
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regulatory parameters of self-healing alginate hydrogel networks prepared via mussel-inspired dynamic chemistry†
Juan V.
Alegre-Requena‡
ab,
Marleen
Häring‡
a,
Raquel P.
Herrera
b and
David
Díaz Díaz
*ac
aInstitute of Organic Chemistry, University of Regensburg, Universitätstrasse. 31, D-93040 Regensburg, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de; Fax: +49 941 943-4121; Tel: +49 941 943-4373
bLaboratorio de Organocatálisis Asimétrica, Departamento de Química Orgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain
cIQAC-CSIC, Jordi Girona 18-26, Barcelona 08034, Spain
First published on 3rd August 2016
Abstract
Several key parameters that influence both the self-healing properties and water retention capacity of biohydrogel networks made of alginate–dopamine (Alg–DA) conjugates have been studied. A judicious combination of biopolymer concentration, dopamine content, pH-dependent cross-linking of catechol groups and the Fe3+-coordination of the networks allowed control over the self-healing ability of these bioinspired materials. Moreover, the thixotropic nature of model hydrogels was confirmed using oscillatory rheological loop tests.
Introduction
Among a plethora of adaptive materials, nature has developed fascinating self-healing bioadhesives in the form of 3,4-dihydroxy-L-phenylalanine (DOPA)-containing proteins that are found in marine mussels such as the blue mussel (Mytulis edulis) (e.g., attachment plaque proteins such as mfp-3 and mfp-5 (mfp = mussel food protein) have been found to have 15–30 mol% of DOPA)1–3 (Fig. 1a). The uniqueness of these adhesives lies in their tethering ability to organic and inorganic surfaces even in wet, saline and turbulent environments.4–6 The exposed part of the byssus thread is covered by a self-healing coating, rich in Fe3+, Ca2+, and DOPA residues that form cohesive bonds with amine or thiol groups of neighboring amino acids and multivalent metal ions.7,8 The byssus is formed from liquid protein precursors whose pH during initial secretion is around 5.8.9 After exposure to sea water, the structure is equilibrated to marine pH around 8.5. Reversible metal coordination between non-oxidized catechol groups from DOPA residues and Fe3+ ions from sea water confers toughness to the adhesive structure because of the high stability constant (i.e., log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KS ≈ 37–40)10–12 and self-healing character of such coordination complexes.7,13,14 Depending on the pH, catechol forms mono- (pH < 5.6), bis- (5.6 < pH < 9.1), and tris-complexes (pH > 9.1) with Fe3+ (Fig. 1b).11,15 Besides their role as an anchor via metal coordination and hydrogel bonding,4,16–19 DOPA residues oxidize easily by both chemical and enzymatic ways to the corresponding DOPA-quinone, which shows a significantly reduced affinity for Fe3+ but is able to act as a covalent cross-linking unit via Michael-type additions or free-radical aryl–aryl couplings.4,20 Other physico-chemical processes, such as π–π stacking,21 and conformational rearrangements following catechol oxidation,1,22–26 are also considered contributors to the adhesion properties shown by marine mussels. As a result of such astonishing properties, intensive research has been conducted during the last few years on exploiting this coordination chemistry in order to mimic these natural structures and develop new functional materials, drug carriers,27 self-healing fibers28,29 and actuators,30,31 among others.32–38
KS ≈ 37–40)10–12 and self-healing character of such coordination complexes.7,13,14 Depending on the pH, catechol forms mono- (pH < 5.6), bis- (5.6 < pH < 9.1), and tris-complexes (pH > 9.1) with Fe3+ (Fig. 1b).11,15 Besides their role as an anchor via metal coordination and hydrogel bonding,4,16–19 DOPA residues oxidize easily by both chemical and enzymatic ways to the corresponding DOPA-quinone, which shows a significantly reduced affinity for Fe3+ but is able to act as a covalent cross-linking unit via Michael-type additions or free-radical aryl–aryl couplings.4,20 Other physico-chemical processes, such as π–π stacking,21 and conformational rearrangements following catechol oxidation,1,22–26 are also considered contributors to the adhesion properties shown by marine mussels. As a result of such astonishing properties, intensive research has been conducted during the last few years on exploiting this coordination chemistry in order to mimic these natural structures and develop new functional materials, drug carriers,27 self-healing fibers28,29 and actuators,30,31 among others.32–38
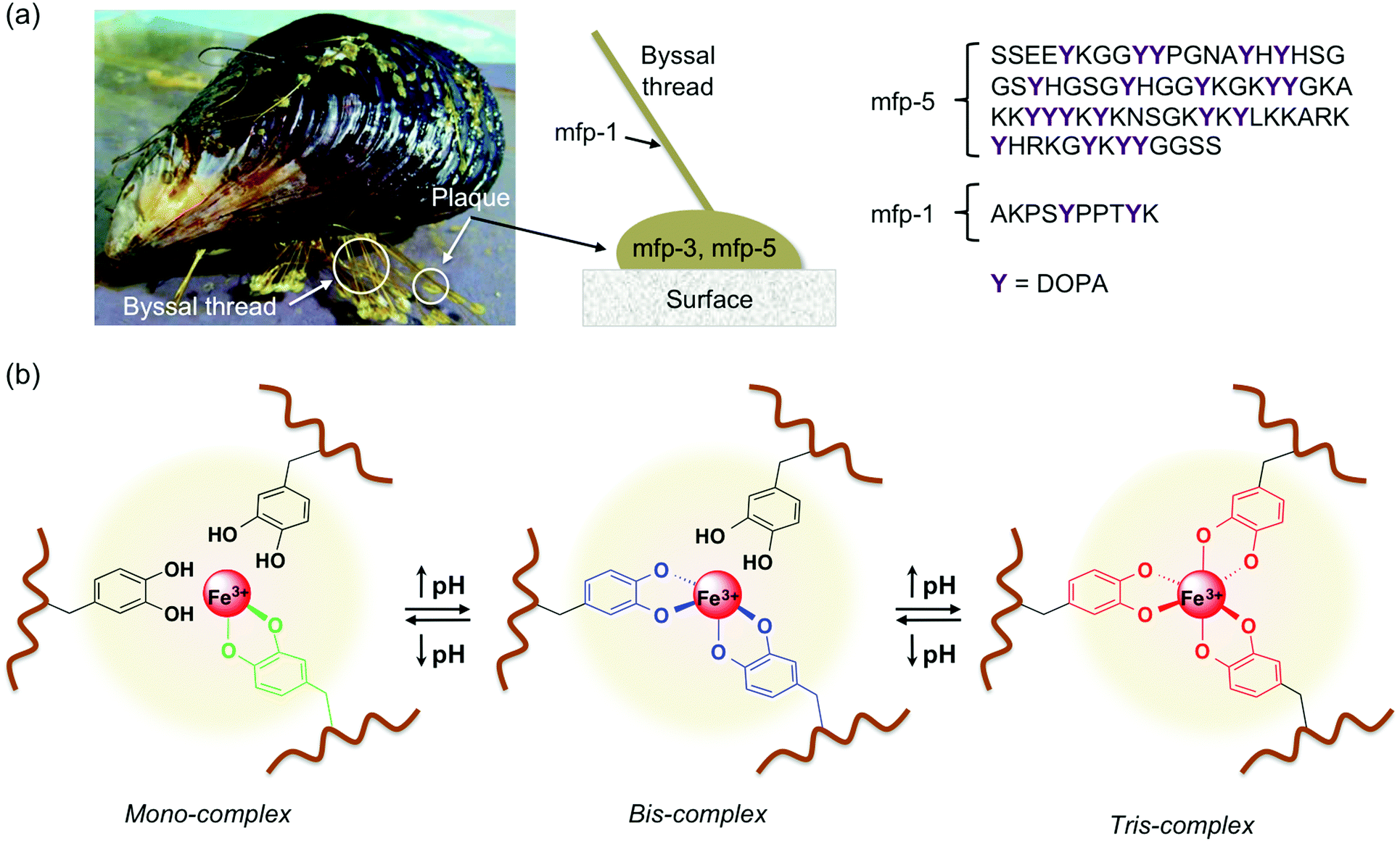 | ||
| Fig. 1 (a) Illustration of a mussel adhered to a surface and representative sequences of mfp-5 and mfp-1 proteins highlighting the content and distribution of DOPA. (b) Scheme of pH-dependent equilibrium between mono-, bis- and tris-complexes between DOPA containing polymers and Fe3+ ions. | ||
In this work we have studied the critical factors that influence the self-healing ability and water-retention capacity of biohydrogel networks made from alginate–dopamine (Alg–DA) conjugates. These systems have been previously reported in some papers for different applications.39–44 Our study focuses on the synergy of different parameters that affect both the self-healing and water retention capacity of these isolated systems, thus aiming to serve as a useful complement to previous investigations in this field, and specifically those dealing with Alg–DA hydrogels. For a better understanding and differentiation between the previous reports in this area, an interconnected overview of the most relevant contributions, embedding our own work, is also provided in the ESI† (Scheme S1).
Experimental
Materials
Alginic acid sodium salt (low viscosity, LOT 051M1864V), dopamine hydrochloride (LOT BCBN7393V), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (97%, LOT BCBP4918V), N-hydroxysuccinimide (NHS) (98%, LOT BCBF6027V), FeCl3 (97%, LOT SZBE0360V), tris(hydroxymethyl)aminomethane hydrochloride (TRIS·HCl) (99%, LOT 111K5405) and HCl 1 M aqueous solution (LOT SZBE0830V) were purchased from Sigma Aldrich. CaCl2·2H2O (99%, LOT 473204168) was purchased from Carl Roth. NaOH pellets (99%, LOT B0881298 309) and phosphate salts were purchased from Merck. Dialysis tubing cellulose membranes (MWCO 14000, LOT 3110) were purchased from Sigma Aldrich. TRIS buffer (50 mM) at pH 8 was prepared by dissolving the appropriate amount of TRIS·HCl in deionized water at room temperature, and adjusting to pH 8 using 1 M NaOH aqueous solution. PBS buffer (100 mM) at pH 5.5 was prepared by dissolving the appropriate amount of dipotassium phosphate and monopotassium phosphate in deionized water at room temperature, and adjusting to pH 5 using 1 M HCl aqueous solution. All chemicals were used as received without further purification.Characterization methods
Fourier transform infrared (FT-IR) spectra were recorded at room temperature using an Excalibur FTS 3000 FT-IR spectrometer (Biorad) equipped with a single reflection ATR (attenuated total reflection) accessory (Golden Gate, Diamond). Xerogel samples for FT-IR measurements were prepared by freeze-drying the corresponding hydrogels. The substitution degrees of different Alg–DA samples were determined by means of spectrophotometry,39 using a Varian Cary 50 UV spectrophotometer and quartz-glass cuvettes of 0.5 cm thickness. UV-vis spectroscopy was also used to confirm the oxidation of catechol moieties under given conditions. Proton nuclear magnetic resonance (1H NMR) spectra were recorded at 25 °C on a Bruker Avance-300 instrument using D2O as the solvent. Elemental analyses were carried out on Mikro-Rapid CHN (Heraeus). The swelling degree (Sw, %) was determined by monitoring the weight change of fresh gel samples immersed into TRIS·HCl (50 mM, pH 7.0) at room temperature. Sw was calculated applying the equation [(ws − wi)/wi] × 100, where wi is the initial weight of the sample and ws the weight at a given time. Gels reached their swelling equilibrium within 10 h. Differential scanning calorimetry (DSC) was performed in a Perkin-Elmer DSC7 under N2 (20 mL min−1) at a heating rate of 5 °C min−1). Oscillatory rheology was performed using an AR 2000 Advanced rheometer (TA Instruments) equipped with a Julabo C cooling system. A 1000 μm gap setting and a torque setting of 40000 dynes per cm2 at 25 °C were used for the measurements in a plain-plate (40 mm, stainless steel). The following experiments were carried out for each sample: (a) dynamic strain sweep (DSS): variation of G′ (storage modulus) and G′′ (loss modulus) with strain (from 0.01 to 100%); (b) dynamic frequency sweep (DFS): variation of G′ and G′′ with frequency (from 0.1 to 10 Hz at 0.1% strain); (c) dynamic time sweep (DTS): variation of G′ and G′′ with time keeping the strain and frequency values constant and within the linear viscoelastic regime as determined by DSS and DFS measurements (strain = 0.1% strain; frequency = 1 Hz). The loss tangent (tanδ, where d is the phase angle between stress and strain) or damping factor (G′′/G′) values were reproducible from batch to batch and the materials were considered gels if G′ > G′′. The self-healing behavior of model gels was investigated using a 3-step loop experiment: (1) application of a low shear strain as established by previous DTS experiments (0.1% strain, 0.1 Hz, 10 min; gel state, G′ > G′′), (2) increase of the shear strain until the gel fractures (1000% strain, 0.1 Hz, 5 min; viscous material, G′ < G′′), and (3) return at the same rate to the initial strain % value (0.1% strain, 0.1 Hz, 30 min; the recovered gel phase, G′ > G′′).
Synthesis of dopamine-modified alginate (Alg–DA)
Alg–DA was synthesized via standard EDC/NHS chemistry according to the procedure described by Wu and co-workers with slight modifications.45 Briefly, EDC (968 mg, 5.05 mmol) and NHS (582 mg, 5.05 mmol) were added to a solution of sodium alginate (1.0 g) in 100 mL PBS buffer solution (100 mM, pH 5.5). After stirring the reaction mixture at room temperature for 45 minutes, an appropriate amount of dopamine hydrochloride (1.92 g, 10.1 mmol, for the 9.78% substituted Alg–DA or 488 mg, 2.5 mmol, for the 5.56% substituted Alg–DA) was added, and the mixture was stirred overnight at room temperature under a N2 atmosphere to suppress uncontrolled oxidation and subsequent self-polymerization. The product was precipitated with ethanol three times, washed with water (6 × 10 mL) and EtOH (6 × 10 mL), filtered and dried under high vacuum, obtaining 1.05 g of 9.78% substituted Alg–DA and 803 mg of 5.56% substituted Alg–DA, respectively (Fig. 2b). The materials were obtained as gray fibrous solids that turned colourless upon dissolution in water within 36 h. The appearance of peaks around 6.4–6.7 ppm in 1H NMR (ESI,† Table S1) and a positive Arnow test47 supported the successful grafting of DOPA molecules.39 Moreover, the characteristic new absorbance at 280 nm was used with the aid of a calibration curve to quantify the conjugation of DOPA onto the biopolymer (the absence of peaks at >400 nm indicates no oxidation products).20 In agreement with previous studies,45 the characteristic IR peaks of the G and M residues of the alginate backbone in the region 810–950 cm−1 as well as those around 1120 cm−1 and 1610 cm−1 (stretching of hydroxyl and carboxylic groups, respectively) were also present in the Alg–DA samples. Moreover, a new peak at around 1080 cm−1 emerged upon functionalization and was attributed to the C–N stretching vibration. | ||
| Fig. 2 (a) Structure of alginate, a water-soluble biopolymer obtained from brown algae and composed of (1 → 4)-β-D-mannuronic acid (M) and (1 → 4)-α-L-guluronic acid (G) units in the form of homopolymeric or heteropolymeric sequences. Formation of hydrogels from alginates occurs upon coordination with Ca2+ ions through a well-established “egg-box” model. (b) General synthesis of Alg–DA conjugates via EDC/NHS coupling. The attachment of DOPA is known to occur preferentially to the M residues.35 | ||
Preparation of hydrogels
Alginate-based hydrogels can be formed by ionic cross-linking of the G residues with many divalent or multivalent cations, especially Ca2+, via an “egg-box” model (Fig. 2a).46 Thus, Alg–DA hydrogels were prepared by the dialysis of Alg–DA solutions against CaCl2 solutions. Typical procedures are described next: (A) conventional Alg and Alg–DA hydrogels were prepared by dissolving Alg or Alg–DA (1.5% w/w = 150 mg of Alg–DA in 10 mL of H2O) in PBS buffer (10 mL, 100 mM) with pH adjusted to 5.5, and dialyzed against 500 mL of aqueous 1 M CaCl2 solution for 48 h at room temperature. (B) Self-polymerized Alg–DA hydrogels were obtained following the above described dialysis protocol but adding a catechol alkaline pH-induced self-polymerization step before the dialysis. In this step, Alg–DA (1.5% w/w) was dissolved in TRIS·HCl buffer (10 mL, 50 mM) with pH adjusted to 8, and was allowed to react under stirring for 24 h at room temperature. In good agreement with previous studies,45 completed polymerization was observed after this time as judged by no further changes in the UV-spectra. Moreover, the solutions gradually darkened in color over the polymerization process and ultimately become black. (C) Fe3+-containing hydrogels were prepared following the above described protocol but adding appropriate concentrations ranging from 0.1–100 mM of FeCl3 to the corresponding CaCl2 dialysis solutions (Fig. 3). | ||
| Fig. 3 (a) Digital photographs of selected alginate-based gel monoliths prepared as described above. Herein, the term “polymerized” refers to the polymerization of the catechol units. See the ESI† for additional pictures, Fig. S5. (b) Plausible general structures of non-polymerized Alg–DA and polymerized Alg–DA with green spheres symbolizing Ca2+ ions and black lines representing the alginate backbone as in Fig. 2. Polymerization of catechol units is indicated by the formation of the new bond marked in red. | ||
The successful formation of a more compact network due to the polymerization of Alg–DA was also supported by (1) a glass transition (Tg) of 102 ± 2 °C, which is ca. 20 °C higher than the non-polymerized material, (2) a higher mechanical strength (vide infra) and (3) a lower aqueous solubility and swelling degree (ca. 90%) compared to the non-polymerized material (ca. 130%) (ESI,† Fig. S1). From a mechanistic point of view, the most widely accepted polymerization mechanism is the oxidative polymerization of the catechol units via quick formation of short-lived o-quinone-type intermediates,48 which was also evidenced by UV-vis spectroscopy (ESI,† Fig. S2).
Results and discussion
Preparation of materials and targeted parameters
Among different variables that can influence to a great extent the properties of DA-containing biohydrogels, the most relevant are concentration, DA content, polymerization of catechol groups and metal coordination of the networks.43,45 In order to study how all these factors affect the potential self-healing properties of non-cytotoxic39 Alg–DA hydrogels,44,45,49 monolith samples (ca. 4–5 cm length × 1.5 cm diameter) with different compositions were prepared. For convenience, Alg–DAs are described as Alg–DA (x/y), where x represents the concentration of Alg–DA in water (% w/w) = 0.75, 1.5 or 3.5, and y is the degree of substitution (DS, % of accessible functional groups = 9.78 or 5.56). The library of prepared hydrogels included (a) a conventional alginate hydrogel as a control material (AlgCONT); (b) a hydrogel based on Alg–DA (1.5/9.78); (c) a hydrogel based on Alg–DA (1.5/5.56); (d) a hydrogel based on catechol-polymerized Alg–DA (x/9.78); (e) a hydrogel based on catechol-polymerized Alg–DA (x/5.56); and (f) samples (a), (d) and (e) containing Fe3+. The synthesis of different hydrogels and their spectroscopic characterization35 were carried out following a dialysis procedure against 1 M CaCl2 solution. Chemical analyses, spectroscopic data and the obtained DS values were also consistent with those previously reported39,42,45 (see Experimental section and ESI† for more details, Fig. S2–S5 and Table S1). It is noteworthy to mention that the DS values calculated using elemental analysis45 were observed to be higher than those obtained using UV-vis spectroscopy, presumably due to the hygroscopic nature of the biopolymers and/or the suppression of solubility problems in the former method. However, this discrepancy does not affect relative comparisons between the synthesized materials. Therefore, in order to facilitate the characterization we used the data calculated by UV-vis as previously reported by other groups.39,42,45As far as we are aware, this is the first report that focuses on the self-healing properties of low-functionalized Alg–Da bulk gels and their regulation. Previous reports have dealt with bioapplications (usually involving in situ gelation),39,45,46,49 different Alg–phenolic conjugates,50,51 and non-covalent conjugates,52 in which either Fe3+-coordination studies and/or gel formation have not been included (ESI,† Scheme S1).
Effects of catechol cross-linking on the self-healing properties of Alg–DA gels
In order to assess the influence of catechol cross-linking on the self-healing behavior of the gels, polymerized Alg–DA hydrogels with different DS (i.e., catechol-polymerized Alg–DA (1.5/9.78), catechol-polymerized Alg–DA (1.5/5.56)) were compared with non-polymerized Alg–DA analogues ((i.e., Alg–DA (1.5/9.78), Alg–DA (1.5/5.56)) and unmodified alginate gels (AlgCONT). Monolithic hydrogels were cut into several pieces with a razor blade (ca. 1 cm length × 1.5 cm diameter) and then gently pressed them back together and were fused within 1 min. In principle, all gels were able to form a bridge over two vials after cutting and rejoining the pieces (Fig. 4). However, only the catechol-polymerized Alg–DA gel bridges resisted vigorous manual shaking when stored inside glass vials, whereas the rest of the samples broke easily into their original pieces. These results are consistent with the higher cross-linking density of the polymerized samples, which may enhance the local hydrogen-bonding capacity of the network. | ||
| Fig. 4 Representative pictures showing the self-healing ability of catechol-polymerized Alg–DA gel (DS = 9.78%; 1.5% w/w). The monolith was cut into pieces that were subsequently rejoined. The pieces fused within 1 min and the bulk material could be suspended between two vials and lifted with tweezers. The self-healing behaviour was also observed after several cycles of disconnection and reconnection of the pieces. See the ESI† for additional pictures, Fig. S5. | ||
Effects of concentration on the self-healing properties of catechol-polymerized Alg–DA gels
Subsequently, we studied the influence of concentration on the self-healing of polymerized Alg–DA gels. Catechol-polymerized Alg–DA with 2 different DS (i.e., 9.78% and 5.56%) and prepared at 3 different concentrations (i.e., 0.75%, 1.5% and 3.5% w/w) were first compared as described in the previous section. The results indicated that the gels prepared at 0.75–1.5% w/w concentration showed better self-healing properties in terms of resistance to manual shaking after being cut into pieces and rejoined. On the other hand, the gels prepared at 3.5% w/w concentration were too brittle and the corresponding bridges were easily destroyed upon hand shaking. This could be explained by the formation defects at the interface above 1.5% w/w concentration. Nevertheless, it is noteworthy that all these catechol-polymerized Alg–DA gels were more brittle than the control gels prepared with unmodified alginate, even when the controls were prepared at higher concentration (i.e., 4% w/w). Therefore, we proceed further with our studies using catechol-polymerized Alg–DA hydrogels prepared at 0.75% w/w concentration. It is worth mentioning that despite the low DS of these gels (i.e., AlgCONT, Alg–DA (0.75/9.78)), their bridges formed after cutting and rejoining were able to support ca. 1.2 times their own weight (Fig. 5). | ||
| Fig. 5 (a) Easy disruption of a bridged gel made from non-polymerized Alg–DA (DS = 9.78%; 1.5% w/w) upon shaking. (b) Stable bridge made from catechol-polymerized Alg–DA gel (DS = 9.78%; 1.5% w/w) after cutting the monolith into pieces and rejoining back together. (c) Load-bearing ability of the previous bridge (1.13 times its own weight). See ESI† for additional pictures, Fig. S5. | ||
Influence of Fe3+ coordination on the self-healing and water retaining properties of the gels
One of the major drawbacks observed for the gels (including the control gel made with unmodified alginate) prepared at 0.75% and 1.5% w/w concentration was a gradual loss of water over time. Since an increase in the rigidity achieved by increasing 2-fold the concentration did not solve this problem, we hypothesized that increasing the cross-linking degree of the gel network by catechol–metal coordination could help to retain the water. As described in the introduction, Fe3+ has been shown to form strong coordination bonds with catechol groups, where the number of ligands bound to the ferric ion depends on the pH.14,53 Hence, different amounts of FeCl3 were added to the CaCl2 dialysis solutions during the synthesis of the corresponding hydrogels as described in the Experimental section. Preliminarily, we used Alg–DA with a substitution degree of 9.78% and we employed FeCl3 concentrations ranging from 0.1 to 100 mM. Furthermore, a control gel using the unmodified alginate and 20 mM FeCl3 was also prepared for comparison. The incorporation of Fe3+ ions into the gel networks was confirmed using FT-IR spectroscopy of the corresponding xerogels. Absorption bands centered at 3488, 3444 and 3220 cm−1 shifted to 3481, 3477 and 4216 cm−1, respectively (it should be noted that in this region –OH and –NH stretching bands are also overlapped). These shifts are usually attributed to the formation of catechol–Fe3+ coordination bonds and alginate–Fe3+ interactions. Specifically, the former is responsible for the unique blue shift of the band centered at 3488 cm−1, which is not observed in the spectra of unmodified alginates (ESI,† Fig. S3 and S4).Table 1 shows the relative water loss of different gels over 1 week. The addition of 10 mM FeCl3 practically suppressed the water loss of Alg–DA hydrogels (entry 6), and also the addition of 20 mM FeCl3 showed the same effect in the hydrogels made of unmodified alginate (entry 5). The use of ≤1 mM FeCl3 did not provide any benefit in terms of water retention compared to the control samples (entries 1–4). It is noteworthy that the gel formed with 1 mM FeCl3 solution slowed down significantly the initial loss of water (during the first 30 min) in comparison to the equivalent gel generated in the absence of Fe3+. However, the total amount of water lost after 1 week in both cases was approximately the same.
| Entry | Gel (concentration (% w/w)/DS (%)) | [FeCl3] (mM) | Relative water loss over 1 week (%) |
|---|---|---|---|
| 1 | AlgCONT (0.75/—) | — | 35 ± 2 |
| 2 | Alg–DA (0.75/9.78) | — | 30 ± 2 |
| 3 | Alg–DA (0.75/9.78) | 0.1 | 30 ± 2 |
| 4 | Alg–DA (0.75/9.78) | 1.0 | 32 ± 2 |
| 5 | AlgCONT (0.75/—) | 20.0 | <1 |
| 6 | Alg–DA (0.75/9.78) | ≥10 | <1 |
To our delight, the self-healing properties were preserved in the gels formed using between 10 and 20 mM FeCl3 in the dialysis solution. Thus, these gels did not show water loss and could form stable bridges over two vials after being cut and rejoined (Fig. 6), without falling apart after manual shaking. These gels showed load-bearing capacity of ca. 3 times their own weight after the self-healing process (ESI,† Fig. S5). The reversibility of Fe3+ cross-linking was confirmed by the treatment of the gel made in the presence of FeCl3 (Table 1, entry 6) with 100 mM EDTA solution (pH 5) for 10 min. After liquid decantation, the monolithic sample showed again ca. 30% water loss over 1 week due to competitive complexation of Fe3+ with EDTA. It is noteworthy to mention that Fe3+ does not induce the oxidation of DOPA.54
 | ||
| Fig. 6 Hydrogel prepared from catechol-polymerized Alg–DA gel (DS = 9.78%; 0.75% w/w) and 20 mM FeCl3. No appreciable water loss was observed over a week for this material. A stable bridge was formed after cutting the monolith into pieces and subsequent reconnection by simple physical contact. The self-healing behaviour was also observed after several cycles of disconnection and reconnection of the pieces. | ||
FeCl3 concentrations higher than 20 mM lead to very brittle gels and broke apart very easily (Fig. 7a). These observations are consistent with previous studies on catechol–Fe3+ systems, in which the gels achieved greatest consistency at the maximum of a Gaussian bell curve, and became weaker as the Fe3+:catechol ratio moved away from this point.55 To prove this theory, we prepared hydrogels from Alg–DA with 5.56% DS and using 10 mM and 20 mM FeCl3 in the dialysis solutions. Under these conditions, the gels have higher Fe3+:catechol ratios compared to the gels formed using Alg–DA with 9.78% DS. As expected, the former gels (5.56% DS) showed poor consistency, and they broke apart very easily as a result of the high Fe3+:catechol ratio. Even though these gels presented self-healing character and water retention, their mechanical weakness made them less useful than their analogues prepared at lower Fe3+:catechol ratios. The effect of pH on the mechanical strength of the materials was evidenced by soaking the gel (i.e., DS = 9.78%, 20 mM FeCl3 – used as a model system for this experiment) for 36 h in aqueous solutions at different pH values. Maximum strength was obtained at pH 9–10 where metal-coordination and oxidation are favoured and dominated by bis- and tris-catecholate moieties. In contrast, lower pH made the gel softer by reversing the metal-coordination cross-linking and forming monomeric species (Fig. 7b). At the same time, the polymer charge is increased as the pH decreases leading to higher polymer hydrophilicity and interpolymer chain repulsion.
 | ||
| Fig. 7 (a) Gaussian bell dependence of mechanical consistency of Alg–DA hydrogels with a Fe3+:catechol ratio. The gels were prepared at 0.75% w/w concentration using catechol-polymerized Alg–DA. (b) Effects of pH on the storage modulus (G′) and swelling degree (Sw) of the hydrogel prepared in the presence of 20 mM FeCl3 (DS = 9.78%). Inset pictures show a colour change from dark yellow to reddish with increasing pH due to higher content of Fe3+ coordination bonds. Dried materials (xerogels) were prepared and fragmented into small pieces in order to visualize better the difference in colour. | ||
Finally, the apparent good correlation between the observed mechanical strength, water retention and mechanical properties of the bulk gels was also confirmed using oscillatory rheological experiments (i.e., DSS, DFS and DTS) (ESI,† Fig. S6). In each case, the storage modulus was found to be approximately one order of magnitude higher than for the loss modulus and presented a low dependence on the frequency, which is typical of viscoelastic networks. Overall, both the storage modulus and the tan δ values showed that the modified alginate gel formed using 20 mM FeCl3 (1) displayed superior mechanical properties (stiffer network) than its equivalent gel prepared in the absence of Fe3+ (2) and two control gels generated with unmodified alginate (ESI,† Fig. S5). One control gel (3) was prepared at the same concentration and conditions (i.e., stirring in TRIS·HCl buffer at pH 8 for 24 h, dialysis using a 1 M CaCl2/20 mM FeCl3 solution) and the other one (4) without FeCl3 (Fig. 8a). The improvement of the mechanical properties is associated with the increase in the cross-linking density of the gel through the formation of sacrificial coordination bonds between the catechol groups and Fe3+ ions to dissipate energy during mechanical deformation. Although it was out of the scope of this study, previous reports have demonstrated that the ratio between covalent bonds (i.e., polymerization of dopamine moiety) vs. physical bonds (i.e., coordination bonds) in similar gel systems41 can be determined by evaluating the dependence of tanδ or moduli values on the frequency as well as the relaxation times of the networks during step-strain measurements.56,57
 | ||
| Fig. 8 (a) Representative frequency sweep experiments of model alginate gel samples (0.75% w/w) prepared either in the presence of 20 mM FeCl3 (1) or in the absence of Fe3+ (2). The equivalent control gels prepared using unmodified alginate are also included for comparison (i.e., (3) containing Fe3+ and (4) without Fe3+). (b) Thixotropic loop test of the gel prepared from catechol-polymerized Alg–DA (0.75/9.78) and 20 mM FeCl3. See ESI† for additional rheological plots, Fig. S5 and S6 (ESI†). tanδ values for each sample (estimated error ± 0.005): 0.103 (1); 0.199 (2); 0.106 (3); 1.198 (4). | ||
In general, the bulk gels also exhibited thixotropic (i.e., self-healing) properties as confirmed by a three-step loop tests (see Experimental section). The application of a large strain (i.e., 1000%) to the sample caused the collapse of the gel phase (G′ < G′′), which could be reverted to the original gel state (G′ > G′′) upon decrease of the strain to the initial value (i.e., 0.1%) (Fig. 8b). The presence of Fe3+ in catechol-polymerized Alg–DA hydrogels was not indispensable for this behavior, although it allowed for a faster recovery of the storage modulus. For instance, ca. 95% of the initial G′ was recovered within 1 min in the case of the Fe3+-containing gel, whereas the equivalent gel without Fe3+ exhibited ca. 74% recovery in the same period (ESI,† Fig. S7).
Conclusions
A great deal of biomimetic research has revealed a captivating adhesion mechanism based on 3,4-dihydroxy-L-phenylalanine (DOPA)-containing proteins existing in marine mussels. Over the years, this has inspired scientists in the development of new functional materials for different applications. In this contribution, we have synthesized DOPA–alginate conjugates and prepared their corresponding hydrogels using a dialysis protocol in the presence of Ca2+ ions. Hydrogel monoliths showed self-healing properties after cutting them into several pieces and rejoining back together. This self-healing ability was tuned by means of biopolymer concentration, dopamine content, cross-linking of catechol groups (by alkaline treatment) and Fe3+-coordination of the networks. The results showed that catechol cross-linked hydrogels prepared at 0.75% w/w concentration and in the presence of at least 10 mM FeCl3 were the best systems in terms of mechanical strength and water retention capacity. Maximum strength was obtained at pH 9–10 where metal-coordination and oxidation are favoured and dominated by bis- and tris-catecholate moieties. The thixotropic nature of the gels was also confirmed by rheological loop tests. Further studies on the influence of other metal salts as well as other cross-linking methods on the self-healing behaviour of these and other biohydrogels based on dynamic chemistry are currently underway in our laboratories.Acknowledgements
Financial support from Universität Regensburg and Deutsche Forschungsgemeinschaft (DFG 9209720) is gratefully acknowledged. J. V. A.-R. thanks the Diputación General de Aragón (DGA) for a predoctoral fellowship. D. D. D. thanks DFG for the Heisenberg Professorship Award.References
- J. H. Waite and X. Qin, Biochemistry, 2001, 40, 2887–2893 CrossRef CAS PubMed.
- D. J. Rubin, A. Miserez and J. H. Waite, Adv. Insect Physiol., 2010, 38, 75–133 Search PubMed.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed.
- J. H. Waite, Int. J. Adhes. Adhes., 1987, 7, 9–14 CrossRef CAS.
- S. W. Taylor, D. B. Chase, M. H. Emptage, M. J. Nelson and J. H. Waite, Inorg. Chem., 1996, 35, 7572–7577 CrossRef CAS.
- H. Zenga, D. S. Hwang, J. N. Israelachvili and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12850–12853 CrossRef PubMed.
- N. Holten-Andersen, G. E. Fantner, S. Hohlbauch, J. H. Waite and F. W. Zok, Nat. Mater., 2007, 6, 669–672 CrossRef CAS PubMed.
- M. J. Harrington, A. Masic, N. Holten-Andersen, J. H. Waite and P. Fratzl, Science, 2010, 328, 216–220 CrossRef CAS PubMed.
- J. Yu, W. Wei, E. Danner, R. K. Ashley, J. N. Israelachvili and J. H. Waite, Nat. Chem. Biol., 2011, 7, 588–590 CrossRef CAS PubMed.
- A. Avdeef, S. R. Sofen, T. L. Bregante and K. N. Raymond, J. Am. Chem. Soc., 1978, 100, 5362–5370 CrossRef CAS.
- S. W. Taylor, G. W. Luther III and J. H. Waite, Inorg. Chem., 1994, 33, 5819–5824 CrossRef CAS.
- M. J. Sever, J. T. Weisser, J. Monahan, S. Srinivasan and J. J. Wilker, Angew. Chem., Int. Ed., 2004, 43, 448–450 CrossRef CAS PubMed.
- A. Miserez, T. Schneberk, C. Sun, F. W. Zok and J. H. Waite, Science, 2008, 319, 1816–1819 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- M. J. Sever and J. J. Wilker, Dalton Trans., 2006, 813–822 RSC.
- J. H. Waite, R. A. Jensen and D. E. Morse, Biochemistry, 1992, 31, 5733–5738 CrossRef CAS PubMed.
- V. V. Papov, T. V. Diamond, K. Biemann and J. H. Waite, J. Biol. Chem., 1995, 270, 20183–20192 CrossRef CAS PubMed.
- J. Wang, M. N. Tahir, M. Kappl, W. Tremel, N. Metz, M. Barz, P. Theato and H.-J. Butt, Adv. Mater., 2008, 20, 3872–3876 CrossRef CAS.
- S.-C. Li, L.-N. Chu, X.-Q. Gong and U. Diebold, Science, 2010, 328, 882–884 CrossRef CAS PubMed.
- L. A. Burzio and J. H. Waite, Biochemistry, 2000, 39, 11147–11153 CrossRef CAS PubMed.
- T. H. Anderson, J. Yu, A. Estrada, M. U. Hammer, J. H. Waite and J. N. Israelachvili, Adv. Funct. Mater., 2010, 20, 4196–4205 CrossRef CAS PubMed.
- M. J. McWhirter, P. J. Bremer, I. L. Lamont and A. J. McQuillan, Langmuir, 2003, 19, 3575–3577 CrossRef CAS.
- R. J. Stewart, T. C. Ransom and V. Hlady, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 757–771 CrossRef CAS PubMed.
- J. Yu, W. Wei, E. Danner, J. N. Israelachvili and J. H. Waite, Adv. Mater., 2011, 23, 2362–2366 CrossRef CAS PubMed.
- J. D. Whiteand and J. J. Wilker, Macromolecules, 2011, 44, 5085–5088 CrossRef.
- H. G. Upritchard, J. Yang, P. J. Bremer, I. L. Lamont and A. J. McQuillan, Langmuir, 2011, 27, 10587–10596 CrossRef CAS PubMed.
- B. J. Kim, H. Cheong, B. H. Hwang and H. J. Cha, Angew. Chem., Int. Ed., 2015, 54, 7318–7322 CrossRef CAS PubMed.
- B. J. Kim, S. Kim, D. X. Oh, A. Masic, H. J. Cha and D. S. Hwang, J. Mater. Chem. B, 2015, 3, 112–118 RSC.
- M. Krogsgaard, A. Andersen and H. Birkedal, Chem. Commun., 2014, 50, 13278–13281 RSC.
- B. P. Lee and S. Konst, Adv. Mater., 2014, 26, 3415–3419 CrossRef CAS PubMed.
- B. P. Lee, M.-H. Lin, A. Narkar, S. Konst and R. Wilharm, Sens. Actuators, B, 2015, 206, 456–462 CrossRef CAS.
- Z. Shafiq, J. Cui, L. Pastor-Pérez, V. San Miguel, R. A. Gropeanu, C. Serrano and A. del Campo, Angew. Chem., Int. Ed., 2012, 51, 4332–4335 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- H. Lee, J. Rho and P. B. Messersmith, Adv. Mater., 2009, 21, 431–434 CrossRef CAS PubMed.
- H. Ceylan, S. Kocabey, A. B. Tekinay and M. O. Guler, Soft Matter, 2012, 8, 3929–3937 RSC.
- S. M. Kang, N. S. Hwang, J. Yeom, S. Y. Park, P. B. Messersmith, I. S. Choi, R. Langer, D. G. Anderson and H. Lee, Adv. Funct. Mater., 2012, 22, 2949–2955 CrossRef CAS PubMed.
- J. Sedó, J. Saiz-Poseu, F. Busqué and D. Ruiz-Molina, Adv. Mater., 2013, 25, 653–701 CrossRef PubMed.
- E. Faure, C. Falentin-Daudré, C. Jérôme, J. Lyskawa, D. Fournier, P. Woisel and C. Detrembleur, Prog. Polym. Sci., 2013, 38, 236–270 CrossRef CAS , and references therein.
- C. Lee, J. Shin, J. S. Lee, E. Byun, J. H. Ryu, S. H. Um, D.-I. Kim, H. Lee and S.-W. Cho, Biomacromolecules, 2013, 14, 2004–2013 CrossRef CAS PubMed.
- J. Longo, T. Garnier, M. Mateescu, F. Ponzio, P. Schaaf, L. Jierry and V. Ball, Langmuir, 2015, 31, 12447–12454 CrossRef CAS PubMed.
- S. H. Hong, M. Shin, J. Lee, J. H. Ryu, S. Lee, J. W. Yang, W. D. Kim and H. Lee, Adv. Healthcare Mater., 2016, 5, 75–79 CrossRef CAS PubMed.
- M.-H. Ryou, J. Kim, I. Lee, S. Kim, Y. K. Jeong, S. Hong, J. H. Ryu, T.-S. Kim, J.-K. Park, H. Lee and J. W. Choi, Adv. Mater., 2013, 25, 1571–1576 CrossRef CAS PubMed.
- J. Hou, C. Li, Y. Guan, Y. Zhang and X. X. Zhu, Polym. Chem., 2015, 6, 2204–2213 RSC.
- C. J. Kastrup, M. Nahrendorf, J. L. Figueiredo, H. Lee, S. Kambhampati, T. Lee, S.-W. Cho, R. Gorbatov, Y. Iwamoto, T. T. Dang, P. Dutta, J. H. Yeon, H. Cheng, C. D. Pritchard, A. J. Vegas, C. D. Siegel, S. MacDougall, M. Okonkwo, A. Thai, J. R. Stone, A. J. Coury, R. Weissleder, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21444–21449 CrossRef CAS PubMed.
- X. Wang, Z. Jiang, J. Shi, C. Zhang, W. Zhang and H. Wu, Ind. Eng. Chem. Res., 2013, 52, 14828–14836 CrossRef CAS.
- P. Sikorski, F. Mo, G. Skjåk-Bræk and B. T. Stokke, Biomacromolecules, 2007, 8, 2098–2103 CrossRef CAS PubMed.
- L. E. Arnow, J. Biochem., 1937, 118, 531–534 CAS.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS PubMed.
- L. García-Fernández, J. Cui, C. Serrano, Z. Shafiq, R. A. Gropeanu, V. San Miguel, J. I. Ramos, M. Wang, G. K. Auernhammer, S. Ritz, A. A. Golriz, R. Berger, M. Wagner and A. del Campo, Adv. Mater., 2013, 25, 529–533 CrossRef PubMed.
- R. Bitton and H. Bianco-Peled, Macromol. Biosci., 2008, 8, 393–400 CrossRef CAS PubMed.
- R. Bitton, E. Josef, I. Shimshelashvili, K. Shapira, D. Seliktar and H. Bianco-Peled, Acta Biomater., 2009, 5, 1582–1587 CrossRef CAS PubMed.
- J. Zhao, C. Fang, Y. Zhu, G. He, F. Pan, Z. Jiang, P. Zhang, X. Cao and B. Wang, J. Mater. Chem. A, 2015, 3, 19980–19988 CAS.
- D. G. Barrett, D. E. Fullenkamp, L. He, N. Holten-Andersen, K. Y. C. Lee and P. B. Messersmith, Adv. Funct. Mater., 2013, 23, 1111–1119 CrossRef CAS PubMed.
- H. Ceylan, M. Urel, T. S. Erkal, A. B. Tekinay, A. Dana and M. O. Guler, Adv. Funct. Mater., 2013, 23, 2081–2090 CrossRef CAS.
- Z. Guo, K. Ni, D. Wie and Y. Ren, RSC Adv., 2015, 5, 37377–37384 RSC.
- N. Holten-Andersen, A. Jaishankar, M. J. Harrington, D. E. Fullenkamp, G. DiMarco, L. He, G. H. McKinley, P. B. Messersmith and K. Y. C. Lee, J. Mater. Chem. B, 2014, 2, 2467–2472 RSC.
- X. Ding, G. K. Vegesna, H. Meng, A. Winter and B. P. Lee, Macromol. Chem. Phys., 2015, 216, 1109–1119 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic data, expanded literature flow diagrams, additional rheological plots and pictures. See DOI: 10.1039/c6nj02367c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |