
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlation between structural, physical and chemical properties of three new tetranuclear NiII clusters†
Marina
Cindrić
*a,
Gordana
Pavlović
b,
Damir
Pajić
c,
Krešo
Zadro
c,
Dominik
Cinčić
a,
Tomica
Hrenar
a,
Edislav
Lekšić
d,
Ana
Belen Pinar Prieto
e,
Predrag
Lazić
f and
Dubravka
Šišak Jung
*g
aUniversity of Zagreb, Faculty of Science, Department of Chemistry, Horvatovac 102a, HR-10000 Zagreb, Croatia. E-mail: marina@chem.pmf.hr; Fax: +385 1 4606 341
bUniversity of Zagreb, Faculty of Textile Technology, Department of Applied Chemistry, Prilaz baruna Filipovića 28a, HR-10000 Zagreb, Croatia
cUniversity of Zagreb, Faculty of Science, Department of Physics, Bijenička cesta 32, HR-10000 Zagreb, Croatia
dPLIVA Croatia, TAPI R&D, Prilaz Baruna Filipovića 25, HR-10000 Zagreb, Croatia
eETH Zurich, Laboratory for Crystallography, Vladimir-Prelog-Weg 5, 8093 Zurich, Switzerland
fInstitute Ruđer Bosković, Department for Theoretical Physics, Bijenička cesta 54, 10000 Zagreb, Croatia
gDECTRIS Ltd, Täfernweg 1, 5405 Baden-Dättwil, Switzerland. E-mail: dubravka.sisak@dectris.com
First published on 31st May 2016
Abstract
Based on a new family of NiII cubane-like clusters, this work addresses current challenges in the synthesis, analysis and dynamics of single-molecule-magnet (SMM) systems. Investigation of various synthetic routes and desorption–sorption processes yielded a series of isomorphous compounds: [Ni4L4(CH3OH)4]·xsolv, [Ni4L4(CH3OH)4] and [Ni4L4]. In order to analyze these deceivingly simple materials, several analytical and quantum mechanical methods had to be used. This revealed materials with mixed lattice solvents, statistical disorder of the solvent, and disordered [Ni4L4] cores which offered an insight into the risks of the self-assembly process and interconversion dynamics of the investigated NiII family. These findings also allowed structural–magnetic relationships to be established, and the outcomes were exploited in two ways: first, the effect of the coordinated and lattice solvent on the magnetic properties was examined, and second, magnetic properties were used to facilitate crystal structure determination.
Introduction
Single-molecule magnets (SMM) are a specific class of metallo-organic complexes whose magnetic behavior depends on the intrinsic magnetic properties of the individual molecules, i.e. it is a direct consequence of inter- and intramolecular interactions in the system. In general, the choice of ligands will govern the topology of the system, as well as the number and type of intra- and intermolecular interactions.1–3 This, in turn, depends on the choice of the synthetic route, and synthetic parameters such as temperature, pH and metal-to-ligand ratio play an important role in the synthesis of the targeted product. As these systems found their potential application in data storage, memory devices, switches and sensors, the targeted synthesis of SMMs became increasingly important.4 Significant progress in synthesis was made by implementation of various “bottom-up” approaches based on the sensible choice of ligands and metal ions. These facilitated the formation of clusters with well-defined geometries and related magnetic properties.5–7 In the last two decades, a series of compounds based on polynuclear complexes of Mn, Fe, Ni, V and Co, or mixed metals have been reported,8–10 of which cubane-like magnetic clusters with a [Ni4(μ3-O)4] core are especially a well-studied class.11–21 Particular interest in this class of compounds arises from their specific core shape and magnetic exchange pathways, governed by μ3-atoms. Consequently, extensive investigations of the [Ni4(μ3-O)4] core provided an insight into the delicate relationship between magnetic and structural properties.22–24 The importance of the exact geometry of the [Ni4O4] core is emphasized in a series of studies, as the symmetry and the corresponding Ni–μ3-O–Ni angles play a crucial role in the intramolecular magnetic interactions.25–27 Moreover, fine details of the cluster structure, such as the type and the amount of solvent, are extremely important, as these factors can determine the magnetic properties of SMM systems. This solvent-effect was first reported in 1999 by Khan and Larinova, who introduced the concept of a “magnetic sponge” to describe magnetic materials that can reversibly release and reabsorb both coordinated and non-coordinated solvent molecules.28 Nowadays, solvent effects are becoming increasingly interesting. In the latest investigation of [Ni4L4(solv)4] type of complexes it was shown that at ambient temperature, these compounds show reversible exchange of the coordinated solvent molecules (MeOH vs. H2O), causing the spin to switch the ground state from S = 4 to S = 0.29 Even more interesting is the recent research of desolvation of SMM materials (Mn12,30 FeIII4,31 metal–organic frameworks32) reporting a serious effect of lattice (non-coordinated) solvents and experimental conditions on magnetic properties. All these findings inspired us to study the dynamics of exchange of coordinated and non-coordinated solvents in a new family of NiII cubane-like clusters, comprising [Ni4L4(CH3OH)4]·xsolv, [Ni4L4(CH3OH)4] and [Ni4L4] compounds. Investigation of synthetic paths, desorption–sorption studies, stability and interconversion processes of these closely related compounds offered a more detailed insight into the outcome of the self-assembly processes. Structural analyses of the obtained compounds, coupled with quantum mechanical studies, were correlated with magnetic measurements. The findings were exploited in two ways. First, the effect of the type and the amount of the solvent in the structure on the magnetic properties was examined, and second, magnetic properties were used to facilitate crystal structure determination.Results and discussion
Synthesis
Depending on the synthetic strategy, deep green plates [Ni4L4(CH3OH)4]·0.32CH3OH·0.32H2O (1) or green hexagonal prisms [Ni4L4(CH3OH)4]·0.63CH3OH (2) were obtained from the reaction mixture (Scheme 1). | ||
| Scheme 1 Synthesis of two green clusters [Ni4L4(CH3OH)4]·0.32CH3OH·0.32H2O (1) and [Ni4L4(CH3OH)4]·0.63CH3OH (2) and one red cluster of the formula [Ni4L4] (3). | ||
Compound 1 was prepared by solvothermal reaction of Ni(O2CMe)2·4H2O and the tridentate Schiff base N-(2-hydroxy-5-methylphenyl)salicylideneimine (H2L) in methanol at 110 °C. However, only slight modifications of the original solvothermal reaction in methanol at 110 °C resulted in different products. The change in temperature from 110 °C to room temperature yielded green hexagonal prisms of compound 2. Furthermore, adding water to the mixture (2.5 MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O, volume ratio) resulted in green plates 1, while adding acetic anhydride to the reaction mixture yielded compound 2. In both cases, it was noticed that the synthesis path is not sensitive to the molar ratio of the precursors. Under the same synthetic conditions, both 1:1 and 1:2 precursor ratios yielded the same products. Although these experiments showed that 1 and 2 products are kinetically and thermodynamically favored, it also became apparent that water molecules compete with methanol for a place in the crystal lattice, and that the outcome can depend on the temperature and solvent mixture. This flexibility of the systems was further supported by stability and sorption studies. When exposed to room conditions, both materials 1 and 2 changed the colour from green to orange-red. While these conversions resulted in similar products 1a and 2a, their respective kinetics differed. The products 1a and 2a have the same molecular formula, [Ni4L4(CH3OH)4], but their structures are slightly different. Conversion 1 → 1a took one day, and 2 → 2a only a few hours, indicating different types of intermolecular interactions (in 1 and 2) between the NiII complex and the non-coordinated solvent. Considering that the reactions resulted in single crystal species, a subtle structural change upon the loss of the non-coordinated solvent could be proposed. In contrast to the room temperature observations described above, heating of clusters 1 and 2 up to 180 °C resulted in the red polycrystalline sample 3 (Scheme 2).
:1 H2O, volume ratio) resulted in green plates 1, while adding acetic anhydride to the reaction mixture yielded compound 2. In both cases, it was noticed that the synthesis path is not sensitive to the molar ratio of the precursors. Under the same synthetic conditions, both 1:1 and 1:2 precursor ratios yielded the same products. Although these experiments showed that 1 and 2 products are kinetically and thermodynamically favored, it also became apparent that water molecules compete with methanol for a place in the crystal lattice, and that the outcome can depend on the temperature and solvent mixture. This flexibility of the systems was further supported by stability and sorption studies. When exposed to room conditions, both materials 1 and 2 changed the colour from green to orange-red. While these conversions resulted in similar products 1a and 2a, their respective kinetics differed. The products 1a and 2a have the same molecular formula, [Ni4L4(CH3OH)4], but their structures are slightly different. Conversion 1 → 1a took one day, and 2 → 2a only a few hours, indicating different types of intermolecular interactions (in 1 and 2) between the NiII complex and the non-coordinated solvent. Considering that the reactions resulted in single crystal species, a subtle structural change upon the loss of the non-coordinated solvent could be proposed. In contrast to the room temperature observations described above, heating of clusters 1 and 2 up to 180 °C resulted in the red polycrystalline sample 3 (Scheme 2).
 | ||
| Scheme 2 Transformation of cluster 1 in the heat/MeOH and water/MeOH cycle. | ||
When exposed to methanol, the colour of the material changed back to green, indicating that sample 3 is most likely to have the formula Ni4L4. Additionally, sorption experiments conducted on compounds 1 and 2 with water vapors showed that water molecules can be incorporated as solvents in the crystal lattice (Fig. S1–S18, ESI†). These processes (sorption/desorption, ageing, and heating) yielded a variety of materials, with different types and amounts of coordinated and crystalline solvents, which required a multitude of analytical techniques to be used: single crystal X-ray diffraction (SCXRD), X-ray powder diffraction (XRPD), thermogravimetry (TG), infrared spectroscopy, chromatography and magnetic measurements. A combination of these techniques with quantum mechanical calculations allowed for characterization of the obtained materials, and thus structure–magnetic relationships to be proposed.
Thermogravimetric analysis (TG)
In order to examine the solvent content and thermal stability of clusters 1, 2 and 3 the samples were heated in an oxygen atmosphere from 25 to 600 °C. Clusters 1 and 2 displayed one-step weight loss, occurring between 25 and 180 °C, and 40 and 140 °C, respectively. Such behaviour was ascribed to the combined loss of lattice and coordinated solvent molecules. However, in the case of compound 1, the relative amounts of water and methanol could not be discerned. The further weight loss occurring between 340 and 430 °C (for 1), and 300 and 430 °C (for 2) was associated with cluster degradation (Fig. S19 and S20, ESI†). Compound 3 was found to undergo a one-step weight loss in the temperature range 302–540 °C, which confirmed the absence of both lattice and coordinated solvent molecules. Thermal stability of the samples obtained by sorption/desorption or by ageing of materials 1 and 2 showed that the combined loss of lattice and coordinated solvent molecules occurred in one step up to 160 °C and 130 °C. This is very similar to that described for compounds 1 and 2, and the small differences could be a consequence of different contents of solvent molecules.Structural studies
Single-crystal X-ray structure analysis revealed that compounds 1, 1a, 2 and 2a are tetranuclear [Ni4(μ3-O)4] clusters with a distorted cubane topology. NiII and the μ3-O oxygen atoms of the methylphenolato moiety from Schiff base ligands (L2− = C6H5(O)–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–(O)C6H4CH3) occupy alternate vertices of the cube, resulting in interpenetrating two concentric tetrahedra, one made by the nickel centres and another by the asymmetrically spanning μ3-O oxygen atoms. Each NiII ion is six-coordinated by five oxygen atoms and one imino nitrogen atom. Three of five oxygen atoms are oxygen atoms of the methylphenolato moiety which span three nickel centres, one is from the neutral MeOH monodentate ligand and the fifth is the oxygen atom from the salicylideneimine part of the ligand (Fig. 1). In the case of compound 1, methanol and water were present as lattice solvents, while in compound 2, only methanol solvent was found, resulting in general formulae [Ni4L4(CH3OH)4]·0.32CH3OH·0.32H2O (1) and [Ni4L4(CH3OH)4]·0.63CH3OH (2). The solvent molecules, MeOH/H2O and MeOH, in the unit cell of compounds were highly disordered and their respective amounts could not be derived from SCXRD data only. These values were obtained by combining chromatographic and SCXRD data. Coulometric titration and head-space gas chromatography were used for the determination of water and methanol in 1 and 2, respectively. The unit cell volumes of 1 and 2 differ by 32 Å3 which is 1% of the unit cell volume. As expected, the crystal structures of compounds 1a and 2a showed a similar NiII core, with no solvent incorporated into the lattice (Fig. S21, ESI†). The difference in the unit cell volumes of 1a and 2a is not negligible (less than 1% of unit cell volume), and it is an indication of possible small structural differences between 1a and 2a.
N–(O)C6H4CH3) occupy alternate vertices of the cube, resulting in interpenetrating two concentric tetrahedra, one made by the nickel centres and another by the asymmetrically spanning μ3-O oxygen atoms. Each NiII ion is six-coordinated by five oxygen atoms and one imino nitrogen atom. Three of five oxygen atoms are oxygen atoms of the methylphenolato moiety which span three nickel centres, one is from the neutral MeOH monodentate ligand and the fifth is the oxygen atom from the salicylideneimine part of the ligand (Fig. 1). In the case of compound 1, methanol and water were present as lattice solvents, while in compound 2, only methanol solvent was found, resulting in general formulae [Ni4L4(CH3OH)4]·0.32CH3OH·0.32H2O (1) and [Ni4L4(CH3OH)4]·0.63CH3OH (2). The solvent molecules, MeOH/H2O and MeOH, in the unit cell of compounds were highly disordered and their respective amounts could not be derived from SCXRD data only. These values were obtained by combining chromatographic and SCXRD data. Coulometric titration and head-space gas chromatography were used for the determination of water and methanol in 1 and 2, respectively. The unit cell volumes of 1 and 2 differ by 32 Å3 which is 1% of the unit cell volume. As expected, the crystal structures of compounds 1a and 2a showed a similar NiII core, with no solvent incorporated into the lattice (Fig. S21, ESI†). The difference in the unit cell volumes of 1a and 2a is not negligible (less than 1% of unit cell volume), and it is an indication of possible small structural differences between 1a and 2a.
 | ||
| Fig. 1 Mercury-rendered ORTEP view of the molecular structure of cluster 1 (color scheme applied: green – nickel, red – oxygen, blue – nitrogen, grey – carbon, light grey – hydrogen). The displacement ellipsoids are drawn at the 50% probability level at 150 K. The CH3OH and H2O molecules present as lattice solvents are omitted for clarity. | ||
All four crystal structures (1, 1a, 2, and 2a) exhibit intramolecular hydrogen bonds between hydroxyl groups of coordinated MeOH molecules and phenolate oxygen atoms. However, no intermolecular classical hydrogen bonds were observed. In all investigated structures (1, 1a, 2, and 2a), the tetrameric unit is cross-linked by C–H⋯O/N intramolecular hydrogen bonds of expected geometries. The basic supramolecular motif that is common for all structures at either room or low temperature is the dimerization of cluster units via the C–H⋯O type of the weak hydrogen bond (Fig. 2). C⋯O and H⋯O separations are approximately 3.4 and 2.5 Å, respectively, in the low-temperature structures 1 and 2 and 3.5 and 2.6 Å in the room-temperature structures 1a and 2a. The H⋯O separations fall in the long distance range for H⋯A contacts, according to T. Steiner.33 The pertinent structural parameters are listed in Tables S1–S6 in the ESI† in addition to a detailed description of the molecular and crystal structures (Scheme S1 and Fig. S22–S24, ESI†).
 | ||
| Fig. 2 Pov–Ray34 (v.3.62) rendered drawing of formation of the centrosymmetric dimers (shown for 1a) via the weak C411–H411⋯O31 intermolecular hydrogen bond between the phenyl C411–H411 group of the 4-methylphenolato moiety and the phenolate oxygen atom O31 (broken magenta lines). The metrical parameters are given in Table S3 (ESI†). | ||
Compounds 1 and 1a, and on the other side, 2 and 2a, differ in C–H⋯O intermolecular hydrogen bonds formed with non-coordinated MeOH/H2O molecules and –C6H5 phenyl groups of the ligand. Their geometries belong to the long distance range of approx. 3.6 Å (for C⋯O) indicating that the solvent molecules are weakly bound to the tetranuclear cluster unit. A detailed inspection of the metrical parameters of these hydrogen bonds reveals that H⋯O distances in 1 are generally shorter (2.30–2.75 Å) than in 2 (2.86 Å) (Table S3, see hydrogen bonds with the O1W and O5 atoms acting as proton-acceptors in 1vs. one hydrogen bond with the O5A atom as a proton-acceptor in 2, ESI†).
Structural analysis of cluster 3, a red polycrystalline material obtained by thermal treatment of cluster 1 (or 2), was carried out using XRPD data, as described in detail in the Experimental section. At this point, it is important to differentiate between the ideal structure that corresponds to the energetically ideal case, as found by DFT-optimizations, and the average structure of the investigated material, which was found to be disordered (XRPD pattern) (Fig. 3).
 | ||
| Fig. 3 Molecular structures of compound 3. Starting models: (a) obtained by removing methanol molecules from structure 1 and (b) after DFT-optimization. Final models: (c) molecular structure obtained from XRPD data (average structure) superimposed on the DFT-optimized structure (ideal structure). | ||
As expected, the ideal structure of compound 3 featured two Ni4L4 species with distorted cubane-type geometry, where each Ni atom exhibits square planar coordination: three oxygens from methylphenolato moieties and one imino nitrogen atom. All corresponding Ni–O–Ni bond angles are found to be larger than 90° (Table S7, ESI†). Considering that Bertrand and co-workers already suggested in 1971 that the deviation of ±14° from 90° angles can be tolerated before the direct ferromagnetic exchange terms cease to be dominant, this structure suggested diamagnetic behaviour of the molecule.35 Each Ni4L4 cluster exhibits two intramolecular C–H⋯O bonds, formed between the oxygen and carbon from methylphenolato moieties. One C–H⋯O bond is formed between two parallelly aligned ligands and the other connects two neighbouring perpendicular ligands. The structure of cluster 3 features a series of intra- and intermolecular C–H⋯π hydrogen bonds, established between the methyl group and the aromatic chelate six-membered ring of the coordinated ligand. In particular, the intramolecular C–H⋯π bond is formed between one set of parallelly aligned ligands. Intermolecular bonds of the same type showed a much richer network: C–H⋯π interactions were established between parallelly and perpendicularly oriented neighbouring ligands, forming a rich network across the crystal. This description of the ideal structure of cluster 3 allowed diamagnetic behaviour to be proposed. However, examination of the peak widths in the XRPD pattern (Fig. 4) suggested that the material exhibits both crystalline and disordered domains, while a high background suggested that species with low-range order could also be present in the material. Therefore, the average structure of the material and its influence on magnetic properties required further analysis.
 | ||
| Fig. 4 Observed (black), calculated (red) and difference (blue) profiles for the Rietveld refinement of compound 3. Reflection positions are marked as vertical bars. | ||
Considering the facts that (i) cluster 3 was obtained by thermal treatment of cluster 2a (1a), (ii) the molecular structure is stabilized by C–H⋯O and C–H⋯π interactions and (iii) the crystal structure of cluster 3 is governed mostly by weak C–H⋯π interactions, the following explanation can be proposed. In order to remove coordinated methanol from material 2a (1a), the molecule needs to adopt a suitable geometry. Four ligands in the Ni4L4 cluster need to be slightly shifted and rotated, which results in increased Ni–O–Ni angles. While this increase generates square-planar geometry of the Ni4L4 cluster, the resulting Ni–O–Ni angles do not need to be increased isotropically. Due to the rich inter- and intramolecular interactions of C–H⋯O and C–H⋯π type, Ni–O–Ni angles can adopt a wide range of values. Consequently, such a range of molecular geometries can exhibit slightly different magnetic properties. Moreover, ambiguities in unit cell determination and the possible presence of species with a local structure also need to be taken into account. First, a limited number of peaks and anisotropic peak broadening allowed only average (most probable) values of unit cell parameters to be determined: existence of other, slightly different cells or supercells cannot be excluded. Second, the possible local structure will also contribute to the magnetic behaviour. Thus, deviations from ideal magnetic values can be expected. However, although the material features disordered domains, it appears to be thermodynamically stable. Comparison of the initial XRPD pattern with the pattern collected after two months, using the same setup, revealed no differences.
Sorption and stability studies
In a series of sorption and ageing studies, the stability of synthesised compounds, desolvation–solvation processes, and possibilities of interconversions of non-coordinated solvents were investigated.At 90% RH, both complexes 1 and 2 showed that water sorption resulted in an increase of mass of less than 0.1%. The corresponding desorption curve did not match the absorption curve, and showed a decrease in mass of about 2.5% and 4% for compounds 1 and 2, respectively. Considering that both structures 1 and 2 feature dynamical disorder (0.32H2O and 0.32CH3OH for structure 1, and 0.63CH3OH in compound 2) these values indicate that during sorption, water molecules are incorporated into the structure until voids in the crystal are fully occupied by water, that is, until every unit cell is occupied by solvent molecules. The morphology of both the clusters was held but strong surface erosion was observed in cluster 2 (Fig. S27, ESI†).
 | ||
| Fig. 5 (a) PXRD patterns for 1 obtained after ageing of 1 in water (water–MeOH cycle) vapor in the 1st (1 → 3w), 2nd (11 → 3w1) and 3rd (12 → 3w2) cycle; (b) PXRD pattern for 1 obtained after annealing of 1 at 180 °C and after 7 days of ageing in MeOH vapor (heat–MeOH cycle) in the 1st (1 → 3), 2nd (11 → 31) and 3rd (12 → 32) cycle. | ||
Magnetic studies
In order to establish magneto-structural correlations, two series of magnetic investigations were carried out for compounds 1, 2 and 3 (Fig. S28–S33, ESI†). The first series (heat–MeOH cycle) temperature dependence of the molar magnetic susceptibility of (a) clusters 3 → 33 obtained after annealing of 1 → 13 and (b) clusters 11 → 13 obtained by exposing clusters 3 → 33 to MeOH vapors is shown in Fig. 6 (for 2 Fig. S32, ESI†).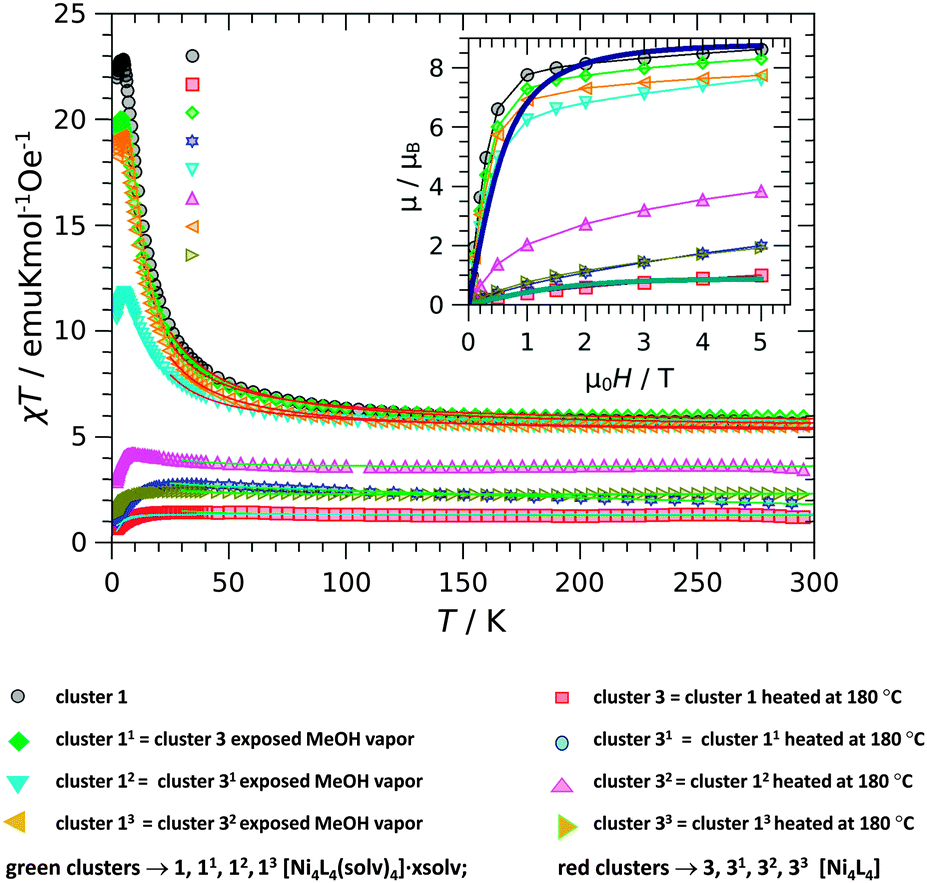 | ||
| Fig. 6 Temperature dependence of molar magnetic susceptibility measured in the field of 0.1 T multiplied with temperature, for cluster 1 series (heated compound 3, [Ni4L4] obtained by annealing of 1 and recycled by exposing to methanol vapors in turn). Lines are fitting curves. Inset: field dependence of magnetization per cubane unit measured at the temperature of 2 K. | ||
The second series (water–MeOH cycle) changes of magnetic behavior are induced by exposure of 1 (or 2) to water and methanol vapors alternatively (Fig. 7). The return to the initial state is almost equally effective in both cycles after exposure to methanol vapors. Small differences in the efficacy are connected with small differences in the dynamics of solvent release observed in other experiments.
 | ||
| Fig. 7 Temperature dependence of molar magnetic susceptibility measured in the field of 0.1 T multiplied with temperature, for cluster 1 series, measured successively after exposure to water vapor and recycling in methanol vapor alternately. Lines are fitting curves. Inset: Field dependence of magnetization per cubane unit measured at the temperature of 2 K. | ||
The Curie constant C of the first series clusters 1 → 3 → 11 → 31 → 12 → 32 → 13 → 33, in the heat–MeOH cycle, has a mean value of 5.1(3) K emu mol−1 Oe−1. This value confirms that in the starting green cluster 1 and recycled compounds (also green clusters 11, 12 and 13) the four spin 1 nickel ions per formulae unit are magnetically uncoupled at the high temperatures. The obtained g-factors (2.26, 2.21, 2.19 and 2.19) are in agreement with the expected values for Ni ions in octahedral coordination, within the 2% error of the measurement.12,15,25 Below ∼100 K, the susceptibility starts to deviate from the Curie–Weiss law, and the χT product increases, reaching max. at 3.8 K, suggesting the ferromagnetic coupling within the cubane. The red cluster 3 (and also red clusters 31, 32, and 33) of the formula [Ni4L4] has completely different magnetic properties; its effective magnetic moment per formula unit is considerably smaller than in [Ni4L4(CH3OH)4]·0.32CH3OH·0.32H2O, 1. Modelling the temperature behaviour of the susceptibility with the Curie–Weiss law gives the C values in the range from 1.4 to 3.4 K emu mol−1 Oe−1. This decrease indicates the partial transition, between 24 and 64% (sample dependent), of NiII ions from spin 1 to the diamagnetic state. The explanation of this effective magnetic moment reduction is possible only with the transition of some NiII ions from spin 1 to spin 0 state. Namely, the obtained C constant is too small to originate from the four spins 1 per cubane unit, and because of the structural improbability of the large antiferromagnetic coupling surviving in the whole temperature range, reduction of the number of magnetic units is the obvious prerequisite. It is known that spin 1 to spin 0 transition for nickel ions occurs with the change of coordination when the electron levels of t2g and eg orbitals change their order when compared to the octahedral coordination in the original complexes. The green–red–green interconversions observed in the series of conducted experiments (1 → 3 → 11…) were assigned to the reversible release and reabsorption of the solvent and to reversible change of coordination around NiII from six to four. However, not all NiII ions come to the diamagnetic state, possibly due to the remaining disorder around some nickel ions preventing against the ideal surrounding needed for transition to low spin. The observed downturn of χT with the lowering of the temperature is similar in all compounds. This decrease is coming from the zero-field splitting.29 Susceptibility of a series of samples obtained after successive exposure to water vapors and recycling in methanol vapors alternately is shown in Fig. 7 (for 2 Fig. S33, ESI†).
In the case of the water–methanol cycle, 1 → 3w → 11 → 3w1 → 12 → 3w2 → 13 → 3w3, the decrease of Curie constant and Weiss parameter values after exposure to water vapors is less expressed than in the case of the heat–methanol cycle. Exposure of 1 to water vapors leads to some more complex mixed state in the sense that it is not saturated, but magnetization is still higher than expected for independent ions. Quantitative analysis of these data is not practical since there are many possible combinations of partial transitions within cubanes, making the fitting procedure over-parameterized and impossible to perform. However, the observations are informative enough to help in explaining the magnetic changes within the light of solvatomagnetic effects.
Effective exchange interaction parameters J, g-factors, and percentage p of the remaining S = 1 ions for cluster 1 (for 2 Table S10, ESI†) in both cycles are given in Table 1. The obtained values of J are in accordance with similar structures,29 where similar corresponding angles within the Ni4 core were reported. Since the maximum of susceptibility is reached at 3.8 K resulting from the exchange coupling of cubane, and the M(H) dependence is reversible and linear for small fields down to 2 K, there is no possibility to observe SMM phenomena under investigated conditions, as was the case in other Ni4.29
| Step | Heat–methanol cycle of cluster 1 | Water–methanol cycle of cluster 1 | ||||
|---|---|---|---|---|---|---|
| J/K | G | p/% | J/K | g | p/% | |
| 1 | 4.53(5) | 2.31(1) | 100 fixed | 4.53(5) | 2.31 fixed | 99.7(2) |
| 3/3w | 1.2(2) | 2.33 fixed | 23.4(6) | 2.1(1) | 2.31 fixed | 91.8(8) |
| 11/11 | 3.55(5) | 2.46(1) | 100 fixed | 3.1(1) | 2.31 fixed | 95.2(3) |
| 31/3w1 | 0.3(1) | 2.33 fixed | 50.5(8) | 2.6(1) | 2.31 fixed | 76.9(4) |
| 12/12 | 2.70(4) | 2.29(1) | 100 fixed | 6.0(2) | 2.31 fixed | 84.1(3) |
| 32/3w2 | 0.8(1) | 2.33 fixed | 64(1) | 2.4(2) | 2.31 fixed | 71.7(4) |
| 13/13 | 3.71(6) | 2.26(1) | 100 fixed | 4.4(2) | 2.31 fixed | 83.8(2) |
| 33/3w3 | 0.8(1) | 2.33 fixed | 40.1(8) | 2.8(3) | 2.31 fixed | 33.9(2) |
More details about modelling of magnetic behaviour as well as the corresponding results of the equivalent series of compound 2 are presented in the ESI.†
Quantum chemical calculations
Geometry optimizations for the [Ni4L4(CH3OH)4] cluster and its modeled derivative were performed using the density functional theory with two hybrid functionals B3LYP and MN12SX. Calculations started from the experimentally determined crystal structures of compounds 1a and 2a (C2 point group of symmetry). In each case, all calculated geometrical parameters were very similar to the experimental ones, thus confirming the validity of the methods chosen. In order to investigate how the lattice solvent affects the magnetic properties of the compound, models of the [Ni4L4(CH3OH)4] cluster with a toluene molecule placed near the phenyl ring of the ligand (L2− = C6H5(O)–CN–(O)C6H4CH3) were constructed (Fig. 8). Since the only experimentally observable difference between unit cells of 1 and 1a in comparison with 2 and 2a was essentially a slight change in the C–H–π distance (Fig. 2), a toluene molecule was placed in two different positions, mimicking the crystal structures of 1 and 2 (Fig. 8). After a series of geometry optimizations, no significant geometrical change in the (NiII)4 core could be observed in either of the models. Natural bond orbital (NBO) analyses performed on each structure revealed that the change in the Ni-solvent distance has a negligible effect on the NBO occupancies of nickel atoms (Tables S11–S14, ESI†). Consequently, the observed differences in magnetic properties are most probably not caused by minor changes within the unit cells of 1 and 1a in comparison with 2 and 2a, but are due to the different solvent disorder acting as an outer electrostatic potential for the (NiII)4 core.
 | ||
| Fig. 8 Optimized geometries of (a) [Ni4L4(CH3OH)4] (C2 point group of symmetry) and (b) the complex of [Ni4L4(CH3OH)4] with toluene (B3LYP/6-31G(d) level of theory); interatomic distances frozen during optimizations are given using dashed lines and the solvent accessible surface is displayed.36 The other complex with slightly shifted toluene molecules is not presented here. | ||
Conclusions
Two starting compounds, [Ni4L4(CH3OH)4]·0.32CH3OH·0.32H2O (1) and [Ni4L4(CH3OH)4]·0.63CH3OH (2), L2− (N-(2-hydroxy-5-methylphenyl)salicylideneiminato anion), and various transition paths were used to generate a range of Ni4L4 clusters, differing in the type and amount of incorporated solvent. (Inter)conversions of coordinated and non-coordinated solvents and their effect on the magnetic and structural properties were examined. Exposing clusters (1) and (2) to room temperature resulted in a slight colour change, caused by the loss of non-coordinated solvates (1a and 2a). Annealing of these compounds yielded cluster 3, [Ni4L4]. Loss of the coordinated solvent caused the colour to be changed to red, and a partial transition of the spin of NiII ions from the ground state from S = 1 to S = 0. This partial transition could be assigned to temperature-induced disorder of the Ni4L4 core in the material. Several cycles of annealing–MeOH sorption experiments showed that the process is not fully reversible. Namely, the geometry of the N4L4 core was shown to vary, and materials obtained by sorption processes were also found to differ, probably in the amount of non-coordinated solvent. Similarly, a series of experiments where compounds 1 and 2 were consecutively exposed to H2O and CH3OH vapors showed differences in their powder diffraction patterns. This indicated that the number and amount of coordinated and non-coordinated solvents can vary, and are not easy to predict. Moreover, according to the quantum-mechanical calculations, it is the disorder that plays an important role in determining magnetic properties of the material. Existence of mixed solvates, and statistical and dynamic disorder of solvent molecules presented here indicate that there are multiple risks in the design of materials with desired magnetic properties: (i) fully controlled synthesis, (ii) establishing the exact magneto-structural relationship, (ii) water/methanol ambiguity, and (iii) stability of synthesised products. Detailed interpretation of the structural and magnetic correlation remains a challenge for future studies of these systems.Experimental
Synthesis and materials
All chemicals obtained from commercial sources were analytically pure and were used without further purification. The Schiff base N-(2-hydroxy-5-methylphenyl)salicylideneimine was prepared according to the literature data.37(a) Cluster 1 was prepared from Ni(O2CMe)2·4H2O (1 mmol) and Schiff base (1 mmol) in methanol (20 mL). The reactants were sealed in a 50 mL Teflon-lined, stainless-steel bomb. The bomb was heated at 110 °C for 1.5 hour. The obtained green plate-like single crystals were collected by filtration and washed with a minimum amount of cold methanol. Yield 51%: anal. calcd (without lattice solvent) (%) for C60H60N4Ni4O12 (1a): C, 57.02; H, 4.78; N, 4.43; Ni, 18.57. Found: C, 56.92; H, 4.82; N, 4.01; Ni, 18.19.
(b) The same crystalline product 1 was obtained using a similar procedure as in (a), in the solvent mixture of methanol (20 mL) and water (5 mL). The green plate-like crystalline product of 1 was not obtained if acetic anhydride (1 mL) was added to the reaction mixture of Ni(O2CMe)2·4H2O (1 mmol) and Schiff base (1 mmol) in dry methanol (20 mL) and using a similar procedure as in (a).
(c) Cluster 2 was prepared by mixing the methanolic solutions of Ni(O2CMe)2·4H2O (1 mmol in 10 mL) and Schiff base (1 mmol in 10 mL) at room temperature. After two days, green, prismatic single crystals were obtained. Yield 58%: anal. calcd (without lattice solvent) (%) for C60H60N4Ni4O12 (2a): C, 57.02; H, 4.82; N, 4.43; Ni, 18.57. Found: C, 56.70; H, 4.80; N, 4.29; Ni, 18.50.
(d) The same crystalline product 2 was obtained using a similar procedure as in (a) in the mixture of dry methanol (20 mL) and acetic anhydride (1 mL).
(e) Cluster 3, as dark red prisms or plates, was obtained after removal of the solvent molecules (coordinated and solvated) of 1 or 2 at 180 °C. Anal. calcd (%) for C56H44N4Ni4O8 (3): C, 59.22; H, 3.91; N, 4.93; Ni, 20.67. Found: C, 59.10 H, 3.79; N, 4.64; Ni, 20.82.
Cluster 1: 3406(b,m), 3056(m), 3007(m), 2917(m), 2793(m), 1604(vs), 1531(s), 1490(vs), 1378(m), 1305(s), 1226(s), 1127(s), 1034(m), 825(s), 750(s), 522(m).
Cluster 2: 3385(w), 3006(m), 2920(m), 2792(m), 2341(w), 2189(w), 1603(vs), 1531(s), 1490(vs), 1378(m), 1306(s), 1223(s), 1145(s), 1031(m), 824(s), 751(s), 524(m).
Cluster 3: 3054(m), 2915(m), 2359(m), 1599(vs), 1532(s), 1464(s), 1377(m), 1280(s), 1221(s), 1147(s), 1125(m), 828(s), 751(s), 668(m).
Calcd for cluster 1: CH3OH 10.85; H2O 0.45%; found: CH3OH 9.89; H2O 0.36%.
Calcd for cluster 2: CH3OH 11.63; found: CH3OH 11.10%.
:0.32 in 1 and 0.63 in 2), which have been refined as free variables, but at the final least-square refinement cycle, in order to obtain the final molecular formulae, the occupancies were fixed. By using SUMP instructions the sum of the occupancies of the two possible oxygen positions O5A and O5B in 2 was constrained to 0.63. The hydrogen atoms belonging to the stereochemically different carbon atoms and due to the data collection temperature were placed in the geometrically idealized positions with assigned isotropic displacement parameters and they were constrained to ride on their parent atoms by using the appropriate SHELXL-2014 HFIX instructions. The hydrogen atoms belonging to the coordinated methanol molecules were firstly found in the difference Fourier maps at the final stages of the refinement procedure and then refined by SHELXL-2014 DFIX instructions at 0.82(1) Å and 0.84(1) Å (for 1a and 2a compounds at 296 K and for 1 and 2 at 150 K, respectively) with assigned isotropic displacement parameters being 1.2 times larger than the equivalent isotropic displacement parameters of the parent oxygen atoms. The hydrogen atoms which belong to the methyl group of the solvent methanol molecule (or water) are not modelled. The molecular geometry calculations and graphics were done using ORTEP-III42 integrated in the WinGX software system, PLATON43 programme and Mercury.44 Supplementary crystallographic data (atomic coordinates, thermal parameters, all intramolecular distances and angles for all structures) are given in the ESI† (in the CIF format).
For the purpose of structure determination of cluster 3, a fresh sample of cluster 1a was produced, and then heated up to 180 °C. This sample was first examined under the microscope, which revealed small crystals with cracked surfaces, resembling a deposit of many thin platelets closely stacked together (Fig. S40, ESI†). Grinding the sample in the mortar resulted in paste-like material that was filled in a 1 mm capillary. X-ray powder diffraction (XRPD) data were measured using the MYTHEN detector47 installed at the Swiss Light Source,48 using an X-ray energy of 17.7 keV (0.7012 Å). Any radiation damage was avoided by collecting 120° of data within ten seconds (Table S15, ESI†). The measured XRPD pattern showed only a few sharp peaks and several broad ones, resulting in low data resolution (dmin = 3 Å). The XRPD pattern was compared to the calculated patterns of 1a (2a), and observed similarities led to the conclusion that these structures are correlated. Therefore, unit cell parameters of 1a (2a) were used to facilitate the indexing of the pattern of cluster 3, and the starting model of the [Ni4L4] cluster could be built using the structural information of 1a (2a). Considering that magnetic measurements of cluster 3 revealed antiferromagnetic properties, a planar-square geometry of the [Ni4L4] cluster was expected. In order to construct such a molecular structure, coordinated methanol from the 1a structure was removed (Fig. 3a, (a)), and this model was subjected to a series of energy optimizations49 (for details, see Computational methods). As expected, all calculations resulted in clusters of four square-planar coordinated Ni-centers, with Ni–O–Ni angles larger than 99°, suggesting the antiferromagnetic properties of the molecule (Fig. 3b, (b)).24,29 This molecular structure and unit cell parameters calculated from XRPD data were used as input for the crystal structure determination program FOX, based on direct-space optimization.50 Although all investigated samples (1, 2, 1a, and 2a) exhibited P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) symmetry, direct-space optimization processes were carried out in P1 symmetry without any restraints, in order to minimize model bias. However, this approach resulted in chemically non-reasonable structures. The reason for this failure was sought in the preferred orientation of the crystals, resulting in incorrect assignment of the relative intensities of the peaks. This obstacle was overcome by employing soft geometrical restraints. This way, solutions with chemically reasonable intermolecular distances could be favored. The obtained crystal structure of cluster 3 was subjected to Rietveld refinement, using the XRS suite of programs.51 Considering that insufficient data resolution did not allow for refinement of the atomic coordinates, only non-structural parameters (peak shapes and positions, zero shift, sample displacement, and scale) were refined (Fig. 4 and Table S15, ESI†). Subsequently, the geometry of the resulting crystal structure was optimized using the quantum mechanical approach (Fig. S41a, ESI†). Comparison between the optimized (ideal) molecular structure and the molecular structure obtained from FOX (average) is given in Fig. S41b (ESI†). All discrepancies between two models are considered to be a consequence of disorder in the material.
symmetry, direct-space optimization processes were carried out in P1 symmetry without any restraints, in order to minimize model bias. However, this approach resulted in chemically non-reasonable structures. The reason for this failure was sought in the preferred orientation of the crystals, resulting in incorrect assignment of the relative intensities of the peaks. This obstacle was overcome by employing soft geometrical restraints. This way, solutions with chemically reasonable intermolecular distances could be favored. The obtained crystal structure of cluster 3 was subjected to Rietveld refinement, using the XRS suite of programs.51 Considering that insufficient data resolution did not allow for refinement of the atomic coordinates, only non-structural parameters (peak shapes and positions, zero shift, sample displacement, and scale) were refined (Fig. 4 and Table S15, ESI†). Subsequently, the geometry of the resulting crystal structure was optimized using the quantum mechanical approach (Fig. S41a, ESI†). Comparison between the optimized (ideal) molecular structure and the molecular structure obtained from FOX (average) is given in Fig. S41b (ESI†). All discrepancies between two models are considered to be a consequence of disorder in the material.
Acknowledgements
Financial support for this research was provided by the Ministry of Science and Technology of the Republic of Croatia.References
- (a) C.-M. Ji, H.-J. Yang, C.-C. Zhao, V. Tangoulis, A.-L. Cui and H.-Z. Kou, Cryst. Growth Des., 2009, 9, 4607–4609 CrossRef CAS; (b) A. O. Polyakov, A. H. Arkenbout, J. Baas, R. G. Blake, A. Meetsma, A. Caretta, P. H. M. van Loosdrecht and T. T. M. Palstra, Chem. Mater., 2012, 24, 133–139 CrossRef CAS; (c) S. Osa, T. Kido, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, J. Am. Chem. Soc., 2004, 126, 420–421 CrossRef CAS PubMed.
- H.-Z. Kou, G.-Y. An, C.-M. Ji, B.-W. Wang and A.-L. Cui, Dalton Trans., 2010, 39, 9604–9610 RSC.
- C. Papatriantafyllopoulou, L. F. Jones, T. D. Nguyen, N. Matamoros-Salvador, L. Cunha-Silva, F. A. Paz, J. Rocha, M. Evangelisti, E. K. Brechen and S. P. Perlepes, Dalton Trans., 2008, 3153–3155 RSC.
- E. Pardo, R. Ruiz-García, J. Cano, X. Ottenwaelder, R. Lescouëzec, Y. Journaux, F. Lloret and M. Julve, Dalton Trans., 2008, 2780–2805 RSC.
- A. J. Zhou, J. L. Liu, R. Herchel, J. D. Leng and M. L. Tong, Dalton Trans., 2009, 3182 RSC.
- P. King, R. Clerac, W. Wernsdorfer, E. C. Anson and A. K. Powell, Dalton Trans., 2004, 2670–2767 RSC.
- R.-X. Yao, X. Xu and X.-M. Zhang, Chem. Mater., 2012, 24, 303–310 CrossRef CAS.
- G.-Y. An, C.-M. Ji, A.-L. Cui and H.-Z. Kou, Inorg. Chem., 2011, 50, 1079–1083 CrossRef CAS PubMed.
- I. S. Tidmarsh, E. Scales, P. R. Brearley, J. Wolowska, L. Sorace, A. Caneschi, R. H. Laye and E. J. L. McInnes, Inorg. Chem., 2007, 46, 9743–9753 CrossRef CAS PubMed.
- S. L. Castro, Z. Sun, C. M. Grant, J. C. Bollinger, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 1998, 120, 2365–2375 CrossRef CAS.
- E.-C. Yang, W. Wernsdorfer, S. Hill, R. S. Edwards, M. Nakano, S. Maccagnano, L. N. Zakharov, A. L. Rheingold, G. Christou and D. N. Hendrickson, Polyhedron, 2003, 22, 1727–1733 CrossRef CAS.
- M. Moragues-Cánovas, M. Helliwell, L. Ricard, E. Riviere, W. Wernsdorfer, E. Brechin and T. Mallah, Eur. J. Inorg. Chem., 2004, 2219–2222 CrossRef.
- (a) Y. Guo, L. Li, Y. Liu, Y. Dong and D. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m675–m676 CAS; (b) K. Ayikoe, R. J. Butcher and Y. Gultneh, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, E66, m1487–m1488 Search PubMed.
- P. Chanduri, V. Kataev, B. Büchner, H.-H. Klaus, B. Kersting and F. Meyer, Coord. Chem. Rev., 2009, 253, 2261–2285 CrossRef.
- J. M. Clemente-Juan, B. Chansou, B. Donnadieu and J.-P. Tuchagues, Inorg. Chem., 2000, 39, 5515–5519 CrossRef CAS PubMed.
- M. A. Palacios, A. J. Mota, J. E. Perea-Buceta, F. J. White, E. K. Brechin and E. Colacio, Inorg. Chem., 2010, 49, 10156–10165 CrossRef CAS PubMed.
- M. S. El Fallah, E. Rentschler, A. Caneschi and D. Gatteschi, Inorg. Chim. Acta, 1996, 247, 231–235 CrossRef CAS.
- M. Mikuriya, K. Minowa and N. Nagao, Inorg. Chem. Commun., 2001, 4, 441–443 CrossRef CAS.
- (a) J.-W. Ran, S.-Y. Zhang, B. Xu, Y. Xia, D. Guo, J.-Y. Zhang and Y. Li, Inorg. Chem., 2008, 11, 73–76 CAS; (b) Q. Liang, R. Huang, X. Chen, Z. Li, X. Zhang and B. Sun, Inorg. Chem. Commun., 2008, 11, 73–76 CrossRef; (c) J.-P. Sun, L.-C. Li and X.-J. Zheng, Inorg. Chem. Commun., 2011, 14, 877–881 CrossRef CAS.
- A. Escuer, M. Font-Bardía, S. B. Kumar, X. Solans and R. Vicente, Polyhedron, 1999, 909–914 CrossRef CAS.
- A. Sieber, C. Boskovic, R. Bircher, O. Waldmann, S. T. Ochsenbein, G. Chabonssant, W. Wernsdorfer, A. Neels, H. Stockli-Evans, S. Janssen, F. Juranyi and H. Mutka, Inorg. Chem., 2005, 44, 4315–4325 CrossRef CAS PubMed.
- M. A. Halcrow, J. S. Sun, J. C. Huffman and G. Christou, Inorg. Chem., 1995, 34, 4167–4177 CrossRef CAS.
- S.-Y. Zhang, W.-Q. Chen, B. Hu, Y.-M. Chen, W. Li and Y. Li, Inorg. Chem. Commun., 2012, 16, 74–77 CrossRef CAS.
- J. A. Bertrand, C. Marabella and D. G Vanderveer, Inorg. Chim. Acta, 1978, 26, 113–118 CrossRef CAS.
- W. L. Gladfelter, M. W. Lynch, W. P. Schaefer, D. N. Hendrickson and H. B. Gray, Inorg. Chem., 1981, 20, 2390–2397 CrossRef CAS.
- L. Ballester, E. Coronado, A. Gutierrez, A. Monge, M. F. Perpinan, E. Pinilla and T. Rico, Inorg. Chem., 1992, 31, 2053–2056 CrossRef CAS.
- S. Petit, P. Neugebauer, G. M. Pilet, G. Chastanet, A.-L. Barra, A. B. Autunes, W. Wernsdorfer and D. Lunean, Inorg. Chem., 2012, 51, 6645–6654 CrossRef CAS PubMed.
- (a) O. Kahn and J. V. Larinova, Chem. – Eur. J., 1999, 5, 3443–3449 CrossRef CAS; (b) H. Zheng-Ming and X.-M. Zhang, Dalton Trans., 2011, 40, 2092–2098 RSC.
- (a) A. Das, F. J. Klinke, S. Demeshko, S. Meyer, S. Dechert and F. Meyer, Inorg. Chem., 2012, 51, 8141–8149 CrossRef CAS PubMed; (b) P. S. Perlepe, A. A. Athanasopoulou, K. I. Alexopoulou, C. P. Raptopoulou, V. Psycharis, A. Escuer, S. P. Perlepes and T. C. Stamatatos, Dalton Trans., 2014, 43, 16605–16609 RSC; (c) S. Karmakar and S. Khanra, CrystEngComm, 2014, 16, 2371–2383 RSC; (d) G. Aromi, E. Bouwman, E. Burzuri, C. Carbonera, J. Krzystek, F. Luis, C. Schlegel, J. van Slageren, S. Tanase and S. J. Teat, Chem. – Eur. J., 2008, 14, 11158–11166 CrossRef CAS PubMed.
- G. Redler, C. Lampropoulos, S. Datta, C. Koo, T. C. Stamatatos, N. E. Chakov, G. Christou and S. Hill, Phys. Rev., 2009, B80, 094401 Search PubMed.
- Y.-Y. Zhu, T.-T. Yin, S.-D. Jiang, A.-L. Barra, W. Wernsdorfer, P. Neugebauer, R. Marx, M. Dörfel, B.-W. Wang, Z.-Q. Wu, J. van Slageren and S. Gao, Chem. Commun., 2014, 50, 15090–15093 RSC.
- (a) A. Masello, K. A. Abboud, W. Wernsdorfer and G. Christou, Inorg. Chem., 2013, 52, 10414–10423 CrossRef CAS PubMed; (b) F.-P. Huang, C. Yang, H.-Y. Li, P.-F. Yao, X.-H. Qin, S.-P. Yan and M. Kurmoo, Dalton Trans., 2015, 44, 6593–6599 RSC; (c) X. Zhang, V. Vieru, X. Feng, J.-L. Liu, Z. Zhang, B. Na, W. Shi, B.-W. Wang, A. K. Powell, L. F. Chibotaru, S. Gao, P. Cheng and J. R. Long, Angew. Chem., Int. Ed., 2015, 54, 9861–9865 CrossRef CAS PubMed.
- (a) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS; (b) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, IUCr monographs on crystallography, Oxford University Press, Great Clarendon Street, Oxford OX2 6DP, UK, 2001, pp. 29–120 Search PubMed.
- (a) M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC; (b) S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer (Version 3.0), University of Western Australia, Perth, 2012 Search PubMed; (c) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC; (d) http://www.povray.org .
- (a) J. A. Bertrand, A. P. Ginsberg, R. I. Kaplan, C. E. Kirkwod, R. L. Martin and R. C. Sherwod, Inorg. Chem., 1971, 10, 240–246 CrossRef; (b) E. K. Barefield, D. H. Busch and S. M. Nelson, Q. Rev., 1968, 22, 457–498 RSC.
- http://www.accelrys.com/ .
- M. Cindrić, N. Strukan, V. Vrdoljak, T. Kajfež and B. Kamenar, Z. Anorg. Allg. Chem., 2002, 628(9–10), 2113–2117 CrossRef.
- STAR e Software V.9.01., Mettler Toledo GmbH, 2006.
- Oxford Diffraction Ltd., Xcalibur CCD system, CrysAlis Software system, Versions 1.171.32.24 and 1.171.33.41. Abingdon; Oxfordshire, England, 2008.
- (a) G. M Sheldrick, A short history of SHELX., Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef PubMed; (b) G. M Sheldrick, SHELXL-2014, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8A CrossRef PubMed.
- L. J. Farrugia, WinGX (Ver. 1.80.05) suite for small-molecule single-crystal crystallography, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- L. J. Farrugia, Ortep-3 for Windows (v. 2.02) – a version of the current release of ORTEP-III, which incorporates a Graphical User Interface (GUI), J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. Spek, A Multipurpose Crystallographic Tool, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, Mercury CSD 2.0 – new features for the visualization and investigation of crystal structures, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- Philips X'Pert Data Collector 1.3e, Philips Analytical B. V. Netherlands, 2001.
- Philips X'Pert Plus 1.0, Philips Analytical B. V. Netherlands, 1999.
- B. Schmitt, C. Brönnimann, E. Eikenberry, F. Gozzo, C. Hörmann, R. Horisberger and B. Patterson, Nucl. Instrum. Methods Phys. Res., 2003, A501, 267–272 CrossRef.
- P. R. Willmott, D. Meister, S. J. Leake, M. Lange, A. Bergamaschi, M. Böge, M. Calvi, C. Cancellieri, N. Casati, A. Cervellino, Q. Chen, C. David, U. flechsig, F. gozzo, B. Henrich, S. Jäggi-Spielmann, B. Jakob, I. Kalichava, P. Karvinen, J. Krempasky, A. Lüdeke, R. Lüscher, S. Maag, C. Quitmann, M. L. Reinle-Schmitt, T. Schmidt, B. Schmitt, A. Streun, I. Vartiainen, M. Vitnis, X. Wang and R. Wullschleger, J. Synchrotron Radiat., 2013, 20, 667–682 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734–743 CrossRef CAS.
- C. Baerlocher and A. Hepp, XRS suite of programs, ETH Zurich, Zurich, Switzerland, 1982 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785–789 CrossRef.
- R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14, 16187–16191 RSC.
- (a) J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS; (b) A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS; (c) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS; (d) J. E. Carpenter and F. Weinhold, THEOCHEM, 1988, 46, 41–62 CrossRef CAS; (e) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev., 1993, B47, 558–562 Search PubMed.
- P. E. Blochl, Phys. Rev., 1994, B50, 17953–17980 Search PubMed.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev., 2011, B83, 195131 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1015860–1015863. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6nj00287k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |