
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning and enhancement of the Mizoroki–Heck reaction using polarized Pd nanocomposite carbon aerogels†
Laura
Martín
ab,
Elies
Molins
*b and
Adelina
Vallribera
*a
aDepartment of Chemistry and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Autònoma de Barcelona, Campus UAB, 08193 Bellaterra, Spain. E-mail: adelina.vallribera@uab.cat
bDepartment of Crystallography, Institut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Bellaterra, Spain
First published on 17th October 2016
Abstract
The application of a positive electric field on a Pd(0)–carbon aerogel nanocomposite enhances the reaction rate of the Mizoroki–Heck reaction. By contrast, when a negative potential is employed the reaction is inhibited. On the other hand, chronoamperometry measurements have revealed the polarizing current intensity as an indirect measure of the reaction evolution. The cationic nature of the reaction intermediates was unambiguously confirmed and a polar mechanism has been finally proposed.
In 2002, Vayenas et al.1 described that the rate of many heterogeneous chemical reactions can be modified by altering the potential between the catalyst and a reference electrode. Related to this concept, recently theoretical calculations suggest that external electric fields (EEFs) can affect the results of several chemical reactions even if no metals are involved. It has been theoretically demonstrated that EEFs can control the selectivity of enzymatic-like bond activations,2 as well as chemical reactions that involve bond forming and bond-breaking steps.3 Moreover, the ability to control reaction rates with an applied electric potential gradient has been recently demonstrated for a Diels–Alder reaction using the scanning tunneling microscopy break-junction approach to deliver an electric-field stimulus.4
In our own research, we have extensively used Pd(0)Nps embedded in silica and carbon aerogels5 as recoverable catalysts for cross-coupling reactions, including the Mizoroki–Heck (M–H).6 In continuation of this research, the aim of this work is to answer this question: can an external electric field manipulate an M–H reaction? Taking advantage of the fact that Pd(0)Nps can be fixed on a high surface area carbon aerogel, which is highly porous and electrically conductive (25–100 S cm−1),7 we envisaged applying the original idea of Vayenas to the use of Pd(0)–carbon aerogel nanocomposites (Pd(0)–CAs) as an electrode and a catalyst at the same time. As the M–H reaction is mediated on transition metal atoms in which oxidative addition, alkene insertion and reductive elimination occur,8 we do not contemplate a redox electrocatalytic process between Pd to Pd2+ at all. In fact, the electrochemical oxidation of palladium is not possible at the electric potentials applied along in this work.9
Metal-doped CAs are well-known5c,6,10,11 since the pioneering work of Baumann et al.11b who used sol–gel polymerization of resorcinol derivatives containing ion-exchange moieties, allowing the incorporation of the metal ions uniformly into the aerogel matrix. In this work Pd(0) nanocomposited CAs were synthesized following this methodology.11b Once the organic gel was obtained, a supercritical drying process was performed and a subsequent pyrolysis step gave rise to Pd(0)–CAs. The Pd(0)–CAs presented a high surface area (401 m2 g−1), low bulk density (0.65 g cm−3) and high metal content (39 wt% of Pd). The corresponding powder X-ray diffraction (XRD) patterns were characteristic of a metallic fcc-Pd phase (Fig. 1B) and no presence of PdO was detected. Transmission electron microscopy (TEM, Fig. 1C) images showed spherical nanoparticles with a mean particle size of 17 ± 7 nm (Fig. 1C2).
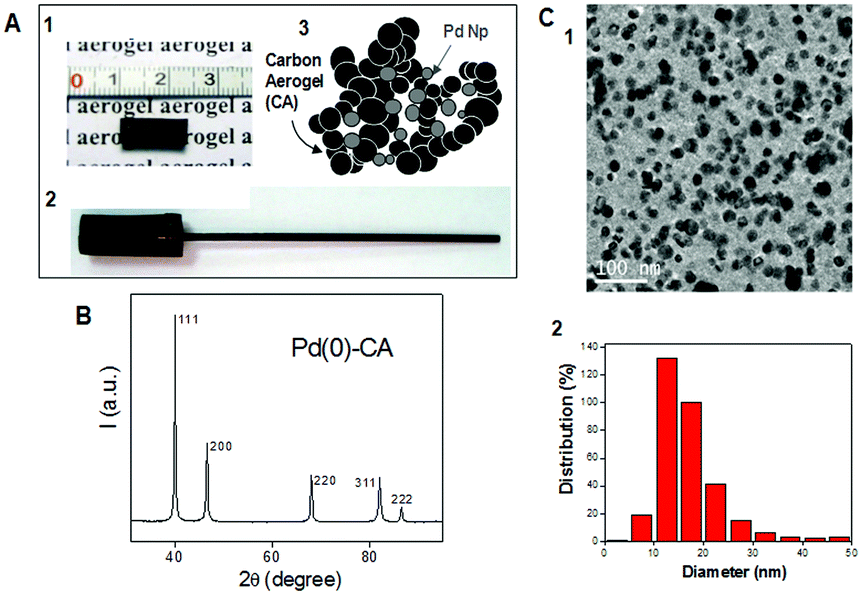 | ||
| Fig. 1 (A1) Photograph image of a piece of Pd(0)–CA; (A2) photograph image of Pd(0)–CAs incorporated into a graphite rod; (A3) schematic representation of the Pd(0)–CA structure with Pd(0)Nps incorporated into the CA matrix; (B) powder XRD pattern; (C1) TEM image; and (C2) particle size distribution. | ||
The electrode-catalyst was obtained through the incorporation of a graphite rod (PGR) into a precursory gel as a current collector (Fig. 1A2). Prior to the drying process and further pyrolysis, the graphite rod was filed off and treated with KOH, thus improving the interaction between its surface and the organic matrix of the gel.12 Commercial pencil leads (CPLs) were also used as current collectors. Cyclic voltammetry measurements comparing different working electrodes were carried out. The results showed that Pd(0)–CA with a PGR presented higher current density values.
The M–H reaction selected was the reaction of iodobenzene, 1, with ethyl acrylate, 4 (Scheme 1). The potentials tested were +1, +0.5, −0.5 and −1 V (vs. Pt), as shown in Table 1 (entries 1, 2, 5 and 6). Unconnected electrodes and 0.0 V reactions were used as comparison references, using the same molar percentage of Pd as the corresponding reaction (entries 3 and 4). According to the results (entries 1–6), positive potentials gave higher conversions (89–94%), whereas negative potentials gave lower ones (45–46%), relative to systems with unconnected electrodes and 0.0 V reactions (55 and 66% yield, respectively). Thus, the reaction is further activated when the potential is positive and inhibited when it is negative. According to these experimental results, our hypothesis is that a change in the Fermi level of the PdNp occurs when an EEF is applied over the Pd(0)–CA. This behavior allows the tuning and enhancement of the reaction rate when a positive potential is applied. An increase in the reaction rate was also observed by Tian and Moeller when applying constant current during the M–H reaction instead of a potential. However, no difference in the reaction rate using the negative or positive current was observed.13 To ensure a minimum current necessary for an EEF stimulus, preliminary M–H experiments were carried out with the addition of tetrabutylammonium tetrafluoroborate (TBABF4) as an electrolyte. We further examined the reaction in the absence of electrolyte and, surprisingly, almost the same behavior was observed (Table 1, compare entries 13 and 14 with entries 1 and 2 and see the ESI† (Tables S1 and S3)). Hence, all the experiments were then performed without the electrolyte. With other tested aryl iodides (2 and 3), an enhancement of the reaction was also achieved using +0.5 V, compared to the results obtained with 0.0 V (Table 1; entries 9–12).
 | ||
| Scheme 1 M–H reactions. | ||
| Entry | Aryl halide | Pd(0)–CA mol% | E (V) vs. Pt | Yields % (3 h) |
|---|---|---|---|---|
| a Conditions: aryl iodide (6 mmol), ethyl acrylate (18 mmol), Et3N (12 mmol) at 90 °C in CH3CN (25 ml), without using electrolyte, and applying the potential indicated in each case. The working electrode was a Pd(0)–CA with a CPL as a current collector. b Using a Pd(0)–CA with a PGR as a current collector. c TBABF4 (0.1 M) was employed as an electrolyte. | ||||
| 1 | 1 | 7 | +1 | 89 |
| 2 | 1 | 7 | +0.5 | 94 |
| 3 | 1 | 7 | 0.0 | 56 |
| 4 | 1 | 7 | Unconnected | 66 |
| 5 | 1 | 7 | −0.5 | 45 |
| 6 | 1 | 7 | −1 | 46 |
| 7 | 1 | 4b | +0.5 | 97 |
| 8 | 1 | 4b | 0.0 | 27 |
| 9 | 2 | 5 | +0.5 | 94 |
| 10 | 2 | 5 | 0.0 | 0 |
| 11 | 3 | 8 | +0.5 | 99 |
| 12 | 3 | 8 | 0.0 | 80 |
| 13c | 1 | 7 | +0.5 | 99 |
| 14c | 1 | 7 | +1 | 92 |
In the reactions using a positive potential and when conversion was close to 100%, deposition of black palladium on the Pt counter electrode Pt(C) and throughout the cell was observed by SEM and EDX analysis (see the ESI†). We discovered that the application of −1 V once the reaction was over, made the Pd deposited come back to the Pd(0)–CA. It is accepted that Pd cross-coupling reactions are in general homogeneous processes, even if heterogeneous catalysts are used,5,6 and the advantage of our method is that the use of this Pd recovery process leads to an excellent recovery of palladium. Compared to our previous published results (leaching about 3 wt% of Pd) for the same catalytic reaction,6a analysis of the crude corresponding to a +0.5 V reaction mixture after the Pd recovery process indicated a low leaching of 0.2 wt% of the palladium present in the Pd(0)–CA. Then, consecutive reactions under positive and negative potentials were carried out using the same batch of Pd(0)–CA. Under these conditions the Pd(0)–CA was reused 5 times with excellent results (99, 98, 98, 98, 92% yield at 3 h reaction time), although a decrease in activity was observed from the 6th cycle (see Table S2 in the ESI† for the results of 9 cycles). TEM images of Pd(0)–CA after 9 cycles showed that palladium was recovered in the fcc-Pd(0) nanoparticle form with a mean particle size of 16 nm. Hence, these results confirm the recyclability of the Pd(0)–CA and that the Pd recovery process preserves the initial characteristics of the palladium supported on the CA.
According to the results discussed so far, activation of palladium is achieved when it is present in any positively charged electrode. We have observed that palladium is always transported to the cathode, from anywhere deposited, when a potential is applied (either positive or negative). Palladium anode deposition has never been observed during the reaction. All of these suggest that cationic palladium intermediates must be generated during the M–H reaction cycle.
Chronoamperometry measurements of the reaction (Table 1, entries 7 and 8) have been carried out at +0.5 V (orange plot, Fig. 2A) and 0.0 V (blue plot, Fig. 2A). At the beginning of the reaction, applying +0.5 V, there were very low current values (between 10 and 20 microamperes when just the potential is applied), but they increased up to a maximum (point A) in parallel with the evolution of the reaction. Then, the current slightly decreases and finally stabilizes when the reaction is almost complete. So, the current appears to be an indirect measure of the reaction evolution. With 0.0 V, the reaction is not enhanced and proceeds slowly with very small current intensities detected. The increase of the current observed in the +0.5 V reaction should be due to the increasing concentration of ionic species that must be generated during the M–H reaction, since at the beginning of the reaction the current values are very low. These species could be the Et3NH+ I− generated at every catalytic cycle and other possible ionic intermediates.14,8a,d The decrease of the current after the maximum (point A, Fig. 2A) indicates a decrease of the concentration of ionic intermediates since the reaction ends, and then, only Et3NH+ I− remains with an almost constant concentration, which should be responsible of the current stabilization. In Fig. 2B the reaction behavior applying +0.5 V and −0.5 V using an electrolyte is represented. When the electrolyte is added, higher current values are observed when a potential is applied. After a complete reaction at +0.5 V, we applied −0.5 V and the Pd deposited on the Pt(C), acting as the anode, led also to the activation of the reaction rate. Moreover, both reactions have the same behavior (Fig. 2B). These results gives further evidence that, when Pd is deposited at the anode, negative and positive potentials achieve activation of Pd and the current appears to be an indirect measure of the reaction evolution.
 | ||
| Fig. 2 Chronoamperometry measurements for the M–H reaction of 1 and 4 (A). The working electrode was a Pd(0)–CA with a PGR as a current collector. The gap after each time control (represented by dots) corresponds to the sampling time. The reactions were carried out by applying constant potentials of +0.5 V and 0.0 V (vs. Pt). (B) Reaction with electrolyte (TBABF4 0.1 M) and applying constant potentials of +0.5 V and −0.5 V (vs. Pt). The working electrode was a Pd–CA(0) with a CPL as a current collector. | ||
Taking into account all the results obtained and those previously published8b,14–17 we propose a mechanistic hypothesis for the EEF accelerated M–H reaction at a positive potential (Fig. 3). Enhancement of the Pd reactivity by the positive potential favors the oxidative reaction of the supported Pd(0) with the aryl halide. The reaction occurs on the surface of the Pd(0) nanoparticles supported on the CA. Once the reaction has started small amounts of palladium are dissolved in the form of Pd(II) soluble complexes (ArPd(CH3CN)(Et3N)I) produced by the oxidative addition of the aryl iodide. Under our conditions, CH3CN and Et3N coordinate to the metal as stabilizing ligands.15 The equilibrium of iodide dissociation leads to the formation of a cationic complex ([ArPd(CH3CN)(Et3N)]+).14,17 Ar–Pd+ intermediates are metal-centered electrophiles attacking the double bond of the ethyl acrylate in a sort of electrophilic addition process. The migratory insertion is the product-forming step of the M–H cycle, in which a C–C bond is formed. Then palladium hydride is eliminated ([HPd(CH3CN)(Et3N)]+) to release the double bond and finally the Pd(0) is released and launches the next cycle. However, when the concentration of the aryl halide is low enough, that is when the reaction finishes, launches of Pd(0) are slower and transport of palladium to the cathode may occur in the form of a cationic hydride complex. This agrees with the observed palladium transport exclusively to the cathode, which can be explained if cationic palladium complexes are generated during the cycle. Besides the cathode, Pd(II) can be reduced to Pd(0) by the action of Et3N and this Pd(0) is deposited on the Pt(C) (cathode). The deposition of palladium throughout the cell could be due to the reduction of Pd(II) by Et3N during its transport to the cathode, and consequent aggregation and precipitation. As mentioned, applying −1 V once the reaction is over, the Pd deposited on Pt(C) and in the cell can be recovered again in the aerogel.
 | ||
| Fig. 3 Proposed mechanism for the enhanced M–H reaction with E > 0. A TEM image has been used to represent the Pd(0)–CA electrocatalyst. W: working electrode; C: counter electrode; and Ref: reference electrode. | ||
The sigmoidal profile of the current vs. time (Fig. 2A, orange curve) can be understood in the Michaelis–Menten kinetics framework as the addition of the contributions of the two main cationic carriers (Et3NH+ as one final product and the palladium intermediate complexes [XPd(CH3CN)2(Et3N)]+) of the M–H reaction.
We then decided to investigate the M–H reaction with an alternating current. This process is carried out by alternating positive and negative potentials in regular periods of time during the course of the reaction, so the positive potential activates the reaction and the negative potential inverts the palladium deposition on the Pt(C), also promoting the M–H reaction (see the ESI†). We have performed only some preliminary assays although they confirm the mechanisms proposed. This process must be studied in more detail to find the optimal conditions just reaching high reaction rates and avoiding palladium deposition.
In conclusion, we have demonstrated enhancement of the M–H reaction using Pd(0)–CAs when a positive electric field is applied. The opposite is observed when using a negative field. Thus, the effect of the electric field on the PdNps appears to be tuning the catalytic activity of Pd. This ability to manipulate chemical reactions with electric fields offers proof-of-principle for other heterogeneous catalysts. Moreover, the current intensity appears to be an indirect measure of the reaction evolution, which is strongly related to the concentration of reaction intermediates. This method, which probably may be extended to other synthetic processes, allows analyzing the kinetics of the reaction, being in the current case of the Michaelis–Menten type. Finally, a mechanistic hypothesis based on cationic palladium intermediates and according to the results observed is proposed.
Experimental section
General procedure for Pd(0)–CA catalyzed M–H reaction under an electric stimulus
A three-electrode system composed of Pd(0)–CA as a working electrode, a platinum cylinder (9 cm2) as a counter electrode and a platinum wire (0.6 mm in diameter) as a quasi reference electrode were used. The reactions were carried out under reflux (90 °C), in the presence of air, magnetic stirring and a constant potential. Ethyl acrylate (18 mmol), aryl iodide (6 mmol) and triethylamine (12 mmol) were dissolved in 25 ml of acetonitrile. When the reaction was over, the solution was decanted and the Pd(0)–CA washed several times with acetonitrile. The combined solvent extracts were evaporated, the residue was dissolved in CH2Cl2 and the solution was washed with aqueous NaHCO3 and a saturated solution of NaCl. The organic extract was evaporated to afford the coupling product. The Pd(0)–CA was always submerged in CH3CN 24 h before its use and kept in the same solvent.Acknowledgements
This research was supported by Spain MICINN (CTQ2011-22649 and ENE2012-36368, ENE2015-63969) and MEC (CTQ2014-53662-P and CTQ2014-51912-REDC) and the DURSI-Generalitat de Catalunya (2014SGR-1643 and 2014SGR-2016). L. M. was funded through a fellowship from the MICINN. We acknowledge helpful discussions with Prof. N. Casañ and J. Llorca.Notes and references
- C. G. Vayenas, S. Bebelis, C. Pliangos, S. Brosda and D. Tsiplakides, Electrochemical Activation of Catalysis, Kluwer Academic, 2002 Search PubMed.
- S. Shaik, S. P. de Visser and D. Kumar, J. Am. Chem. Soc., 2004, 126, 11746 CrossRef CAS PubMed.
- R. Meir, H. Chen, W. Lai and S. Shaik, ChemPhysChem, 2010, 11, 301 CrossRef CAS PubMed.
- A. C. Aragonès, N. L. Haworth, N. Darwish, S. Ciampi, N. J. Bloomfield, G. G. Wallace, I. Diez-Perez and M. L. Cote, Nature, 2016, 531, 88 CrossRef PubMed.
- (a) S. Cacchi, C. L. Cotet, G. Fabrizi, G. Forte, A. Goggiamani, L. Martín, S. Martínez, E. Molins, M. Moreno-Mañas, F. Petrucci, A. Roig and A. Vallribera, Tetrahedron, 2007, 63, 2519 CrossRef CAS; (b) R. Soler, S. Cacchi, G. Fabrizi, G. Forte, L. Martín, S. Martínez, E. Molins, M. Moreno-Mañas, F. Petrucci, R. M. Sebastián and A. Vallribera, Synthesis, 2007, 3068 CAS; (c) L. Martín, E. Molins and A. Vallribera, Tetrahedron, 2012, 68, 6517 CrossRef.
- (a) S. Martínez, A. Vallribera, C. L. Cotet, M. Popovici, L. Martín, A. Roig, M. Moreno-Mañas and E. Molins, New J. Chem., 2005, 29, 1342 RSC; (b) S. Martínez, M. Moreno-Mañas, A. Vallribera, U. Schubert, A. Roig and E. Molins, New J. Chem., 2006, 30, 1093 RSC.
- J. Li, X. Wang, Q. Huang, S. Gamboa and P. J. Sebastian, J. Power Sources, 2006, 158, 784 CrossRef CAS.
- (a) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed; (b) X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047 CrossRef CAS PubMed; (c) X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047 CrossRef CAS PubMed; (d) J. P. Knowles and A. Whiting, Org. Biomol. Chem., 2007, 5, 31 RSC.
- (a) M. Grdeń, M. Łukaszewski, G. Jerkiewicz and A. Czerwiński, Electrochim. Acta, 2008, 53, 7583 CrossRef; (b) A. J. Bard and L. R. Faulkner, Electrochemical Methods. Fundamentals and Applications, John Wiley and Sons Inc., 2nd edn, 2001 Search PubMed.
- A. Vallribera and E. Molins, in Aerogel Supported Nanoparticles in Catalysis in Nanoparticles and Catalysis, ed. D. Astruc, Wiley-VCH, 2008, pp. 161–194 Search PubMed.
- (a) C. Moreno-Castilla and F. J. Maldonado-Hódar, Carbon, 2005, 43, 455 CrossRef CAS; (b) T. F. Baumann, G. A. Fox, J. H. Satcher, Jr., N. Yoshizawa, R. Fu and M. S. Dresselhaus, Langmuir, 2002, 18, 7073 CrossRef CAS; F. J. Maldonado-Hódar, C. Moreno-Castilla and A. F. Pérez-Cadenas, Microporous Mesoporous Mater., 2004, 69, 119 Search PubMed; (c) L. C. Cotet, M. Gich, A. Roig, I. C. Popescu, V. Cosoveanu, E. Molins and V. Danciu, J. Non-Cryst. Solids, 2006, 352, 2772 CrossRef CAS.
- S. Park and J. Kim, J. Colloid Interface Sci., 2000, 232, 311 CrossRef CAS PubMed.
- J. Tian and K. D. Moeller, Org. Lett., 2005, 7, 5381 CrossRef CAS PubMed.
- A. Jutand, Chem. Rev., 2008, 108, 2300 CrossRef CAS PubMed.
- F. Zhao, M. Shirai, Y. Ikushima and M. Arai, J. Mol. Catal. A: Chem., 2002, 180, 211 CrossRef CAS.
- A. Biffis, M. Zecca and M. Basato, Eur. J. Inorg. Chem., 2011, 1131 Search PubMed.
- C. Amatore, B. Godin, A. Jutand and F. Lemaître, Chem. – Eur. J., 2007, 13, 2002 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of Pd(0)–CAs; cyclic voltammetry and chronoamperometry measurements; procedure for M–H reactions under an electric field; TEM, SEM, XRD and EDX characterization of materials; graphics for current intensity evolution; and characterization of 5–7. See DOI: 10.1039/c6nj02279k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |