). A similar synthetic strategy involving the condensation and cyclization of phenylhydrazine with various substituted acetophenones followed by Vilsmeier–Haack reaction was reported by Sun et al.48 (Scheme 9). In this synthetic route, a series of 1,3-diphenyl-N-(phenylcarbamothioyl)-1H-pyrazole-4-carboxamide derivatives (36a–40d) were synthesized via the interaction of substituted cyanic 1,3-diphenyl-1H-pyrazole-4-carboxylic thioanhydride (36–40) and substituted anilines (Table 9).
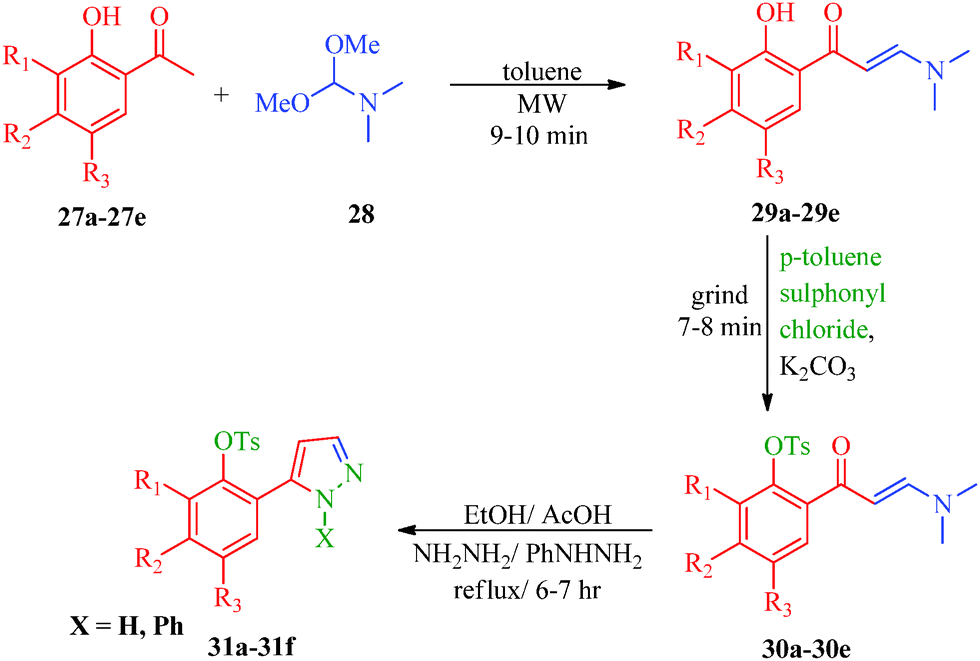
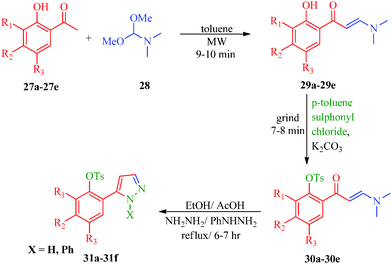 |
| | Scheme 7 Synthesis of 1H-pyrazole derivatives (31a–31f) containing aryl sulphonate moieties acting as COX-2 inhibitors. | |
Table 7 1H-Pyrazole derivatives (31a–31f)
| Entry |
Compound |
R1 |
R2 |
R3 |
| 1 |
|
a
|
H |
H |
H |
| 2 |
b
|
I |
H |
I |
| 3 |
c
|
Br |
H |
Br |
| 4 |
d
|
I |
Me |
Me |
| 5 |
e
|
H |
H |
Cl |
| 6 |
f
|
H |
H |
H |
| 7 |
g
|
I |
H |
I |
| 8 |
h
|
Br |
H |
Br |
| 9 |
i
|
I |
Me |
Me |
| 10 |
j
|
H |
H |
Cl |
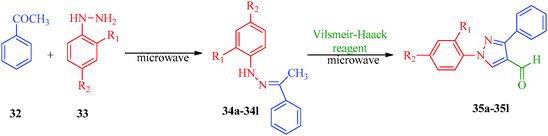 |
| | Scheme 8 Microwave-assisted synthetic route for the preparation of pyrazole-4-carbaldehyde derivatives (35a–35l) via Vilsmeier–Haack reagent. | |
Table 8 Pyrazole-4-carbaldehyde derivatives (35a–35l)
| Entry |
Compound |
R1 |
R2 |
| 1 |
|
a
|
H |
H |
| 2 |
b
|
CH3 |
H |
| 3 |
c
|
H |
CH3 |
| 4 |
d
|
OCH3 |
H |
| 5 |
e
|
H |
OCH3 |
| 6 |
f
|
F |
H |
| 7 |
g
|
H |
F |
| 8 |
h
|
Br |
H |
| 9 |
i
|
H |
Br |
| 10 |
j
|
Cl |
H |
| 11 |
k
|
H |
Cl |
| 12 |
l
|
H |
NHCOCH3 |
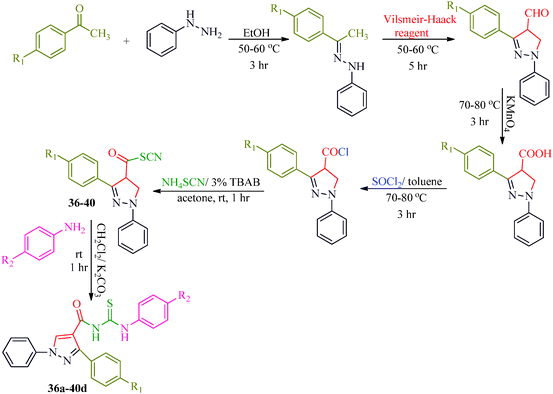 |
| | Scheme 9 Schematic representation of the preparation of a series of 1,3-diphenyl-N-(phenylcarbamothioyl)-1H-pyrazole-4-carboxamide derivatives (36a–40d). | |
Table 9 Structure of 1,3-diphenyl-N-(phenylcarbamothioyl)-1H-pyrazole-4-carboxamide derivatives (36a–40d)
| Entry |
Compound |
R1 |
R2 |
| 1 |
|
36a
|
H |
Me |
| 2 |
36b
|
H |
OMe |
| 3 |
36c
|
H |
F |
| 4 |
36d
|
H |
Cl |
| 5 |
37a
|
Me |
Me |
| 6 |
37b
|
Me |
OMe |
| 7 |
37c
|
Me |
F |
| 8 |
37d
|
Me |
Cl |
| 9 |
38a
|
Cl |
Me |
| 10 |
38b
|
Cl |
OMe |
| 11 |
38c
|
Cl |
F |
| 12 |
38d
|
Cl |
Cl |
| 13 |
39a
|
F |
Me |
| 14 |
39b
|
F |
OMe |
| 15 |
39c
|
F |
F |
| 16 |
39d
|
F |
Cl |
| 17 |
40a
|
OMe |
Me |
| 18 |
40b
|
OMe |
OMe |
| 19 |
40c
|
OMe |
F |
| 20 |
40d
|
OMe |
Cl |
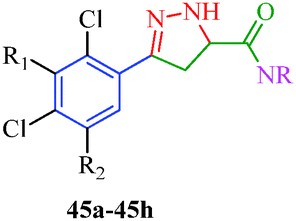
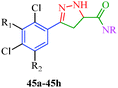
Sidique et al.49 reported the synthesis of pyrazole amides (45) as selective inhibitors of tissue-nonspecific alkaline phosphatases from pyrazole acids (Scheme 10). These acids were obtained by the reaction of acetophenone derivatives with sodium methoxide and dimethyl oxalate to furnish 1,3-diketone. Diketone derivatives (42) were then reacted with hydrazine to give pyrazole esters (43), which gave pyrazole acids (44) upon saponification (Table 10). A multistep synthetic strategy was put forward by Ma et al.50 to synthesize trifluoroethylpyrazole derivatives (51a–51c) with herbicidal activity (Scheme 11). The intermediates 46 were obtained starting from glyoxalic acid and hydroxylamine hydrochloride and was then reacted with trifluoroethylpyrazole ethanethioate (49) followed by treatment with mCPBA to furnish the desired products.
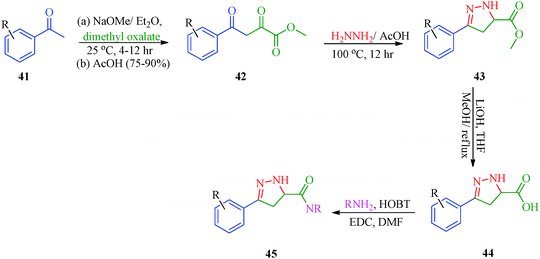 |
| | Scheme 10 Synthesis of pyrazole amides (45) as selective inhibitors. | |
Table 10 Pyrazole amide derivatives (45)
| Entry |
Compound |
R1 |
R2 |
R |
| 1 |
|
45a
|
H |
H |
NH2CH2CH2OH |
| 2 |
45b
|
H |
F |
NH2CH2CH2OH |
| 3 |
45c
|
H |
H |
NH2CH(CH3)2 |
| 4 |
45d
|
H |
H |
NH(CH3)2 |
| 5 |
45e
|
H |
H |
NH2CH2CH2CH2OH |
| 6 |
45f
|
H |
H |
NH2CH2CH2OMe |
| 7 |
45g
|
H |
H |
NH2(CH2CH2OH)2 |
| 8 |
45h
|
Cl |
H |
NH2CH2CH2OH |
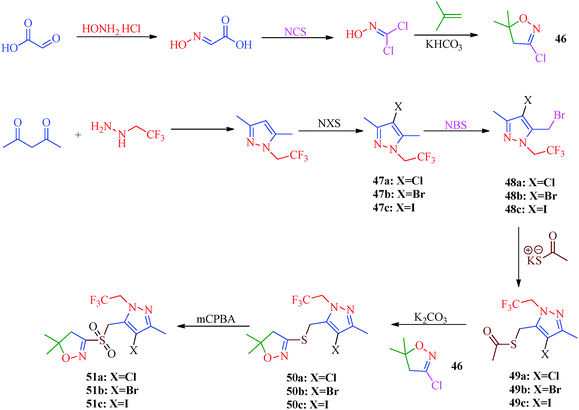 |
| | Scheme 11 Multistep synthetic strategy for the synthesis of trifluoroethylpyrazole derivatives (51a–51c). | |
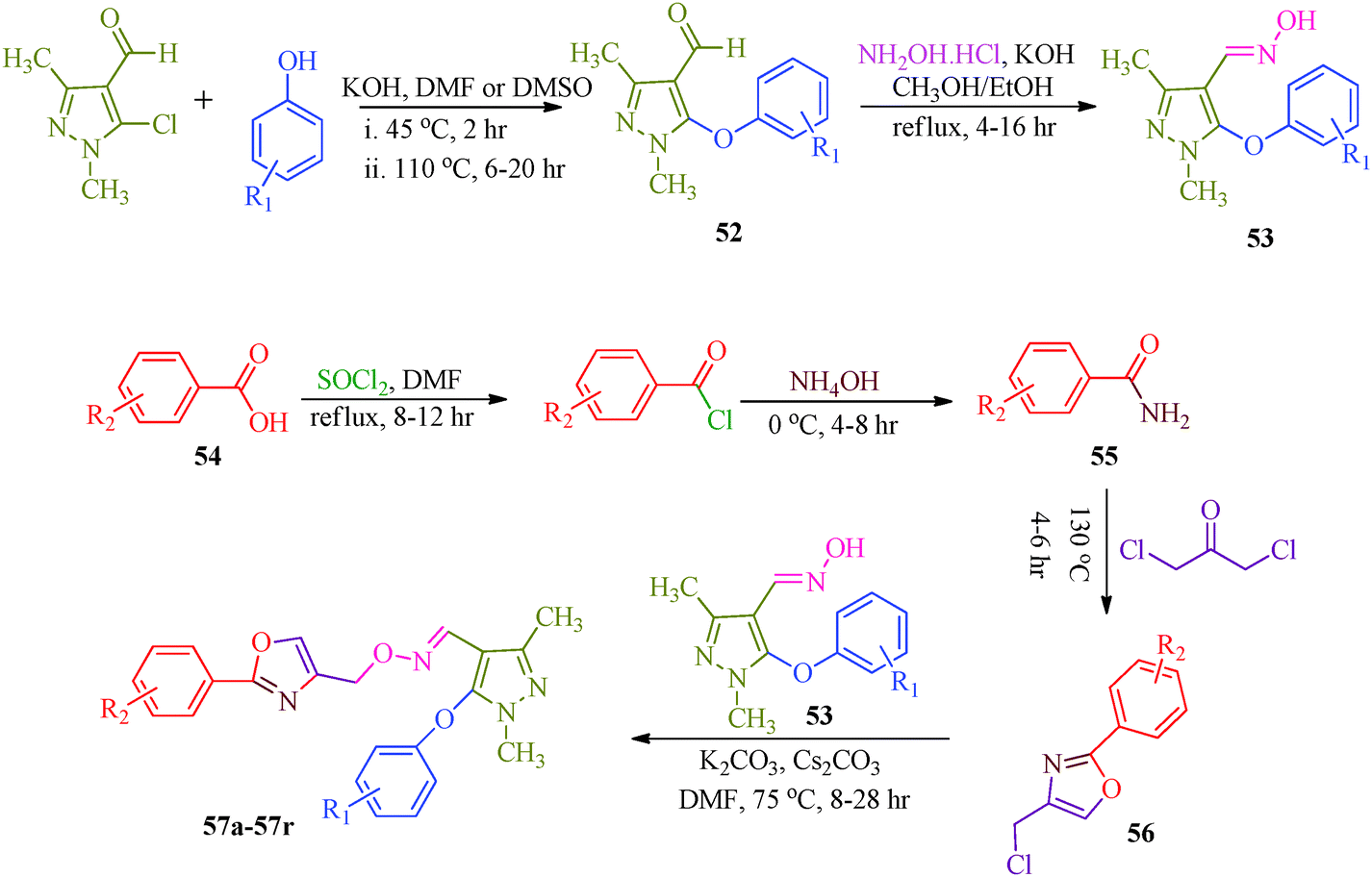
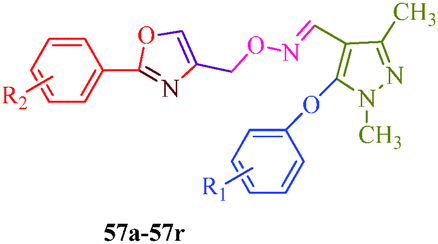
Starting from pyrazole aldehyde,52 Wang et al.51 reported a synthetic route for oxazole ring-containing pyrazole derivatives (57a–57r) that show acaricidal, insecticidal and fungicidal activities (Scheme 12). Aldehyde 52 was directly converted to the key intermediate 53 by treatment with hydroxylamine. Intermediate 4-chloromethyl-2-aryloxazole (56) was successively prepared from substituted benzoic acid (54). Further reaction of 53 with 56 promoted by Cs2CO3 produced the target compound (Table 11).
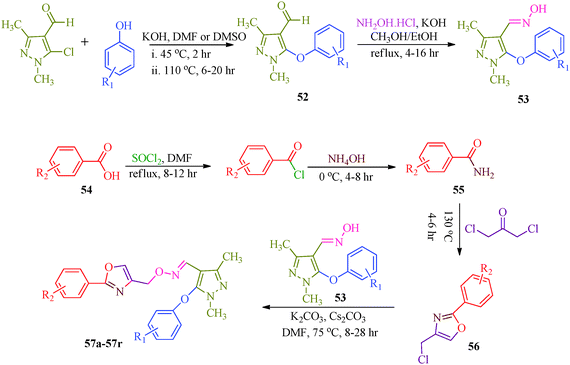 |
| | Scheme 12 Multistep synthetic approach for the synthesis of pyrazole derivatives (57a–57r). | |
Table 11 Pyrazole derivatives (57a–57r)
| Entry |
Compound |
R1 |
R2 |
| 1 |
|
57a
|
4-CH3 |
4-F |
| 2 |
57b
|
4-OCH3 |
4-F |
| 3 |
57c
|
4-F |
4-F |
| 4 |
57d
|
4-Cl |
4-F |
| 5 |
57e
|
2,4-F2 |
4-F |
| 6 |
57f
|
2,4-Cl2 |
4-F |
| 7 |
57g
|
4-F |
4-Cl |
| 8 |
57h
|
4-Cl |
4-Cl |
| 9 |
57i
|
4-Br |
4-Cl |
| 10 |
57j
|
3-F |
4-Cl |
| 11 |
57k
|
3-CF3 |
4-Cl |
| 12 |
57l
|
2,4-Cl2 |
4-Cl |
| 13 |
57m
|
4-F |
2,4-Cl2 |
| 14 |
57n
|
4-Cl |
2,4-Cl2 |
| 15 |
57o
|
4-Br |
2,4-Cl2 |
| 16 |
57p
|
3-CF3 |
2,4-Cl2 |
| 17 |
57q
|
4-OCF3 |
2,4-Cl2 |
| 18 |
57r
|
2,4-F2 |
2,4-Cl2 |
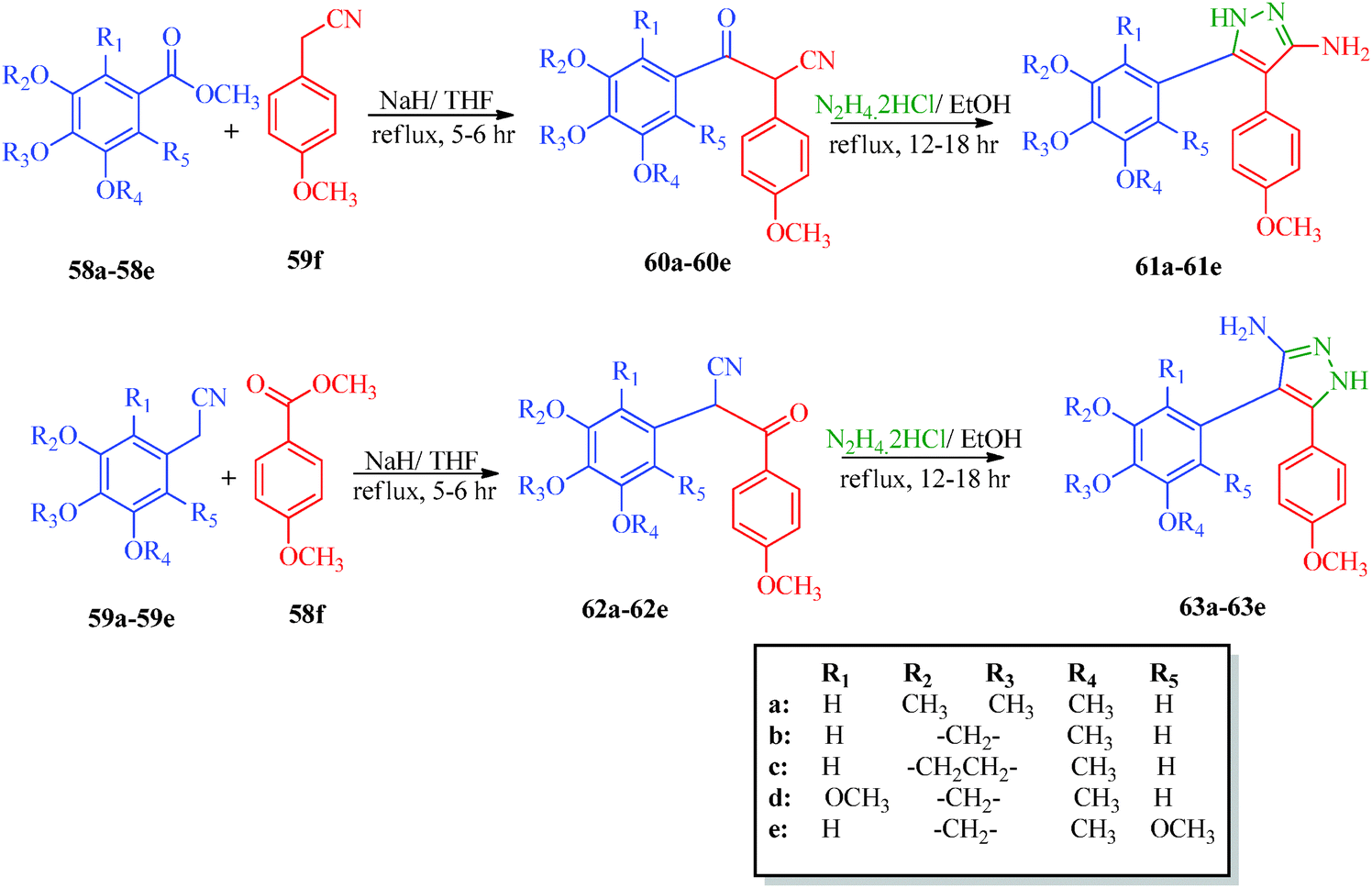
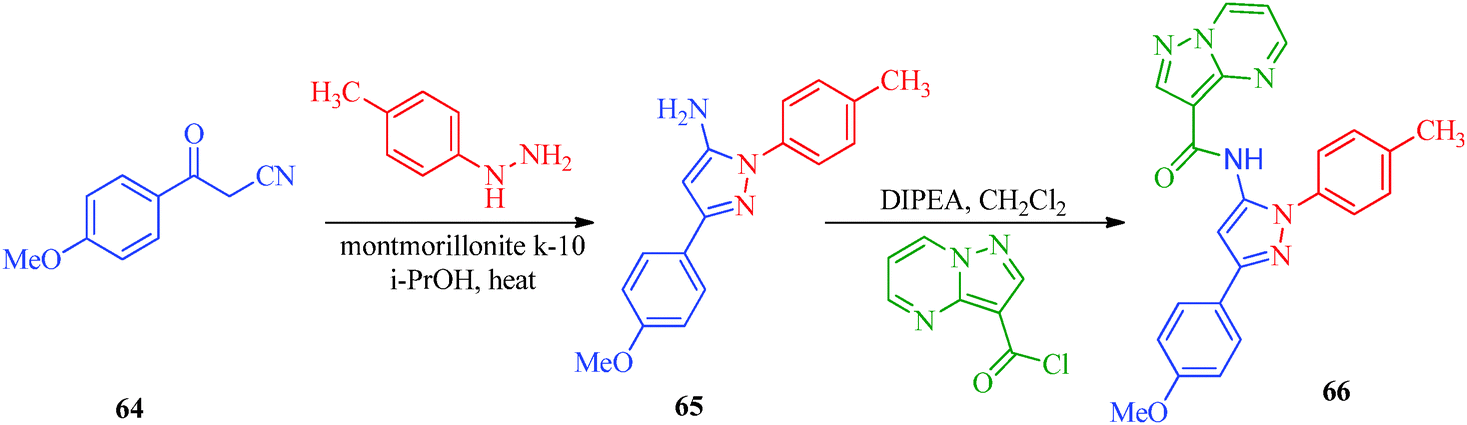
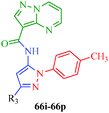
A highly regioselective approach for the synthesis of cis-restricted 3-aminopyrazole has been reported by Tsyganov et al.53 (Scheme 13). Polyalkoxybenzoic acids (58) and aromatic acetonitriles (59) underwent base-mediated condensation to furnish α-ketonitriles 60 and 62, which subsequently cyclized into pyrazoles 61 and 63 with hydrazine hydrochloride. McElroy et al.54 reported the convenient synthesis of amidopyrazoles utilizing ketonitriles (Scheme 14). The α-ketonitriles 64 were condensed with 4-methylphenylhydrazine to form 65.55 Aminopyrazole (65) was then allowed to react with acid chloride to produce the desired product, which shows inhibitory activity against IRAK4 (Table 12).
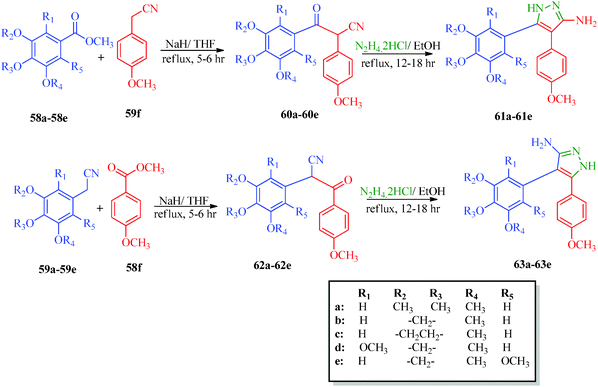 |
| | Scheme 13 Synthesis of 4,5-polyalkoxydiaryl-3-aminopyrazoles 61 and 63. | |
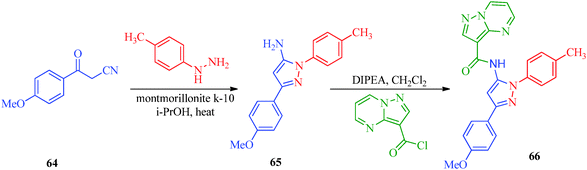 |
| | Scheme 14 Synthetic path for the preparation of amidopyrazoles (66). | |
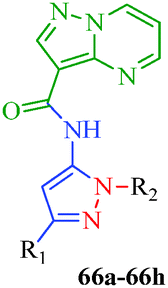
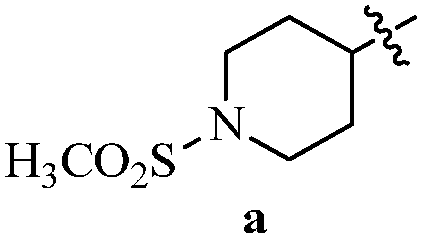
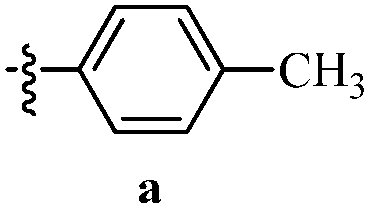
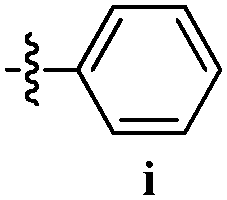
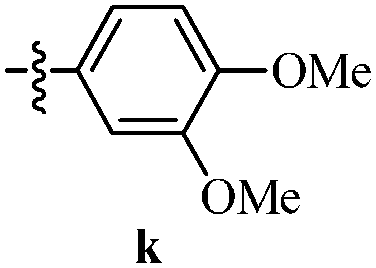
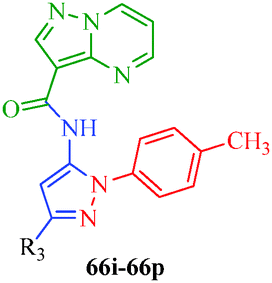
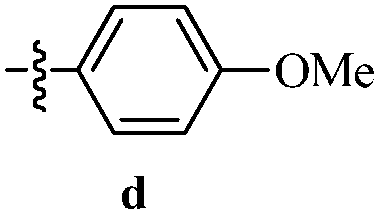
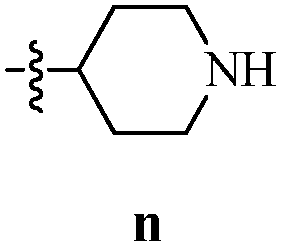
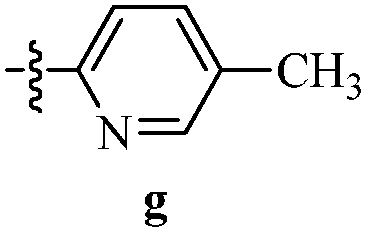
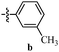
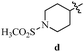
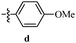
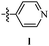
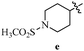
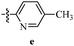
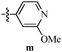
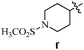
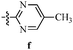
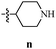
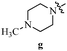
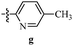
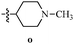
Table 12 Amidopyrazoles derivatives (66a–66h and 66i–66p)
| Entry |
Compound |
R1(a–h) |
R2(a–h) |
R3(i–p) |
| 1 |
|
|
|
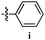
|
| 2 |
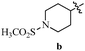
|
|
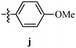
|
| 3 |
|
|
|
| 4 |
|
|
|
|
| 5 |
|
|
|
| 6 |
|
|
|
| 7 |
|
|
|
| 8 |
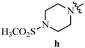
|
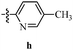
|
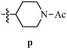
|
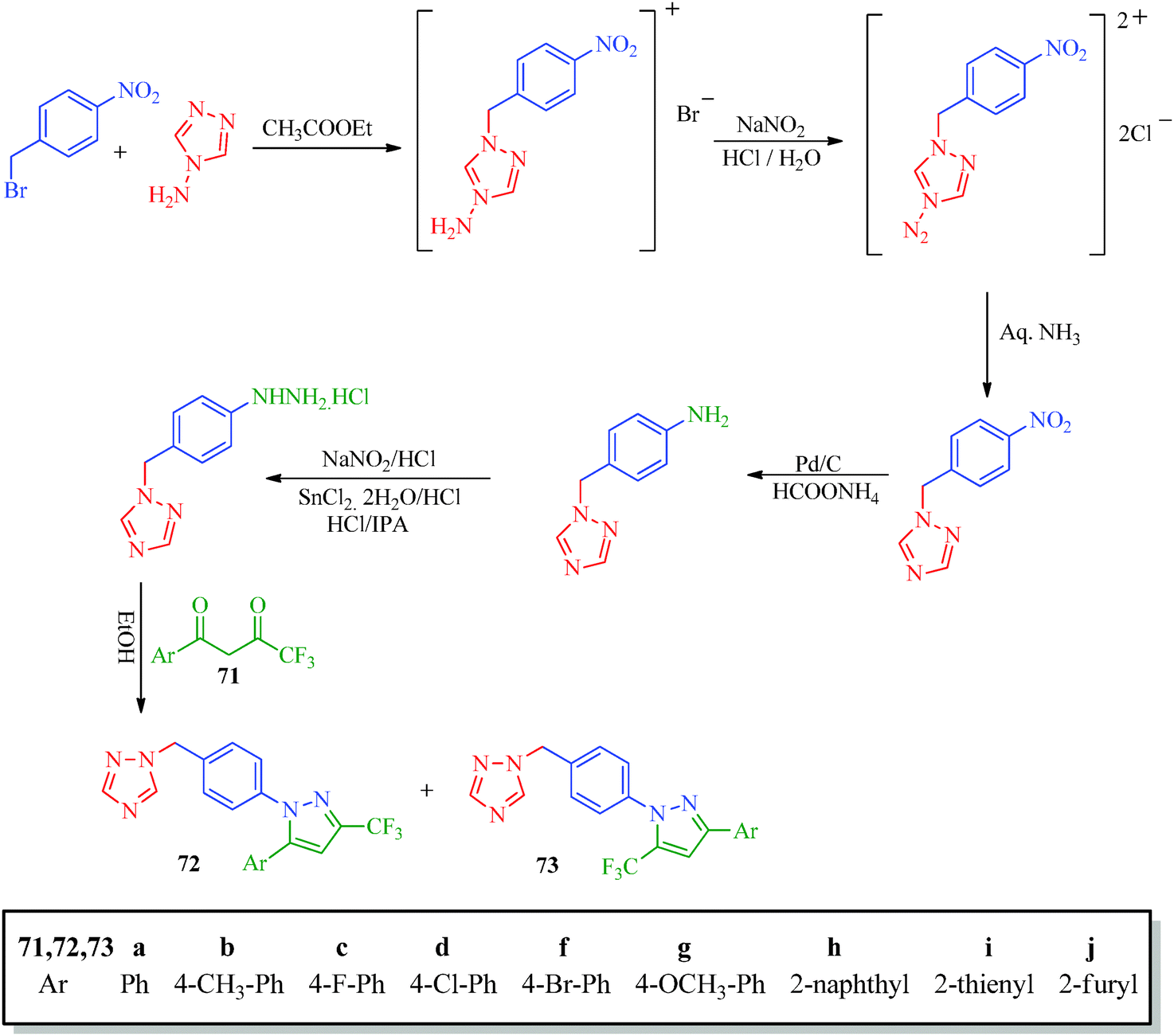
Inceler et al.56 reported a novel synthetic approach to synthesize ester and amide derivatives of 1-phenyl-3-(thiophen-3-yl)-1H-pyrazole-4-carboxylic acid as anticancer agents (Scheme 15). Carboxylic acid derivatives (68) were obtained from hydrazone derivatives (67) when reacted with POCl3 and DMF followed by oxidation. Treatment of 69 with appropriate amines or phenols in the presence of Et3N/CH2Cl2 provides access to the target product (Table 13). An interesting synthesis of pyrazolylbenzyltriazole derivatives (72, 73) as cyclooxygenase inhibitors was developed by Chandna et al.57 using 1-[(4-hydrazinophen-1-yl) methyl]-1H-1,2,4-triazole hydrochloride (Scheme 16). The triazole intermediate was obtained via the condensation of 4-nitrobenzyl bromide and 4-aminotriazole in ethyl acetate followed by diazotization and reduction. The intermediate was then treated with trifluoromethyl-β-diketones (71) to afford the target compounds.
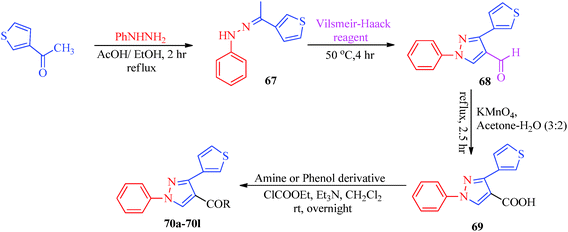 |
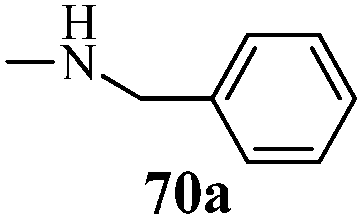
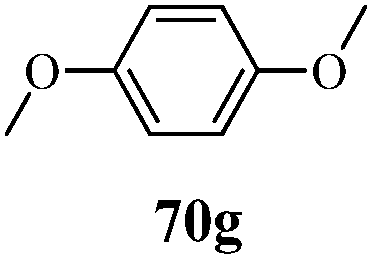
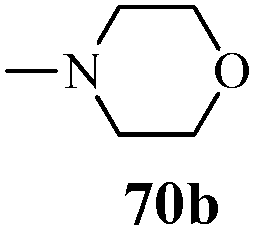
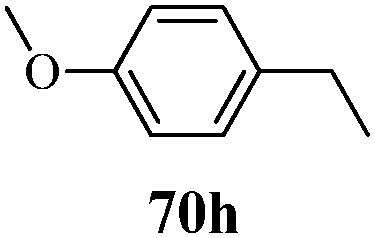
| | Scheme 15 Synthesis of 1-phenyl-3-(thiophen-3-yl)-1H-pyrazole-4-carboxylic acid derivatives (70a–70l). | |
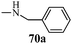
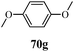
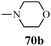
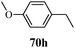
Table 13 1-Phenyl-3-(thiophen-3-yl)-1H-pyrazole-4-carboxylic acid derivatives (70a–70l)
| Entry |
R |
Entry |
R |
| 1 |
|
7 |
|
| 2 |
|
8 |
|
| 3 |
|
9 |
|
| 4 |
|
10 |
|
| 5 |
|
11 |
|
| 6 |
|
12 |
|
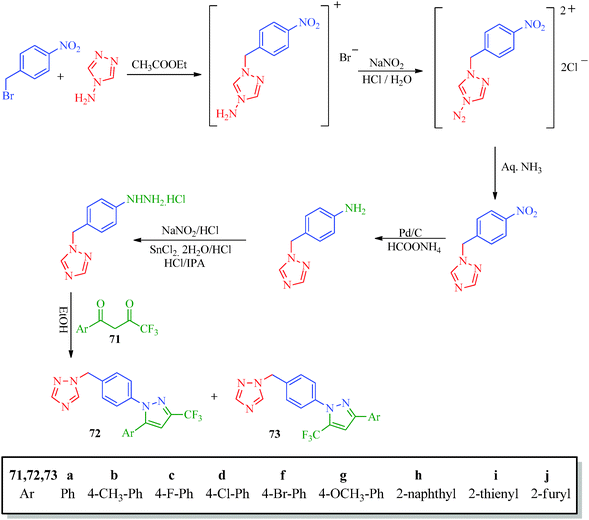 |
| | Scheme 16 Synthesis of pyrazolylbenzyltriazole derivatives (72 and 73). | |
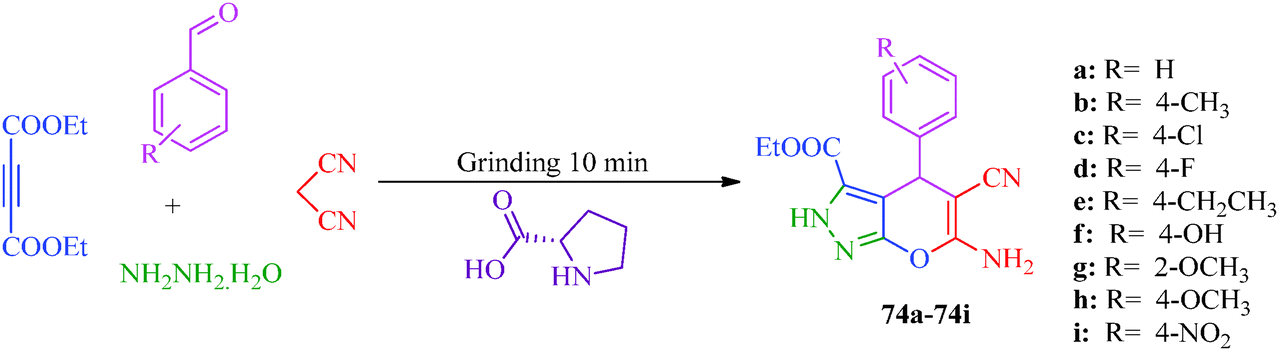
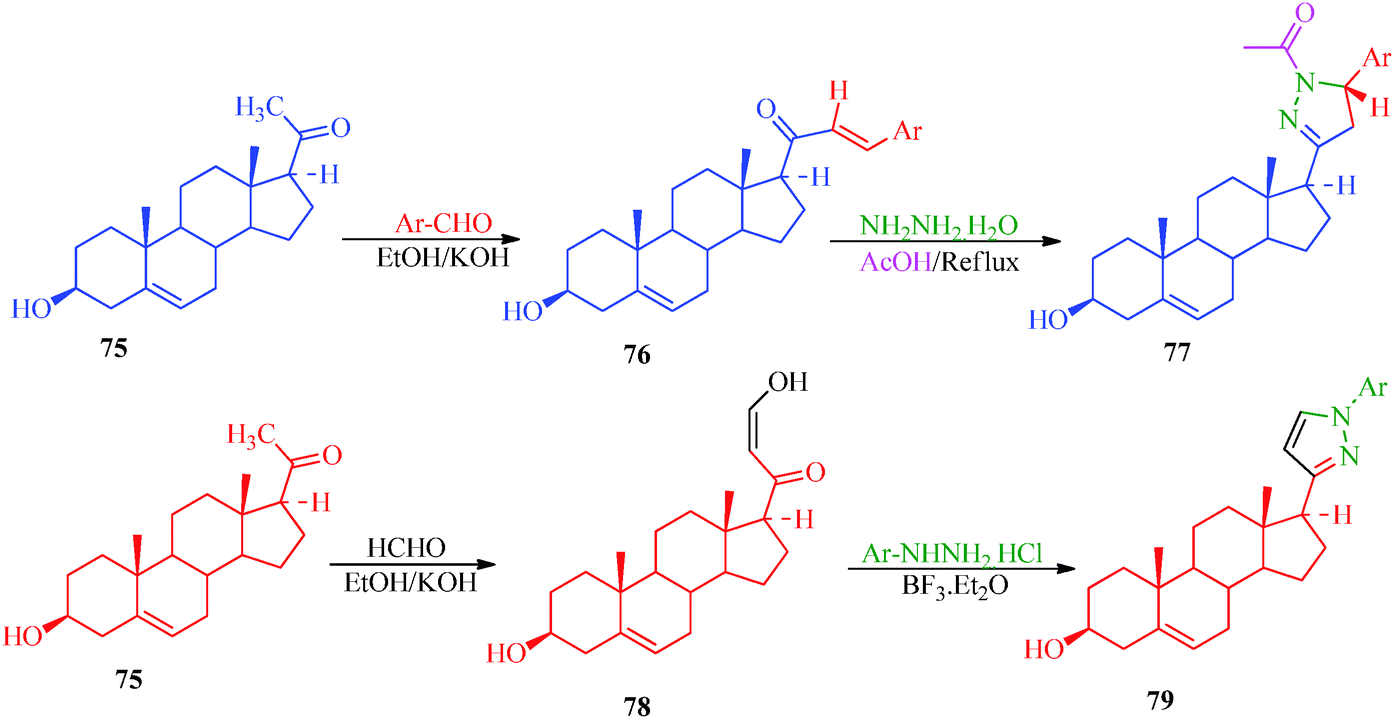
A multicomponent and green synthetic route for the synthesis of pyrano pyrazole derivatives as antioxidants and antimicrobial agents was reported by Ambethkar et al.58 (Scheme 17). The target compounds (74a–74i) were obtained from aryl aldehyde, malononitril, hydrazine hydrate and diethyl acetylenedicarboxylate by a grinding method. The synthesis of some steroidal pyrazolines and pyrazoles was also reported by Banday et al.59 In this synthetic strategy (Scheme 18), pyrazolines (77) were synthesized by the reaction of pregnenolone (75) with aryl aldehyde to obtain 76, which was further treated with hydrazine hydrate in the presence of acetic acid. In the synthetic protocol for pyrazoles (79), prior to treatment with hydrazine, pregnenolones were first converted to 78 (Table 14).60
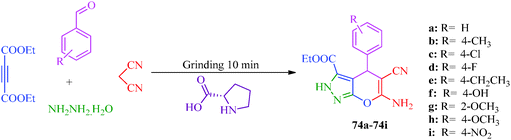 |
| | Scheme 17 One-pot multicomponent and green synthetic route for the preparation of pyrano pyrazole derivatives (74a–74i). | |
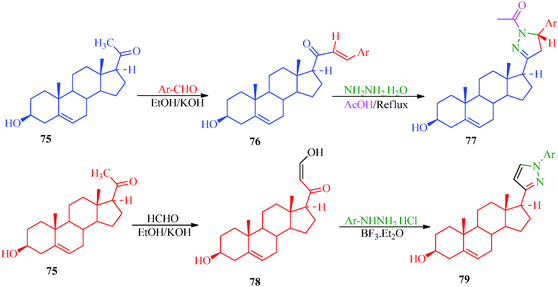 |
| | Scheme 18 Synthetic protocol for the preparation of steroidal pyrazolines and pyrazoles. | |
Table 14 Nature of the aryl group (Ar-) in steroidal pyrazole derivatives (77a–d and 79a–d)
| Entry |
Ar |
Entry |
Ar |
| 1 |
|
5 |
|
| 2 |
|
6 |
|
| 3 |
|
7 |
|
| 4 |
|
8 |
|
Biology
Monoamine oxidase inhibitors
Monoamine oxidase (MAO) is a flavoprotein located at the outer membrane of the mitochondria in neuronal, glial, and other cells. MAO is widely distributed among mammals, plants, prokaryotic and eukaryotic microorganisms and catalyzes the oxidative deamination of monoamine; thus, MAO is a target enzyme for antidepressant drugs. In addition, it is also responsible for the biotransformation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) into 1-methyl-4-phenylpyridinium, a Parkinsonian-producing neurotoxin. MAO exists in two forms, MAO-A and MAO-B, which are characterized by specific substrates and inhibitors. MAO-A oxidizes norepinephrine and serotonin [5-hydroxytrypamine, 5-HT], whereas MAO-B preferentially deaminates 2-phenylethylamine (2-PEA) and benzylamine. These properties determine the clinical importance of MAO inhibitors.61,62 Several N1-thiocarbamoyl-3,5-diaryl-4,5-dihydro-(1H)-pyrazoles (80a–80j) have been synthesized by Palaska et al.63 and assayed as MAO inhibitors against monoamine oxidases isolated and purified from the mitochondrial extracts of rat liver homogenates and human platelets (Fig. 2).
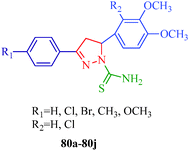 |
| | Fig. 2 Pyrazole derivatives as monoamine oxidase inhibitors. | |
They suggested that these compounds were time-dependent inhibitors of MAO since their inhibitory activities were significantly increased with incubation time, and the inhibition of liver MAO by these compounds was found to be non-competitive and irreversible. However, the N1-thiocarbamoyl-3,5-di(hetero)aryl-4,5-dihydro-(1H)-pyrazole derivatives (4a–4t) synthesized by Chimenti et al.36 (Scheme 1) exhibit inhibitory activity against A and B isoforms of human MAO (hMAO). The authors suggested that the presence of a fluorine atom in the 4′-position of the 5-phenyl substituent on the pyrazoline ring is important for activity. The most active compound of the series is compound 4m (IC50 = 2.75 ± 0.81 μM and selectivity ratio is 25), which has a 4′-fluorophenyl substituent in the 5-position and a fur-2′-yl group in the 3-position of the pyrazoline ring (Table 2). These compounds also show irreversible inhibitory effects.
BRAF inhibitors
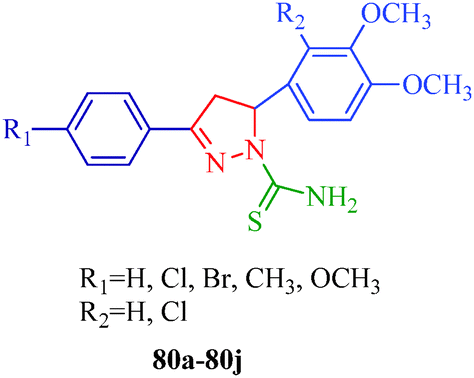
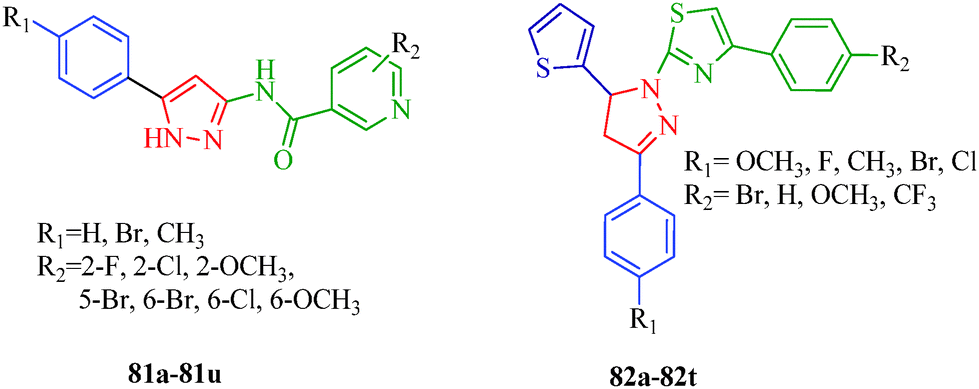
BRAF is a human gene that makes a protein called B-Raf, formally known as serine/threonine protein kinase B-Raf, which is mutated in some human cancers. BRAF belongs to the RAS/RAF/MEK/ERK/MAP kinase pathway, which mediates cellular responses to growth signals. This mitogen-activated protein kinase pathway plays an important role in the transduction of mitogenic signals from the cell membrane to the nucleus to regulate cell growth, survival, differentiation and proliferation in response to external stimuli.64 Various mutations occur in BRAF, and the substitution of valine for glutamic acid at position 600 (V600E; formally identified as V599E) has been identified as the most common mutation in human cancer.65 Targeting the protein kinase pathway through the inhibition of BRAFV600E is important in cancer therapeutics, particularly melanoma. The 4,5-dihydro-2H-pyrazole-2-hydroxyphenyl derivatives synthesized from hydrazine hydrate and chalcones by Liu et al.38 (Scheme 2) showed remarkable antiproliferative effects against BRAFV600E, WM266.5 (human melanoma cell line) and MCF-7 (human breast cancer cell line). Among them, compounds 9d and 9m displayed the strongest inhibitory activity (9d: IC50 = 1.31 μM for MCF-7 and IC50 = 0.45 μM for WM266.5; 9m: IC50 = 0.97 μM for MCF-7 and IC50 = 0.72 μM for WM266.5) (Table 3). The structure–activity relationships (SARs) of these dihydropyrazole derivatives indicate that compounds with para electron-donating substituents (9a–9h) show more potent activities than those with para electron-withdrawing substituents (9i–9t). A series of novel 5-phenyl-1H-pyrazole derivatives with niacinamide moieties (81a–81u) were reported by Wang et al.66 as potential BRAFV600E inhibitors. Among them, compound 81o displayed the strongest inhibitory activity (IC50 = 2.63 μM for WM266.4 and IC50 = 3.16 μM for A375). The authors also suggested that para electron-donating substituents showed more potent inhibitory activity. Zhao et al.67 reported 4,5-dihydro-1H-pyrazole thiazole derivatives (82a–82t) as BRAFV600E inhibitors. The compound containing R1 = Cl and R2 = CF3 (82t) showed the most potent inhibitory activities against BRAFV600E kinase (IC50 = 0.05 μM) and two cancer cell lines, WM266.4 (IC50 = 0.12 μM) and MCF-7 (IC50 = 0.16 μM). In contrast to above, the authors observed that compounds with para electron-withdrawing groups are more active than compounds with para electron-donating groups (Fig. 3).
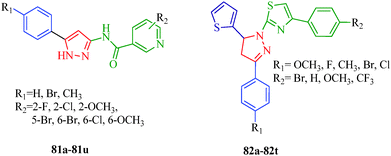 |
| | Fig. 3 Pyrazole derivatives as BRAF inhibitors. | |
DNA gyrase inhibitor
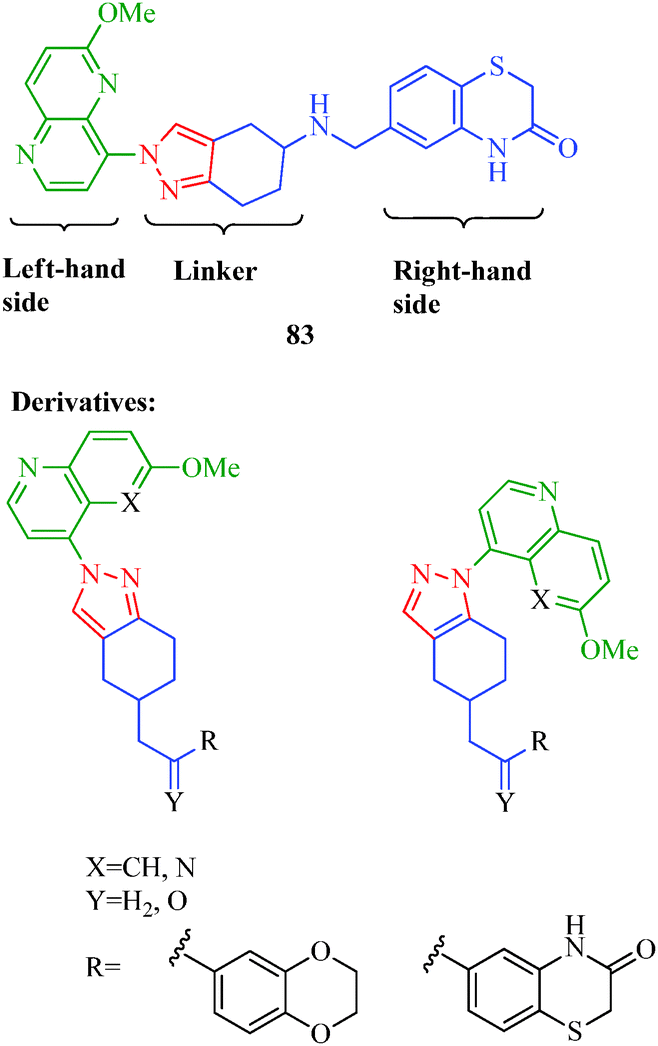
DNA gyrase is an essential bacterial enzyme involved in the vital processes of DNA replication, transcription and recombination. It belongs to a class of enzymes known as topoisomerases, which are involved in the control of topological transitions of DNA. This enzyme possesses a unique ability to catalyze the ATP-dependent negative supercoiling of double-stranded closed-circular DNA.68 The mechanism by which gyrase influences the topological state of DNA molecules is of inherent interest from an enzymological standpoint; hence, gyrase hence receives much attention as a selected target for antibacterial agents.69 Many pyrazole derivatives are acknowledged to possess a wide range of antibacterial bioactivities70 and act as DNA gyrase inhibitors. Gomez et al.71 synthesized a series of novel pyrazole derivatives as DNA gyrase inhibitors that exhibit antibacterial activities against Gram-negative (E. coli) and Gram-positive (S. aureus and S. pneumoniae) bacterial strains. Compound 83 was described as one of the most active compounds, with potent antibacterial activities against both Gram-positive and Gram-negative bacterial strains. The authors highlighted the importance of the linker length and the orientations of the moieties on the right- and left-hand sides in the antibacterial activity (Fig. 4).
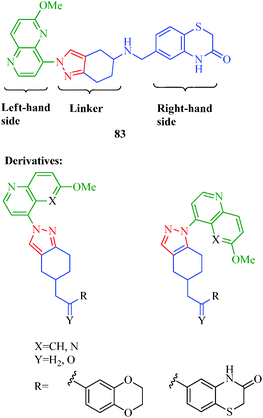 |
| | Fig. 4 Pyrazole derivatives as DNA gyrase inhibitors. | |
The compound synthesized by Liu et al.39 exhibited antibacterial activity (Scheme 3) against two Gram-negative bacterial strains (E. coli and P. aeruginosa) and two Gram-positive bacterial strains (B. subtilis and S. aureus). The compounds were examined against DNA gyrase isolated from B. subtilis and S. aureus; compounds 13b, 13l, and 13r showed poor inhibitory activities, whereas 13a, 13d, 13g, 13h, 13o, 13p, 13s and 13t displayed potent inhibitory activities (Table 4). Among all derivatives, compound 13t displayed broad-spectrum antimicrobial activity against all tested bacterial strains, compound 13d showed the most potent activity (MIC = 0.39 μg mL−1) against both B. subtilis and S. aureus, and compounds 13h and 13t showed the most potent activities (MIC = 0.39 μg mL−1) against P. aeruginosa and E. coli. This demonstrated that the potent anti-Gram-positive bacterial activities of the synthetic compounds were probably correlated to their related DNA gyrase inhibitory activities. The SARs of these dihydro-pyrazole derivatives indicated that compounds with para electron-donating substituents (13a–13h) on the A-ring showed more potent activities than those with electron-withdrawing substituents (13i–13t) on the A-ring. Compounds with F (13i–13l), Cl (13m–13p) and Br (13q–13t) substituents had mostly moderate effects compared to compounds with OCH3 (13e–13h). Moreover, keeping the substituent constant on the A-ring and changing the substituent of the B-ring also affected the activities of these compounds. Compounds with halogen atoms at the 5-position display higher activities than compounds with two halogen atoms at both the 3- and 5-positions, and the activity is generally greater when the substituent at the 5-position is Cl compared to Br. Thus, compounds 13d and 13h, which have a para electron-donating group in the A-ring and Cl in the B-ring, both showed wonderful antibacterial activity. Furthermore, a bioassay of Gram-positive bacteria showed that the order of strength is CH3 > OCH3 (13a–13h) and Cl > Br > F (13i–13t) for para-substituted A-ring compounds. On the other hand, for Gram-negative bacteria, the strength order is OCH3 > CH3 (13a–13h) and Br > Cl > F (13i–13t) for para-substituted A-ring compounds. Thus, compound 13d, which has a para-CH3 group on the A-ring and Cl on the B-ring, showed the best anti-Gram-positive activity. The compounds 13h, with a para-OCH3 group on the A-ring and Cl on the B-ring, and 13t, with Br on the A-ring and Cl on the B-ring, both showed the best anti-Gram-negative activities.
Antihepatotoxicity
The liver is a vital organ and plays a major role in metabolism. The liver has numerous functions in the human body, including the regulation of glycogen storage, decomposition of red blood cells, hormone production and detoxification. The liver is involved in the transformation and clearing of chemicals from the body and therefore is susceptible to toxicity from these agents. This type of liver damage driven by certain chemicals and drugs is known as hepatotoxicity. Although hepatotoxicity can be prevented or treated, if not, it can lead to progressive liver injury and liver fibrosis and ultimately cirrhosis, portal hypertension, liver failure, and in some instances, cancer. Silymarin isolated from seeds of S. marianum, which is commonly known as milk thistle, has been found to be a potent antihepatotoxic agent against a variety of toxicants. Silymarin has been recognized as a mixture of three isomers: silybin, silychristin and silydianin. The main component of silymarin is silybin, which contains a 1,4-benzodioxane ring system and possesses significant antihepatotoxic activity.72 Khalilullah et al.40 synthesized pyrazole derivatives containing 1,4-benzodioxane rings as antihepatotoxic agents (Scheme 4). These compounds were evaluated against CCl4-induced hepatic damage in rats and shown to possess significant antihepatotoxic activity. Among all compounds reported, compounds 19d and 19j demonstrated significant antihepatotoxic activities compared to the standard drug silymarin. Other compounds in the series showed moderate antihepatotoxic activity. Upon administration, the synthesized compounds provided significant protection against hepatocyte injury and promoted normal tissues as neither fatty accumulation nor necrosis was observed.
Antileishmanial
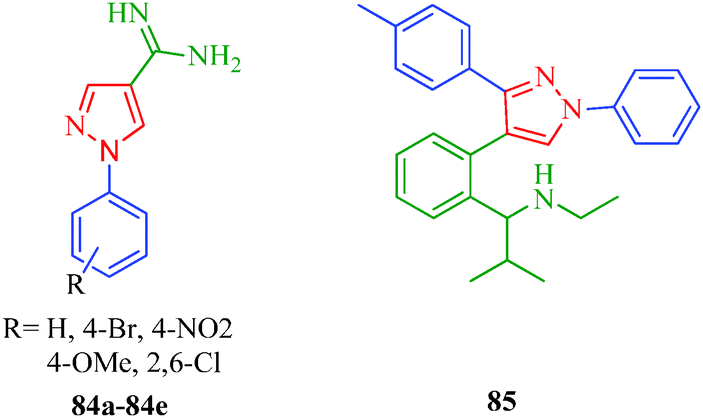
Leishmaniasis is a tropical vector-borne disease caused by protozoan parasites of the genus Leishmania and spread by the bites of infected female Phlebotomine sand flies.73 The insect vector injects promastigotes into the host's skin, and soon after, the parasite transforms to an amastigote. Leishmaniasis represents a complex disease and may be divided into different types: visceral leishmaniasis or kala azar (black fever) is potentially fatal if untreated; cutaneous leishmaniasis is the most serious and most common form, is long-lasting, and causes open sores; diffuse cutaneous leishmaniasis includes disseminated lesions that resemble leprosy and occurs in individuals with defective cell-mediated immune response; and mucocutaneous leishmaniasis produces lesions that can lead to the extensive and disfiguring destruction of the mucous membranes of the nose, mouth and throat cavities.74 The 1-aryl-4-(4,5-dihydro-1H-imidazol-2-yl)-1H-pyrazole derivatives reported by Santos et al.41 (Scheme 5) were tested against the promastigote stages of L. amazonensis, L. infantum and L. braziliensis parasites and established that among them, six derivatives (23a, 23b, 23e, 23g, 24d, and 24f) were more effective against L. amazonensis compared to the other species. The relationship between structure and antileishmanial activity against L. amazonensis revealed that compound 24d, which has chlorine substituents at both meta-positions of the aryl nucleus, was the most active, while a similar structure without the amino group (23d) presented unsatisfactory activity. On the other hand, the pyrazole derivatives 23b (without amino group) and 24b (with amino group) having only one chlorine atom at meta-position of the aryl nucleus shows moderate antileishmanial activity against L. amazonensis. The authors also synthesized 1-aryl-1H-pyrazole-4-carboxamides (84a–84e) that showed antileishmanial activity when tested against L. amazonensis.75 They reported that the compound containing a pyrazole ring with a slightly different substituent than 23 and 24 displayed impressive antileishmanial activity. Dardari et al.76 synthesized 4-[2-(1-(ethylamino)-2-methyl-propyl)phenyl]-3-(4-methylphenyl)-1-phenylpyrazole (84), which showed antileishmanial activity against L. tropica, L. major, and L. infantum. The IC50 values of 85 for the promastigote growth of L. tropica, L. major, and L. infantum were 0.48, 0.63 and 0.40 μg ml−1, respectively. They also indicated that L. infantum was the most sensitive strain. The growth of L. infantum promastigotes was inhibited by 50% after 24 h at a concentration of 0.75 μg mL−1 of 85, whereas the growths of L. major and L. tropica promastigotes were only inhibited by 8% and 9.3%, respectively, at the same concentration. It is also interesting to note that L. infantum causes lethal visceral leishmaniasis in humans and canines and is reported more and more frequently in association with the human immunodeficiency virus (HIV) (Fig. 5).
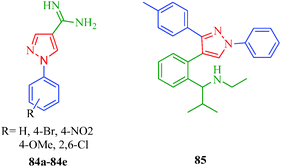 |
| | Fig. 5 Pyrazole derivatives as antileishmanial agents. | |
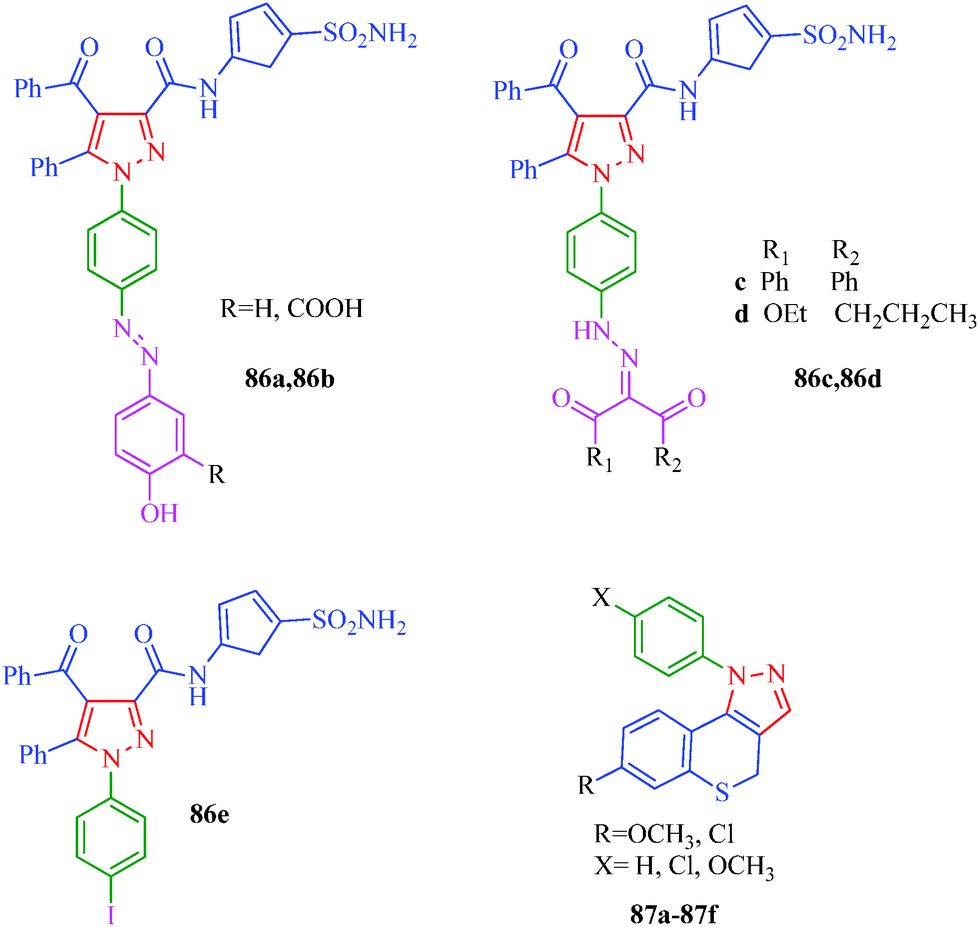
Antiproliferative
Cell proliferation is a process that results in an increase of the number of cells and is one of the significant processes in the overall growth of an individual. Abnormal cell growth and over-proliferation can cause various health problems. Cancers and tumors are some of the most fatal and fast-growing diseases. This group of diseases is characterized by the loss of control over cellular growth and is not easy to combat. Several pyrazolo[3,4-d]pyrimidine derivatives possessing antiproliferative activity were reported by Huang et al.43 (Scheme 6). These compounds show antiproliferative activity against a panel of human cancer cell lines, including lung carcinoma (NCl-H226), nasopharyngeal (NPC-TW01), and T-cell leukemia (Jurkat) cells. SAR studies indicated that the introduction of o-Cl-Ph or p-Br-Ph groups at the N-1 position and p-Me-Ph or p-Cl-Ph groups at the C-3 position in the pyrazole ring enhances bioactivity. Furthermore, the SARs indicated that 1-(quinolin-2-yl)-1H-pyrazolo[3,4-d]pyrimidines are more potent than 1-(pyridin-2-yl)-1H-pyrazolo[3,4-d]pyrimidines. Among all derivatives (Table 6), compound 26h was found to possess the best efficacy against NCl-H226 (IC50 = 18 μM) and NPC-TW01 (IC50 = 23 μM) cancer cells. Mert et al.77 reported a novel series of pyrazole–sulfonamide derivatives (86a–86e) synthesized from 1-(4-aminophenyl)-4-benzoyl-5-phenyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)-1H-pyrazole-3-carboxamide. The reported compound showed antiproliferative activity against HeLa and C6 cell lines and selective effects against C6 cell lines. According to the test results, compound 86d in particular has considerable antiproliferative activity against HeLa and C6 cell lines compared to reference drugs cis-platin and 5-FU. Compounds 86a and 86e have good antiproliferative activities against the C6 cell line at high concentrations, and SAR studies showed that presence of phenolic hydroxyl and iodine in 86a and 86e, respectively, may be responsible for the enhancement in inhibitory activity. Compound 86b also includes a carboxylate group next to the hydroxyl group, and carboxylate groups have been suggested to reduce the antiproliferative activity. A set of novel benzothiopyranopyrazole derivatives (87a–87f) with antiproliferative activity was reported by Via et al.78 These compounds showed antiproliferative activity when tested against HeLa and HL-60 cells; the activities were characterized by the presence of a methoxy substituent at the 7-position and a pendant phenyl, p-chlorophenyl, or p-methoxyphenyl in the 1-position of the heterocyclic moiety, respectively. Compound 87b appeared to be the most active compound against both cell lines (Fig. 6). The investigation of the mechanism of action of this compound highlighted the absence of interaction with DNA, and HeLa cells treated with 87b showed an evident depolarization of the mitochondrial membrane. These findings suggest that the reported compounds activate the apoptotic pathway to damage mitochondrial functions, and this mechanism accounts for the antiproliferative effects on tumor cell lines.
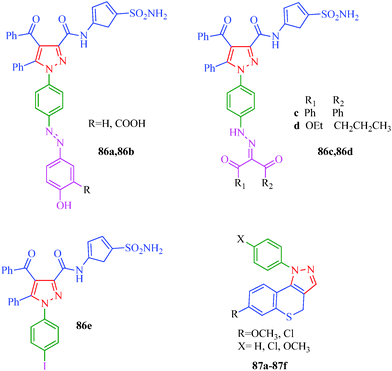 |
| | Fig. 6 Pyrazole derivatives as antiproliferative agents. | |
Anti-inflammatory
Inflammation is a multifactorial process that reflects the responses of the organisms to various stimuli and is related to many disorders such as pain, redness of skin, swelling, and loss of function in the case of acute inflammation; chronic inflammation may lead to arthritis, asthma, and psoriasis.79 Cyclooxygenases (COXs) are key enzymes in the biosynthesis of prostanoids, thromboxanes, and prostacyclins, and are involved in several physiological processes, including inflammation.80 COXs exist in two isoforms: COX-1 is constitutively expressed and is critical for the protection of gastric mucosa, platelet aggregation, and renal blood flow; COX-2 is inducible and expressed during inflammation, pain, and oncogenesis.81 The association of COX-2 with induced inflammation has led to the hypothesis that the selective inhibition of COX-2 over COX-1 might provide a good anti-inflammatory effect with fewer side effects compared to classical non-steroidal anti-inflammatory drugs (NSAIDs). Therefore, selective COX-2 inhibitors with better safety profiles have been marketed and termed as coxibs, including Celecoxib, Rofecoxib, Valdecoxib, and Etoricoxib.82 Numerous pyrazole derivatives have found clinical applications as NSAIDs, and Celecoxib is one bearing a pyrazole moiety. Other examples of pyrazole derivatives as NSAIDs include Mefobutazone, Ramifenazone, and Famprofazone. A series of novel pyrazole derivatives (88–90) were synthesized by Tewari et al.,83 and their anti-inflammatory activities were determined using carrageenan rat paw edema bioassay. Among the reported compounds, 88b, 88c and 89b showed maximum inhibitory effects, while compounds 89c and 89d showed intermediate effects. Molecular modeling showed that pyrazole analogues interact with the COX-2 active site through classical hydrogen bonds, π–π interactions and cation–π interactions, thereby augmenting the anti-inflammatory activities of the reported compounds (Fig. 7).
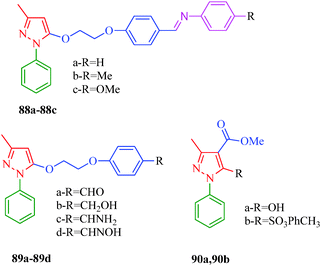 |
| | Fig. 7 Pyrazole derivatives as anti-inflammatory agents. | |
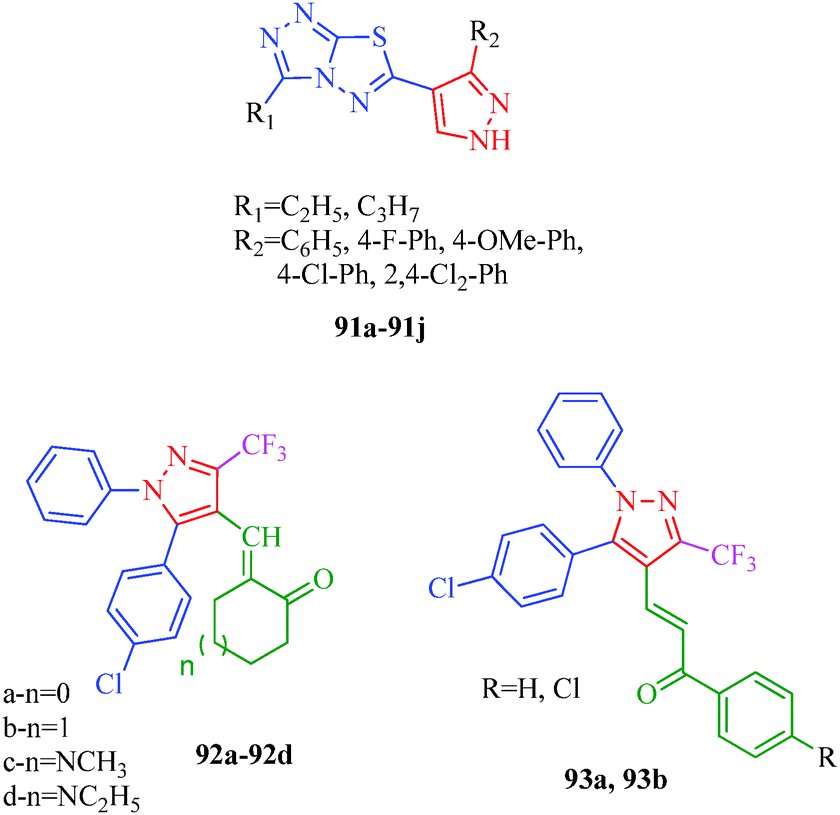
Kendre et al.44 reported some new 1H-pyrazole derivatives containing aryl sulfonate moieties (Scheme 7) with anti-inflammatory effects. The authors suggested that minor changes in the aromatic ring by halogen substitutions profoundly influenced the activity, and 31b was found to show maximum activity (Table 7). Malladi et al.84 synthesized 3,6-disubstituted-1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazoles bearing pyrazole moieties (91a–91i) with anti-inflammatory activities. Among the reported compounds, 91i showed the most significant anti-inflammatory activity (64.7% inhibition) compared to the standard drug Diclofenac sodium, and compounds 91d and 91f showed 56.9% inhibition. The presence of propyl and p-chlorophenyl substituents accounted for the significant activity of 91i. Compound 91d has ethyl and p-chlorophenyl moieties, and 91f has propyl and phenyl moieties, accounting for their moderate activities. El-Sayed et al.85 also synthesized pyrazole derivatives (92 and 93) having anti-inflammatory activities, and compounds 92a and 92d were the most selective among the tested compounds with reasonable inhibitory profiles against COX-2. Based on SAR studies, the authors concluded that the anti-inflammatory activities of 92a–92d were higher than those of 93a and 93b (Fig. 8).
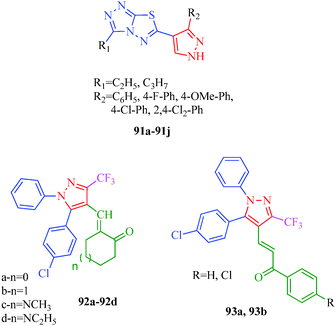 |
| | Fig. 8 Pyrazole derivatives as anti-inflammatory agents. | |
The pyrazole-4-carbaldehyde derivatives synthesized by Selvam et al.47 (Scheme 8) also possess anti-inflammatory activities in addition to being analgesic in nature. Among the synthesised compounds, the para-substituted derivatives (35c, 35e, 35g, 35i, 35k and 35l) showed excellent activities, and 35g, 35i, and 35k showed higher activities compared to other para derivatives (Table 8). This was attributed to the presence of para position halogen moieties. The pyrazolylbenzyltriazoles synthesized (Scheme 16) by Chandna et al.57 exhibited anti-inflammatory activities in addition to acting as antimicrobial agents. Four compounds (72b, 72e, 72g and 72h) were found to be the most potent COX-2 inhibitors, with 72h showing the best COX-2 profile (IC50 = 5.01 μM) followed by 72b (IC50 = 5.25 μM) and 72e (IC50 = 6.69 μM); 72b is a direct analogue of Celecoxib with a benzyltriazole moiety in the place of sulfonamide.
Analgesic
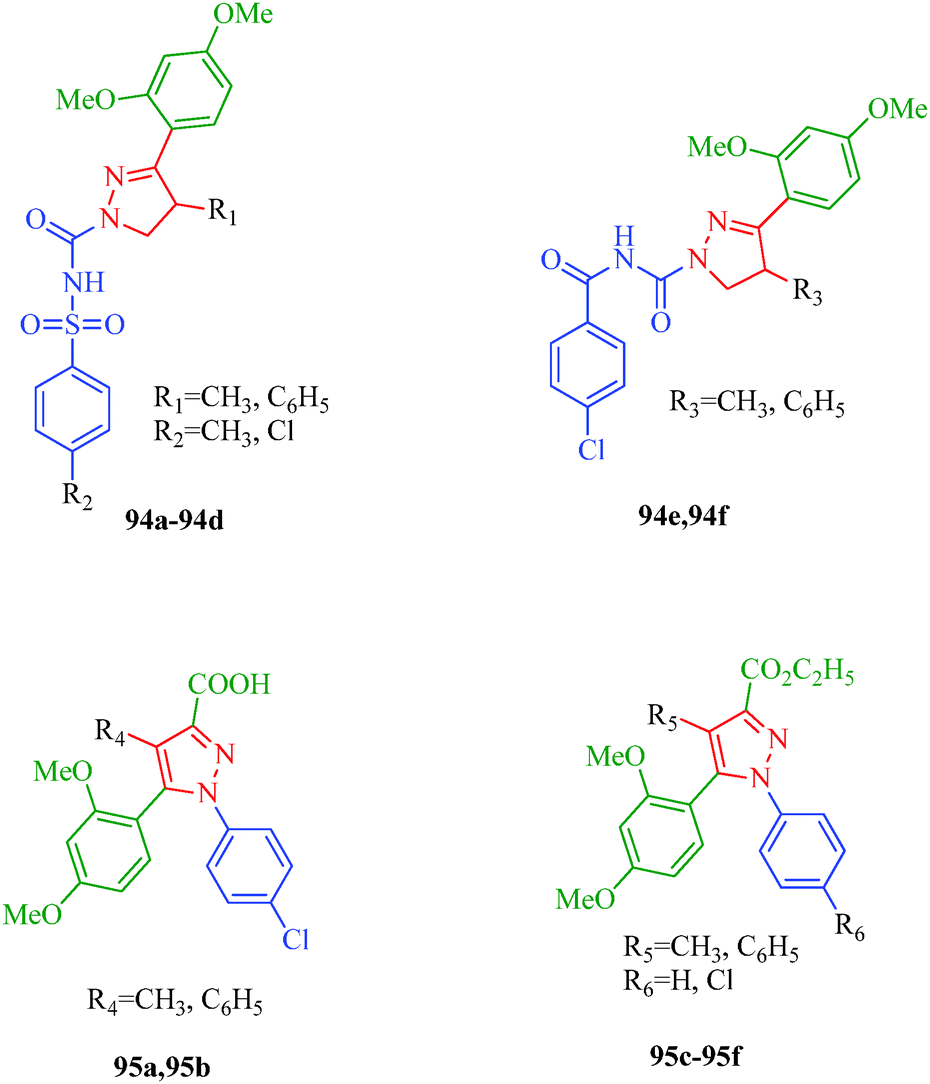
Pain is an unpleasant sensation and it is widely accepted to be one of the most important determinants of quality of life. Individuals with persistent pain suffer four times more from depression and anxiety than healthy persons. The identification of compounds able to treat both acute and chronic pain with limited side effects is one of the most prominent goals in biomedical research. Abd-El Gawad et al.86 designed pyrazole derivatives with analgesic activities (94 and 95) and suggested that the substitution of the pyrazole ring with at least one aryl moiety is essential for activity. All the reported compounds showed analgesic activity except 95d (Fig. 9).
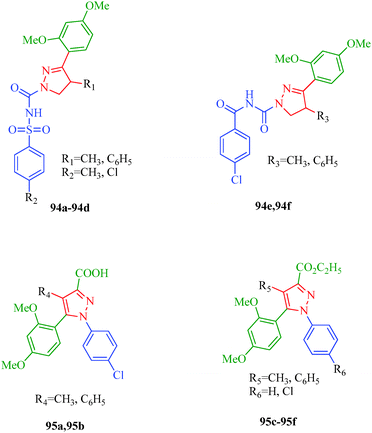 |
| | Fig. 9 Pyrazole derivative as analgesics. | |
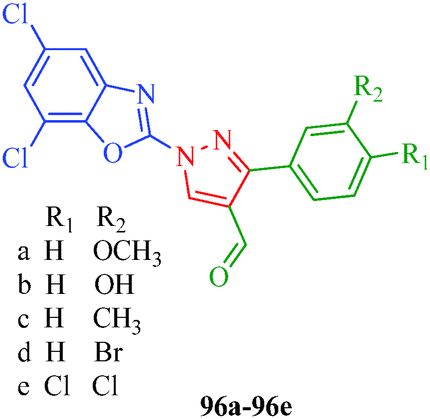
Selvam et al.47 (Scheme 8) reported that compounds 35g, 35i and 35k containing –F, –Br and –Cl substituents at the para position, respectively, showed significant analgesic activities, whereas compounds 35c, 35e and 35l containing –CH3, –OCH3 and –NHCOCH3 substituents, respectively, exhibited weaker analgesic activities (Table 8). This indicates that the electronic nature of the substituent group at the para position in the phenyl ring played a significant role in the bioactivity. A novel series of 1-(5,7-dichloro-1,3-benzoxazol-2-yl)-3-phenyl-1H-pyrazole-4-carbaldehyde derivatives (96a–96e) with analgesic properties were synthesized by Jayanna et al.87 Compounds 96b and 96d with para-hydroxyl and para-bromo substituents, respectively, exhibited potent analgesic activities with 55.78% and 54.63% protection, respectively, compared to acetyl salicylic acid as a standard drug (Fig. 10).
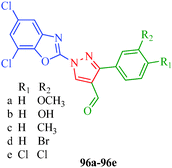 |
| | Fig. 10 Pyrazole derivatives as analgesics. | |
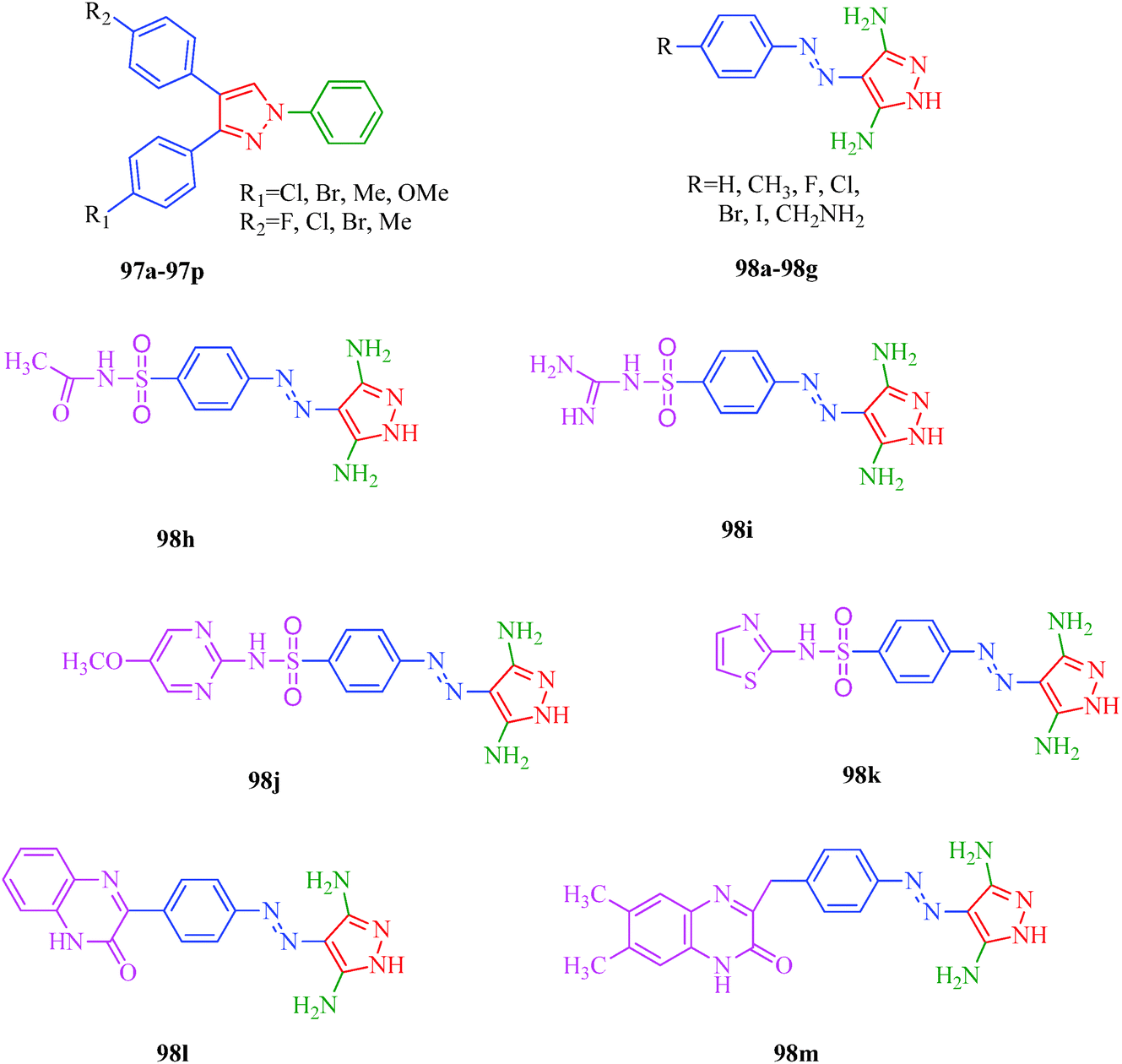
Cyclin-dependent kinase inhibitors
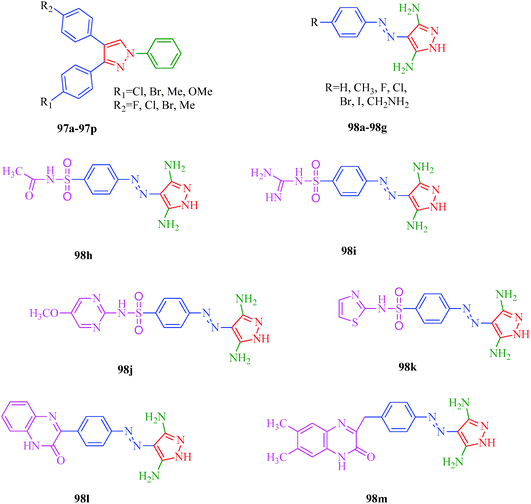
Cyclin-dependent kinases (CDKs) are a family of serine-threonine protein kinases, which govern the initiation, progression and completion of cell cycle events. They are also involved in regulating transcription, mRNA processing and differentiation of nerve cells. The activities of CDKs are critically dependent on the presence of their regulatory partners (cyclins), whose levels of expression are tightly controlled throughout the different phases of the cell cycle.88 Loss of cell cycle control resulting in aberrant cellular proliferation is one of the key characteristics of cancer, and the inhibition of CDKs is expected to be the most effective strategy for controlling tumor growth and hence an effective weapon in cancer therapy.89 The 1,3-diphenyl-N-(phenylcarbamothioyl)-1H-pyrazole-4-carboxamide derivatives synthesized by Sun et al.48 (Scheme 9) exhibited anti-proliferative activities against A549 (carcinomic human alveolar basal epithelial cells) and CDK2,4,6-inhibitory activities. Among the synthesized compounds, 40b demonstrated the most potent inhibitory activity (IC50 = 25, 35 and 25 nM for CDK2, CDK4 and CDK6, respectively) (Table 9). SAR studies showed that the compounds with OMe and Me groups as electron-donating substituents exhibited better activities than those with electron-withdrawing substituents. Huang et al.90 synthesized N-((1,3-diphenyl-1H-pyrazole-4-yl) methyl) aniline derivatives (97a–97p) as CDK inhibitors. These compounds exhibited potent CDK2-inhibitory activities and antiproliferative activities against MCF-7 and B16-F10 cell lines. Among the synthesized compounds, 97a showed the most potent inhibitory activity; 97a inhibited the growths of MCF-7 and B16-F10 cell lines with IC50 values of 1.88 ± 0.11 and 2.12 ± 0.15 μM, respectively, and inhibited CDK2/cyclin E holoenzyme activity with IC50 = 0.98 ± 0.06 μM. Another group of pyrazole derivatives (98a–98m) as CDK inhibitors was reported by Kryštof et al.91 Among the 4-arylazo-3,5-diamino-1H-pyrazole derivatives, 98a is the first representative of a novel group of compounds that can diminish the catalytic activity of CDK2-cyclin E. Compounds 98c–98e have similar inhibitory activities to 98a, while the non-polar 4-methyl-phenyl derivative 98b showed a slightly lower activity. Compounds 98h–98m also showed CDK-inhibitory effects; however, large substituents were found to be responsible for the decrease in CDK-inhibitory effects, probably due to steric hindrance (Fig. 11).
 |
| | Fig. 11 Pyrazole derivatives as CDK inhibitors. | |
TNAP inhibitor
Alkaline phosphatase is a hydrolase enzyme responsible for removing phosphate groups from many types of molecules, including nucleotides, proteins and alkaloids. Humans and most other mammals contain the following alkaline phosphate isozymes: tissue-specific alkaline phosphatases (placental, intestinal and germ cell) and tissue-non-specific alkaline phosphatase (TNAP).
TNAP hydrolyzes extracellular inorganic pyrophosphate (PPi), a potent mineralization inhibitor, to enable the physiological deposition of hydroxyapatite in bones and teeth.92 In this way, a controlled steady-state level of PPi is maintained, thereby sustaining normal bone formation and growth. Its lack of function in hypophosphatasia leads to impaired mineralization, while its overexpression or increased activity induces ectopic calcification. Due to its key function in mineralization, TNAP is an obvious target to prevent ectopic calcification, inducing vascular calcification.93 This has convinced many researchers to search for TNAP inhibitors to be used as a drug to target diseases caused by medial calcification, such as idiopathic infantile arterial calcification, end-stage renal disease and diabetes.94 Sidique et al.49 synthesized a novel series of pyrazole amide derivatives (Scheme 10) as potent and selective inhibitors of TNAP. SAR studies showed that the incorporation of a hydroxyl group on the amide generally increases the potency. The most potent compound in this series was the 2,3,4-trichlorophenyl analogue 45h (Table 10). This compound showed exceptional activity with an in vitro IC50 of 5 nM. Furthermore, compound 45h was inactive (IC50 > 10 μM) against both the related placental alkaline phosphatase (PLAP) isozyme and the housekeeping enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH), indicating a selectivity for TNAP of at least 2000-fold. By performing detailed kinetic studies, the authors demonstrated that the mechanism of action of 45h is competitive with respect to the substrate.
Agrochemicals
Herbicidal.
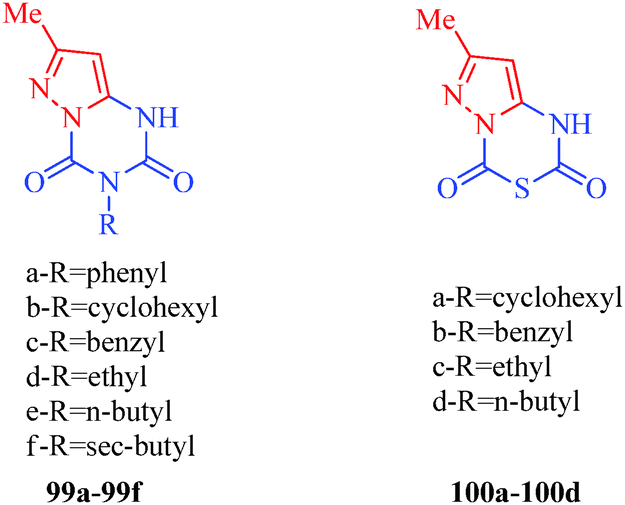
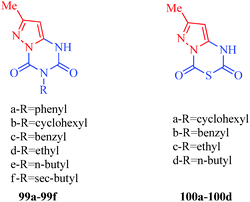
Herbicides (commonly known as weed killers) are pesticides used to remove unwanted plants. These unwanted plants are known as weeds, and they compete with productive crops or pasture, ultimately converting productive land into unusable scrub. Weeds can be poisonous or distasteful, produce burrs or thorns, and/or otherwise interfere with the use and management of desirable plants by contaminating harvests or interfering with livestock. Weeds compete with crops for nutrients, water, and physical space and may harbor insects and pests; thus, weeds are capable of greatly undermining both crop quality and yield. Weed control is the major constraint in food production efficiency. Several herbicides kill specific targets while leaving the desired crops relatively unharmed. With concerns about environmental pollution and the difficulties faced by food and agriculture management systems, several new weed control systems and strategies had been put forward. Many pyrazole derivatives with promising properties have been reported with different principles of action. Among the active principles through which several compounds act as inhibitors of weed growth include photosynthetic electron transport inhibition as shown by pyrazole derivatives 99 and 100.95 Among the pyrazolo[1,5-a][1,3,5]triazine-2,4-dione derivatives, those including butyl (99d) and cyclohexyl (99b) substituents showed maximal activity. Compounds 100c and 100d were more effective than their triazine counterparts. On the other hand, the comparison between compounds 99b–99e and 100a–100d, which have the exact same substituents, strongly suggested that the thiadiazine ring may amplify the inhibitory effect (Fig. 12).
 |
| | Fig. 12 Pyrazole derivatives as herbicidal agents. | |
Ma et al.50 synthesized N-(2,2,2)-trifluoroethylpyrazole derivatives (Scheme 11) as herbicides that work by inhibiting long-chain fatty acid elongase (VLCFAE). The herbicidal tests showed that when the dihydroisoxazole was attached to the 5-position of the N-(2,2,2)-trifluoroethylpyrazole ring linked by thiomethylene or sulfonylmethylene, the corresponding compounds presented good herbicidal activities. It was also found that when the substituent at the 4-position of the pyrazole ring was chlorine, the corresponding compound 51a exhibited the best herbicidal activity. When applied to soil in field trials, compound 51a showed excellent inhibitory activity against both dicotyledonous and monocotyledonous weeds along with good safety to maize and rape.
Insecticidal and acaricidal.
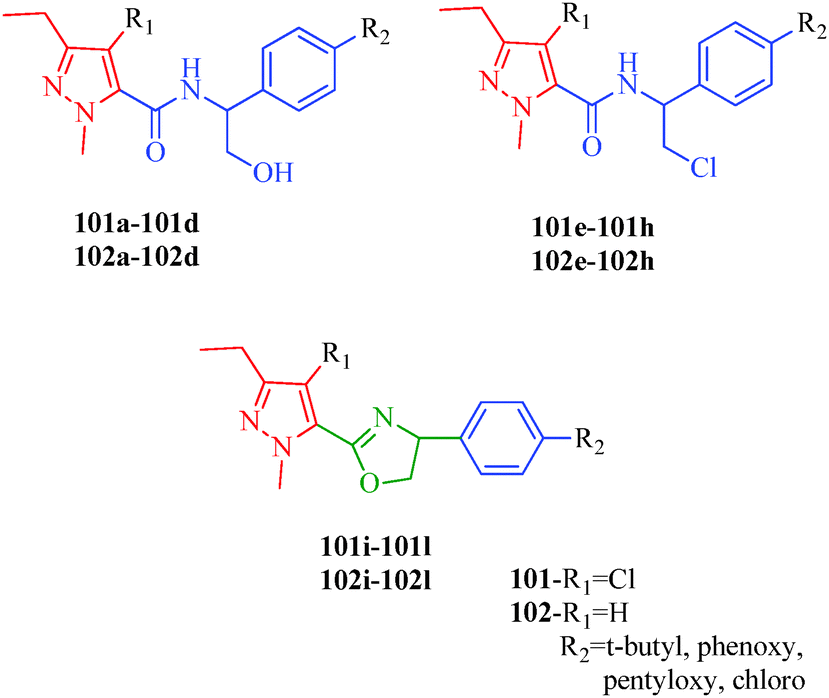
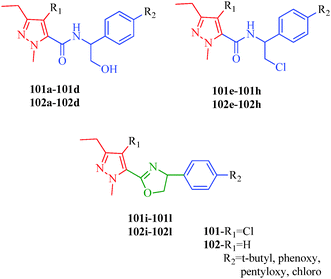
Insecticides are substances used to kill insects, and acaricides are specially used to kill members of the arachnid subclass acari, which includes ticks and mites. Both insecticides and acaricides are used in agriculture and medicine, although their main use is in the field of agriculture and crop protection. Modern insecticides should have a favorable combination of properties, including high levels of insecticidal activity, low application rates, crop tolerance, low levels of toxicity to mammals, and environmental friendliness. Many pyrazole derivatives have been reported as insecticides and acaricides, including a series of novel pyrazoles containing α-hydroxymethyl-N-benzyl carboxamide, α-chloromethyl-N-benzyl carboxamide, and 4,5-dihydrooxazole moieties designed and synthesized by Song et al.96 These compounds (101a–101l and 102a–102l) possessed good to excellent activities against a broad spectrum of insects such as cotton bollworm (H. armigera), bean aphid (A. craccivora), mosquito (Cx. pipiens pallens), and spider mite (T. cinnabarinus). The compounds containing α-chloromethyl-N-benzyl and 4,5-dihydrooxazole moieties exhibited higher activities than the compounds containing α-hydroxymethyl-N-benzyl moieties (Fig. 13).
 |
| | Fig. 13 Pyrazole derivatives as insecticidal and acaricidal agents. | |
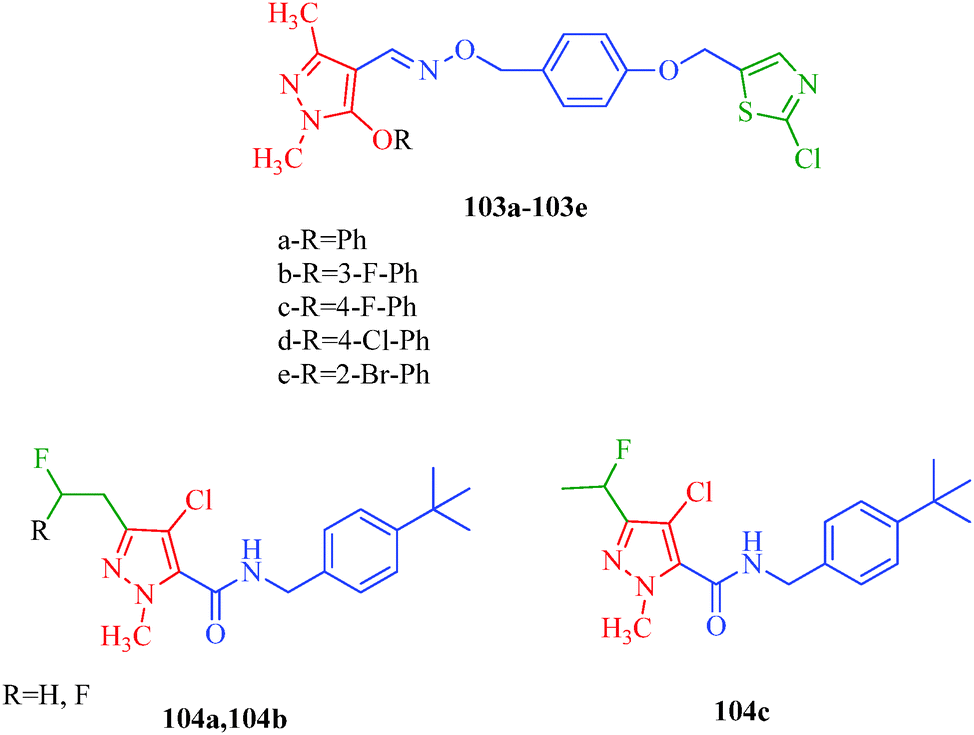
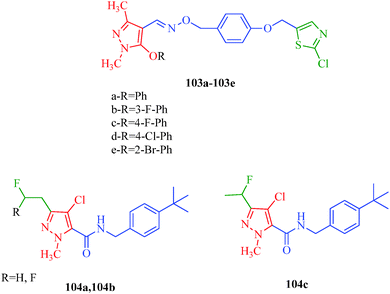
Wang et al.51 reported a series of pyrazole derivatives containing oxazole rings (Scheme 12), as insecticides. Among the synthesized compounds, 57b–57d and 57f–57j achieved 100% inhibition against A. craccivora and 80% inhibition against N. lugens (Table 11). In addition, some of the designed compounds exhibited moderate insecticidal activities against T. cinnabarinus at dosages of 500 mg l−1; in particularly, compounds 57i and 57j exhibited 100% inhibition against T. cinnabarinus. The authors also showed that compound 57c (when R1 and R2 are 4-fluoro atoms) was more potent against N. lugens than other oxime derivatives, and compound 57j (when R1 is a 3-fluoro atom and R2 is the 4-chloro atom) was more active against A. craccivora and T. cinnabarinus than other oxime compounds. Dai et al.97 synthesized a series of pyrazole oxime derivatives (103a–103e) exhibiting promising insecticidal and acaricidal activities against T. cinnabarinus and P. xylostella. Particularly, the 4-fluro-substituted compound 103c and 4-chloro-substituted compound 103d displayed relatively good acaricidal activities against T. cinnabarinus and potential insecticidal activities against P. xylostella compared to other oxime derivatives. Fustero et al.98 synthesized fluorinated Tebufenpyrad analogs (104a–104b) with acaricidal activity. Unfortunately, T. urticae is difficult to control because of its ability to quickly develop resistance to acaricides. The authors suggested that the introduction of fluorine into organic molecules causes significant physiochemical and biological changes. Interestingly, the compounds 104a and 104b cause complete or nearly complete adult mortality (Fig. 14).
 |
| | Fig. 14 Pyrazole derivatives as insecticidal and acaricidal agents. | |
Anticancer
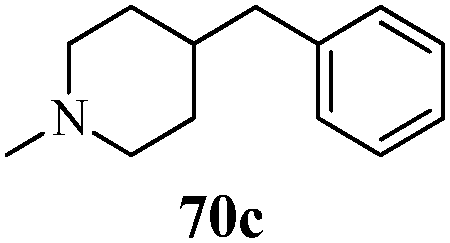
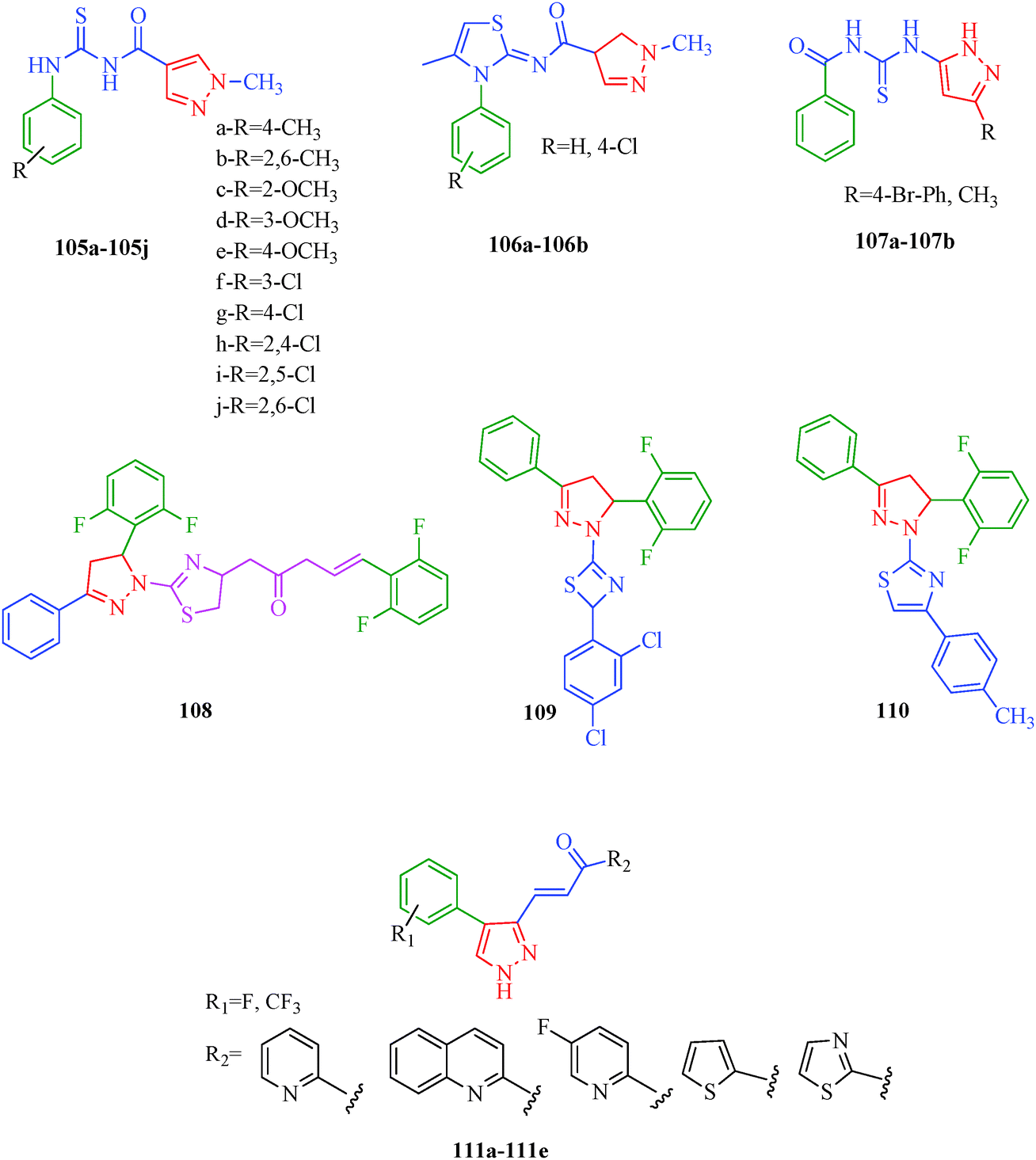
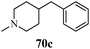
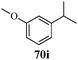
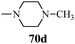
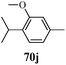
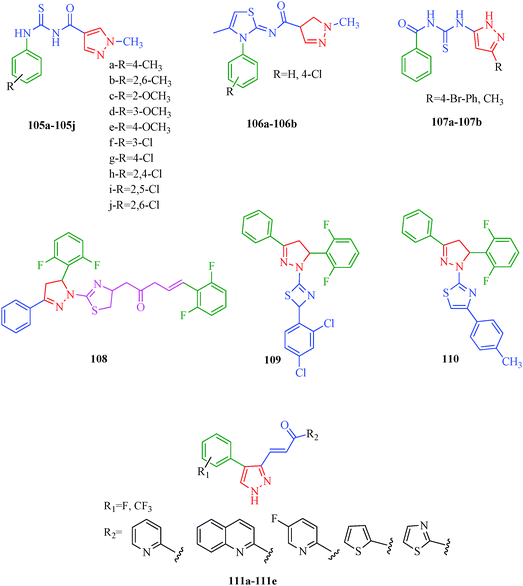
Cancer is a diverse group of diseases characterized by abnormal cell growth with the potential to invade or spread to other parts of the body. There are more than 100 types of cancer, including breast cancer, skin cancer, lung cancer, colon cancer, prostate cancer, and lymphoma. Symptoms vary depending on the type of cancer. The treatment of cancer may include chemotherapy, radiation therapy, and surgery. Cancer is a worldwide health problem and the most frightening disease in humans. The great majority of cancers are caused by environmental factors and carcinogens. Due to increasing pollution and the use of carcinogens, this fatal disease is increasing in prevalence. The majority of cancers are either resistant to chemotherapy or acquire resistance during treatment. As a result, the design and discovery of non-traditional, efficient, and safe chemical classes of agents are the prime targets of contemporary medicinal chemistry. Pyrazoles constitute an important heterocyclic family exhibiting anticancer activity, and many studies have been done to design new and potent anticancer drugs. A key feature of cancerous cells is their uncontrolled proliferation; thus, the inhibition of proliferative pathways is believed to be an effective strategy to fight cancer. The ester and amide derivatives of 1-phenyl-3-(thiophen-3-yl)-1H-pyrazole-4-carboxylic acid (Scheme 15) synthesized by Inceler et al.56 exhibited anticancer activities against various cancer cell lines. Among all the synthesized amide derivatives, compounds 70c and 70f possess the best inhibitory effects on cell growth (Table 13). Compound 70c, which has a benzylpiperidine group in the amide portion, showed significant inhibitory effects with the best results against Raji and HL60 cell lines. Ester derivatives of 1-phenyl-3-(thiophen-3-yl)-1H-pyrazole-4-carboxylic acid (70g–70l) did not affect the growth of HeLa, MCF7, MDA-MB-231, Raji, and HL60 cells. A series of functionally substituted pyrazole compounds have been synthesized (105–107) and evaluated in vitro for their antiproliferative effects on a panel of 60 cell lines by Nitulescu et al.99 Promising results were obtained with N-benzoyl-N′-(3-(4-bromophenyl)-1H-pyrazol-5-yl)-thiourea (107a). SAR studies revealed that substituted pyrazolyl thiourea derivatives are more active than the related N-[(1-methyl-1H-pyrazole-4-yl)]-thiourea derivatives. Prasad et al.100 reported novel 4,5-dihydropyrazole derivatives (108–110) exhibiting excellent anticancer activity compared to the reference drug cisplatin. Compound 109 having 4-chloro substitution showed excellent anticancer activity (IC50 = 4.94 μM) against HeLa (human cervix carcinoma cell lines), which implied that lipophilic and electron-withdrawing halobenzyl groups are beneficial for cytotoxic activity against HeLa cell lines. Sankappa Rai U. et al.101 synthesized a new series of pyrazole chalcones (111a–111e) exhibiting anticancer activities against MCF-7 and HeLa cell lines. Compound 111c showed the highest inhibition in human MCF-7 and HeLa cell lines; its highest activity was attributed to the 4-fluoro-phenyl and 5-fluoro-pyridin moieties (Fig. 15).
 |
| | Fig. 15 Pyrazole derivatives as anticancer agents. | |
The cis-restricted 3-aminopyrazole (Scheme 13) analogues of combretastatins synthesized by Tsyganov et al.53 as anticancer agents selectively target microtubules. Microtubules are responsible for the formation of the mitotic spindle, which is essential for proper chromosomal separation during cell division. The reported compounds possess antiproliferative and microtubule-destabilizing activities. Molecules 61a and 63a, with 3,4,5-trimethoxy groups on ring A, exhibited the highest antimitotic microtubule-destabilizing effect in sea urchin embryo assay. These compounds also showed considerable cytotoxicity against a panel of 60 human cancer cell lines. In addition, both 61a and 63b inhibited the growth of multi-drug-resistant, P-glycoprotein-overexpressing ovarian cancer cells, suggestive of their potential as anticancer agents.
IRAK4 inhibitors
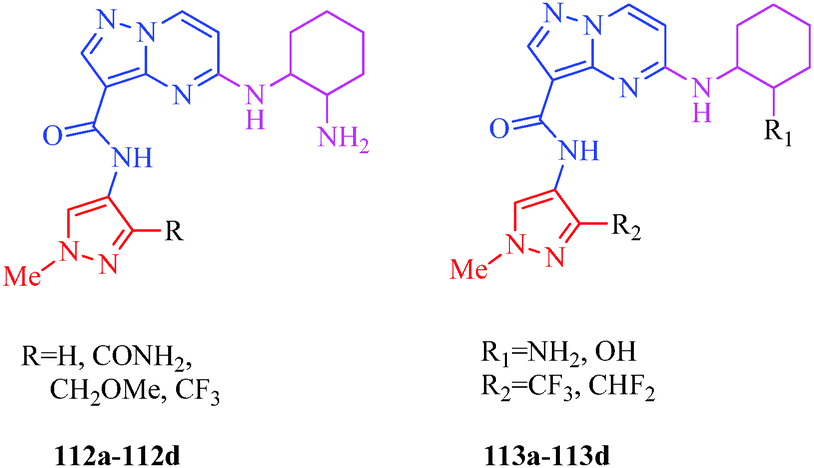
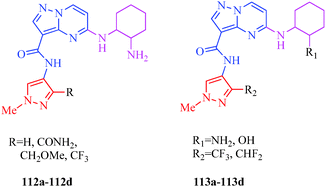
Interleukin receptor-associated kinase 4 (IRAK4) is a member of the IRAK family of intracellular serine-threonine kinase, which includes IRAK1, IRAK2, IRAKM or IRAK3 and IRAK4. IRAK4 is an essential signal transducer downstream of the interleukin-1 receptor (IL1R) and toll-like receptor (TLR) superfamily. IRAK4 mediates innate immune response by regulating the expression of inflammatory genes in multiple target cells. The innate immune system, also known as the non-specific immune system, comprises the cells and mechanism that defend the host from infection by other organisms. IRAK4 signaling is essential in the fundamental pathogenesis of multiple immune-inflammatory diseases and for cell survival and proliferation in hematological tumors. The proximal location of IRAK4 to key immune signaling receptors has generated significant interest in the therapeutic targeting of IRAK4 for autoimmune and inflammatory diseases. A potent and selective IRAK4 inhibitor would be useful to explore cell malignancies as well as for autoimmune diseases such as rheumatoid arthritis, psoriasis, inflammatory bowel disease, and gout.102 The degree of selectivity required of IRAK4 inhibitors is likely to vary depending on the targeted disease. A series of potent, selective, and orally bioavailable pyrazole IRAK4 inhibitors were synthesized by McElroy et al.54 (Scheme 14). SAR studies indicated that piperidine and piperazine substituents at the C-3 position were well-tolerated, and the identity of the nitrogen substituent could be used to modulate potency and solubility. In contrast, alkyl-substituted piperidines and piperazines were highly soluble and very potent. Limited SAR studies indicated that o-fluoro and pyridine substituents at the pyrazole N-1 position provided the best levels of potency (Table 12). A series of 5-amino-N-(1H-pyrazol-4-yl)-pyrazolo[1,5-a]pyrimidine-3-carboxamides (112 and 113) as IRAK4 inhibitors was reported by Lim et al.103 The authors reported that the replacement of the substituents responsible for the poor permeability and improvement in physical properties led to the identification of IRAK4 inhibitors with excellent potency, kinase selectivity, and pharmacokinetic properties suitable for oral dosing (Fig. 16).
 |
| | Fig. 16 Pyrazole derivatives as IRAK4 inhibitors. | |
Antimicrobial
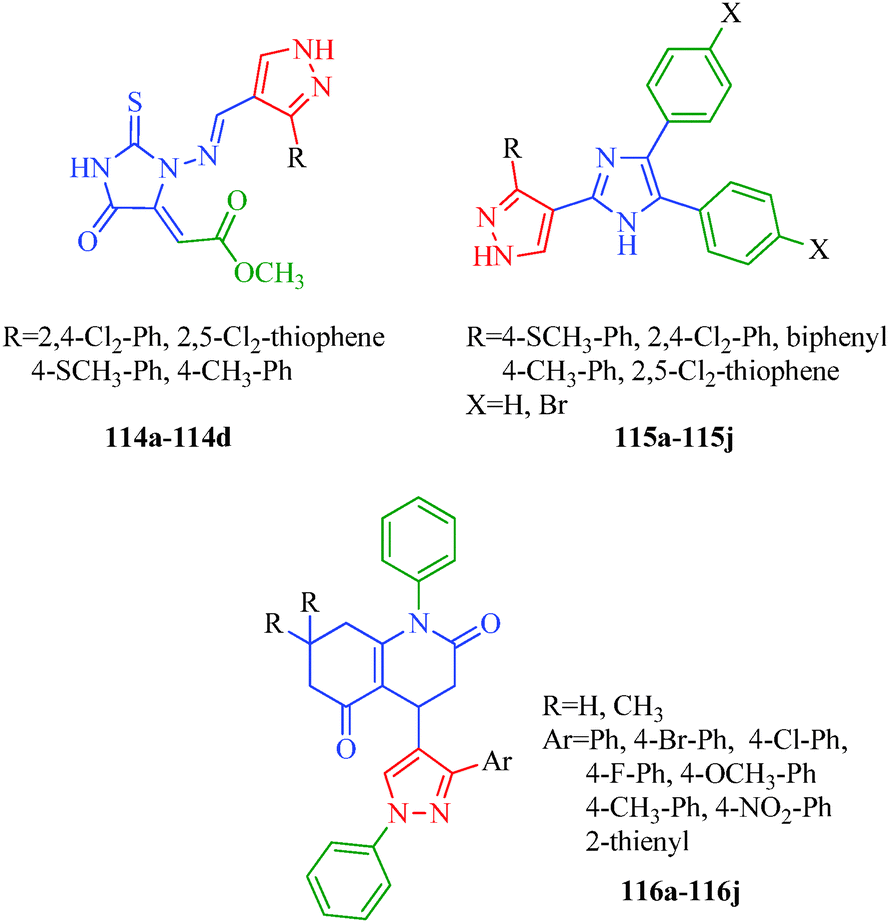
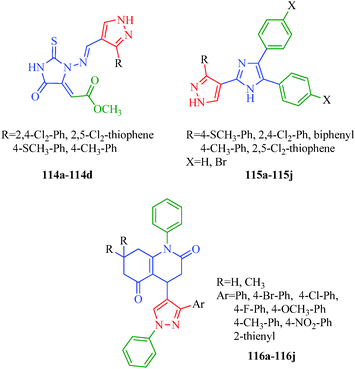
Antimicrobial agents are compounds used to kill microorganisms or inhibit their growth. They can be grouped according to the microorganisms they act primarily against. Many diseases are indeed caused by these microorganisms, and these microbial infections pose a tremendous threat to the human race. The efficacies of many antimicrobial agents are being threatened by a global increase in the numbers of resistant microorganisms that were once susceptible to some of these agents. In spite of the large number of antibiotics and chemotherapeutics available for medicinal use, the emergence of old and new antibiotic-resistant microbial strains in the last decades has generated a substantial need for new classes of antimicrobial agents. Pyrazole and its derivatives have been found to possess various antibacterial, antifungal and antiviral properties. Chandna et al.57 reported pyrazolylbenzyltriazoles (Scheme 16) as antimicrobial agents. All the reported compounds possessed variable antibacterial activities against Gram-positive bacteria (S. aureus and B. subtilis), and some possessed moderate to good activity against Gram-negative bacteria (E. coli and P. aeruginosa). Two compounds (72c and 72e) were found to be the most effective against S. aureus. However, compounds 72a, 72c and 72f demonstrated comparable antibacterial activities to that of ciprofloxacin against B. subtilis. Vijesh et al.104 synthesized pyrazole-incorporated imidazole derivatives (114 and 115) that exhibit antimicrobial properties against various bacterial (E. coli, S. aureus, B. subtilis, S. typhimorium, C. profingens and P. aeruginosa) and fungal strains (A. flavus, A. niger, C. albicans, M. gypseum, and T. rubrum). Among the reported compounds, compound 114b showed excellent antimicrobial activity at concentrations of 1 and 0.5 mg ml−1. Compound 114b has a thioanisyl moiety, which accounted for the enhanced antibacterial activity. The compounds in the second series (115a–115j) showed moderate antimicrobial activities. The imidazole and pyrazole nucleus, which is present in both series, is responsible for the biological activity. However, the presence of other substituents is responsible for the varied biological activities of the compounds. A novel series of substituted 4-pyrazolyl-N-arylquinoline-2,5-dione (116a–116j) derivatives was synthesized as antimicrobial agents by Thumar et al.105 Most of the reported compounds possess antibacterial and antifungal activities against B. subtilis, C. tetani, S. pneumoniae, S. typhi, V. cholerae, E. coli, A. fumigatus and C. albicans. Compounds 116g, 116h and 116k were found to be highly potent in inhibiting the growth of most of the employed strains. A SAR study showed that among all compounds, that containing a para-bromophenyl ring was the most potent against both fungal species (C. albicans and A. fumigatus; Fig. 17).
 |
| | Fig. 17 Pyrazole derivatives as antimicrobial agents. | |
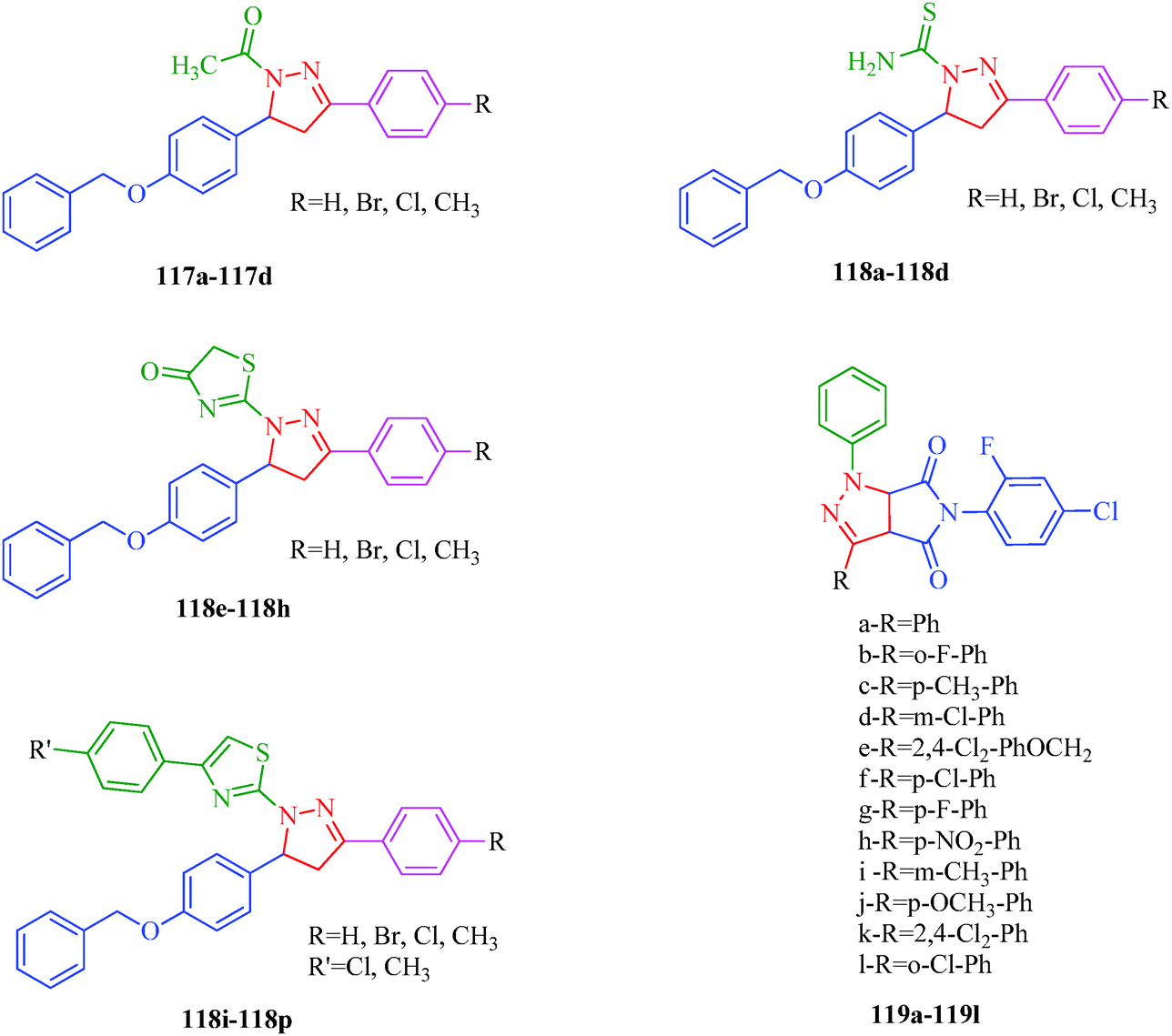
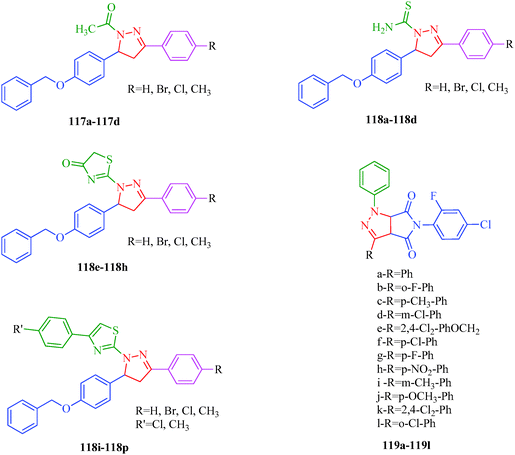
The dihydropyrano[2,3-c]pyrazole derivatives synthesized by Ambethkar et al.58 (Scheme 17) using a grinding method exhibited antimicrobial activities against S. albus, S. pyogenes, K. pneumonia, P. aeruginosa and C. albicans. New N-acetyl (117a–117d) and N-thiocarbamoyl (118a–118d) derivatives of 4,5-dihydropyrazole were synthesized by El-Sabbagh et al.13N-Thiocabamoylpyrazole derivatives were cyclized to afford the novel pyrazolothiazol-4(5H)-ones (118e–118h) and pyrazolothiazoles (118i–118p). The reported compounds exhibited antiviral activities against several viruses like herpes simplex virus type 1 (KOS) [HSV-1 KOS], herpes simplex virus type 2 (G) [HSV-2G], vaccinia virus [VV], vesicular stomatitis virus [VSV], Coxsackie virus B4 [CV-B4], respiratory syncytial virus [RSV] and parainfluenza-3 virus [PI-3V]. A series of cycloadducts-pyrazoles formed via 1,3-dipolar cycloaddition reactions has been reported by Zhang et al.106 Most of the target compounds (119a–119l) exhibited significant antifungal activities against C. cassiicola. Among them, compound 119a had good fungicidal activity against P. syringae pv. Lachrymans. When the benzene ring is substituted by halogen, the compounds (119b and 119g) generally have excellent antifungal activity against C. cassiicola, whereas the activities decreased for di-halogen, alkane, and H substituents (119a, 119c, and 119k; Fig. 18).
 |
| | Fig. 18 Pyrazole derivatives as antimicrobial agents. | |
The 2-(1-phenyl-1H-pyrazol-5-yl)-phenyl-4-methylbenzenesulfonate derivatives synthesized (Scheme 7) by Kendre et al.44 exhibited antimicrobial activities against both Gram-positive and Gram-negative bacterial and fungal strains, in addition to acting as anti-inflammatory agents (Table 7). Among the synthesized compounds, 31e was found to be most active against various pathogens with reference to ampicillin and norcadine.
Antioxidant
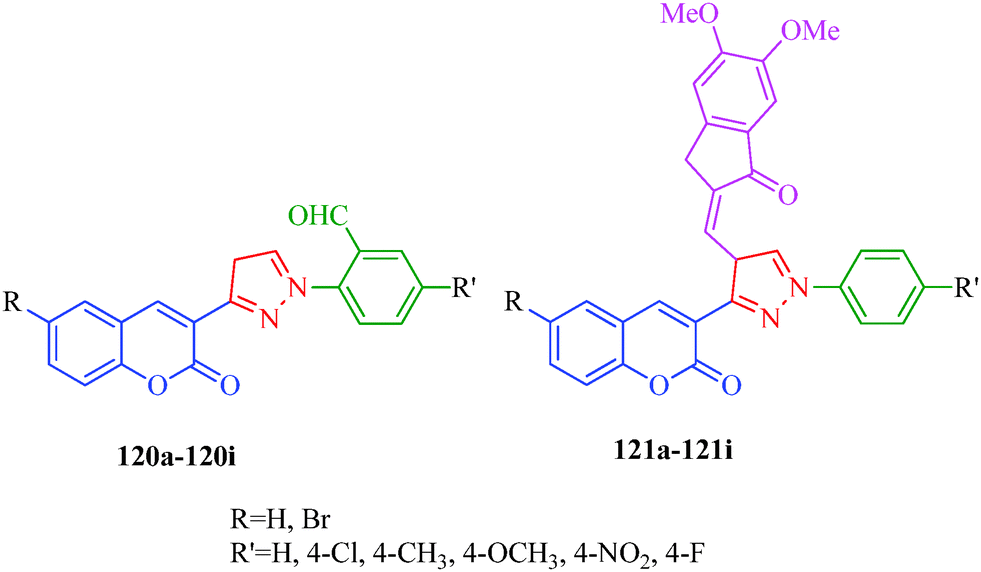
Free radicals are highly reactive chemicals that can potentially harm cells and are capable of attacking the healthy cells of the body, potentially damaging molecules. Free radicals can be hazardous to the body and damage all major components of cells, including DNA, proteins and cell membranes. Antioxidants, also known as free radicals scavengers, are chemicals that interact with and neutralize free radicals, thus preventing them from causing damage. The body makes some antioxidants, which are known as endogenous antioxidants. The capacities of endogenous antioxidants are affected by the age, diet, and health status of the individual. However, the body relies on external (exogenous) sources, primarily the diet, to obtain the rest of the antioxidants it needs. Cells with damaged DNA stagnate and are prone to developing cancer and growths. This kind of damage also accelerates the aging process and diabetes, directly causing wrinkles and age spots and severely taxing the immune system. Free radicals may also responsible for other diseases such as cardiovascular disease, neural disorders, Alzheimer's disease, alcohol-induced liver disease. Therefore, the search for new antioxidants has received much attention. Kenchappa et al.107 synthesized coumarin derivatives containing pyrazole rings (120 and 121) as potent antioxidant agents. They suggested that the presence of the pyrazole ring endows these species with important pharmacological and therapeutic properties. SAR studies revealed that compounds containing electron-withdrawing groups/halogens showed good antioxidant properties (Fig. 19).
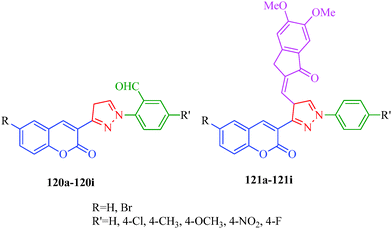 |
| | Fig. 19 Pyrazole derivatives as antioxidants. | |
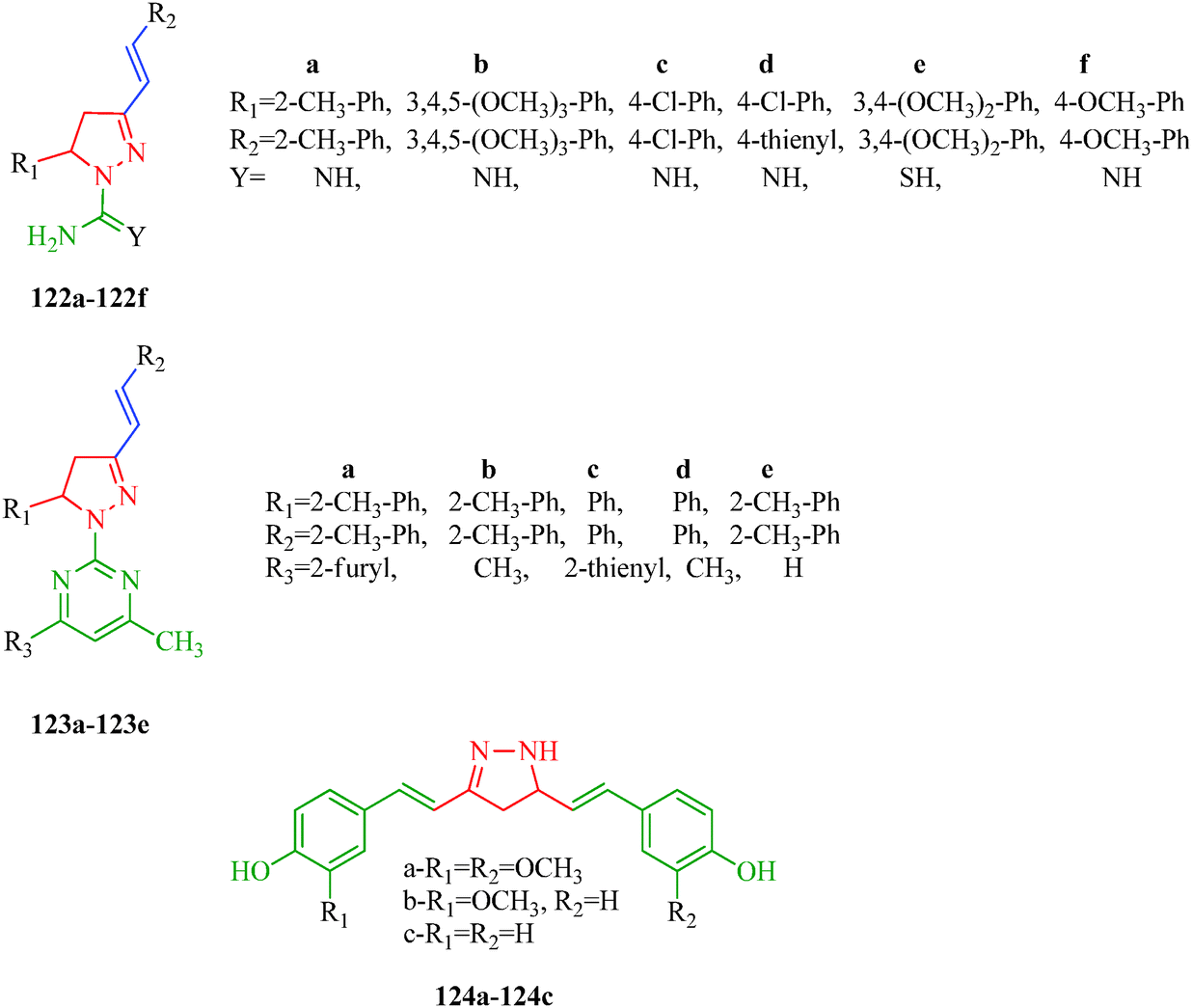
The dihydropyrano[2,3-c]pyrazole derivatives synthesized by Ambethkar et al.58 (Scheme 17) exhibiting antimicrobial activity were screened for their antioxidant activity against 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenger. All the compounds showed radical-scavenging activity. Compound 74f showed better scavenging capacity, and other compounds 74a–74e and 74g–74i showed moderate scavenging ability. The presence of a hydroxyl group in the para position of 74f extends the p-conjugation, stabilizing the produced free radicals. Gressler et al.108 synthesized 2-(4,5-dihydro 1H-pyrazol-1-yl)pyrimidine and 1-carboxamidino-1H-pyrazole derivatives (122 and 123) as antioxidant agents. The authors showed that the presence of thienyl exerts a significant influence on radical stability. On the other hand, the presence of chlorine substituents does not appear to significantly affect the antioxidant properties of the compounds in this series. Selvam et al.109 reported pyrazole derivatives (124a–124c) as analogues of curcumin, a potent antioxidant agent. Compound 124a was found to be more potent than curcumin. The authors showed that o-methoxy substitution enhanced the stability of the radical (Fig. 20).
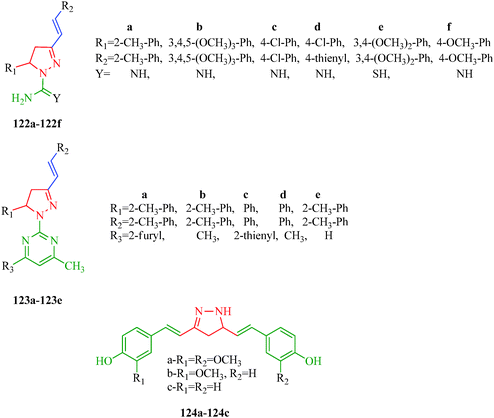 |
| | Fig. 20 Pyrazole derivatives as antioxidants. | |
5α-Reductase inhibitor
5α-Reductases are enzymes that are involved in steroid metabolism. These are also known as 3-oxo-5α-steroid 4-dehydrogenases, which participate in three metabolic pathways: bile acid biosynthesis, androgen/oestrogen metabolism, and prostate cancer. The enzyme is produced in many tissues in both males and females, and there are three isoenzymes of 5α-reductase that vary in different tissues with age. The enzyme steroid 5α-reductase (5AR) catalyzes the NADPH-dependent reductive conversion of testosterone to dihydrotestosterone. Increased 5AR activity leads to increased levels of dihydrotestosterone in the peripheral tissues, which is implicated in the pathogenesis of prostate cancer, acne and pattern baldness. The inhibition of 5α-reductase is most known for preventing the conversion of testosterone to the more potent dihydrotestosterone (DHT) in androgen-associated disorders. The mechanism of 5α-reductase inhibition is complex and involves the binding of NADPH to the enzyme followed by the substrate. Specific substrates include testosterone, progesterone, androstenedione, epitestosterone, cortisol, aldosterone, and deoxycorticosterone. Drugs that inhibit 5α-reductase are used to treat benign prostatic hyperplasia, prostate cancer, and pattern baldness and in hormone replacement therapy. Recently, several steroid-based D-ring heterocyclic analogs have been evaluated as efficient 5α-reductase inhibitors. Banday et al.59 synthesized two series of D-ring-substituted pyrazolinyl and pyrazolyl pregnenolone derivatives (Scheme 18) exhibiting 5α-reductase inhibitory activities. All the compounds reported (Table 14) exhibited promising inhibitory activity, particularly against type-I human 5α-reductase. However, compounds 77b, 77c and 79b were found to best inhibit 5α-reductase enzyme. The authors reported that all of the active derivatives are halogenated, indicating that halogen substitution increases the activity. They also demonstrated that pyrazolines (77a–77d) are more active and selective towards type-I reductase.
Conclusions and perspectives
The remarkable chemical diversity of heterocyclic compounds continues to be of relevance to drug discovery. Drug development has been a chief component in the rapid maturation of the field of medicinal chemistry during the past several decades; during this period, the scientific community has paid significant attention to the development of intriguing and challenging molecular architectures of nitrogen-containing heterocyclic compounds. There is a growing body of evidence that pyrazole and its derivatives provide a viable and valuable area for drug discovery. Here, we have presented an overview of the many efficient, mild, operationally simple and non-conventional synthetic methods to access a library of highly functionalized pyrazole derivatives and a broad range of biological activities displayed by these scaffolds that can optimally present a way to capture their intrinsic value. The SARs of the reported compounds reveal that a greater understanding of the pattern of substituents, including electron-donating, electron-withdrawing, and some heterocyclic moieties, on the basic skeleton plays a key role in regulating the biological potentials of the synthesized compounds. Furthermore the target-oriented or focused-library approach seeks to elaborate structural modifications onto existing, bioactive pyrazole scaffolds in a parallel, systematic fashion to improve the inherent biological activity or drug-like properties. The ability to predict drug-like and lead-like properties along with recent technological advances could be sufficient to revitalize the exploitation of the value of pyrazole derivatives in the quest for new drugs.
Acknowledgements
We sincerely thank Department of Chemistry, Aligarh Muslim University, Aligarh, India, for providing necessary facilities.
References
- B. Eftekhari-Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958–3043 CrossRef CAS PubMed
 .
.
- Y. Ju and R. S. Varma, J. Org. Chem., 2006, 71, 135–141 CrossRef CAS PubMed .
- D. Zárate-Zárate, R. Aguilar, R. I. Hernández-Benitez, E. M. Labarrios, F. Delgado and J. Tamariz, Tetrahedron, 2015, 71, 6961–6978 CrossRef .
- E. M. Gordon, R. W. Barrett, W. J. Dower, S. P. A. Fodor and M. A. Gallop, J. Med. Chem., 1994, 37, 1385–1401 CrossRef CAS PubMed .
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS PubMed .
- X. Li, L. He, H. Chen, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3636–3646 CrossRef CAS PubMed .
- C. T. Walsh, Tetrahedron Lett., 2015, 56, 3075–3081 CrossRef CAS .
- S. M. E. Khalil, J. Coord. Chem., 2003, 56, 1013–1024 CrossRef CAS .
- H. Kiyani, F. Albooyeh and S. Fallahnezhad, J. Mol. Str., 2015, 1091, 163–169 CrossRef CAS .
- P.-C. Lv, J. Sun, Y. Luo, Y. Yang and H.-L. Zhu, Bioorg. Med. Chem. Lett., 2010, 20, 4657–4660 CrossRef CAS PubMed .
- S. A. H. El-Feky, Z. K. Abd El-Samii, N. A. Osman, J. Lashine, M. A. Kamel and H. K. Thabet, Bioorg. Chem., 2015, 58, 104–116 CrossRef CAS PubMed .
- C. B. Sangani, D. C. Mungra, M. P. Patel and R. G. Patel, Chin. Chem. Lett., 2012, 23, 57–60 CrossRef CAS .
- O. I. El-Sabbagh, M. M. Baraka, S. M. Ibrahim, C. Pannecouque, G. Andrei, R. Snoeck, J. Balzarini and A. A. Rashad, Eur. J. Med. Chem., 2009, 44, 3746–3753 CrossRef CAS PubMed .
- B. Insuasty, A. Tigreros, F. Orozco, J. Quiroga, R. Abonia, M. Nogueras, A. Sanchez and J. Cobo, Bioorg. Med. Chem., 2010, 18, 4965–4974 CrossRef CAS PubMed .
- V. Michon, C. H. Penhoat, F. Tombret, J. M. Gillardin, F. Lepage and L. Berthon, Eur. J. Med. Chem., 1995, 30, 147–155 CrossRef CAS .
- D. M. Bailey, P. E. Hansen, A. G. Hlavac, E. R. Baizman, J. Pearl, A. F. DeFelice and M. E. Feigensonf, J. Med. Chem., 1985, 28, 256–260 CrossRef CAS PubMed .
- J. B. Shi, W. J. Tang, X. B. Qi, R. Li and X. H. Liu, Eur. J. Med. Chem., 2015, 90, 889–896 CrossRef CAS PubMed .
- J. Rangaswamy, H. Vijay Kumar, S. T. Harini and N. Naik, Bioorg. Med. Chem. Lett., 2012, 22, 4773–4777 CrossRef CAS PubMed .
- P. Horrocks, M. R. Pickard, H. H. Parekh, S. P. Patel and R. B. Pathak, Org. Biomol. Chem., 2013, 11, 4891–4898 Search PubMed .
- A. M. Vijesh, A. M. Isloor, S. Prashant, S. Sundershan and H. Kun Fun, Eur. J. Med. Chem., 2013, 62, 410–415 CrossRef CAS PubMed .
- M. Mamaghani, R. Hossein Nia, F. Shirini, K. Tabatabaeian and M. Rassa, Med. Chem. Res., 2015, 24, 1916–1926 CrossRef CAS .
- R. C. Khunt, V. M. Khedkar, R. S. Chawda, N. A. Chauhan, A. R. Parikh and E. C. Coutinho, Bioorg. Med. Chem. Lett., 2012, 22, 666–678 CrossRef CAS PubMed .
- S. Mert, R. Kasimogullari, T. Ica, F. Colak, A. Altun and S. Ok, Eur. J. Med. Chem., 2014, 78, 86–96 CrossRef CAS PubMed .
- H. Fu and J. Yao, J. Am. Chem. Soc., 2001, 123, 1434–1439 CrossRef CAS .
- M. Wang, J. Zhang, J. Liu, C. Xu and H. Ju, J. Lumin., 2002, 99, 79–83 CrossRef CAS .
- F. Karci, F. Karci, A. Demirçali and M. Yamaç, J. Mol. Liq., 2013, 187, 302–308 CrossRef CAS .
- X. C. Gao, H. Cao, L. Q. Zhang, B. W. Zhang, Y. Cao and C. H. Huang, J. Mater. Chem., 1999, 9, 1077–1080 RSC .
- J. Burschka, F. Kessler, M. K. Nazeeruddin and M. Gratzel, Chem. Mater., 2013, 25, 2986–2990 CrossRef CAS .
- U. M. Kauhanka and M. M. Kauhanka, Liq. Cryst., 2006, 33, 121–127 CrossRef CAS .
- P.-T. Chou and Y. Chi, Chem. – Eur. J., 2007, 13, 380–395 CrossRef CAS PubMed .
- X. Tu, W. Hao, Q. Ye, S. Wang, B. Jiang, G. Li and S. Tu, J. Org. Chem., 2014, 79, 11110–11118 CrossRef CAS PubMed .
- G. Molteni and P. Del Buttero, Tetrahedron: Asymmetry, 2005, 16, 1983–1987 CrossRef CAS .
- D. Castagnolo, S. Schenone and M. Botta, Chem. Rev., 2011, 111, 5247–5300 CrossRef CAS PubMed .
- P. Singh, K. Paul and W. Holzer, Bioorg. Med. Chem., 2006, 14, 5061–5071 CrossRef CAS PubMed .
- V. Kumar, K. Kaur, G. K. Gupta and A. K. Sharma, Eur. J. Med. Chem., 2013, 69, 735–753 CrossRef CAS PubMed .
- F. Chimenti, S. Carradori, D. Secci, A. Bolasco, B. Bizzarri, P. Chimenti, A. Granese, M. Yanez and F. Orallo, Eur. J. Med. Chem., 2010, 45, 800–804 CrossRef CAS PubMed .
- F. Chimenti, E. Maccioni, D. Secci, A. Bolasco, P. Chimenti, A. Granese, O. Befani, P. Turini, S. Alcaro, F. Ortuso, R. Cirilli, F. La Torre, M. C. Cardia and S. Distinto, J. Med. Chem., 2005, 48, 7113–7122 CrossRef CAS PubMed .
- J. J. Liu, H. Zhang, J. Sun, Z. C. Wang, Y. S. Yang, D. D. Li, F. Zhang, H. Bin Gong and H. L. Zhu, Bioorg. Med. Chem., 2012, 20, 6089–6096 CrossRef CAS PubMed .
- J.-J. Liu, J. Sun, Y.-B. Fang, Y.-A. Yang, R.-H. Jiao and H.-L. Zhu, Org. Biomol. Chem., 2014, 12, 998–1008 Search PubMed .
- H. Khalilullah, S. Khan, M. Jawed Ahsan and B. Ahmed, Bioorg. Med. Chem. Lett., 2011, 21, 7251–7254 CrossRef CAS PubMed .
- M. S. dos Santos, M. L. V. Oliveira, A. M. R. Bernardino, R. M. de Léo, V. F. Amaral, F. T. de Carvalho, L. L. Leon and M. M. Canto-Cavalheiro, Bioorg. Med. Chem. Lett., 2011, 21, 7451–7454 CrossRef CAS PubMed .
- C. C. Cheng and R. K. Robins, J. Org. Chem., 1956, 21, 1240–1256 CrossRef CAS .
- Y.-Y. Huang, L.-Y. Wang, C.-H. Chang, Y.-H. Kuo, K. Kaneko, H. Takayama, M. Kimura, S.-H. Juang and F. F. Wong, Tetrahedron, 2012, 68, 9658–9664 CrossRef CAS .
- B. V. Kendre, M. G. Landge, W. N. Jadhav and S. R. Bhusare, Chin. Chem. Lett., 2013, 24, 325–328 CrossRef CAS .
- B. R. Patil, S. R. Bhusare, R. P. Pawar and Y. B. Vibhute, Tetrahedron Lett., 2005, 46, 7179–7181 CrossRef CAS .
- S. B. Patwari, M. A. Baseer, Y. B. Vibhute and S. R. Bhusare, Tetrahedron Lett., 2003, 44, 4893–4894 CrossRef CAS .
- T. Panneer Selvam, P. Vijayaraj Kumar, G. Saravanan and C. Rajaram Prakash, J. Saud. Chem. Soc, 2014, 18, 1015–1021 CrossRef .
- J. Sun, X. H. Lv, H. Y. Qiu, Y. T. Wang, Q. R. Du, D. D. Li, Y. H. Yang and H. L. Zhu, Eur. J. Med. Chem., 2013, 68, 1–9 CrossRef CAS PubMed .
- S. Sidique, R. Ardecky, Y. Su, S. Narisawa, B. Brown, J. L. Millán, E. Sergienko and N. D. P. Cosford, Bioorg. Med. Chem. Lett., 2009, 19, 222–225 CrossRef CAS PubMed .
- H.-J. Ma, Y.-H. Li, Q.-F. Zhao, T. Zhang, R.-L. Xie, X.-D. Mei and J. Ning, J. Agric. Food Chem., 2010, 58, 4356–4360 CrossRef CAS PubMed .
- S. L. Wang, Y. J. Shi, H. B. He, Y. Li, Y. Li and H. Dai, Chin. Chem. Lett., 2015, 26, 672–674 CrossRef CAS .
- H. J. Park, K. Lee, S. J. Park, B. Ahn, J. C. Lee, H. Cho and K. I. Lee, Bioorg. Med. Chem. Lett., 2005, 15, 3307–3312 CrossRef CAS PubMed .
- D. V. Tsyganov, L. D. Konyushkin, I. B. Karmanova, S. I. Firgang, Y. A. Strelenko, M. N. Semenova, A. S. Kiselyov and V. V. Semenov, J. Nat. Prod., 2013, 76, 1485–1491 CrossRef CAS PubMed .
- W. T. McElroy, Z. Tan, G. Ho, S. Paliwal, G. Li, W. M. Seganish, D. Tulshian, J. Tata, T. O. Fischmann, C. Sondey, H. Bian, L. Bober, J. Jackson, C. G. Garlisi, K. Devito, J. Fossetta, D. Lundell and X. Niu, ACS Med. Chem. Lett., 2015, 6, 677–682 CrossRef CAS PubMed .
- G. J. Reddy, D. Latha and K. S. Rao, Org. Prep. Proced. Int., 2004, 36, 494–498 CrossRef CAS .
- N. Inceler, A. Yilmaz and S. N. Baytas, Med. Chem. Res., 2013, 22, 3109–3118 CrossRef CAS .
- N. Chandna, J. K. Kapoor, J. Grover, K. Bairwa, V. Goyal and S. M. Jachak, New J. Chem., 2014, 38, 3662–3672 RSC .
- S. Ambethkar, V. Padmini and N. Bhuvanesh, J. Adv. Res., 2015, 6, 975–985 CrossRef CAS PubMed .
- A. H. Banday, S. A. Shameem and S. Jeelani, Steroids, 2014, 92, 13–19 CrossRef CAS PubMed .
- J. Wölfling, É. A. Oravecz, D. Ondré, E. Mernyák, G. Schneider, I. Tóth, M. Szécsi and J. Julesz, Steroids, 2006, 71, 809–816 CrossRef PubMed .
- R. M. Geha, K. Chen, J. Wouters, F. Ooms and J. Shih, J. Biol. Chem., 2002, 277, 17209–17216 CrossRef CAS PubMed .
- P. Foley, M. Gerlach, M. B. H. Youdim and P. Riederer, Parkinsonism Relat. Disord., 2000, 6, 25–47 CrossRef CAS PubMed .
- E. Palaska, F. Aydin, G. Uçar and D. Erol, Arch. Pharm. Chem. Life Sci., 2008, 341, 209–215 CrossRef CAS PubMed .
- C. Peyssonnaux, A. Eychène, R. Rap and B. Ksr, Biol. Cell., 2001, 93, 53–62 CrossRef CAS PubMed .
- A. Gorden, I. Osman, W. Gai, D. He, W. Huang, A. Davidson, A. N. Houghton, K. Busam and D. Polsky, Cancer Res., 2003, 63, 3955–3957 CAS .
- S.-F. Wang, Y.-L. Zhu, P.-T. Zhu, J. A. Makawana, Y.-L. Zhang, M.-Y. Zhao, P.-C. Lv and H.-L. Zhu, Bioorg. Med. Chem., 2014, 22, 6201–6208 CrossRef CAS PubMed .
- M. Y. Zhao, Y. Yin, X. W. Yu, C. B. Sangani, S. F. Wang, A. M. Lu, L. F. Yang, P. C. Lv, M. G. Jiang and H. L. Zhu, Bioorg. Med. Chem., 2015, 23, 46–54 CrossRef CAS PubMed .
- R. J. Reece and A. Maxwell, Crit. Rev. Biochem. Mol. Biol., 1991, 26, 335–375 CrossRef CAS PubMed .
- T. P. Tran, E. L. Ellsworth, J. P. Sanchez, B. M. Watson, M. A. Stier, H. D. H. Showalter, J. M. Domagala, M. A. Shapiro, E. T. Joannides, S. J. Gracheck, D. Q. Nguyen, P. Bird, J. Yip, A. Sharadendu, C. Ha, S. Ramezani, X. Wu and R. Singh, Bioorg. Med. Chem. Lett., 2007, 17, 1312–1320 CrossRef CAS PubMed .
- A. A. Bekhit and T. Abdel-Aziem, Bioorg. Med. Chem., 2004, 12, 1935–1945 CrossRef CAS PubMed .
-
L. Gomez, M. D. Hack, J. Wu, J. J. M. Wiener, H. Venkatesan, A. Santillán, D. J. Pippel, N. Mani, B. J. Morrow, S. T. Motley, K. J. Shaw, R. Wolin, C. A. Grice and T. K. Jones, Bioorg. Med. Chem. Lett., 2007, 17, 2723–2727 Search PubMed .
- S. A. Khan, B. Ahmad and T. Alam, Pak. J. Pharm. Sci., 2006, 19, 290–294 CAS .
- U. Sharma and S. Singh, J. Vector Borne Dis, 2008, 45, 255–272 Search PubMed .
- P. Desjeux, Comp. Immunol. Microbiol. Infect. Dis., 2004, 27, 305–318 CrossRef CAS PubMed .
- M. S. dos Santos, A. O. Gomes, A. M. R. Bernardino, M. C. de Souza, M. A. Khan, M. A. de Brito, H. C. Castro, P. A. Abreu, C. R. Rodrigues, R. M. M. de Leo, L. L. Leonf and M. M. C. Cavalheiro, J. Braz. Chem. Soc., 2011, 22, 352–358 CrossRef CAS .
- Z. Dardari, M. Lemrani, A. Sebban, A. Bahloul, M. Hassar, S. Kitane, M. Berrada and M. Boudouma, Arch. Pharm. Chem. Life Sci., 2006, 339, 291–298 CrossRef CAS PubMed .
- S. Mert, A. Ş. Yaǧlioǧlu, I. Demirtas and R. Kasimoǧullari, Med. Chem. Res., 2014, 23, 1278–1289 CrossRef CAS .
- L. Dalla Via, A. M. Marini, S. Salerno, C. La Motta, M. Condello, G. Arancia, E. Agostinelli and A. Toninello, Bioorg. Med. Chem., 2009, 17, 326–336 CrossRef CAS PubMed .
- P. Eleftheriou, A. Geronikaki, D. Hadjipavlou-Litina, P. Vicini, O. Filz, D. Filimonov, V. Poroikov, S. S. Chaudhaery, K. K. Roy and A. K. Saxena, Eur. J. Med. Chem., 2012, 47, 111–124 CrossRef CAS PubMed .
- M. Hamberg and B. Samuelsson, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 899–903 CrossRef CAS .
- H. R. Herschman, Biochim. Biophys. Acta, 1996, 1299, 125–140 CrossRef .
- C. Charlier and C. Michaux, Eur. J. Med. Chem., 2003, 38, 645–659 CrossRef CAS PubMed .
- A. K. Tewari, V. P. Singh, P. Yadav, G. Gupta, A. Singh, R. K. Goel, P. Shinde and C. G. Mohan, Bioorg. Chem., 2014, 56, 8–15 CrossRef CAS PubMed .
- S. Malladi, A. M. Isloor, P. Shetty, H. K. Fun, S. Telkar, R. Mahmood and N. Isloor, Med. Chem. Res., 2012, 21, 3272–3280 CrossRef CAS .
- M. A.-A. El-Sayed, N. I. Abdel-Aziz, A. a-M. Abdel-Aziz, A. S. El-Azab, Y. a. Asiri and K. E. H. Eltahir, Bioorg. Med. Chem., 2011, 19, 3416–3424 CrossRef CAS PubMed .
- N. M. Abd-El Gawad, G. S. Hassan and H. H. Georgey, Med. Chem. Res., 2012, 21, 983–994 CrossRef CAS .
- N. D. Jayanna, H. M. Vagdevi, J. C. Dharshan, R. Raghavendra and S. B. Telkar, Med. Chem. Res., 2013, 22, 5814–5822 CrossRef CAS .
-
P. G. Wyatt, A. J. Woodhead, V. Berdini, J. A. Boulstridge, M. G. Carr, D. M. Cross, D. J. Davis, L. A. Devine, T. R. Early, R. E. Feltell, E. J. Lewis, R. L. McMenamin, E. F. Navarro, M. A. O'Brien, M. O'Reilly, M. Reule, G. Saxty, L. C. A. Seavers, D.-M. Smith, M. S. Squires, G. Trewartha, M. T. Walker and A. J.-A. Woolford, J. Med. Chem., 2008, 51, 4986–4999 Search PubMed .
- G. Castanedo, K. Clark, S. Wang, V. Tsui, M. Wong, J. Nicholas, D. Wickramasinghe, J. C. Marsters and D. Sutherlin, Bioorg. Med. Chem. Lett., 2006, 16, 1716–1720 CrossRef CAS PubMed .
- X.-F. Huang, X. Lu, Y. Zhang, G.-Q. Song, Q.-L. He, Q.-S. Li, X.-H. Yang, Y. Wei and H.-L. Zhu, Bioorg. Med. Chem., 2012, 20, 4895–4900 CrossRef CAS PubMed .
- V. Kryštof, P. Cankař, I. Fryšová, J. Slouka, G. Kontopidis, P. Džubák, M. Hajdúch, J. Srovnal, W. F. de Azevedo Jr., M. Orság, M. Paprskářová, J. Rolčík, A. Látr, P. M. Fischer and M. Strnad, J. Med. Chem., 2006, 49, 6500–6509 CrossRef PubMed .
- Á. Sebastián-Serrano, L. De Diego-garcía, C. Martínez-Frailes, J. Ávila, H. Zimmermann, J. Luis, M. T. Miras-portugal and M. Díaz-hernández, Comput. Struct. Biotechnol. J., 2015, 13, 95–100 CrossRef PubMed .
- J. Debray, L. Chang, S. Marques, S. Pellet-Rostaing, D. Le Duy, S. Mebarek, R. Buchet, D. Magne, F. Popowycz and M. Lemaire, Bioorg. Med. Chem., 2013, 21, 7981–7987 CrossRef CAS PubMed .
- W. N. Addison, F. Azari, E. S. Sorensen, M. T. Kaartinen and M. D. McKee, J. Biol. Chem., 2007, 282, 15872–15883 CrossRef CAS PubMed .
- C. B. Vicentini, D. Mares, A. Tartari, M. Manfrini and G. Forlani, J. Agric. Food Chem., 2004, 52, 1898–1906 CrossRef CAS PubMed .
- H. J. Song, Y. X. Liu, L. X. Xiong, Y. Q. Li, N. Yang and Q. M. Wang, J. Agric. Food Chem., 2012, 60, 1470–1479 CrossRef CAS PubMed .
- H. Dai, Y. S. Xiao, Z. Li, X. Y. Xu and X. H. Qian, Chin. Chem. Lett., 2014, 25, 1014–1016 CrossRef CAS .
- S. Fustero, R. Román, J. F. Sanz-Cervera, A. Simón-Fuentes, J. Bueno and S. Villanova, J. Org. Chem., 2008, 73, 8545–8552 CrossRef CAS PubMed .
- G. M. Nitulescu, C. Draghici and A. V. Missir, Eur. J. Med. Chem., 2010, 45, 4914–4919 CrossRef CAS PubMed .
- Y. Rajendra Prasad, G. V. S. Kumar and S. M. Chandrashekar, Med. Chem. Res., 2013, 22, 2061–2078 CrossRef CAS .
- U. Sankappa Rai, A. M. Isloor, P. Shetty, K. S. R. Pai and H. K. Fun, Arabian J. Chem., 2015, 8, 317–321 CrossRef CAS .
- D. Chaudhary, S. Robinson and D. L. Romero, J. Med. Chem., 2015, 58, 96–110 CrossRef CAS PubMed .
- J. Lim, M. D. Altman, J. Baker, J. D. Brubaker, H. Chen, Y. Chen, T. Fischmann, C. Gibeau, M. A. Kleinschek, E. Leccese, C. Lesburg, J. K. F. Maclean, L. Y. Moy, E. F. Mulrooney, J. Presland, L. Rakhilina, G. F. Smith, D. Steinhuebel and R. Yang, ACS Med. Chem. Lett., 2015, 6, 683–688 CrossRef CAS PubMed .
- A. M. Vijesh, A. M. Isloor, S. Telkar, S. K. Peethambar, S. Rai and N. Isloor, Eur. J. Med. Chem., 2011, 46, 3531–3536 CrossRef CAS PubMed .
- N. J. Thumar and M. P. Patel, Med. Chem. Res., 2012, 21, 1751–1761 CrossRef CAS .
- C.-Y. Zhang, X.-H. Liu, B.-L. Wang, S.-H. Wang and Z.-M. Li, Chem. Biol. Drug Des., 2010, 75, 489–493 Search PubMed .
- R. Kenchappa, Y. D. Bodke, A. Chandrashekar, M. Aruna Sindhe and S. K. Peethambar, Arabian J. Chem. DOI:10.1016/j.arabjc.2014.05.029 .
- V. Gressler, S. Moura, A. F. C. Flores, D. C. Flores, P. Colepicolo and E. Pinto, J. Braz. Chem. Soc., 2010, 21, 1477–1483 CrossRef CAS .
- C. Selvam, S. M. Jachak, R. Thilagavathi and A. K. Chakraborti, Bioorg. Med. Chem. Lett., 2005, 15, 1793–1797 CrossRef CAS PubMed .
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.