
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hexahalometallate salts of trivalent scandium, yttrium and lanthanum: cation–anion association in the solid state and in solution†
Martin J. D.
Champion
,
William
Levason
,
David
Pugh
and
Gillian
Reid
*
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 20th June 2016
Abstract
The hexahalide salts, [NnBu4]3[LaCl6], [BMPYRR]3[LaCl6] (BMPYRR = 1-butyl-1-methylpyrrolidinium), [EMIM]3[MX6] (EMIM = 1-ethyl-3-methylimidazolium; M = La, X = Cl, Br, I; M = Sc, Y, Ce, X = Cl) and [EDMIM]3[MX6] (EDMIM = 1-ethyl-2,3-dimethylimidazolium; M = Y, X = Cl; M = La, X = Cl, I) have been prepared and X-ray crystal structures determined for several of them, with a view to probing the effect of varying the trivalent metal ion, the halide and the counter-cation on the structures adopted in the solid state. The crystal structures of the EMIM and EDMIM salts show extensive H-bonding between the halide ligands and organic cations; based upon the H-bonding distances, this appears to be strongest for the [EMIM]3[MCl6] salts, becoming progressively weaker for heavier metal ion or halide. In terms of the cations, changing from EMIM to EDMIM also reduces the strength of the H-bonding. The strength of the cation–anion pairing in solution has also been probed in solution via NMR spectroscopy where possible (45Sc, 89Y and 189La) and, for the EMIM salts, via the shift of δ(H2) relative to [EMIM]Cl at a standard concentration. The trends observed in solution mirror those determined in the solid state.
Introduction
In the course of our work on electrodeposition of metals and semiconductors from unusual low polarity solvents, such as CH2Cl21 and supercritical CH2F2,2 we have established that halometallates can often offer both the solubility and extent of dissociation into discrete cations and anions to be highly effective as reagents for delivering the elements of choice. Taking germanium as an example, we have shown that the cation, halide and anion charge all play an important role in determining the extent of cation–halogermanate anion association both in the solids and in solution.3 While we have often used quaternary ammonium salts for this purpose, recent work on the electrodeposition of elemental germanium from supercritical CH2F2 has established that using the 1-ethyl-3-methylimidazolium (EMIM) cation leads to improved quality of the electrodeposited germanium,4 and hence this and other substituted imidazolium cation salts, which are also used extensively for electrodeposition from ionic liquids,5 have become of greater importance in this area of work.In order to gain a better understanding of the cation–anion association in halometallate salts, we describe here a study of the [MX6]3− trianions incorporating a range of trivalent metal ions from group 3 and the lanthanides, in the anticipation that the trianionic charge would give rise to significant cation–anion association both in the solid state and in low polarity solvents. We report the synthesis of a series of halometallate anion salts of scandium, yttrium and several lanthanides. All complexes have been characterised by NMR spectroscopy (1H, 45Sc, 89Y and 139La as appropriate), IR spectroscopy, elemental analysis and, in several cases, single-crystal X-ray diffraction. To gain further insight into the cation–anion pairing in these species, we have explored the effects that systematic changes in the counter-cation, metal ion and halide co-ligands have on the solid-state structures adopted and the spectroscopic behaviour of these halometallate salts in MeCN (and where possible, CH2Cl2) solution.
A small number of earlier studies report structural data on halometallate salts of the Ln(III) ions. The examples most pertinent to the present study are [EMIM]3[LnCl6] (Ln = La, Pr, Nd, Sm, Eu) grown from hydrated LnCl3 in [EMIM]Cl at 110 °C,6,7 [MPPYRR]3[NdI6] (MPPYRR = 1-methyl-1-propylpyrrolidinium),8 and [SEt3]3[LnI6] (Ln = Nd, Sm), obtained from reaction of LnI2 with the ionic liquid [SEt3][NTf2] (NTf2 = bis-(trifluoromethanesulfonyl)imide).9 A very recent study has described the structures of a series of [BMIM]3[CeX6] salts (BMIM = 1-butyl-3-methylimidazolium), also obtained directly from CeX3 in the [BMIM]X ionic liquids, which are reported to exhibit intense Ce3+ based 5d–4f-centred emission.10
Results and discussion
Preparations
The hexahalide salts, [NnBu4]3[LaCl6], [BMPYRR]3[LaCl6] (BMPYRR = 1-butyl-1-methylpyrrolidinium, [EMIM]3[MX6] (M = La, X = Cl, Br, I; M = Sc, Y, Ce, X = Cl) and [EDMIM]3[MX6] (EDMIM = 1-ethyl-2,3-dimethylimidazolium; M = Y, X = Cl; M = La, X = Cl, I), were selected for study on the basis that they would allow us to probe the effects that the cation (extent of H-bonding or not), the metal ion size and the halide type have upon the solid state and solution properties. The salts were readily prepared as powdered solids in good yield by treatment of anhydrous MX3 with three (or slightly greater) molar equivalents of the appropriate halide salt of the organic cation, [NnBu4]+, [BMPYRR]+, [EMIM]+ or [EDMIM]+) in MeCN, acetone or CH2Cl2 solution, and their formulations were confirmed by microanalyses. The isolated salts are moderately soluble in MeCN and, in some cases, in CH2Cl2 solution. Crystals of several examples with EMIM and EDMIM cations were obtained by vapour diffusion of Et2O into a concentrated MeCN solution of the relevant salt.Varying the halide
In order to assess the effect of varying the halide on the nature of the hydrogen bonding interactions, the [EMIM]3[LaX6] system (X = Cl, Br, I) was chosen. [EMIM]3[LaCl6] had previously been synthesised and crystallographically characterised.6,7 Crystals of the bromide (Fig. 1) and iodide (Fig. 2) analogues were obtained by slow diffusion of Et2O into an MeCN solution. Surprisingly these are rare examples of discrete [LaX6]3− anions with organic cations: [2,4,6-trimethylpyridinium]4[LaCl6]Cl11 and [BMPYRR]4[LaI6][NTf2]12 are the only other structurally authenticated complexes.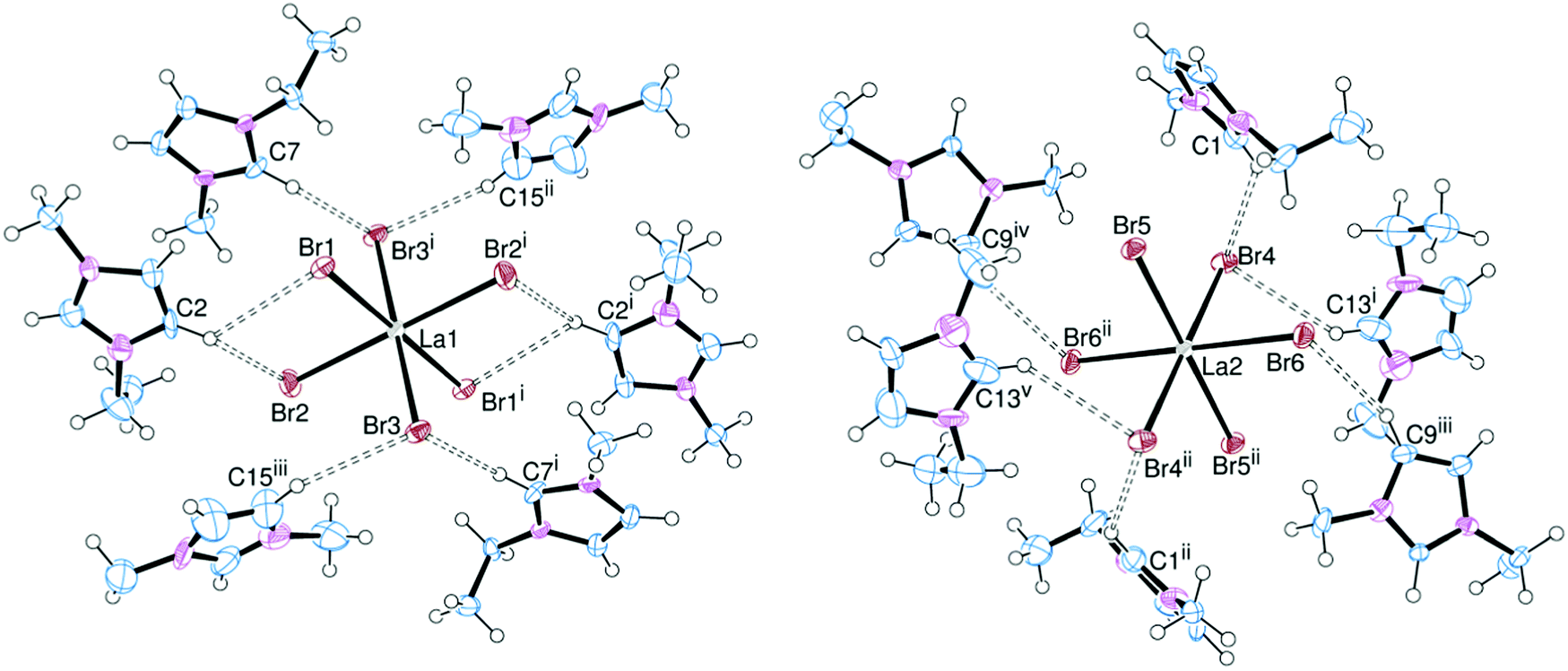 | ||
| Fig. 1 ORTEP representation of [EMIM]3[LaBr6] showing the hydrogen bonding (dotted lines) surrounding the La1-centred cation (left) and the La2-centred cation (right). Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å): La1–Br1 2.9371(13), La1–Br2 2.9296(16), La1–Br3 2.9534(13), La2–Br4 2.9477(15), La2–Br5 2.9484(14), La2–Br6 2.9524(13). Symmetry codes: (i) 2 − x, 1 − y, 1 − z; (ii) 1 − x, 1 − y, −z. | ||
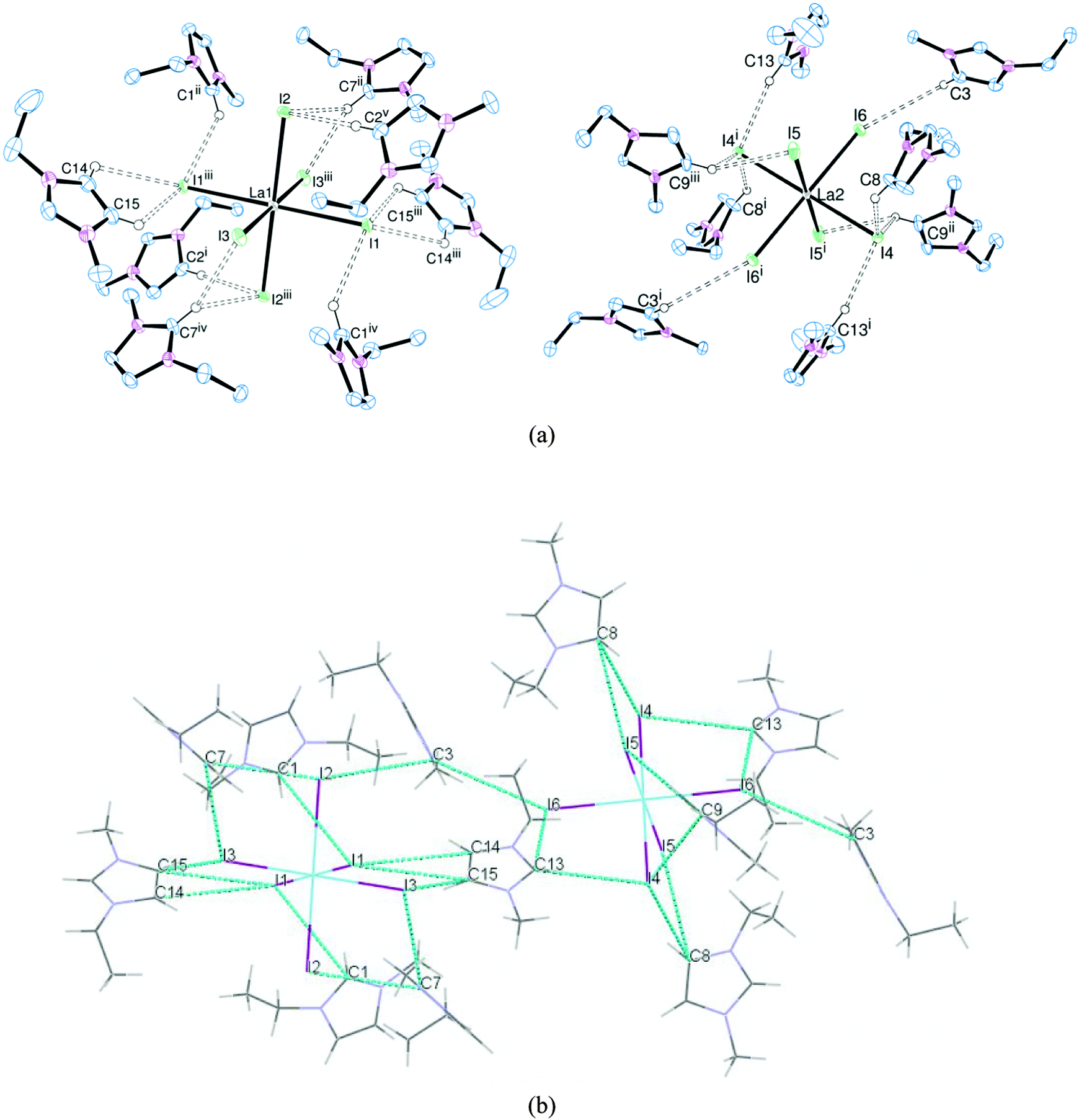 | ||
| Fig. 2 (a) ORTEP representation of [EMIM]3[LaI6] showing the hydrogen bonding (dotted lines) surrounding the La1-centred cation (left) and the La2-centred cation (right). Displacement ellipsoids are drawn at the 50% probability level and H atoms not involved in hydrogen bonding are omitted for clarity. Selected bond lengths (Å): La1–I1 3.1557(5), La1–I2 3.1448(4), La1–I3 3.1930(8), La2–I4 3.1988(5), La2–I5 3.1663(5), La2–I6 3.1733(10). Symmetry codes: (i) 1 − x, 3 − y, 1 − z; (ii) −x, 2 − y, 1 − z; (iii) x, 2.5 − y, 1.5 − z; (iv) 1 − x, 0.5 − y, 1 − z; (v) 1 − x, 0.5 − y, 1.5 − z. (b) Representation of [EMIM]3[LaI6] showing the extended structure generated by the hydrogen bonding (dotted lines). | ||
[EMIM]3[LaCl6] and [EMIM]3[LaBr6] are isomorphous and isostructural, whereas [EMIM]3[LaI6] crystallises with a different unit cell, presumably reflecting the increased steric requirements of I− over Br− and Cl−. Nonetheless, in each case the [LaX6]3− anion is octahedrally coordinated with six halides comprising the primary coordination sphere. Two crystallographically independent [LaX6]3− anions were observed in the solid state with different arrangements of hydrogen bonding cations, including bifurcated hydrogen bonding, accounting for the difference. Most of the hydrogen bonding occurs through the acidic NCN proton of the imidazolium ring but interactions through the NCCN backbone protons (which are less acidic) were also observed, resulting in an extended structure in the solid state (Fig. 2(b)).
Here it should be noted that in our experience, the criteria used for assigning the strength of hydrogen bonds13 are not always applicable where metal complexes with heavy halogens are acting as acceptors. Other important factors such as crystal packing effects and π-bonding interactions can have a significant effect on the donor–acceptor distances and donor–hydrogen–acceptor (DHA) angles which are used to classify ‘strong’, ‘moderate’ or ‘weak’ hydrogen bonds. Typically, these DHA interactions are quantified via the crystallographically determined D⋯A (C⋯X) distances. The radii of Br− and especially I− also have an effect on the C⋯X bond lengths. Nonetheless, for a given halide, the shortest C⋯X distances for the [EMIM]3[LaX6] series of complexes (Table 1) occur where the NCN proton (H2) acts as the donor, consistent with it being the most acidic proton on the imidazolium ring. The C⋯X distances where the backbone NCCN protons (H4/H5) act as donors are typically 0.1–0.2 Å longer, implying weaker hydrogen bonding. In all cases the DHA angles are also generally greater than 130°, hence we assign these as ‘moderate’ hydrogen bonds.
| [EMIM]3[LaCl6]a | [EMIM]3[LaBr6] | [EMIM]3[LaI6] | ||||
|---|---|---|---|---|---|---|
| Length (Å) | Angle (°) | Length (Å) | Angle (°) | Length (Å) | Angle (°) | |
| a Data from ref. 7. | ||||||
| C⋯X (H2) | 3.557(8) | 152.2 | 3.579(12) | 134.2 | 3.942(5) | 139.8 |
| 3.479(6) | 131.6 | 3.659(10) | 153.8 | 3.758(5) | 134.0 | |
| 3.393(4) | 120.2 | 3.765(17) | 134.7 | 3.864(5) | 134.9 | |
| 3.612(4) | 150.2 | 3.825(5) | 160.4 | |||
| C⋯X (H4/5) | 3.635(5) | 150.3 | 3.723(12) | 121.6 | 3.974(5) | 136.7 |
| 3.614(6) | 148.4 | 3.683(11) | 139.8 | 3.972(5) | 113.5 | |
| 3.692(5) | 146.7 | 3.744(12) | 118.3 | 3.796(5) | 143.2 | |
| 3.632(5) | 145.9 | 3.755(11) | 146.8 | 3.894(5) | 119.0 | |
| 3.564(6) | 137.5 | 3.708(12) | 145.1 | 3.830(5) | 124.0 | |
| 3.693(12) | 153.7 | 4.210(5) | 172.5 | |||
| 3.708(16) | 143.9 | |||||
Varying the metal
Another variable to assess in this series of complexes is the nature of the metal ion. By choosing a range of trivalent metals for [EMIM]3[MCl6] (M = Sc, Y, La, Ce) it is possible to vary the ionic radius, and hence the charge![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :radius ratio, and observe the effect on the hydrogen bonding. Addition of [EMIM]Cl to MCl3 led to the formation of crystalline samples of [EMIM]3[ScCl6] (Fig. 3), [EMIM]3[YCl6] (Fig. 4) and [EMIM]3[CeCl6] (Fig. 5).
:radius ratio, and observe the effect on the hydrogen bonding. Addition of [EMIM]Cl to MCl3 led to the formation of crystalline samples of [EMIM]3[ScCl6] (Fig. 3), [EMIM]3[YCl6] (Fig. 4) and [EMIM]3[CeCl6] (Fig. 5).
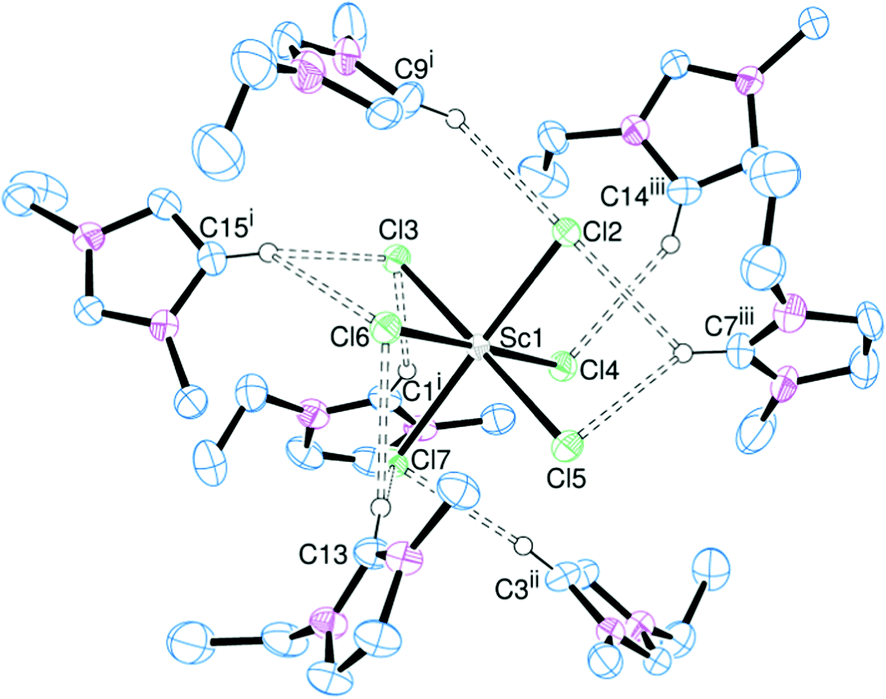 | ||
| Fig. 3 ORTEP representation of [EMIM]3[ScCl6] showing the hydrogen bonding (dotted lines). Displacement ellipsoids are drawn at the 50% probability level and hydrogen atoms not involved in hydrogen bonding are omitted for clarity. Selected bond lengths (Å): Sc–Cl2 2.479(1), Sc–Cl3 2.513(1), Sc–Cl4 2.5066(7), Sc–Cl5 2.520(1), Sc–Cl6 2.4735(8), Sc–Cl7 2.522(1). Symmetry codes: (i) 1 − x, −y, z − 0.5; (ii) x − 0.5, 0.5 − y, z; (iii) x, y − 1, z. | ||
 | ||
| Fig. 4 ORTEP representation of [EMIM]3[YCl6] showing the hydrogen bonding (dotted lines). Displacement ellipsoids are drawn at the 50% probability level and hydrogen atoms not involved in hydrogen bonding are omitted for clarity. Selected bond lengths (Å): Y–Cl1 2.6541(12), Y–Cl2 2.614(1), Y–Cl3 2.6185(12), Y–Cl4 2.640(1), Y–Cl5 2.6515(14), Y–Cl6 2.6524(14). Symmetry codes: (i) 1 − x, 1 − y, z − 0.5; (ii) 0.5 − x, y − 0.5, z − 0.5; (iii) x, y − 1, z; (iv) x, y, z − 1. | ||
 | ||
| Fig. 5 ORTEP representation of [EMIM]3[CeCl6] showing the hydrogen bonding (dotted lines) surrounding the Ce1-centred cation (left) and the Ce2-centred cation (right). Displacement ellipsoids are drawn at the 50% probability level and H atoms not involved in hydrogen bonding are omitted for clarity. Selected bond lengths (Å): Ce1–Cl1 2.7872(16), Ce1–Cl2 2.7514(17), Ce1–Cl3 2.7738(16), Ce2–Cl4 2.7755(17), Ce2–Cl5 2.7752(17), Ce2–Cl6 2.7722(17). Symmetry codes: (i) −x, −y, −z; (ii) −1 − x, −y, 1 − z; (iii) −x, 0.5 + y, 0.5 − z; (iv) x, −0.5 − y, 0.5 + z; (v) −x, y − 0.5, 0.5 − z. | ||
The Sc and Y complexes are isostructural and isomorphous, with a slight increase in cell parameters owing to the increase in metal ionic radius (Sc = 0.745 Å, Y = 0.90 Å for 6-coordinate complexes).14 The known lanthanide examples are structurally different, but within the series (including La, Eu, Gd, Nd, Pr and Sm analogues) they are all isomorphous and isostructural7 and the cell parameters for [EMIM]3[CeCl6] fit well with this trend.
In the solid state all the metal centres are six-coordinate regular octahedra. There is only one metal environment in the asymmetric unit of [EMIM]3[ScCl6] and [EMIM]3[YCl6] and the hydrogen bonding interactions are very similar (Table 2). Despite the decreased charge:radius ratio for Y over Sc, the hydrogen bonds through H4/5 are identical within experimental error. However, there is a small increase in the hydrogen bond lengths involving H2 for [EMIM]3[YCl6] compared to [EMIM]3[ScCl6]. These are notably shorter than the equivalent bonds in [EMIM]3[LaCl6], continuing the same trend. Trifurcated hydrogen bonding is present in [EMIM]3[CeCl6] through C13 (Fig. 5).
| [EMIM]3[ScCl6] | [EMIM]3[YCl6] | [EMIM]3[CeCl6] | ||||
|---|---|---|---|---|---|---|
| Length (Å) | Angle (°) | Length (Å) | Angle (°) | Length (Å) | Angle (°) | |
| C⋯X (H2) | 3.506(3) | 149.5 | 3.547(4) | 146.6 | 3.547(7) | 150.8 |
| 3.496(3) | 131.3 | 3.531(5) | 130.0 | 3.425(7) | 132.4 | |
| 3.407(3) | 138.1 | 3.422(5) | 135.9 | 3.333(7) | 117.1 | |
| 3.604(3) | 135.2 | 3.650(4) | 134.6 | 3.569(8) | 141.9 | |
| 3.548(3) | 146.8 | 3.588(5) | 145.0 | |||
| C⋯X (H4/5) | 3.586(3) | 166.7 | 3.584(5) | 165.0 | 3.568(7) | 150.4 |
| 3.692(3) | 152.5 | 3.695(4) | 153.1 | 3.583(7) | 143.2 | |
| 3.604(3) | 135.6 | 3.604(5) | 135.8 | 3.622(8) | 142.7 | |
| 3.434(3) | 131.7 | 3.459(4) | 128.6 | 3.542(8) | 147.2 | |
| 3.383(3) | 135.1 | 3.365(5) | 136.2 | 3.512(7) | 139.7 | |
Varying the cation
Altering the nature of the cation can have a marked effect on the structural properties of the complexes. In order to probe this effect the [CATION]3[LaCl6] system was initially chosen (CATION = EMIM, EDMIM, NnBu4 and BMPYRR). Complexes were prepared by adding a slight excess of [CATION]Cl to LaCl3 in MeCN, resulting in a series of compounds where the hydrogen bonding interactions should be moderate (EMIM), weak (EDMIM) and non-existent (NnBu4, BMPYRR). The difference between the [EDMIM]+ cation and the [EMIM]+ cation is the presence of a methyl group at the C2 position, thus the most acidic protons are now on the backbone of the imidazolium ring. This was expected to significantly influence the hydrogen bonding. The structure of [EDMIM]3[LaCl6] is shown in Fig. 6.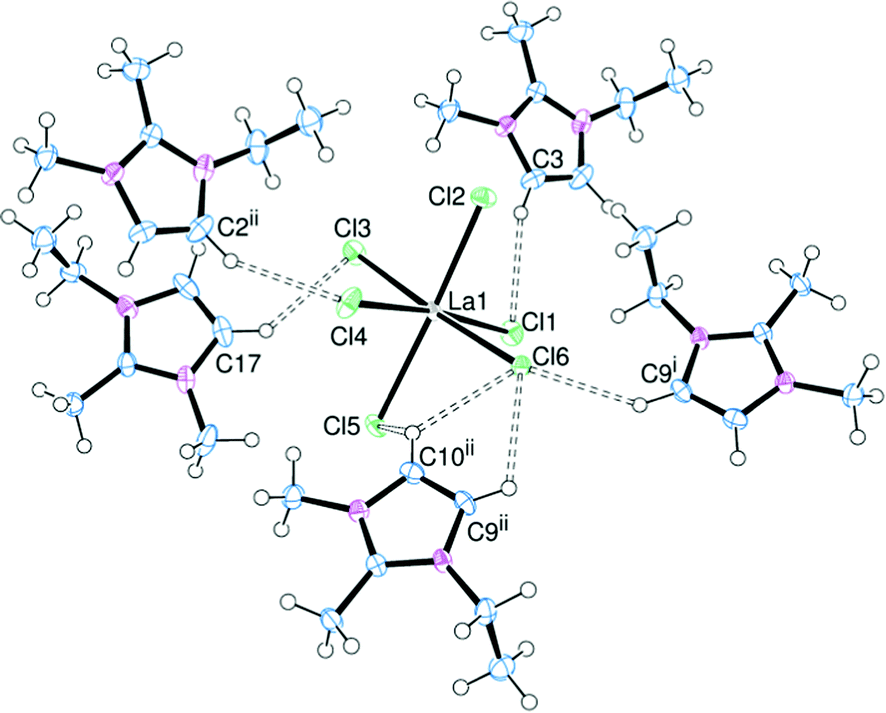 | ||
| Fig. 6 ORTEP representation of [EDMIM]3[LaCl6] showing the hydrogen bonding (dotted lines). Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å): La–Cl1 2.7963(7), La–Cl2 2.7582(6), La–Cl3 2.8029(6), La–Cl4 2.7950(7), La–Cl5 2.8262(6), La–Cl6 2.7742(5). Symmetry codes: (i) 1 − x, 1 − y, 1 − z; (ii) x − 1, y, z. | ||
Structural characterisation of [EDMIM]3[LaCl6] revealed that the [LaCl6]3− trianion was stabilised by hydrogen bonding through five associated [EDMIM]+ cations. The C⋯Cl distances were slightly shorter than the corresponding C⋯Cl distances for [EMIM]3[LaCl6], suggesting that hydrogen bonding through the backbone protons may be slightly stronger when no H2 proton is present. However, 1H NMR data (below) show that the chemical shifts for H4/5 are consistent with complete dissociation of the molecule in CD3CN solution. The La–Cl bond lengths are comparable to those observed for [EMIM]3[LaCl6].
A comparison between the [EMIM]+ and [EDMIM]+ cations can also be drawn for the [YCl6]3− and [LaI6]3− salts. The structures of [EDMIM]3[YCl6] and [EDMIM]3[LaI6] are shown in Fig. 7 and 8, respectively.
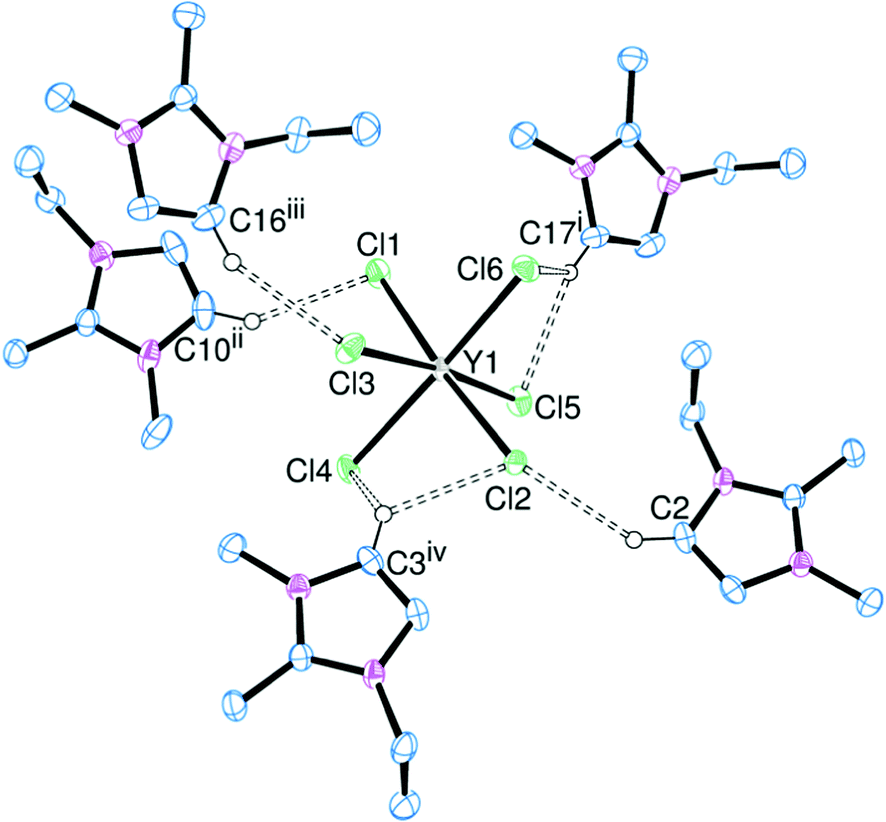 | ||
| Fig. 7 ORTEP representation of [EDMIM]3[YCl6] showing the hydrogen bonding (dotted lines). Displacement ellipsoids are drawn at the 50% probability level and hydrogen atoms not involved in hydrogen bonding are omitted for clarity. Selected bond lengths (Å): Y–Cl1 2.6580(5), Y–Cl2 2.6398(5), Y–Cl3 2.6496(5), Y–Cl4 2.6800(5), Y–Cl5 2.6464(5), Y–Cl6 2.6167(5). Symmetry codes: (i) −x, 2 − y, −z; (ii) x + 0.5, 1.5 − y, z − 0.5; (iii) −x, 1 − y, −z; (iv) 1 − x, 1 − y, −z. | ||
 | ||
| Fig. 8 ORTEP representation of [EDMIM]3[LaI6] showing the hydrogen bonding (dotted lines) surrounding the La1-centred cation (left) and the La2-centred cation (right). Displacement ellipsoids are drawn at the 50% probability level and H atoms not involved in hydrogen bonding are omitted for clarity. Selected bond lengths (Å): La1–I1 3.1800(7), La1–I2 3.1545(6), La1–I3 3.1903(7), La2–I4 3.1779(5), La2–I5 3.1566(5), La2–I6 3.1870(5). Symmetry codes: (i) 1 − x, −y, 1 − z; (ii) −x, −1 − y, −z; (iii) x, y − 1, z; (iv) −x, −1 − y, 1 − z; (v) 1 − x, −1 − y, 1 − z; (vi) x, y, 1 + z; (vii) x, 1 + y, z. | ||
[EDMIM]3[YCl6] and [EDMIM]3[LaCl6] are isomorphous and isostructural, whereas [EMIM]3[YCl6] and [EMIM]3[LaCl6] crystallise in different space groups. This suggests that the hydrogen bonding interactions play a greater role in the crystal packing arrangements for the [EMIM]+ salts. The C⋯Cl (H4/H5) distances (Table 3) are not notably different between [EMIM]3[YCl6] and [EDMIM]3[YCl6].
| [EDMIM]3[YCl6] | [EDMIM]3[LaCl6] | [EDMIM]3[LaI6] | ||||
|---|---|---|---|---|---|---|
| Length (Å) | Angle (°) | Length (Å) | Angle (°) | Length (Å) | Angle (°) | |
| C⋯X (H4/5) | 3.542(2) | 142.4 | 3.580(2) | 149.6 | 3.948(3) | 118.8 |
| 3.693(2) | 123.8 | 3.515(2) | 136.6 | 3.956(3) | 118.5 | |
| 3.388(2) | 129.8 | 3.521(2) | 148.9 | 3.933(3) | 139.2 | |
| 3.482(2) | 145.1 | 3.686(2) | 122.5 | 3.775(3) | 125.2 | |
| 3.520(2) | 149.9 | 3.455(2) | 125.8 | 3.883(3) | 124.3 | |
| 3.544(2) | 150.2 | 3.601(2) | 112.0 | 3.892(3) | 124.8 | |
| 3.577(2) | 122.6 | 3.524(2) | 143.0 | 3.883(3) | 115.3 | |
For [EDMIM]3[LaI6] two different [LaX6]3− environments are apparent, with one iodine acting as an acceptor to five different donor hydrogens. However, the C⋯I distances (Table 3) are not notably different between the EMIM and EDMIM compounds.
Spectroscopic characterisation and comparisons
In keeping with our previous observations for [GeCl6]2− anions,3 the chemical shift of the H2 proton in the 1H NMR spectrum was used to indicate the strength of the hydrogen bonding in solution (Table 4).| Compound | δ(1H)a/ppm | |
|---|---|---|
| N–CH–N | Δ | |
| a 6.8 mM solutions in CD3CN. b Change in shift compared with [EMIM]Cl at the same concentration in CD3CN. | ||
| [EMIM]3[LaCl6] | 9.27 | +0.45 |
| [EMIM]3[LaBr6] | 8.93 | +0.11 |
| [EMIM]3[LaI6] | 8.61 | −0.21 |
| [EMIM]3[CeCl6] | 9.77 | +0.95 |
| [EMIM]3[YCl6] | 9.26 | +0.44 |
| [EMIM]3[ScCl6] | 9.41 | +0.59 |
| [EMIM]Cl | 8.82 | — |
| [EMIM]Br | 8.84 | — |
From the NMR data it is apparent that the chemical shift of the signal associated with the H2 proton shifts to low frequency for the heavier halides. This correlates with decreasing strength of the solution phase hydrogen bonding interactions upon changing from Cl to I. The chemical shifts of the less acidic H4/5 protons are not affected by changing the halide, suggesting that the salts are completely dissociated in solution. This is consistent with the trend observed for [EMIM][GeX3] (X = Cl, Br, I).3
For the [MX6]3− salts, 45Sc, 89Y and 139La NMR spectra were also obtained (Table 5). All complexes were soluble in MeCN at approximate 10–15 mM concentration. The [NnBu4]+ salt was soluble at a similar concentration in CH2Cl2, whereas the [EMIM]+, [BMPYRR]+ and [EDMIM]+ salts were poorly soluble in CH2Cl2. For the [LaCl6]3− salts a small high frequency shift in δ139La occurs as the cation changes along the series: [NnBu4]+ → [BMPYRR]+ → [EDMIM]+ → [EMIM]+ in both MeCN and CH2Cl2 solution, but given the large 139La NMR chemical shift range, these differences may not be significant. The 139La NMR resonance shifts to high frequency as the halide becomes heavier; the [LaI6]3− system showing a very broad resonance at ∼1400 ppm (W1/2 ∼ 12000 Hz). The yttrium salts show no 89Y NMR resonance at room temperature in MeCN, presumably due to chloride exchange. However, cooling to 233 K reveals a sharp singlet for each compound.
| Salt | δ (ppm) | W 1/2 (Hz) | δ (ppm) | W 1/2 (Hz) |
|---|---|---|---|---|
| MeCN | CH2Cl2 | |||
| a Spectrum recorded at 233 K. b n.o. = not observed due to low solubility. | ||||
| [NnBu4]3[LaCl6] | 846 | 800 | 872 | 1200 |
| [BMPYRR]3[LaCl6] | 855 | 500 | 880 | 300 |
| [EDMIM]3[LaCl6] | 864 | 600 | n.o.b | — |
| [EMIM]3[LaCl6] | 867 | 400 | 892 | 300 |
| [EMIM]3[LaBr6] | 1095 | 1200 | n.o.b | — |
| [EMIM]3[LaI6] | ∼1400 | 12000 |
n.o.b | — |
| [EMIM]3[YCl6]a | 407 | — | — | — |
| [EDMIM]3[YCl6]a | 401 | — | — | — |
| [EMIM]3[ScCl6] | 254 | 170 | — | — |
Conclusions
All of the salts are based upon six-coordinate [MX6]3− units, and for those containing EMIM and EDMIM cations, significant C–H⋯X hydrogen-bonding interactions to the substituted imidazolium cations are evident from crystallographic studies. The shortest C⋯X distances for the [EMIM]3[MX6] series involve the NCN (H2) proton, with much weaker interactions to the H4/H5 protons. Despite the decreased charge:radius ratio along the series from Sc to Y to La, the hydrogen bonds through H4/5 are unaffected. However, there is a small increase in the hydrogen bond lengths involving H2 along the same series.
Substitution of the H2 proton with a Me group in the EDMIM cation has a significant influence on the cation–anion interactions in the solid state. The structure of [EDMIM]3[LaCl6] revealed five associated [EDMIM]+ cations and the C⋯Cl distances involving H4/H5 were shorter than those for [EMIM]3[LaCl6].
The solution 1H NMR trends indicate that the hydrogen bonding to the EMIM (H2) cations becomes weaker as the halide becomes heavier, while the EDMIM salts appear to be completely dissociated in solution.
Experimental
All preparations were carried out using standard Schlenk techniques under a N2 atmosphere. Anhydrous MCl3 (M = La, Ce) were obtained by reacting hydrated MCl3 (Sigma) with excess SOCl2, then drying in vacuo. Anhydrous ScCl3, YCl3, LaBr3 and LaI3 were obtained from Alfa and used as received. [EMIM]X (X = Cl, Br, I), [EDMIM]Cl, [NnBu4]Cl and [BMPYRR]Cl were obtained from Sigma, dried in vacuo at 100 °C for 4 hours and stored in a glove box. [EDMIM]I was prepared by stirring a solution of [EDMIM]Cl and NaI in water for 4 hours, then extracting into CH2Cl2 followed by drying in vacuo at 100 °C for 4 hours. MeCN and CH2Cl2 were dried prior to use from CaH2. Infrared spectra were recorded as Nujol mulls between CsI plates using a Perkin-Elmer Spectrum 100 spectrometer over the range 4000–200 cm−1. Raman spectra were recorded from powdered solid samples using a Perkin-Elmer FT-Raman 2000R with a Nd:YAG laser. 1H NMR spectra were recorded in CD2Cl2 and CD3CN using a Bruker DPX400 spectrometer and are referenced to the residual protio solvent. 45Sc, 89Y and 139La NMR spectra were also recorded in CD2Cl2 or CD3CN on a Bruker DPX400 spectrometer and are referenced to 0.1 mol dm−3 solutions of the chlorides in D2O solutions at pH = 1. For yttrium-89 NMR spectra a 3 s pulse delay was used and TEMPO (2,2,6,6-tetramethylpiperidine-oxyl) was added as relaxation agent. Microanalyses were undertaken by Medac Ltd or London Metropolitan University.Preparations
| Compound | [EMIM]3[ScCl6] | [EMIM]3[YCl6] | [EMIM]3[CeCl6] | [EMIM]3[LaBr6] |
|---|---|---|---|---|
| a Common items: wavelength (Mo-Kα) = 0.71073 Å; θ(max) = 27.5°. b R 1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑wFo4]1/2. | ||||
| Formula | C18H33Cl6N6Sc | C18H33Cl6N6Y | C18H33CeCl6N6 | C18H33Br6LaN6 |
| M/g mol−1 | 591.16 | 635.11 | 686.32 | 951.87 |
| Temp./K | 150(2) | 100(2) | 100(2) | 100(2) |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic | Monoclinic |
| Space group (no.) | Pna21 (33) | Pna21 (33) | P21/c (14) | P21/c (14) |
| a/Å | 19.411(2) | 19.580(2) | 15.492(3) | 15.925(5) |
| b/Å | 13.059(1) | 13.036(2) | 12.549(2) | 12.787(4) |
| c/Å | 11.046(1) | 11.166(1) | 14.708(3) | 15.252(5) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 90 | 90 | 90.482(3) | 90.588(5) |
| γ/° | 90 | 90 | 90 | 90 |
| U/Å3 | 2800.0(4) | 2849.9(5) | 2859.2(9) | 3106(2) |
| Z | 4 | 4 | 4 | 4 |
| μ(Mo-Kα) mm−1 | 0.853 | 2.626 | 2.170 | 9.111 |
| F(000) | 1224 | 1296 | 1372 | 1800 |
| Total reflections | 15098 |
14268 |
12086 |
14407 |
| Unique reflections | 5189 | 6374 | 5811 | 6318 |
| R int | 0.027 | 0.037 | 0.122 | 0.092 |
| Goodness-of-fit on F2 | 1.023 | 0.796 | 0.962 | 1.145 |
| R 1 [Io > 2σ(Io)] | 0.026 | 0.028 | 0.063 | 0.082 |
| R 1 (all data) | 0.029 | 0.043 | 0.090 | 0.116 |
| wR2b [Io > 2σ(Io)] | 0.063 | 0.045 | 0.167 | 0.134 |
| wR2 (all data) | 0.064 | 0.048 | 0.187 | 0.148 |
| Compound | [EMIM]3[LaI6] | [EDMIM]3[YCl6] | [EDMIM]3[LaCl6] | [EDMIM]3[LaI6] |
|---|---|---|---|---|
| Formula | C18H33I6LaN6 | C21H39Cl6N6Y | C21H39Cl6LaN6 | C21H39I6LaN6 |
| M/g mol−1 | 1233.81 | 677.19 | 727.19 | 1275.89 |
| Temp./K | 120(2) | 100(2) | 100(2) | 100(2) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group (no.) | P21/c (14) | P21/n (14) | P21/n (14) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2) (2) |
| a/Å | 18.803(7) | 9.9170(8) | 10.027(2) | 10.347(2) |
| b/Å | 11.133(2) | 16.274(1) | 16.306(2) | 11.075(3) |
| c/Å | 17.017(2) | 19.028(2) | 19.133(3) | 16.577(3) |
| α/° | 90 | 90 | 90 | 92.594(3) |
| β/° | 102.162(7) | 99.973(1) | 99.265(3) | 90.255(3) |
| γ/° | 90 | 90 | 90 | 106.930(4) |
| U/Å3 | 3482(1) | 3024.6(4) | 3087.4(9) | 1815.1(7) |
| Z | 4 | 4 | 4 | 2 |
| μ(Mo-Kα) mm−1 | 6.562 | 2.480 | 1.924 | 6.299 |
| F(000) | 2232 | 1392 | 1464 | 1164 |
| Total reflections | 34033 |
16980 |
26415 |
16557 |
| Unique reflections | 7929 | 6878 | 7055 | 8311 |
| R int | 0.059 | 0.027 | 0.020 | 0.028 |
| Goodness-of-fit on F2 | 1.073 | 1.028 | 1.078 | 0.984 |
| R 1 [Io > 2σ(Io)] | 0.031 | 0.028 | 0.023 | 0.019 |
| R 1 (all data) | 0.036 | 0.039 | 0.026 | 0.021 |
| wR2b [Io > 2σ(Io)] | 0.075 | 0.064 | 0.055 | 0.041 |
| wR2 (all data) | 0.078 | 0.067 | 0.057 | 0.042 |
Structure solution and refinement were straightforward using WinGX and software packages within,16 except as detailed below. Although Q-peaks corresponding to the location of protons were observed in the Fourier difference map, hydrogen atoms were placed in geometrically assigned positions with C–H distances of 0.95 Å (CH), 0.98 Å (CH3) or 0.99 Å (CH2) and refined using a riding model, with Uiso(H) = 1.2Ueq(C) (CH, CH2) or 1.5Ueq(C) (CH3). Mercury17and enCIFer18 were used to prepare material for publication.
All the crystals of [EMIM]3[CeCl6] looked at in this study were multiply twinned and all attempts at multi-component twin integration failed. A satisfactory dataset was obtained by integrating on just the major component and ignoring the minor overlaps from the other domains. This caused a couple of large Q-peaks to appear close to the cerium atoms (3.2 e Å−3 at ∼1.04 Å distance from Ce) as well as two large holes (−5.9 e Å−3 at ∼0.84 Å from Ce). This explains the level A and B CheckCIF errors PLAT971_ALERT_2_A (holes) and PLAT971_ALERT_2_B (Q-peaks). The crystals of [EMIM]3[LaBr6] were all also multi-component twins and treated in a similar fashion.
Acknowledgements
This work was funded by EPSRC through a Programme Grant (EP/I033394/1) and also through EP/K039466/1. The SCFED Project (http://www.scfed.net) is a multidisciplinary collaboration of British universities investigating the fundamental and applied aspects of supercritical fluids.References
- P. N. Bartlett, S. L. Benjamin, C. H. de Groot, A. L. Hector, R. Huang, A. Jolleys, G. P. Kissling, W. Levason, S. J. Pearce, G. Reid and Y. Wang, Mater. Horiz., 2015, 2, 420 RSC; P. N. Bartlett, D. A. Cook, C. H. de Groot, A. L. Hector, R. Huang, A. Jolleys, G. P. Kissling, W. Levason, S. J. Pearce and G. Reid, RSC Adv., 2013, 3, 15645 RSC.
- J. Ke, W. Su, S. M. Howdle, M. W. George, D. Cook, M. Perdjon-Abel, P. N. Bartlett, W. Zhang, F. Cheng, W. Levason, G. Reid, J. Hyde, J. Wilson, D. C. Smith, K. Mallik and P. Sazio, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14768 CrossRef CAS PubMed; J. Ke, P. N. Bartlett, D. Cook, T. L. Easun, M. W. George, W. Levason, G. Reid, D. Smith, W. Su and W. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 1517 RSC; P. N. Bartlett, J. Burt, D. A. Cook, C. Y. Cummings, M. W. George, A. L. Hector, M. M. Hasan, J. Ke, W. Levason, D. Pugh, G. Reid, P. W. Richardson, D. C. Smith, J. Spencer, N. Suleiman and W. Zhang, Chem. – Eur. J., 2016, 22, 302 CrossRef PubMed; C. Cummings, P. N. Bartlett, D. Pugh, G. Reid, W. Levason, M. M. Hasan, A. L. Hector, J. Spencer and D. C. Smith, J. Electrochem. Soc., 2014, 162, D619 CrossRef.
- P. N. Bartlett, C. Y. Cummings, W. Levason, D. Pugh and G. Reid, Chem. – Eur. J., 2014, 20, 5019 CrossRef CAS PubMed.
- C. Y. Cummings, P. N. Bartlett, D. Pugh, G. Reid, W. Levason, M. M. Hasan, A. L. Hector, J. Spencer, D. C. Smith, S. Marks and R. Beanland, ChemElectroChem, 2016, 3, 679 CrossRef.
- H. Ohno, Electrochemical Aspects of Ionic Liquids, Wiley, New York, 2005 Search PubMed; Electrodeposition from Ionic Liquids, ed. F. Endres, A. P. Abbot and D. R. Macfarlane, Wiley, New York, 2008 Search PubMed.
- K. Matsumota, T. Tsuda, T. Nohira, R. Hagiwara, Y. Ito and O. Tamada, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m186 Search PubMed.
- C. C. Hines, D. B. Cordes, S. T. Griffin, S. I. Watts, V. A. Cocalia and R. D. Rogers, New J. Chem., 2008, 32, 872 RSC.
- A. Babai and A.-V. Mudring, Inorg. Chem., 2006, 45, 4874 CrossRef CAS PubMed.
- A. Babai and A.-V. Mudring, Inorg. Chem., 2005, 44, 8168 CrossRef CAS PubMed.
- K. Pohako-Esko, T. Wehner, P. S. Schulz, F. W. Heinemann, K. Muller-Buschbaum and P. Wasserscheid, Eur. J. Inorg. Chem., 2016, 1333 CrossRef CAS.
- J. Hallfeldt, PhD thesis, University of Hannover, 2003.
- A. Babai and A.-V. Mudring, J. Alloys Compd., 2006, 418, 122 CrossRef CAS.
- (a) G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed; (b) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- Rigaku Corporation, Tokyo, Japan, 2011.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
- F. H. Allen, O. Johnson, G. P. Shields, B. R. Smith and M. Towler, J. Appl. Crystallogr., 2004, 37, 335 CrossRef CAS.
Footnote |
| † CCDC 1472564 ([EMIM]3[ScCl6]), 1472565 ([EMIM]3[YCl6]), 1472566 ([EMIM]3[CeCl6]), 1472567 ([EMIM]3[LaBr6]), 1472568 ([EMIM]3[LaI6]), 1472569 ([EDMIM]3[YCl6]), 1472570 ([EDMIM]3[LaCl6]) and 1472571 ([EDMIM]3[LaI6]). For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6nj01068g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |