
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel yttrium and zirconium catalysts featuring reduced Ar-BIANH2 ligands for olefin hydroamination (Ar-BIANH2 = bis-arylaminoacenaphthylene)†
Alessandro
Cimino
 a,
Filippo
Moscatelli
a,
Francesco
Ferretti
a,
Fabio
Ragaini
*a,
Stéphane
Germain
b,
Jérôme
Hannedouche
b,
Emmanuelle
Schulz
*b,
Lapo
Luconi
c,
Andrea
Rossin
c and
Giuliano
Giambastiani
*cd
a,
Filippo
Moscatelli
a,
Francesco
Ferretti
a,
Fabio
Ragaini
*a,
Stéphane
Germain
b,
Jérôme
Hannedouche
b,
Emmanuelle
Schulz
*b,
Lapo
Luconi
c,
Andrea
Rossin
c and
Giuliano
Giambastiani
*cd
aDipartimento di Chimica, Università degli Studi di Milano, via C. Golgi 19, 20133 Milano, Italy. E-mail: fabio.ragaini@unimi.it
bInstitut de Chimie Moléculaire et des Matériaux d'Orsay, UMR CNRS 8182, Université Paris-Sud, F-91405, Orsay Cedex, France. E-mail: emmanuelle.schulz@u-psud.fr
cInstitute of Chemistry of Organometallic Compounds ICCOM-CNR and Consorzio INSTM, Via Madonna del Piano 10, 50019 Sesto F.no Florence, Italy. E-mail: giuliano.giambastiani@iccom.cnr.it
dKazan Federal University, 420008 Kazan, Russian Federation
First published on 21st October 2016
Abstract
The novel class of bis-arylaminoacenaphthylenes (Ar-BIANH2) was employed for the preparation of zirconium and yttrium complexes to be used as catalysts for cyclohydroamination of a number of primary and secondary aminoalkenes. The complex [(2,6-iPr2C6H3-BIAN)Zr(NMe2)2(η1-NHMe2)] was isolated and completely characterized, including X-ray diffraction analysis. Despite its easy and almost quantitative isolation, it showed only moderate catalytic performance in the intramolecular hydroamination, irrespective of the cyclization precursor used. On the other hand, in situ generated YIII complexes obtained using the same class of ligands were found to be very active, leading to the hydroamination of substrates including those normally reluctant in undergoing cyclization such as those featuring an internal non-activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. Electron donating substituents and especially steric hindrance on the ligand improve the performance of the catalysts, allowing us to decrease the catalyst loading to 1 mol% in the latter case.
C double bond. Electron donating substituents and especially steric hindrance on the ligand improve the performance of the catalysts, allowing us to decrease the catalyst loading to 1 mol% in the latter case.
Introduction
Direct addition of amines to non-activated carbon–carbon double bonds, the so-called olefin hydroamination, is one of the most straightforward atom-economical processes for the synthesis of valuable nitrogen-containing compounds from relatively low-cost and ubiquitous starting materials.1 Over the years, time and efforts have led to the development of a plethora of metal and metal-free catalysts to promote this highly desirable transformation inter- and intramolecularly. Despite some tremendous achievements in terms of catalytic efficiency, (stereo)selectivities and scope of applications, some challenges still need to be addressed.2 One of the main unsolved ones is undoubtedly the relatively poor reactivity of 1,2-dialkyl-substituted olefins compared to terminal olefins concomitantly with the problematic control of the regio- and enantioselectivity of the direct amine addition.3 Recently, some formal hydroamination procedures have been elegantly reported as alternative strategies to circumvent some of these issues.4,5 Nevertheless and despite their brilliant results, these surrogates are far from ideal from an atom-economical point of view; so there is still a need for the development of novel catalysts to directly tackle some of the remaining issues of the classical hydroamination reaction. Herein, we report our latest work demonstrating the preparation of new zirconium- and yttrium based complexes and their evaluation as catalysts in the cyclohydroamination of classical benchmark substrates and challenging ones.Some of us have recently reported on the properties of a class of bis-arylaminoacenaphthylene compounds (Ar-BIANH2) derived from the reduction of easily available bis-aryliminoacenaphthenes (Ar-BIAN, Scheme 1. Numbering refers to compounds prepared and employed in the present work).6
 | ||
| Scheme 1 General reactivity of Ar-BIANH2. | ||
Complexes of the formal dianion of Ar-BIANH2 are well-known in the literature,7 but their preparation usually requires the reaction of the parent diimine with a strongly reducing metal (e.g. Na, Li, Ca), possibly followed by transmetalation of the alkaline or alkaline-earth intermediate to the targeted metal. However, alkaline and alkaline-earth complexes are extremely air- and moisture-sensitive; thus, their practical handling for the synthesis of new compounds as well as their long-term storage is troublesome. On the other hand, Ar-BIANH2 compounds are only moderately air-sensitive in the solid state and can be stored indefinitely under a dinitrogen atmosphere. Moreover, the use of isolated Ar-BIANH2 ligands holds important synthetic advantages that widen the ligand applicability range and facilitate the use of related complexes in catalysis. Indeed, new complexes can be prepared by ligand deprotonation or transamination instead of the classical reduction/transmetalation protocol. Finally, no alkaline or alkaline earth-based salts are generated as reaction by-products throughout the complexation procedure, so that the obtained complexes may be generated in situ and employed without any interference from any ionic product cogenerated during the synthesis.
Hill and coworkers have recently reported on the use of “dearomatized” Ar-BIAN complexes of alkaline earth metals as highly active catalysts for the intramolecular hydroamination of aminoalkenes.3a,b Given our interest in the development of new group 3 and 4 metals as hydroamination catalysts, we thought about exploiting our potential bis-amido ligands for the preparation of new yttrium and zirconium derivatives. In this regard, the ability of the isolated Ar-BIANH2 to undergo ligand deprotonation in the presence of metal-alkyl or metal-amido precursors has paved the way for the preparation of new highly electrophilic compounds.8
Results and discussion
Ligands synthesis
Among the Ar-BIANH2 ligands employed, Ph-BIANH2 (1) and 4-MeOC6H4-BIANH2 (2) have already been prepared by some of us.6 They had been obtained by reduction of the parent Ar-BIAN ligands by NaBH4 in methanol. In the original report, the reaction was carried out at reflux for 3 hours. However, on repetition of the same reaction with NaBH4 batches obtained by different suppliers, yields and purities were not always reproducible. We now developed an improved methodology by working at room temperature, which afforded the products reproducibly independent of the origin of the reducing agent. This novel procedure was also employed to prepare 3,5-(CF3)2C6H3-BIANH2 (3).9The protocol based on NaBH4 was tested on the sterically hindered 2,4,6-Me3C6H2-BIAN and 2,6-iPr2C6H3-BIAN. However, in neither case the reduction proceeded to an appreciable extent even at reflux temperature. The reduced 2,4,6-Me3C6H2-BIANH2 (4) has never been reported before in the literature, whereas 2,6-iPr2C6H3-BIANH2 (5) had been isolated by slow diffusion of water vapors into a THF solution of (2,6-iPr2C6H3-BIAN)Mg(THF)3,10 which is not, in our view, a synthetically convenient approach. Compound 5 was alternatively prepared in situ by the same group by treating a solution of the aforementioned magnesium complex with methanol.11 Reduction by metallic sodium in place of magnesium was also mentioned,10 but the procedure was not described. Taking advantage of these scattered data, we now obtained pure 4 and 5 by reducing the corresponding Ar-BIAN compounds in THF with a slight excess of metallic sodium, followed by the addition of methanol to the so-obtained sodium complexes and without the need to isolate the latter (Scheme 2).
 | ||
| Scheme 2 Preparation of (2,4,6-Me3C6H2)BIANH2 (4) and (2,6-iPr2-C6H3)BIANH2 (5). | ||
Synthesis and characterization of (2,6-iPr2C6H3-BIAN)Zr(NMe2)2(η1-HNMe2) complex (5a)
The bulky ligand 5 was preliminarily scrutinized for the isolation of a ZrIV complex. The transamination reaction between the metal precursor Zr(NMe2)4 and the bis-amino ligand 2,6-iPr2C6H3-BIANH2 (5) proceeded smoothly already at room temperature in benzene, with complete conversion after 3 h and the evolution of only one equivalent of dimethylamine (Scheme 3). Solvent evaporation provided green crystals of 5a as air- and moisture-sensitive microcrystalline solids in about 95% yield. The isolated complex is highly soluble in aromatic and aliphatic hydrocarbons and its spectroscopic and diffractometric characterization (vide infra) are consistent with a five-coordinated metal ion. As a distinctive spectral feature, the 1H NMR spectrum at room temperature (298 K) shows two singlets at 2.74 and 1.70 ppm, ascribed to the methyl protons of the two residual dimethylamido groups and the Me2NH η1-coordinated to the metal center, respectively. The X-ray diffraction analysis of 5a confirmed the coordination sphere at the metal ion. A perspective view of the complex structure is given in Fig. 1 along with a list of selected bond lengths and angles.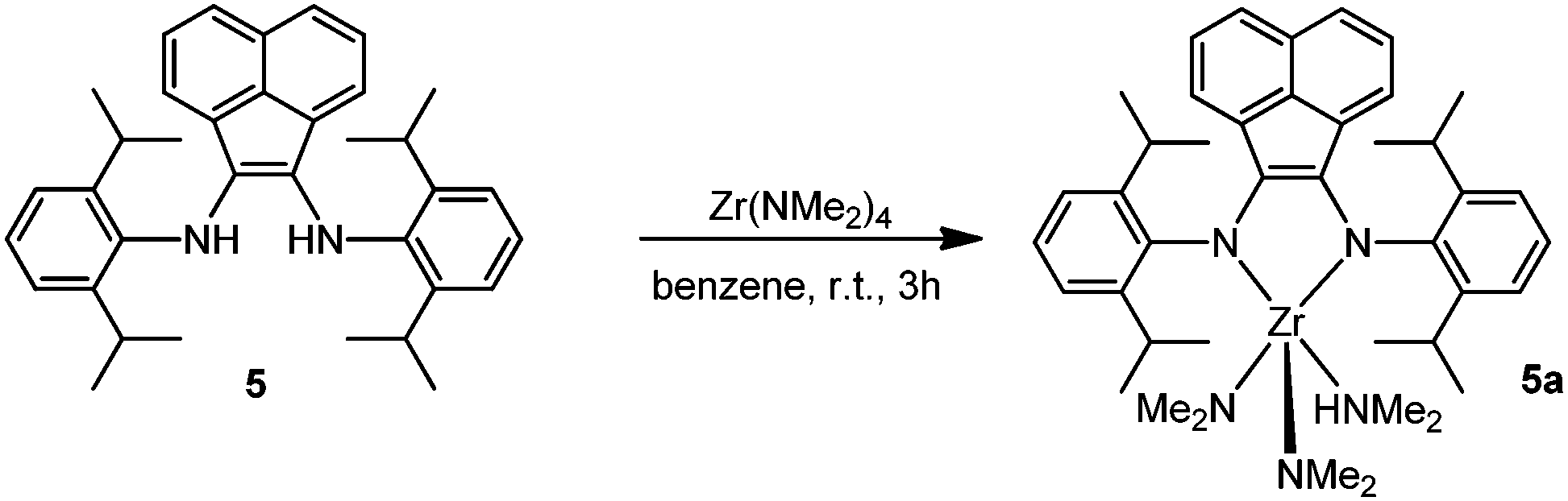 | ||
| Scheme 3 Synthesis of zirconium complex 5a. | ||
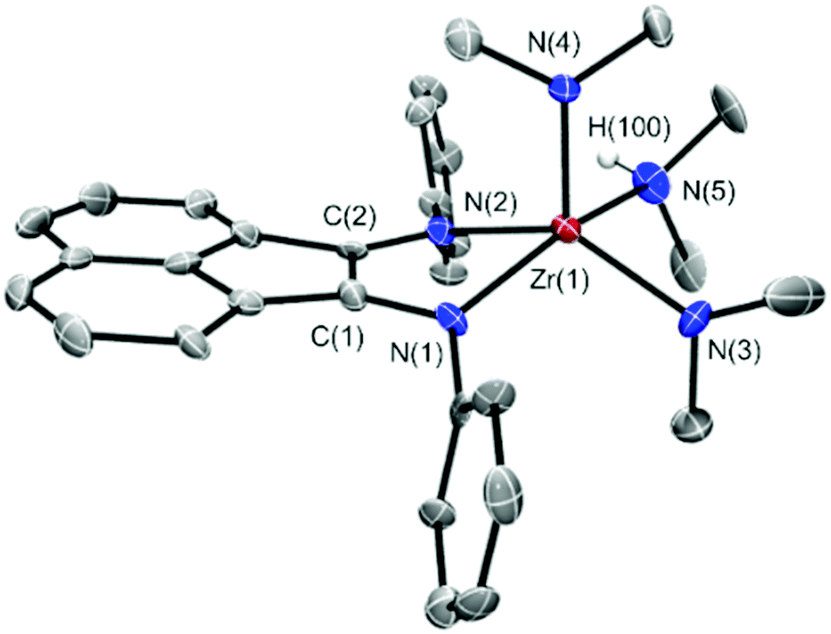 | ||
| Fig. 1 ORTEP drawing of the crystal structure of 5a. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms on the ligands (apart from the –NHMe2 group) and the iPr substituents on the phenyl rings attached to N are omitted for clarity. Selected bond distances (Å) and angles (°): Zr(1)–N(1) 2.133(4), Zr(1)–N(2) 2.207(4), Zr(1)–N(3) 2.065(5), Zr(1)–N(4) 2.029(4), Zr(1)–N(5) 2.406(4), N(5)–H(100) 0.90(7); N(1)–Zr(1)–N(2) 78.15(18), N(1)–Zr(1)–N(4) 101.69(18), N(1)–Zr(1)–N(3) 97.1(2), N(1)–Zr(1)–N(5) 153.5(2). | ||
5a crystallizes in the P212121 orthorhombic space group, with four molecules per unit cell. The Zr ion is five-coordinated, in a distorted square pyramidal coordination geometry (τ = 0.25). The equatorial plane is defined by the two ligand N atoms, one –NMe2 and the –NHMe2 group, while the last dimethylamido substituent occupies the pyramidal apex. The Zr–N distances and the N–Zr–N angles fall in the same range as those found in similar five-coordinated Zr amido complexes, with the Zr–N(3) or Zr–N(4) bond being shorter than the Zr–N(5) bond, as expected when passing from an anionic to a neutral donor. The metal ion lies slightly above the N(1)–N(2)–N(3)–N(5) plane [d(Zr–plane) = 0.60 Å]. Tables S1–S4 in the ESI† collect all the main structural parameters and refinement details of 5a. Crystals of 5a can be conveniently stored for months under an inert atmosphere (N2) and at low temperature (−30 °C) without any apparent alteration. On the other hand, a progressive complex decomposition with the formation of intractable side-products takes place at room temperature in aromatic hydrocarbons (benzene or toluene) as well as in chlorinated ones (CH2Cl2). Thus, the original bright green solution turns dark brown just upon keeping the complex solution at room temperature in the NMR tube for a few hours. Unfortunately, all our attempts to obtain more robust alkyl derivatives of 5a failed. The reaction of 5a with an excess of Me3Al (10 eq.) under controlled experimental conditions gave only a complex mixture of unidentified intermediates along with a relatively high amount of the free ligand. At odds with that observed with Zr(NMe2)4, attempts to react the ligand with Zr(Bn)4 led to the complete recovery of the reagents even after heating the reaction mixture to reflux for hours. Attempts were also made to prepare the ZrIV amido complex starting from other Ar-BIANH2 ligands. As for ligand 4, NMR evidence suggests that 4a is generated as a single complex. However, all our attempts to isolate it as a pure compound failed and its use as in situ prepared catalyst for intramolecular hydroamination gave only very modest results.
The less sterically hindered ligands 1 and 3 have also been scrutinized for the isolation of the corresponding ZrIV amido complexes. Unfortunately, both ligands gave mixtures of inseparable compounds, which have no longer been processed or employed in catalysis. Overall, it can be inferred that the higher the ligand bulkiness, the highest the purity and unicity of the resulting complex. Indeed, the steric hindrance generated by the aryl substituents is expected to facilitate the generation of complexes with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal to ligand ratio, thus avoiding the generation of over-coordinated metal ions or complex mixtures.
:1 metal to ligand ratio, thus avoiding the generation of over-coordinated metal ions or complex mixtures.
Catalytic performance of 5a in the intramolecular hydroamination of aminoalkenes
The freshly prepared bis-amido complex 5a was exploited as a homogeneous catalyst for the intramolecular hydroamination/cyclization of model primary and secondary amines tethered to monosubstituted alkenes (Table 1). Two different reaction temperatures were used so as to differentiate the catalyst performance and improve the reaction conversion.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Substrate | T (°C) | t (h) | Conv.b (%) |
| a Reaction conditions: toluene solvent (2.5 mL), substrate = 0.21 mmol, catalyst = 5 mol%. b Conversions were determined by GC. | |||||
| 1 | Zr(NMe2)4 | Ia | 60 | 2 | 55 |
| 2 | Zr(NMe2)4 | Ia | 60 | 10 | 77 |
| 3 | 5a | Ia | 60 | 2 | 17 |
| 4 | 5a | Ia | 60 | 10 | 26 |
| 5 | 5a | Ia | 100 | 10 | 47 |
| 6 | 5a | IIa | 60 | 10 | 20 |
| 7 | 5a | IIIa | 60 | 10 | 16 |
| 8 | 5a | IVa | 100 | 20 | 25 |

As shown in Table 1, catalyst 5a is active for the targeted reaction on both primary and secondary aminoalkenes. However, irrespective of the cyclization precursor used and the adopted reaction conditions (Table 1, entries 3–8 vs. 1–2), the chemical conversion was lower compared to that obtained using the plain Zr(NMe2)4 precursor.
Neither Thorpe–Ingold effects due to the substitution pattern of the cyclization precursor nor any other stereo-electronic effect can be reasonably invoked to justify the observed moderate conversions. On the other hand, these outcomes are in line with the moderate stability observed for 5a in solution. Indeed, a progressive decomposition of the catalyst along with the formation of intractable solid by-products takes place in a few hours.
In situ generation of yttrium complexes
Given the instability problems generally encountered in the synthesis and isolation of the ZrIV complexes based on the Ar-BIANH2 ligands, the YIII derivatives were prepared and employed in situ in the homogeneous hydroamination catalysis. This procedure was also dictated by the typically higher moisture and oxygen sensitivity of organolanthanides compared with their ZrIV analogues.Some of us had previously described a straightforward procedure for the generation of amido alkyl ate complexes by the in situ combination of the yttrium ate precursor [Li(THF)4][Y(CH2TMS)4]12 and enantiopure chiral N-substituted binaphthyldiamine ligands in toluene.3m,13 The as-prepared mixture was successfully employed to promote the enantioselective cyclisation of tethered amines to mono- or 1,2-disubstituted alkenes in valuable yields and enantioselectivities. A similar in situ approach had been adopted to obtain binaphthylamido yttrium complexes starting from a homoleptic yttrium source, [Y(CH2TMS)3(THF)2].14 These neutral complexes were particularly active for the transformation of substrates bearing secondary amines.15 Taking into account these results, the same experimental procedures using both YIII precursors have been applied in the presence of the pure Ar-BIANH2 ligands (Scheme 4). A slight excess of ligand was introduced into the reaction mixture and exchange occurred, with each yttrium species, at room temperature, accompanied by a significant color change from deep red/dark violet for the ligands to green for all complexes, except for 3b, bluish-violet. Solutions were stirred for 30 minutes and then directly engaged with the targeted substrate to check the catalytic ability. It should be stressed that the in situ preparation of the catalysts was found to be completely reproducible and the same outcome (differences in conversion values within 2%) was observed for catalytic reactions repeated under the same experimental conditions.
 | ||
| Scheme 4 In situ preparation of Ar-BIAN alkyl ate and neutral yttrium complexes (only the main species formed are shown). | ||
However, the in situ generated catalyst was most often not a single pure complex. Scheme 4 reports the main species present in solution, whose identification was supported by literature precedents (vide supra). However, only in the case of 5c and by working with a 1:1 molar ratio of yttrium precursor/5 was a 1H NMR spectrum obtained that is strongly indicative of a single species with the composition shown (see the ESI†). In other cases, minor components are also observable by NMR spectroscopy that makes a clear assignment of all signals not possible. All attempts to isolate and precisely characterize the so-obtained complexes remained unsuccessful.
Catalytic performance of the YIII complexes
Structurally diverse aminoalkene cyclization precursors were engaged in the yttrium catalyzed cyclohydroamination reaction. In particular, the efficiency of the in situ prepared complexes was studied with respect to a number of more or less challenging substrates towards cyclization (Scheme 5). As these reactions are all benchmark transformations, the catalyst efficiencies were evaluated by NMR spectroscopy, following substrate conversion. In all cases, only the expected products were obtained (see the ESI† for details). | ||
| Scheme 5 Intramolecular hydroamination with Ar-BIAN yttrium complexes. | ||
The catalytic activity of ate complexes 1b–5b was preliminarily scrutinized in combination with non-sterically demanding substrates (such as IIa and IIIa) in order to evaluate the influence of the ligand structure and bulkiness on the complex reactivity. A complete list of the catalytic outcomes is given in Table 2. The catalyst performance of the yttrium tetra-alkyl precursor in the cyclization of both primary aminoalkenes is also reported for the sake of comparison. As Table 2 shows, IIa was conveniently transformed into the corresponding cyclization product with high conversion and in a relatively short time, using 6 mol% of the tetra-alkyl precursor [Li(THF)4][Y(CH2TMS)4] at room temperature (Table 2, entry 1). However, the cyclization of IIIa occurred with more difficulty under these conditions (Table 2, entry 9) likely due to a less relevant Thorpe–Ingold effect. All the in situ prepared YIIIAr-BIAN ate complexes (1b–5b) proved to catalyze these transformations, albeit with variable efficiency. In the case of ate complexes bearing non-sterically hindered Ar-BIANH2 ligands (1b and 2b), only a moderate catalytic performance was obtained (Table 2, entries 2–3 and 10–11). A similar trend was observed with complex 3b, where the presence of electron withdrawing (EWG) trifluoromethyl substituents on the N-phenyl rings (Table 2, entries 4 vs. 2 and 12 vs. 10) translated into a poorly active system for the targeted reaction. On the contrary, the use of bulky ligands decorated with electron donating groups (EDG) clearly led to a strong acceleration of the cyclization process with almost complete substrate conversions in less than 1 h (Table 2, entries 5–6 vs. 2 and 13–14 vs. 10). Noteworthy, catalyst 5b offered the highest performance, leading to complete cyclization of both aminoalkene precursors to the corresponding pyrrolidines in a relatively short time (between 1 and 3 h) at room temperature and with a catalyst loading as low as 1 mol% (Table 2, entries 8 and 16).
| Entry | Catalyst | Substrate | t (h) | Conv.b (%) |
|---|---|---|---|---|
| a Reactions were carried out in C7D8 under argon at room temperature using 6 mol% catalyst unless otherwise stated. b Determined by in situ1H NMR spectroscopy. c 2 mol% catalyst. d 1 mol% catalyst. | ||||
| 1 | [Li(THF)4][Y(CH2TMS)4] | IIa | 2 | 83 |
| 2 | 1b | IIa | 17 | 75 |
| 3 | 2b | IIa | 17 | 75 |
| 4 | 3b | IIa | 16 | 63 |
| 5 | 4b | IIa | 0.75 | >95 |
| 6 | 5b | IIa | 0.25 | >95 |
| 7 | 5b | IIa | 0.4 | >95 |
| 8 | 5b | IIa | 1 | >95 |
| 9 | [Li(THF)4][Y(CH2TMS)4] | IIIa | 18 | <10 |
| 10 | 1b | IIIa | 88 | 46 |
| 11 | 2b | IIIa | 88 | 50 |
| 12 | 3b | IIIa | 45 | 75 |
| 13 | 4b | IIIa | 1.5 | >95 |
| 14 | 5b | IIIa | 0.25 | >95 |
| 15 | 5b | IIIa | 0.5 | >95 |
| 16 | 5b | IIIa | 3 | >95 |
Overall, it can be inferred that the use of sterically hindered diamine ligands, preferably bearing EDG as aryl substituents, favors the generation of the targeted amido alkyl ate complexes and avoids the formation of tetra-amido species, generally recognized to be less catalytically active for promoting the hydroamination reaction.16
The catalytic performance of the most representative complexes from this series was then evaluated in the cyclization of the model secondary aminoalkene Va (2,2-dimethyl-pent-4-enyl)-methyl-amine (Scheme 5 and Table 3). Again, all the in situ prepared ate and neutral complexes were active towards the cyclization of this secondary amine. Interestingly, and as a general comment, in the presence of ate complexes cyclization occurred more rapidly compared to the transformation of the primary aminoalkene analogue IIIa (Table 3, entries 1–2 vs.Table 2, entries 9–10). As observed with substrates IIa and IIIa, complexes possessing EDG as aryl-ligand substituents caused an increase in the reaction rate (Table 3, entries 3–5). This effect was even more pronounced for the sterically hindered complex 5b, where almost complete cyclization of Va takes place in very short reaction times, also at very low catalyst loadings (Table 3, entries 5–7). The neutral YIII complexes were highly active species, with almost complete conversion in about half an hour (entries 8–10). As already demonstrated,15 neutral complexes are particularly active in the hydroamination/cyclization of secondary amines. Nevertheless, substrate Va was not sterically demanding enough to allow for an accurate evaluation of the ligand effects on the reactivity of the yttrium complexes in the hydroamination reaction. For this reason, the more challenging precursors VIa and VIIa were engaged to complete the study (Scheme 5 and Table 4). While the former is the precursor for less favorable 6-membered heterocycles (piperidine derivatives), the latter presents a less reactive internal double bond. To obtain the cyclization products, all reactions had to be performed at higher temperatures (50 °C for VIa and 70 °C for VIIa, respectively) while keeping the catalyst loading at 6 mol%.
| Entry | Catalyst | t (h) | Conv.b (%) |
|---|---|---|---|
| a Reactions were carried out in C7D8 under argon at room temperature using 6 mol% catalyst unless otherwise stated. b Determined by in situ1H NMR spectroscopy. c 2 mol% catalyst. d 1 mol% catalyst. | |||
| 1 | [Li(THF)4][Y(CH2TMS)4] | 16 | 88 |
| 2 | 1b | 68 | >95 |
| 3 | 2b | 0.8 | 92 |
| 4 | 4b | 4 | 91 |
| 5 | 5b | 0.5 | >95 |
| 6 | 5b | 0.5 | >95 |
| 7 | 5b | 1 | 92 |
| 8 | [Y(CH2TMS)3(THF)2] | 0.25 | >95 |
| 9 | 4c | 0.5 | 92 |
| 10 | 5c | 0.5 | 92 |
| Entry | Catalyst | Substrate | t (h) | Conv.b (%) |
|---|---|---|---|---|
| a Reactions were carried out in C7D8 under argon at 50 °C for the transformation of VIa and 70 °C for VIIa using 6 mol% catalyst unless otherwise stated. b Determined by in situ1H NMR spectroscopy. c 2 mol% catalyst. | ||||
| 1 | [Li(THF)4][Y(CH2TMS)4] | VIa | 24 | 35 |
| 2 | 2b | VIa | 21 | 10 |
| 3 | 4b | VIa | 20 | 80 |
| 4 | 5b | VIa | 17 | >95 |
| 5 | [Y(CH2TMS)3(THF)2] | VIa | 24 | <10 |
| 6 | 4c | VIa | 18 | 10 |
| 7 | 5c | VIa | 18 | 34 |
| 8 | [Li(THF)4][Y(CH2TMS)4] | VIIa | 24 | 15 |
| 9 | 2b | VIIa | 24 | 20 |
| 10 | 4b | VIIa | 24 | >95 |
| 11 | 5b | VIIa | 24 | >95 |
| 12 | 5b | VIIa | 24 | 43 |
| 13 | [Y(CH2TMS)3(THF)2] | VIIa | 24 | 42 |
| 14 | 4c | VIIa | 24 | 35 |
| 15 | 5c | VIIa | 24 | 88 |
As Table 4 shows, all catalysts promoted the cyclization reactions of VIa and VIIa, albeit with a reduced reactivity compared with the previously reported cyclization precursors. Once again, complexes bearing more sterically hindered ligands offered the best performance with both aminoalkenes. Complete conversion of both substrates was obtained with the 4b and 5b ate complexes (Table 4, entries 4, 10–11). Cyclization of VIIa up to 88% was also obtained with the neutral complex 5c (Table 4, entry 15). Neutral complexes bearing less hindered ligands (i.e.4c) or no ligands did not seem to resist the high temperatures required to promote the cyclization process (Table 4, entries 13–14). Overall, the amido alkyl ate complex 5b and (to a lesser extent) the neutral yttrium complex 5c are rare examples of active species that can promote hydroamination in cases of challenging substrates like those possessing internal non-activated double bonds. A non-negligible activity was maintained even at low catalyst loadings (Table 4, entry 12).3a,m,13,16,17
Conclusions
In this paper, we have investigated the reactivity of ZrIV and YIII complexes prepared from different Ar-BIANH2 compounds in the intramolecular hydroamination of primary and secondary aminoalkenes. A new and convenient synthetic methodology was described for the preparation and isolation of the bis-anilido ligands from the respective bis-imino precursors (Ar-BIAN). The effectiveness of the proposed methodology for the generation of the Ar-BIANH2 ligand class has allowed for the isolation and characterization of a bis-amido zirconium complex (5a) by means of a transamination reaction. The same bis-anilido ligands have also been successfully employed for the in situ synthesis of YIII complexes, to be tested for the intramolecular hydroamination reaction. While 5a has shown only moderate catalyst performance, the in situ prepared organoyttrium complexes have been found to be excellent catalyst candidates for hydroamination with a number of substrates, including those normally reluctant in undergoing cyclization. Complexes prepared from the sterically hindered ligand 5 (containing two ortho isopropyl groups on each aryl ligand bound to N) showed the highest activity, in combination with both zirconium and yttrium. This is in line with that observed when Ar-BIAN compounds are used as ligands in the palladium- or nickel catalyzed polymerization of olefins,18 but in contrast to that observed when the same ligands are employed to generate catalysts for several reactions like Pd-catalyzed olefin/CO copolymerization, Ru-catalyzed allylic aminations by nitroarenes, etc., with steric hindrance inhibiting the reactions in these cases.19 Such a behavior is indicative of an active role of ligands in preventing catalyst deactivation through aggregation or over-coordination and highlights the importance of steric effects in the hydroamination reactions here described. Overall, the successful approach to the synthesis of early transition metal and rare-earth complexes based on Ar-BIANH2 ligands paves the way for future exploitation of these bis-amino bidentate systems in combination with late transition metals; studies in this direction are currently ongoing in our labs.Experimental section
General procedure
Unless otherwise stated, all the manipulations concerning the ligand synthesis were performed under a nitrogen atmosphere by using standard Schlenk techniques. As for the investigation of the reactivity of zirconium compounds, all air- and/or moisture-sensitive reactions were performed under an inert atmosphere in flame-dried flasks using standard Schlenk-type techniques or in a nitrogen-filled dry box. Catalytic reactions were performed under an inert atmosphere (N2) in a 10 mL round bottom flask and the reaction course was monitored by GC-MS analysis. As for the investigation of yttrium compounds, all manipulations were carried out under an argon atmosphere by using standard Schlenk or glove box techniques.Ar-BIANs were synthesized as previously reported (see the ESI† for the full procedure and NMR characterization).20 Amino alkenes Ia,21IIa,16IIIa,22IVa,23Va,24VIa,25VIIa,16 [Li(THF)4][Y(CH2SiMe3)4]12 and Y(CH2SiMe3)3(THF)214 were prepared according to reported procedures. When employed in yttrium catalyzed reactions, aminoalkenes were dried over calcium hydride, transferred under vacuum and further dried for at least 1 h over 3 Å molecular sieves with a few drops of toluene-d8 prior to use. Methanol was freshly distilled under nitrogen over Mg(OMe)2. Distilled water was degassed by ultrasound irradiation under nitrogen in a cleaning bath. Benzene and toluene were purified by distillation from sodium/triglyme benzophenone ketyl and stored over activated 4 Å molecular sieves. Benzene-d6 was dried over sodium/benzophenone ketyl and condensed in vacuo over activated 4 Å molecular sieves prior to use. Toluene-d8 was dried over sodium benzophenone ketyl, transferred under vacuum and stored over activated 3 Å molecular sieves. All other reagents and solvents were used as purchased from commercial suppliers without further purification (unless otherwise stated). 1D (1H and 13C) and 2D (COSY H,H, HETCOR H,C) NMR spectra were recorded either on a Bruker Avance 300 MHz instrument (300.13 and 75.47 MHz for 1H and 13C, respectively) or on Bruker AM250, Bruker AV300 and AV360 and DRX400 NMR spectrometers, operating at 250, 300, 360 and 400 MHz, respectively. Chemical shifts are reported in ppm (δ) relative to TMS, referenced to the chemical shifts of residual solvent resonances (1H and 13C). The multiplicity of the 13C{1H} NMR spectra was determined on the basis of the 13C{1H} JMOD sequence and quoted as: CH3, CH2, CH and C for primary, secondary, tertiary and quaternary carbon atoms, respectively. The C, H, and N elemental analyses were performed on a Thermo FlashEA 1112 Series CHNS-O elemental analyzer. The GC-MS analyses were performed on a Shimadzu QP2010S apparatus equipped with a SUPELCO SPB-1 fused silica capillary column (30 m length, 0.53 mm i.d., 15 μm film thickness).
General synthesis of Ar-BIANH2 using NaBH4 as a reducing agent
The synthesis is a modification of that previously reported by some of us.6 Ar-BIAN (1 mmol) and freshly distilled methanol (7 mL) were added to a Schlenk flask under a nitrogen atmosphere. Solid NaBH4 (3 mmol) was then added. The color of the solution changed from yellow/orange to red/purple. The reaction was maintained under stirring overnight. The mixture was then concentrated to half volume under vacuum. The precipitate was collected by filtration on a glass frit, washed with degassed water (3 × 10 mL), and dried under vacuum. Satisfactory elemental analysis values could not be obtained due to the air sensitivity of the Ar-BIANH2 derivatives, as also previously observed.6
General synthesis of sterically crowded Ar-BIANH2 using Na as a reducing agent
Metallic sodium cut into small pieces (85 mg, 3.7 mmol) was placed in a Schlenk flask under a nitrogen atmosphere, washed with THF twice (2 × 3 mL) and then suspended in THF (25 mL). The sterically crowded Ar-BIAN (1.5 mmol) was then added as a solid and the reaction mixture was stirred for 5 h. The color of the mixture progressively turned from orange/yellow to red and finally dark green. MeOH (2 mL) was added and the mixture was stirred until the color turned purple. The solvent was evaporated under vacuum. The residue was suspended in MeOH (20 mL), collected by filtration on a glass frit, washed with water (3 × 10 mL) and dried under vacuum.Synthesis of 5a
To a solution of 5 (0.100 g, 0.20 mmol) in dry and degassed benzene (3 mL), a solution of tetrakis(dimethylamido)zirconium (0.053 g, 0.20 mmol) in dry and degassed benzene (2 mL) was added. The reaction mixture was maintained at room temperature, under stirring, for 3 h and then concentrated in vacuo to obtain the crude mixture as a dark-green solid. The crude sample was washed with pentane and filtered to afford analytically pure green crystals of 5a in 95% isolated yield. Crystals suitable for X-ray diffraction analysis were grown from a concentrated toluene solution at −30 °C. Crystals of 5a are indefinitely stable under nitrogen atmosphere at −30 °C. On the other hand, a progressive complex decomposition and formation of intractable side-products takes place in aromatic hydrocarbons already at room temperature. Such an undesired side effect occurs irrespective of the concentration of 5a in solution.1H NMR (300 MHz, C6D6, 293 K): δ 1.23 (d, 3JHH = 6.8 Hz, 12H, CH(CH3ACH3B, H15,18)), 1.29 (d, 3JHH = 6.8 Hz, 12H, CH(CH3ACH3B, H16,19)), 1.70 (br s, 6H, HN(CH3)2, H21), 2.74 (br s, 12H, N(CH3)2, H20), 3.86 (sept, 3JHH = 6.8 Hz, 4H, CH(CH3)2, H14,17), 6.15 (d, 3JHH = 6.9 Hz, 2H, H3), 6.89 (dd, 3JHH = 8.4 Hz and 6.9 Hz, 2H, H4), 7.09 (d, 3JHH = 8.4 Hz, 2H, H5), 7.28 (m, 6H, H10,11,12). 13C{1H} NMR (75 MHz, C6D6, 293 K, selected data): δ 24.6 (CH(CH3ACH3B)), 26.1 (CH(CH3ACH3B)), 28.2 (CH(CH3)2, C14,17), 38.6 (HN(CH3), C21), 41.7 (N(CH3), C20), 119.2 (C3), 123.7 (C5), 123.9 (C10,12), 125.2 (C11), 127.1 (C4), 137.1 (C2), 144.7 (C9,13), 145.0 (C1), 148.4 (C8). Anal. calcd (%) for C42H55N5Zr (725.18): C, 69.56; H, 8.20; N, 9.66. Found: C, 69.62; H, 8.25; N, 9.60.
General procedure for zirconium catalyzed intramolecular hydroamination reactions of aminoalkenes Ia–IVa
All catalytic tests were set-up in an inert atmosphere in a N2-filled drybox. The amido precursor 5a was tested as a neutral catalyst in the intramolecular hydroamination reaction in a two-necked 10 mL round-bottom flask equipped with a magnetic stirring bar, a glass stopper and a septum. In a typical procedure, a solution of the catalyst (5 mol%) in dry and degassed toluene (1 mL) was treated in one portion with a solution of the aminoalkene (0.21 mmol) in dry and degassed toluene (1.3 mL) and ferrocene as an internal standard (0.2 mL of a stock 0.17 M ferrocene solution in toluene). Toluene was used as a reaction solvent and the catalyst content was fixed to 5 mol% for each run. Afterwards, the system was heated at the final temperature and the reaction course was periodically monitored by analyzing the sample mixtures by GC-MS analysis at fixed times.General procedure for in situ preparation of complexes 1b to 5b
In a glovebox, a solution of [Li(THF)4][Y(CH2TMS)4] (0.042 mmol) in C7D8 (1 mL) was stirred for few minutes. The considered Ar-BIANH2 ligand (0.050 mmol) was solubilized in C7D8 (1 mL) and the corresponding solution was slowly added dropwise into the [Li(THF)4][Y(CH2TMS)4] solution under vigorous stirring. As the ligand was slowly added, the dark violet to red reaction mixture (according to the ligand structure) turned to a dark green to blue colored solution. The homogeneous reaction solution was then allowed to stir 30 min at ambient temperature.General procedure for in situ preparation of complexes 4c and 5c
The same procedure was used starting from a solution of Y(CH2SiMe3)3(THF)2 (0.042 mmol) and the Ar-BIANH2 ligands (0.050 mmol).General procedure for the hydroamination catalytic reactions using yttrium complexes
In a glovebox, an aliquot (see the molar ratios indicated in tables) of the in situ prepared complex mixtures in C7D8 was taken off by a micropipette and transferred to a vial containing the appropriate amino-alkene (0.23 mmol), previously dried on 4 Å molecular sieves with a few drops of toluene-d8 for at least 1 h at room temperature. The reaction mixture was then introduced into a screw tap or a J. Young tap NMR tube and, if appropriate, placed in an oil bath heated at the required temperature. The conversion of the reaction was monitored by comparative integration of the signal relative to the olefinic protons of the substrate and the signal relative to the protons of the product.X-ray data measurements
Single crystal X-ray data were collected at low temperature (100 K) on an Oxford Diffraction Xcalibur PX diffractometer equipped with a CCD area detector using Cu Kα radiation (λ = 1.5418 Å). The CrysAlis CCD 1.171 program was used for the data collection. Data reduction was carried out using the program CrysAlis RED 1.171 and the absorption correction was applied using the program ABSPACK 1.17. Direct methods implemented in Sir97 were used to solve the structures and refinements were performed by full-matrix least-squares against F2 implemented in SHELX97. All the non-hydrogen atoms were refined anisotropically, while the hydrogen atoms were fixed in calculated positions and refined isotropically using a thermal factor depending on the one of the atoms to which they are bound (riding model). Disorder on some methyl groups of the –NMe2 substituents was not explicitly treated during the refinement, since no significant improvement of the R factor could be achieved. Molecular plots were produced by using the program ORTEP3.Acknowledgements
A. C. and F. M. thank the Erasmus Project for a fellowship that allowed their stay in France. F. F. thanks the University of Milan for a PostDoc fellowship. G. G., L. L. and A. R. thank the Fondazione Cariplo (“Crystalline Elastomers” project) for funding this research activity. S. G., J. H. and E. S. thank MENSR, CNRS, and the Groupe de Recherche International (GDRI) “Homogeneous Catalysis for Sustainable Development” for financial support.References
- For general and more specialized reviews on hydroamination, see: (a) V. Rodriguez-Ruiz, R. Carlino, S. Bezzenine-Lafollee, R. Gil, D. Prim, E. Schulz and J. Hannedouche, Dalton Trans., 2015, 44, 12029–12059 RSC; (b) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed; (c) A. K. Gupta and K. L. Hull, Synlett, 2015, 1779–1784 CAS; (d) S. M. Coman and V. I. Parvulescu, Org. Process Res. Dev., 2015, 19, 1327–1355 CrossRef CAS; (e) E. Bernoud, C. Lepori, M. Mellah, E. Schulz and J. Hannedouche, Catal. Sci. Technol., 2015, 5, 2017–2037 RSC; (f) A. L. Reznichenko, A. J. Nawara-Hultzsch and K. C. Hultzsch, in Stereoselective Formation of Amines, ed. W. Li and X. Zhang, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 191–260, DOI:10.1007/128_2013_500; (g) J. Hannedouche and E. Schulz, Chem. – Eur. J., 2013, 19, 4972–4985 CrossRef CAS PubMed; (h) K. D. Hesp and M. Stradiotto, ChemCatChem, 2010, 2, 1192–1207 CrossRef CAS; (i) G. Zi, Dalton Trans., 2009, 9101–9109 RSC; (j) S. R. Chemler, Org. Biomol. Chem., 2009, 7, 3009–3019 RSC; (k) T. E. Muller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed; (l) A. L. Reznichenko and K. C. Hultzsch, Organic Reactions, John Wiley & Sons, Inc., 2015, vol. 88 Search PubMed.
- For some work by our groups in this field, see: (a) D. M. Lyubov, L. Luconi, A. Rossin, G. Tuci, A. V. Cherkasov, G. K. Fukin, G. Giambastiani and A. A. Trifonov, Chem. – Eur. J., 2014, 20, 3487–3499 CrossRef CAS PubMed; (b) L. Luconi, A. Rossin, G. Tuci, S. Germain, E. Schulz, J. Hannedouche and G. Giambastiani, ChemCatChem, 2013, 5, 1142–1151 CrossRef CAS; (c) L. Luconi, A. Rossin, A. Motta, G. Tuci and G. Giambastiani, Chem. – Eur. J., 2013, 19, 4906–4921 CrossRef CAS PubMed; (d) L. Luconi, J. Klosin, A. J. Smith, S. Germain, E. Schulz, J. Hannedouche and G. Giambastiani, Dalton Trans., 2013, 42, 16056–16065 RSC.
- For scarce examples of catalytic systems with the ability to react with 1,2-dialkyl-substituted olefins, see: (a) M. Arrowsmith, M. S. Hill and G. Kociok-Kohn, Organometallics, 2014, 33, 206–216 CrossRef CAS; (b) M. Arrowsmith, M. S. Hill and G. Kociok-Kohn, Organometallics, 2011, 30, 1291–1294 CrossRef CAS; (c) K. Manna, W. C. Everett, G. Schoendorff, A. Ellern, T. L. Windus and A. D. Sadow, J. Am. Chem. Soc., 2013, 135, 7235–7250 CrossRef CAS PubMed; (d) E. Chong, S. Qayyum, L. L. Schafer and R. Kempe, Organometallics, 2013, 32, 1858–1865 CrossRef CAS; (e) F. Lauterwasser, P. G. Hayes, W. E. Piers, L. L. Schafer and S. Bräse, Adv. Synth. Catal., 2011, 353, 1384–1390 CrossRef CAS; (f) Y. Chapurina, H. Ibrahim, R. Guillot, E. Kolodziej, J. Collin, A. Trifonov, E. Schulz and J. Hannedouche, J. Org. Chem., 2011, 76, 10163–10172 CrossRef CAS PubMed; (g) L. D. Julian and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 13813–13822 CrossRef CAS PubMed; (h) K. D. Hesp, S. Tobisch and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 413–426 CrossRef CAS PubMed; (i) L. J. E. Stanlake and L. L. Schafer, Organometallics, 2009, 28, 3990–3998 CrossRef CAS; (j) Z. Liu and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 1570–1571 CrossRef CAS PubMed; (k) J.-S. Ryu, T. J. Marks and F. E. McDonald, J. Org. Chem., 2004, 69, 1038–1052 CrossRef CAS PubMed; (l) J.-S. Ryu, T. J. Marks and F. E. McDonald, Org. Lett., 2001, 3, 3091–3094 CrossRef CAS PubMed; (m) Y. Chapurina, J. Hannedouche, J. Collin, R. Guillot, E. Schulz and A. Trifonov, Chem. Commun., 2010, 46, 6918–6920 RSC; (n) L. H. Lühning, C. Brahms, J. P. Nimoth, M. Schmidtmann and S. Doye, Z. Anorg. Allg. Chem., 2015, 641, 2071–2082 CrossRef.
- (a) J. Zheng, J. Qi and S. Cui, Org. Lett., 2016, 18, 128–131 CrossRef CAS PubMed; (b) Y. Xi, T. W. Butcher, J. Zhang and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 776–780 CrossRef CAS PubMed; (c) Y. Yang, S.-L. Shi, D. Niu, P. Liu and S. L. Buchwald, Science, 2015, 349, 62–66 CrossRef CAS PubMed; (d) M. Villa and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2015, 54, 11906–11908 CrossRef CAS PubMed; (e) J. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886–891 CrossRef CAS PubMed; (f) T. M. Nguyen, N. Manohar and D. A. Nicewicz, Angew. Chem., Int. Ed., 2014, 53, 6198–6201 CrossRef CAS PubMed; (g) T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588–9591 CrossRef CAS PubMed.
- For seminal works and reviews on CuH-catalysed formal hydroamination, see: (a) M. T. Pirnot, Y.-M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48–57 CrossRef CAS PubMed; (b) S. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 15913–15916 CrossRef CAS PubMed; (c) S. Zhu, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746–15749 CrossRef CAS PubMed; (d) Y. Miki, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 10830–10834 CrossRef CAS PubMed.
- M. Viganò, F. Ferretti, A. Caselli, F. Ragaini, M. Rossi, P. Mussini and P. Macchi, Chem. – Eur. J., 2014, 20, 14451–14464 CrossRef PubMed.
- (a) I. L. Fedushkin, A. A. Skatova, V. A. Chudakova and G. K. Fukin, Angew. Chem., Int. Ed., 2003, 42, 3294–3298 CrossRef CAS PubMed; (b) I. L. Fedushkin, O. V. Kazarina, A. N. Lukoyanov, A. A. Skatova, N. L. Bazyakina, A. V. Cherkasov and E. Palamidis, Organometallics, 2015, 34, 1498–1506 CrossRef CAS; (c) I. L. Fedushkin, O. V. Maslova, E. V. Baranov and A. S. Shavyrin, Inorg. Chem., 2009, 48, 2355–2357 CrossRef CAS PubMed.
- While this work was in progress, Trifonov also reported on the catalytic activity of yttrium complexes bearing a different Schiff base for hydroamination reactions: (a) A. A. Kissel, T. V. Mahrova, D. M. Lyubov, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, I. Del Rosal and L. Maron, Dalton Trans., 2015, 44, 12137–12148 RSC; (b) A. A. Kissel, D. M. Lyubov, T. V. Mahrova, G. K. Fukin, A. V. Cherkasov, T. A. Glukhova, D. Cui and A. A. Trifonov, Dalton Trans., 2013, 42, 9211–9225 RSC.
- While writing this paper we become aware that 3 has also been obtained by Fedushkin and coworkers by reducing 3,5-(CF3)2C6H3-BIAN with zinc in undried pyridine. However, the latter procedure requires heating at 110 °C for 10 h and affords a lower yield than ours: I. L. Fedushkin, A. A. Skatova, N. L. Bazyakina, V. A. Chudakova, N. M. Khvoinova, A. S. Nikipelov, O. V. Eremenko, A. V. Piskunov, G. K. Fukin and K. A. Lyssenko, Russ. Chem. Bull., 2013, 62, 1815–1828 CrossRef CAS.
- I. L. Fedushkin, V. A. Chudakova, G. K. Fukin, S. Dechert, M. Hummert and H. Schumann, Russ. Chem. Bull., 2004, 53, 2744–2750 CrossRef CAS.
- I. L. Fedushkin, N. M. Khvoinova, A. Y. Baurin and G. K. Fukin, Russ. Chem. Bull., 2006, 55, 451–456 CrossRef CAS.
- B. Wang, D. Wang, D. Cui, W. Gao, T. Tang, X. Chen and X. Jing, Organometallics, 2007, 26, 3167–3172 CrossRef CAS.
- Y. Chapurina, H. Ibrahim, R. Guillot, E. Kolodziej, J. Collin, A. Trifonov, E. Schulz and J. Hannedouche, J. Org. Chem., 2011, 76, 10163–10172 CrossRef CAS PubMed.
- F. Estler, G. Eickerling, E. Herdtweck and R. Anwander, Organometallics, 2003, 22, 1212–1222 CrossRef CAS.
- C. Queffelec, F. Boeda, A. Pouilhes, A. Meddour, C. Kouklovsky, J. Hannedouche, J. Collin and E. Schulz, ChemCatChem, 2011, 3, 122–126 CrossRef CAS.
- I. Aillaud, J. Collin, C. Duhayon, R. Guillot, D. Lyubov, E. Schulz and A. Trifonov, Chem. – Eur. J., 2008, 14, 2189–2200 CrossRef CAS PubMed.
- I. Aillaud, K. Wright, J. Collin, E. Schulz and J.-P. Mazaleyrat, Tetrahedron: Asymmetry, 2008, 19, 82–92 CrossRef CAS.
- (a) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1203 CrossRef CAS PubMed; (b) D. J. Tempel, L. K. Johnson, R. L. Huff, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2000, 122, 6686–6700 CrossRef CAS; (c) J. Merna, Z. Host'alek, J. Peleska and J. Roda, Polymer, 2009, 50, 5016–5023 CrossRef CAS; (d) A. Meduri, T. Montini, F. Ragaini, P. Fornasiero, E. Zangrando and B. Milani, ChemCatChem, 2013, 5, 1170–1183 CrossRef CAS.
- (a) A. Scarel, M. R. Axet, F. Amoroso, F. Ragaini, C. J. Elsevier, A. Holuigue, C. Carfagna, L. Mosca and B. Milani, Organometallics, 2008, 27, 1486–1494 CrossRef CAS; (b) F. Ragaini, S. Cenini and S. Tollari, J. Mol. Catal., 1993, 85, L1–L5 CrossRef CAS; (c) F. Ragaini, S. Cenini, S. Tollari, G. Tummolillo and R. Beltrami, Organometallics, 1999, 18, 928–942 CrossRef CAS; (d) F. Ragaini, S. Cenini and M. Gasperini, J. Mol. Catal. A: Chem., 2001, 174, 51–57 CrossRef CAS.
- (a) M. Gasperini, F. Ragaini and S. Cenini, Organometallics, 2002, 21, 2950–2957 CrossRef CAS; (b) R. van Asselt, C. J. Elsevier, W. J. J. Smeets, A. L. Spek and R. Benedix, Recl. Trav. Chim. Pays-Bas, 1994, 113, 88–98 CrossRef CAS.
- S. W. Hong, S. Tian, M. V. Metz and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 14768–14783 CrossRef CAS PubMed.
- J. Y. Kim and T. Livinghouse, Org. Lett., 2005, 7, 1737–1739 CrossRef CAS PubMed.
- B. D. Stubbert and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 4253–4271 CrossRef CAS PubMed.
- P. D. Knight, I. Munslow, P. N. O'Shaughnessy and P. Scott, Chem. Commun., 2004, 894–895 RSC.
- D. V. Gribkov, K. C. Hultzsch and F. Hampel, J. Am. Chem. Soc., 2006, 128, 3748–3759 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The synthetic procedure and spectroscopic data for Ar-BIAN, full NMR spectra for new Ar-BIANH2, spectroscopic data for zirconium and yttrium complexes and for organic hydroaminated products, and full crystallographic data. CCDC 1431252. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6nj02199a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |