
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Calixarenes with naphthalene units: calix[4]naphthalenes and hybrid[4]arenes†
T.
Boinski
a,
A.
Cieszkowski
a,
B.
Rosa
a,
B.
Leśniewska
b and
A.
Szumna
*a
aInstitute of Organic Chemistry, Polish Academy of Science, Kasprzaka 44/52, 01-244 Warsaw, Poland. E-mail: agnieszka.szumna@icho.edu.pl
bInstitute of Physical Chemistry, Polish Academy of Science, Kasprzaka 44/52, 01-244 Warsaw, Poland
First published on 20th September 2016
Abstract
Macrocycles consisting of naphthalene units connected via methylene bridges offer certain advantages as macrocylic scaffolds as compared with parent calixarenes. These advantages originate from their electron rich and enlarged cavities. Additionally, macrocycles containing 1,3-bridged naphthalene units are dissymmetric and therefore they present interesting stereochemical features, including inherent chirality. We describe here a facile, one-step synthesis of two new calix[4]naphthalenes, by the condensation of 1,6-dimethoxynaphthalene with formaldehyde catalyzed by Brønsted acid (TFA). Additionally, we show the first example of hybrid[n]arene containing a 1,6-dimethoxynaphthalene unit and 1,3-dimethoxybenzene units obtained by a simple, one-pot condensation. The conformational and complexation properties were also studied for the resulting hybrid macrocycles.
Introduction
The emergence of a new versatile macrocyclic building block that enables build-up of the functional architecture on its semi-rigid scaffold often ignites new research directions in supramolecular chemistry and many related areas. Versatile macrocycles like calix[n]arenes, cucurbit[n]urils, cyclodextrines, cyclotriveratrylenes and calixpyrroles have been discovered decades ago and widely exploited since then.1 For years chemists concentrated on the functionalization of the existing scaffolds rather than on hunting for new ones, because the existing scaffolds offered almost unlimited possibilities. However, with the discovery of pillar[5]arene, a completely new, yet simple and very useful macrocylic building block, by the group of Ogoshi and Nakamoto in 2008,2 the quest for new polyphenolic macrocycles has received a new inspiration. Since then the group of Stoddart obtained a family of electron-rich macrocycles known as asar[n]arenes.3 We4 and others5–10 have synthesized a family of hybrid[n]arenes containing different polyphenols embedded within one macrocycle. Many new non-phenolic methylene-bridged macrocycles have also been synthesized recently, for example calix[3]carbazoles.11 Great interest in new macrocycles and their almost immediate applications indicate that new semi-rigid supramolecular scaffolds are still sought-after.Calix[n]naphthalenes, i.e. macrocycles consisting of naphthalene units connected via methylene bridges at their meta-positions, may offer some advantages as macrocylic scaffolds originating from enlarged cavity with electron rich aromatic surfaces. Additionally, due to naphthalene geometry, the resulting macrocycles are dissymmetric and therefore they may present interesting stereochemical features, including inherent chirality. Indeed, extended cavity features of calix[n]naphthalenes have been exploited for the complexation of fullerenes.12 However, the number of the family members of calix[n]naphthalenes is small. They include macrocycles obtained from 1-hydroxynaphththalene13 and 2-hydroxynaphthalene14 and various macrocycles obtained from 2,7-dialkoxynaphthalenes15,16 or chromotropic acid.17
Recently, we have found that methylene bridged polyphenolic macrocycles, for example pillar[5]arenes and hybrid[4]arenes, can be synthesized by a Brønsted acid-mediated direct reaction of alkoxybenzenes with formaldehyde.4,18 The important finding was that the reaction is reversible when it is catalysed by a Brønsted acid (TFA in that case). It enabled an efficient solvent-templated synthesis of deca-O-alkylpillar[5]arenes and rearrangements of already-formed homo-macrocycles to thermodynamically preferred hybrid macrocycles. In the current paper we show that such thermodynamically controlled reactions can be used not only for substrates containing single aromatic rings but also for the formation of new naphthalene-based macrocycles.
Results and discussion
In order to obtain hybrid macrocycles containing naphthalene units we have used a one pot TFA-catalysed macrocylization reaction of formaldehyde with 1,6-dimethoxynaphthalene 1b and 1,3-dimethoxybenzene 1a (Fig. 1). Our initial approach concentrated on macrocycles having the same number of units of each type, therefore we have used a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of polyphenolic building blocks. However, in this case we observed the preferential formation of a macrocycle that contains one 1,6-dimethoxynaphthalene unit and three 1,3-dimethoxybenzene units, as it was established based on the NMR spectrum and mass spectrometry. 1H–13C HSQC and 1H–13C HMBC spectra indicate that methylene bridges link the macrocycle at 2,4 positions of the naphthalene unit, suggesting structure 2. Considering the [1+3] stoichiometry of product 2 we adjusted the stoichiometry of reagents accordingly. This way hybrid [1+3] macrocycle 2 was obtained in 47% yield.
:1 ratio of polyphenolic building blocks. However, in this case we observed the preferential formation of a macrocycle that contains one 1,6-dimethoxynaphthalene unit and three 1,3-dimethoxybenzene units, as it was established based on the NMR spectrum and mass spectrometry. 1H–13C HSQC and 1H–13C HMBC spectra indicate that methylene bridges link the macrocycle at 2,4 positions of the naphthalene unit, suggesting structure 2. Considering the [1+3] stoichiometry of product 2 we adjusted the stoichiometry of reagents accordingly. This way hybrid [1+3] macrocycle 2 was obtained in 47% yield.
 | ||
| Fig. 1 Synthesis of hybrid[4]arene 2. | ||
Since 1,6-dimethoxynaphthalene 1b has not been previously used in macrocylization reactions with formaldehyde, we have also tested it under the conditions that may lead to the formation of homo-macrocycles. The reaction of 1,6-dimethoxynaphthalene 1b with formaldehyde in DCE catalysed by TFA gave two main products in similar yields, both of them being tetramers, as indicated by mass spectrometry. One of the products exhibits highly symmetric 1H and 13C NMR spectra with only a single 1,6-dimethoxynaphthalene unit being symmetrically independent. 1H–13C HSQC and 1H–13C HMBC spectra suggest that methylene bridges link the macrocycle at 2,4 positions of the 1,6-dimethoxynaphthalene unit, thus the product has C4-symmetric structure 3 (Fig. 2). In contrast, 1H and 13C NMR spectra of the second tetrameric product show that all 1,6-dimethoxynaphthalene units are symmetrically non-equivalent. Analysis of 2D NMR spectra suggests a C1-symmetric structure, 4, for the second tetrameric product (Fig. 2). Calix[4]naphthalene 4 is a regioisomer of calix[4]naphthalene 3 and differs by the orientation of a single naphthalene unit. The optimization of the synthetic procedure by changing the concentration, solvent, reaction time and amount of catalyst resulted in obtaining 3 and 4 in a maximum yield of 15% each (Table 1).
 | ||
| Fig. 2 Synthesis of calix[4]naphthalenes 3 and 4. | ||
| Entry | Solvent | C of 1b/M | Time/h | TFA/% | Yield of 3/% | Yield of 4/% |
|---|---|---|---|---|---|---|
| 1 | DCE | 0.1 | 18 | 5 | 12 | 13 |
| 2 | DCE | 0.1 | 4 | 5 | 14 | 12 |
| 3 | DCE | 0.1 | 2 | 5 | 15 | 15 |
| 4 | DCE | 0.5 | 2 | 5 | 8 | 9 |
| 5 | DCE | 0.025 | 2 | 25 | 14 | 15 |
| 6 | DCE | 0.1 | 1 | 1.25 | 8 | 7 |
| 7 | CHCl3 | 0.1 | 2 | 5 | 13 | 15 |
| 8 | DCE | 0.1 | 2 | 40 | 10 | 12 |
| 9 | DCE | 0.1 | 2 | 20 | 12 | 13 |
| 10 | DCE | 0.1 | 2 | 2 | 11 | 12 |
| 11 | DCE | 0.1 | 2 | 1 | 5 | 6 |
Calix[n]arenes exhibit various degrees of conformational flexibility that depend mainly on steric hindrance within macrocyclic rings and on additional interactions (e.g. stabilizing intramolecular hydrogen bonding). For calixarenes existing in fixed or slowly interconverting (at the NMR timescale) cone conformations the bridging methylene protons are found as a pair of doublets in the NMR spectra. Here, for macrocycles 2–4 the methylene bridges are found as broad singlets in their NMR spectra at 303 K (ESI†). Moreover, at 218 K the signals get broader but they do not split (ESI†). It indicates that the macrocyclic rings either: (i) exhibit conformations rapidly inverting on the NMR time scale or (ii) exhibit static conformations with geminal protons related by symmetry. Due to a substitution pattern of the building units and the fact that a planar structure of the macrocycle is not possible, we conclude that symmetry-based relationship between geminal protons is not possible. Therefore, the macrocyclic 16-membered rings reported here exhibit dynamic conformations in solution, similar to other calixarenes that are devoid of stabilizing hydrogen bonding interactions, i.e. OH-depleted calix[4]arenes and per-O-alkylated resorcinarenes. The current findings are also in agreement with our previous conclusions for hybrid[n]arenes, which claimed considerable conformational flexibility of 1,3-bridged units within medium-sized macrocyclic rings.4 Conformational lability of macrocycles 2–4 determines their stereochemical properties. Due to dissymmetric naphthalene substitution patterns, macrocycles 2–4 have a possibility to exhibit inherent chirality. However, inherent chirality additionally requires hindered rotation about single bonds in order to block eversion of macrocyclic rings.19 For macrocycles 2–4 the rotation about single bonds is not hindered, so the current products are achiral. However, in the presence of a proper guest or after a chemical modification that will hinder the rotation, macrocycles 2–4 can become chiral.
We were able to grow single crystals of 4 suitable for X-ray analysis. In the solid state 4 exhibits a boat conformation with two opposite naphthalene rings positioned orthogonally to the main macrocyclic plane and almost parallel to each other (Fig. 3). Interestingly, two methyl substituents from the parallel naphthalene units point towards the cavity and towards the opposite lying aromatic rings. This CH⋯π interaction seems to be favourable and stabilizing for this conformation. The other two naphthalene rings are positioned roughly within the plane of the macrocyclic ring, however, they are substantially distorted.
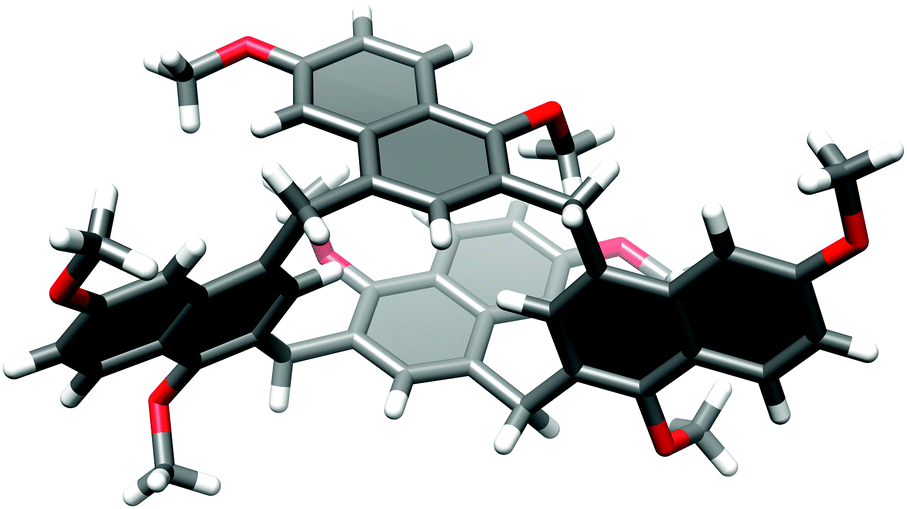 | ||
| Fig. 3 X-ray structure of 4 (for the ORTEP plot see Fig. S1, ESI†). | ||
Even though macrocycle 4 possesses a very small cavity in the boat conformation found in the solid state, the dynamic character of the macrocyclic ring in solution may enable guest complexation. The recognition properties of macrocycles 2–4 were tested using guests rac-5 and 6 containing imidazolium and pyridinium ions respectively (Fig. 4). The guest molecules were chosen using the criterion of importance (the vast presence of imidazolium and pyridinium fragments in bioactive substances and in structural motifs of topologically bound molecules) and possible favourable non-covalent interactions between electron-rich aromatic walls and organic cations (cation–π interactions). Initial screening was done using equimolar mixtures of the host and the guest in CDCl3. For both guests rac-5 and 6 a complexation induced shift was detected upon interaction with hosts 2–4 (Fig. 5–7). For guest rac-5 the changes were the most pronounced during the interaction with all hosts. In all cases Hc protons of rac-5 exhibited a downfield shift, while protons Ha and Hb exhibited upfield shifts. The downfield shift of protons is usually observed during the formation of hydrogen bonds or during interactions with edges of π-surfaces, while the upfield shift is often observed for interactions with central parts of π-surfaces. In the current case hydrogen bonding is less likely and we suggest that the observed changes come mainly from the interactions between the electron-rich π-surfaces of the hosts and the electron-poor π-surface of rac-5.
 | ||
| Fig. 4 Complexation studies: (a) chemical structures of the guests; (b) titration of 3 by rac-5 (CDCl3, 298 K, C(3) = 15.5 mM); (c) titration of 3 by 6 (CDCl3, 298 K, C(3) = 14.4 mM). Points represent experimental data, lines represent fitted curves. | ||
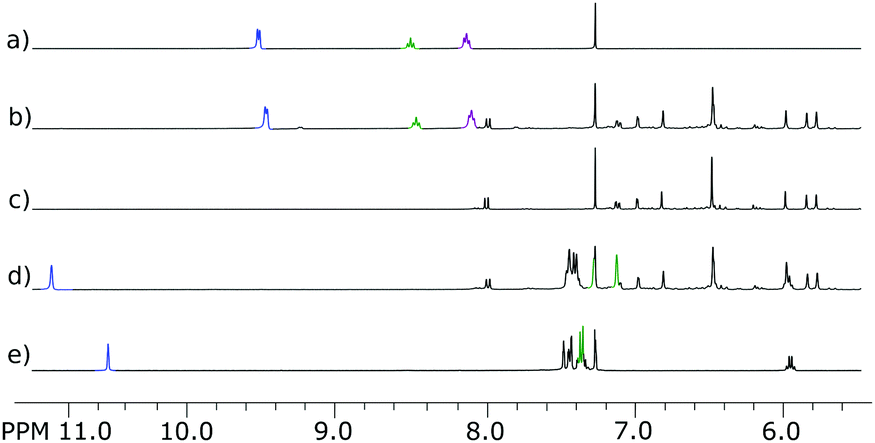 | ||
| Fig. 5 Complexation properties of hybrid[4]arene 2: (a) 6; (b) 2 + 6 (C(2) = 14.41 mM, C(6) 15.64 mM); (c) 2; (d) 2 + 5 (C(2) = 15.49 mM, C(5) = 15.63 mM); (e) 5 (all in CDCl3 at 298 K). | ||
 | ||
| Fig. 6 Complexation of 5 by calix[4]naphthalenes: (a) 4; (b) 4 + 5 (C(4) = 5.36 mM, C(5) = 5.23 mM); (c) 5; (d) 3 + 5 (C(3) = 15.49 mM, C(5) = 15.49 mM); (e) 3 (all in CDCl3 at 298 K). | ||
 | ||
| Fig. 7 Complexation of 6 by calix[4]naphthalenes: (a) 4; (b) 4 + 6 (C(4) = 7.03 mM, C(6) = 6.99 mM); (c) 6; (d) 3 + 6 (C(3) = 14.43 mM, C(6) = 15.63 mM); (e) 3 (all in CDCl3 at 298 K). | ||
Interactions between electron-rich and electron-poor aromatic surfaces may involve the formation of a charge-transfer complex and induce substantial changes in the UV/Vis spectra. Additionally, due to the presence of a 1,6-dimethoxynaphthalene unit the macrocycles exhibit fluorescence. The UV/Vis and fluorescence spectra were recorded for macrocycles 2–4 (Fig. 8), their complexes with rac-5 and 6 (Fig. S36–S42, ESI†) and 1b as a reference. Examination of the possible appearance of new CT bands, shifts of the existing bands in UV/Vis and fluorescence spectra, changes in molar absorption coefficients or quantum yields did not show considerable changes upon complexation. These results indicate that charge transfer processes are not pronounced for current complexes.
 | ||
| Fig. 8 UV/Vis (a) and fluorescence (b) spectra of macrocycles 2–4 and reference compound 1b (all in CHCl3 at 298 K). | ||
Association constants were determined by 1H NMR titration followed by a non-linear curve fitting as implemented in HYPNMR.20 All signals that exhibited detectable changes in chemical shifts were fitted simultaneously. Reasonable fits have only been obtained by assuming the formation of 1:1 and 1:2 (H:G) complexes. The results indicate that in all cases macrocycles 2–4 exhibited considerable binding constants towards positively charged organic ions (Table 2), albeit with moderate selectivity.
| rac-5 | 6 | |||
|---|---|---|---|---|
| logβ1 |
logβ2 |
logβ1 |
logβ2 |
|
| 2 | 1.55 | 3.97 | 2.32 | 4.92 |
| 3 | 1.99 | 3.93 | 1.53 | 3.96 |
| 4 | 3.08 | 4.50 | 1.63 | 4.88 |
Experimental
2
To a solution of 1a (7.5 mmol, 1.035 g) and 1b (2.5 mmol, 0.47 g) in DCE (100 ml), paraformaldehyde (10 mmol, 0.3 g) and TFA (5 ml) were added. The solution was refluxed for 12 h. During the reaction the colour changed from colourless to dark purple. The reaction was cooled down to rt and excess of Na2CO3 was added in order to neutralize TFA. The mixture was filtered off and evaporated to dryness. The product was purified by chromatography (30 g silica gel, CHCl3/ethyl acetate from 100:0 to 100:2). Yield: 47% (0.764 g). 1H NMR (CDCl3, 400 MHz, 298 K): 7.99 (d, 1H, 3J = 9 Hz), 7.11 (dd, 1H, 3J = 9 Hz, 4J = 2 Hz), 6.98 (d, 1H, 4J = 2 Hz); 6.81 (s, 1H), 6.47 (s, 3H), 5.98 (s, 1H), 5.84 (s, 1H), 5.77 (s, 1H), 4.07 (s, 2H), 3.95 (s, 2H), 3.90 (s, 3H), 3.89 (s, 3H), 3.86 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 3.74 (s, 3H), 3.65 (s, 2H), 3.64 (s, 3H), 3.60 (s, 2H), 3.22 (s, 3H). 13C NMR (CDCl3, 100 MHz, 298 K): 157.02, 156.93, 156.88, 155.99, 155.96, 155.88, 155.48, 153.15, 133.82, 132.81, 131.53, 131.22, 129.20, 128.67, 125.27, 123.88, 123.40, 121.54, 120.80, 120.63, 119.81, 119.61, 119.53, 117.28, 104.01, 95.15, 94.49, 94.34, 62.06, 55.81, 55.70, 55.65, 55.64, 55.48, 55.34, 55.08, 31.82, 28.72, 28.54, 28.16. MS (m/z): HRMS (ESI) calcd for C40H42O8Na ([M + Na]+) 673.2777, found 673.2767.
3 and 4
To a solution of 1b (5 mmol, 0.94 g) in CHCl3 (50 ml), paraformaldehyde (5 mmol, 0.15 g) and TFA (5 ml) were added. The reaction was refluxed for 12 h. During the reaction the colour changed from colourless to dark purple. The reaction was cooled down to rt and excess of Na2CO3 was added in order to neutralize TFA. The mixture was filtered off and evaporated to dryness. The products were isolated by chromatography (30 g silica gel, CHCl3/ethyl acetate from 100:0 to 100:2). Yield: 3: 15% (150 mg) and 4: 15% (152 mg). 3: 1H NMR (400 MHz, 298 K): 7.97 (d, 4H, 3J = 9 Hz), 7.16 (d, 4H, 4J = 2 Hz), 7.15(dd, 4H, 3J = 9 Hz, 4J = 2 Hz), 6.69 (s, 4H), 4.37 (s, 8H), 3.76 (s, 12H), 3.44 (s, 12H). 13C NMR (100 MHz, 298 K): 157.61, 152.72, 133.45, 131.42, 129.49, 124.75, 124.21, 123.27, 117.78, 103.55, 61.76, 55.22, 32.16 MS (m/z): HRMS (ESI) calcd for C52H48O8Na ([M + Na]+) 823.3231, found 823.3247.
4
1H NMR (400 MHz, 298 K): 8.13 (d, 1H, 3J = 9 Hz), 8.10 (d, 1H, 3J = 9 Hz), 7.82 (d, 1H, 3J = 9 Hz), 7.77 (d, 1H, 3J = 9 Hz), 7.36 (m, 2H), 7.23 (m, 2H), 6.97 (m, 1H), 6.96 (s, 1H), 6.90(d, 1H, 4J = 2 Hz), 6.86 (s, 1H), 6.83 (d, 1H, 4J = 2 Hz), 6.31 (s, 1H), 6.22 (s, 1H), 4.52 (s, 2H), 4.35 (s, 2H), 4.29 (s, 2H), 4.21 (s, 2H), 4.01 (s, 3H), 3.99 (s, 3H), 3.94 (s, 3H), 3.93 (s, 3H), 3.59 (s, 3H), 3.50 (s, 3H), 3.04 (s, 3H), 2.82 (s,3H). 13C NMR (100 MHz, 298 K): 157.74, 157.71, 157.58, 157.28, 153.52, 153.42, 152.60, 151.79, 133.87, 133.30, 133.24, 133.08, 131.98, 131.78, 131.23, 130.73, 130.23, 128.34, 127.16, 126.34, 125.79, 125.38, 124.78, 124.40, 124.38, 124.27, 124.25, 124.03, 123.41, 123.30, 123.14, 122.34, 117.95, 117.80, 117.42, 117.20, 104.23, 104.17, 102.62, 102.61, 99.99, 61.66, 61.62, 61.43, 55.34, 55.28, 55.22, 55.03, 35.33, 32.85, 31.61, 27.88. MS (m/z): HRMS (ESI) calcd for C52H48O8Na ([M + Na]+) 823.3231, found 823.3239.Crystal data for 4
C52H48O8·CH2Cl2, M = 885.83, 0.25 × 0.07 × 0.07 mm3, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2), a = 12.8554(6) Å, b = 13.0685(6) Å, c = 13.4125(6) Å, α = 85.321(4)°, β = 83.005(4)°, γ = 82.019(4)°, V = 2210.08(18) Å3, Z = 2, Dc = 1.331 g cm−3, F000 = 932, SuperNova, Dual, Cu at zero, Eos, CuKα radiation, λ = 1.54178 Å, T = 173(2) K, 2θmax = 144.6°, 33685 reflections collected, 8567 unique (Rint = 0.0493). Final GooF = 1.033, R1 = 0.0616, wR2 = 0.1655, R indices based on 5928 reflections with I > 2σ(I) (refinement on F2), 576 parameters, 0 restraints. Lp and absorption corrections applied, μ = 1.784 mm−1. CCDC 1474998.
(No. 2), a = 12.8554(6) Å, b = 13.0685(6) Å, c = 13.4125(6) Å, α = 85.321(4)°, β = 83.005(4)°, γ = 82.019(4)°, V = 2210.08(18) Å3, Z = 2, Dc = 1.331 g cm−3, F000 = 932, SuperNova, Dual, Cu at zero, Eos, CuKα radiation, λ = 1.54178 Å, T = 173(2) K, 2θmax = 144.6°, 33685 reflections collected, 8567 unique (Rint = 0.0493). Final GooF = 1.033, R1 = 0.0616, wR2 = 0.1655, R indices based on 5928 reflections with I > 2σ(I) (refinement on F2), 576 parameters, 0 restraints. Lp and absorption corrections applied, μ = 1.784 mm−1. CCDC 1474998.
Conclusions
There is a continuing need for new semi-rigid macrocyclic compounds for the construction of new supramolecular systems and for their further modifications and application as “biased” architectures.21,22 Following these needs we have demonstrated the synthesis of two new regioisomeric calix[4]naphthalenes, by a one-step condensation of 1,6-dimethoxynaphthalene with formaldehyde catalysed by Brønsted acid (TFA). Additionally, we have also shown the first example of a hybrid[n]arene containing a 1,6-dimethoxynaphthalene unit and 1,3-dimethoxybenzene units obtained by a simple, one-pot condensation. The newly obtained macrocycles exhibit pronounced affinities for organic cations.Acknowledgements
This work was supported by the National Science Center (grant UMO-2013/09/N/ST5/00907).Notes and references
- Supramolecular Chemistry: From Molecules to Nanomaterials, ed. J. W. Steed and P. A. Gale, Wiley, 2012 Search PubMed.
- T. Ogoshi, S. Kanai, S. Fujunami, T. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022 CrossRef CAS PubMed.
- S. T. Schneebeli, C. Cheng, K. J. Hartlieb, N. L. Strutt, A. A. Sarjeant, C. L. Stern and J. F. Stoddart, Chem. – Eur. J., 2013, 19, 3860 CrossRef CAS PubMed.
- T. Boinski, A. Cieszkowski, B. Rosa and A. Szumna, J. Org. Chem., 2015, 80, 3488 CrossRef CAS PubMed.
- Z. Zhang, J. M. Lim, M. Ishida, V. V. Roznyatovskiy, V. M. Lynch, H. Y. Gong, X. Yang, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2012, 134, 4076 CrossRef CAS PubMed.
- J. L. Sessler, W. S. Cho, V. Lynch and V. Kral, Chem. – Eur. J., 2002, 8, 1134 CrossRef CAS.
- G. Cafeo, F. H. Kohnke, G. L. La Torre, M. F. Parisi, R. P. Nascone, A. J. P. White and D. J. Williams, Chem. – Eur. J., 2002, 8, 3148 CrossRef CAS.
- M. Y. Song, H. K. Na, E. Y. Kim, S. J. Lee, K. I. Kim, E. M. Baek, H. S. Kim, D. K. Ana and C. H. Lee, Tetrahedron Lett., 2004, 45, 299 CrossRef CAS.
- J. Cho, S. Lee, S. Hwang, S. H. Kim, J. S. Kim and S. Kim, Eur. J. Org. Chem., 2013, 4614 CrossRef CAS.
- F. Troisi, L. Mogavero, C. Gaeta, E. Gavuzzo and P. Neri, Org. Lett., 2007, 9, 915 CrossRef CAS PubMed.
- P. Yang, Y. Jian, X. Zhou, G. Li, T. Deng, H. Shen, Z. Yang and Z. Tian, J. Org. Chem., 2016, 81, 2974 CrossRef CAS PubMed.
- M. Ashram, S. Mizyed and P. E. Georghiou, J. Org. Chem., 2001, 66, 1473 CrossRef CAS PubMed; S. Mizyed, P. R. Tremaine and P. E. Georghiou, J. Chem. Soc., Perkin Trans. 2, 2001, 3 RSC; P. E. Georghiou, S. Mizyed and S. Chowdhury, Tetrahedron Lett., 1999, 40, 611 CrossRef.
- P. E. Georghiou, M. Ashram, Z. Li and S. G. Chaulk, J. Org. Chem., 1995, 60, 7284 CrossRef CAS.
- S. Chowdhury and P. E. Georghiou, J. Org. Chem., 2002, 67, 6808 CrossRef CAS PubMed.
- B. J. Shorthill, R. G. Granucci, D. R. Powell and T. E. Glass, J. Org. Chem., 2002, 67, 904 CrossRef CAS PubMed.
- B. J. Shorthill and T. E. Glass, Org. Lett., 2001, 3, 577 CrossRef CAS PubMed.
- B.-L. Poh, C. S. Lim and K. S. Khoo, Tetrahedron Lett., 1989, 30, 1005 CrossRef CAS.
- T. Boinski and A. Szumna, Tetrahedron, 2012, 68, 9419 CrossRef CAS.
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274 RSC.
- C. Frassineti, L. Alderighi, P. Gans, A. Sabatini, A. Vacca and S. Ghelli, Anal. Bioanal. Chem., 2003, 376, 1041 CrossRef CAS PubMed.
- J. L Atwood, L. J. Barbour and A. Jerga, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4837 CrossRef PubMed.
- D. A. Fowler, A. S. Rathnayake, S. Kennedy, H. Kumari, C. M. Beavers, S. J. Teat and J. L. Atwood, J. Am. Chem. Soc., 2013, 135, 12184 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and ESI MS spectra, titration experiments. CCDC 1474998. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6nj01736c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |