
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Melt-cast materials: combining the advantages of highly nitrated azoles and open-chain nitramines†‡
Thomas M.
Klapötke
*,
Alexander
Penger
,
Carolin
Pflüger
and
Jörg
Stierstorfer
Department of Chemistry, Ludwig Maximilian University, Munich, Butenandtstr. 5-13, 81377 Munich, Germany. E-mail: tmk@cup.uni-muenchen.de
First published on 25th April 2016
Abstract
Numerous efforts to substitute TNT as the melt-cast matrix in explosive charges are ongoing due to its low performance and security issues. In this study the syntheses and full structural as well as spectroscopic characterizations of 2-nitrazapropyl substituted polynitroazoles, as potential melt-cast explosives, are presented. This straightforward method of derivatizing the heterocyclic N–H function by introducing a further energetic group improved the stability and energetic properties of the products. X-ray crystallographic measurements were performed for all compounds and afforded insights into structural characteristics such as strong intermolecular interactions. All compounds were characterized in terms of their sensitivities towards impact, friction and electrostatic discharge, and their thermal stabilities. The energetic properties were calculated with the EXPLO5 6.02 program.
Introduction
Melt-cast explosives are used in mortars, grenades and artillery shells and also in civil applications, e.g. for mining and demolition. Nowadays, the melt-cast technology is based on 2,4,6-trinitrotoluene (TNT), 2,4-dinitroanisole (DNAN) and 1,3,3-trinitroazetidine (TNAZ), the structures of which are depicted in Fig. 1. In general, the melt-cast explosives are molten in kettles heated at 80 to 120 °C with hot water or steam ingredients.1 Therefore, an ideal melt-cast explosive or its formulations should have a low melting point (70–120 °C), a sufficient separation of melting and decomposition processes, a low vapor pressure to diminish the inhalation toxicity and a higher density and better performance than the explosives already used. Furthermore, for processing, the melt-cast explosives should show no shrinking or cracking on cooling and no separation from the shell or casing.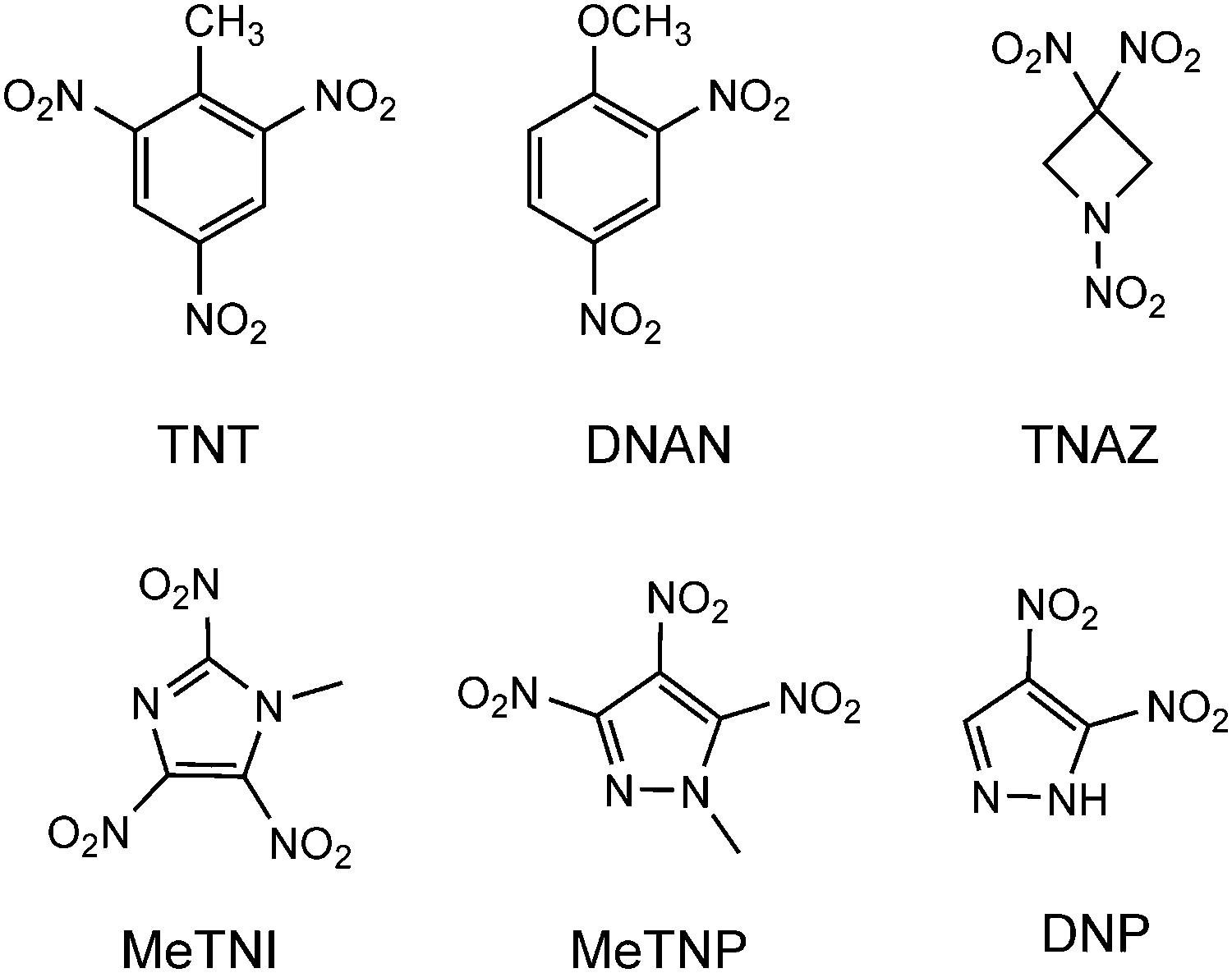 | ||
| Fig. 1 Melt-cast explosives. | ||
The drawbacks of TNT include its low performance (Vdet = 7300 m s−1) and the fact that its formulations are weak, brittle and prone to cracking, which increases impact sensitivities and exudation, and leads to dimensional instability with regard to thermal cycling.2 DNAN does not show a toxicity drawback, which the manufacturing of TNT also possesses, and it is less sensitive than TNT, but its performance is lower. Formulations of TNAZ show issues concerning sensitivities and the ability of the melt-cast process to perform crack-free and tension-free castings.1 Therefore, the development of new melt-cast explosives displaying wide-ranging improvements is still pursued. Polynitrated azoles such as 1-methyl-2,4,5-trinitroimidazole (MeTNI)3 and 1-methyl-3,4,5-trinitropyrazole (MeTNP)4 as well as 3,4-dinitropyrazole (DNP)5 (Fig. 1) show promising properties as replacements for TNT in melt-cast explosives due to their high positive heats of formation, which result from their large numbers of C–N and N–N bonds as well as from ring/cage strain.6 However, the nitro groups lead to increased acidity of the heterocyclic proton, which may result in problems concerning compatibility and storage. A common strategy to avoid the acidity and diminish the hygroscopicity is the N-alkylation of the nitro groups by methylation or the introduction of alkyl chains, which leads to better stabilities and compatibilities in formulations of explosive charges.4a,7
Alkylation using nitramine-containing side groups is an interesting strategy proposed for improving the energetic properties because the nitramine group may take part in intermolecular interactions as the acceptor and donor for hydrogen bonds as well as for dipolar N⋯O and C⋯O interactions, which should result in higher densities in comparison to the methylated derivatives.8 The linkage of two equal azoles by nucleophilic substitution of 1,3-dichloro-2-nitrazapropane with the corresponding potassium salts of nitrated azoles has been investigated in different research groups,7c,i,9 based on to studies of Bottaro and Highsmith.10 The resulting open-chain nitramines showed high thermal stabilities, high detonation performances and varying sensitivities towards impact and friction but unfortunately they did not melt before decomposition. Therefore, it was concluded that a nitramine function should be introduced without linking the azoles. Preliminary results using 1-chloro-2-nitrazapropane and the in situ double-deprotonated 3-nitro-1,2,4-triazol-5-one as well as various deprotonated tetrazole derivatives (as azole building blocks) revealed promising properties, especially in regard to thermal behavior, melting before decomposing.9,11
We now present an extended study of our current work combining the advantages of highly nitrated azoles and nitramines by alkylation of the N–H function with 1-chloro-2-nitrazapropane to obtain potential melt-cast explosives.
Results and discussion
Syntheses
1-Chloro-2-nitrazapropane was synthesized in two steps as shown in Scheme 1.11 After the nitration of 1,3,5-trimethyl-hexahydro-1,3,5-triazine, the conversion of 2-nitro-2-azapropyl acetate to 1-chloro-2-nitrazapropane was carried out by heating under reflux with thionyl chloride in dichloromethane and with catalytic amounts of acetic acid and sulfuric acid.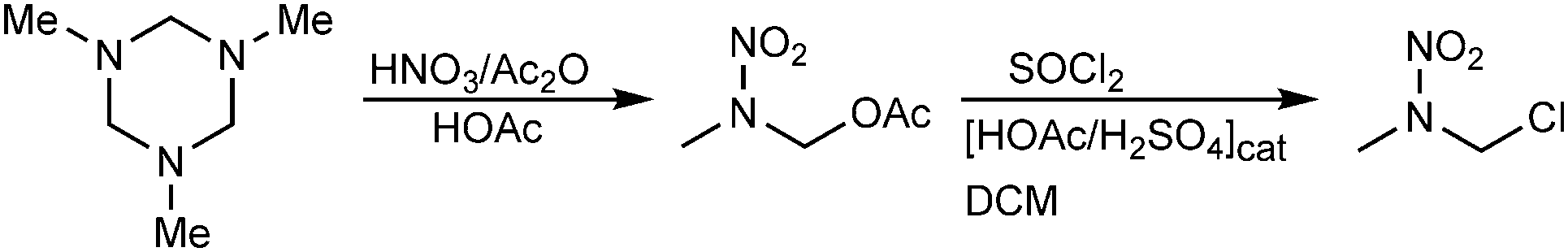 | ||
| Scheme 1 Synthesis of 1-chloro-2-nitrazapropane starting from 1,3,5-trimethyl-hexahydro-1,3,5-triazine. | ||
The appropriate nitrogen-rich heterocycles were obtained from literature-reported procedures4a,10b,12 and were generally converted to the corresponding potassium salts by using potassium hydroxide. The alkylation of the potassium salts with 1-chloro-2-nitrazapropane in aprotic solvents such as acetone or acetonitrile yielded the desired energetic nitramines as depicted in Scheme 2.
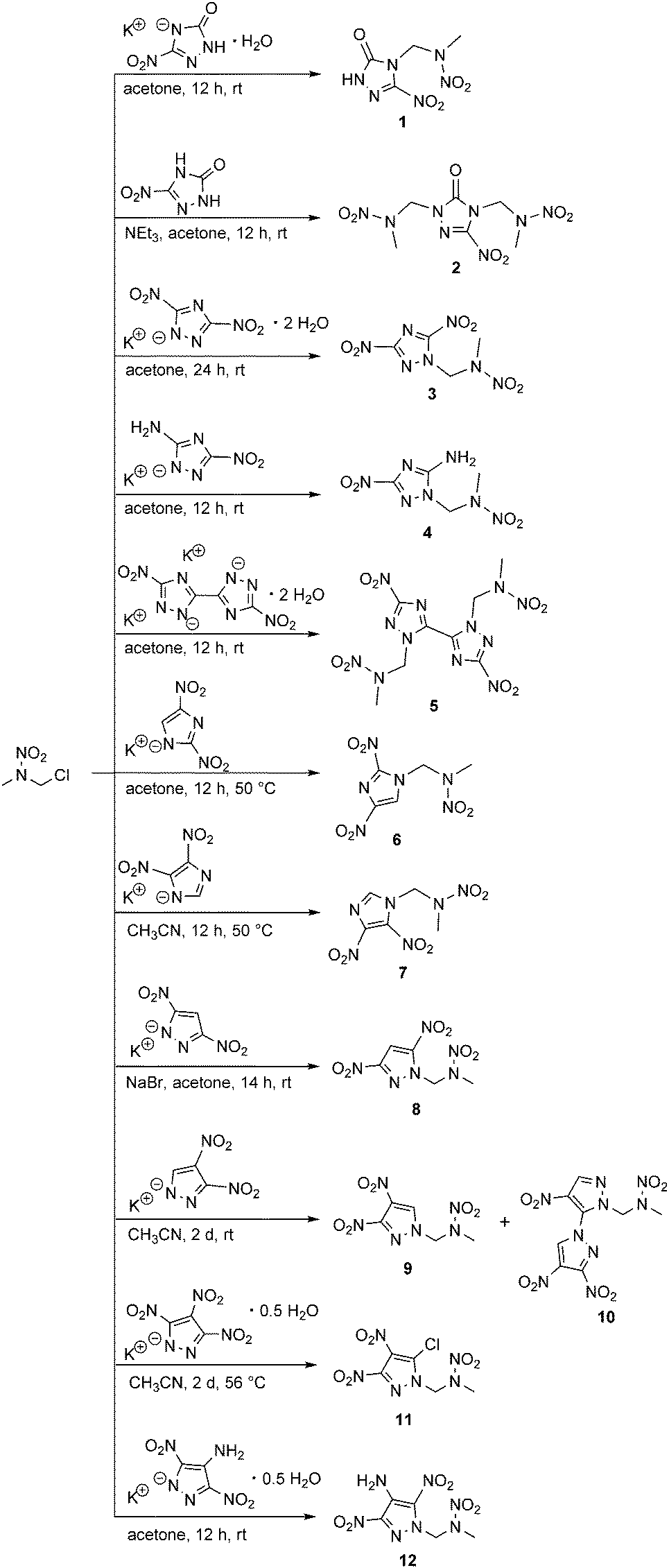 | ||
| Scheme 2 Syntheses of energetic nitramines starting from 1-chloro-2-nitrazapropane. | ||
In contrast to 1-(3-nitro-1H-1,2,4-triazol-5-on-4-yl)-2-nitrazapropane (1), which was synthesized from the mono potassium salt, the twice alkylated 2 was obtained from the in situ deprotonated free acid 3-nitro-1,2,4-triazol-5-one (NTO) using triethylamine as the base.
Various studies concerning the chemical stability of alkylated dinitrotriazoles show elimination of nitrite and formation of NTO derivatives and bistriazolyl systems under basic reaction conditions.13 Thus, the alkylation of potassium 3,5-dinitro-1,2,4-triazolate was carried out at ambient temperature and in stoichiometric amounts to avoid the elimination of nitrite. The nucleophilic substitution of 1-chloro-2-nitrazapropane with the potassium salt of 5-amino-3-nitro-1,2,4-triazole (ANTA) took place at the endocyclic nitrogen atom to yield nitramine 4, selectively. Potassium 3,3′-dinitro-5,5′-bi(1,2,4-triazolate) was alkylated twice at the nitrogen atoms N1/N1′ to afford nitramine 5.
Furthermore, the symmetric and asymmetric dinitroimidazole and symmetric dinitropyrazole derivatives were successfully alkylated to yield nitramines 6, 7, and 8, respectively. The reaction velocity of the synthesis of 8 was optimized by a Finkelstein halogen-exchange using sodium bromide.
Two different substituted nitramines were formed by the alkylation of the potassium 3,4-dinitropyrazolate. The alkylation at N1 of 3,4-dinitropyrazole afforded the desired nitramine 9. The formation of the other bipyrazolyl based nitramine (10) is caused by the high electrophilicity of the C3 position, which leads to a loss of the nitro group.14 The supposed intermediate, the existing 3-chloro-4-nitropyrazolate,15 reacted with another equivalent of potassium 3,4-dinitropyrazolate, thereby forming the 3,4,4′-trinitro-1,3′-bipyrazolyl system of nitramine 10. The dependence of the regioselectivity of the alkylation of 3,4-dinitropyrazole on the reaction temperature was studied, revealing that the alkylation at N1 is preferred at low temperatures. The relative ratio of nitramines 9 and 10 was determined by the intensities of the 1H NMR spectra with a reaction time of 24 h at 25 °C (84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16) and 82 °C (66:34). The isolated yields are not related to the reported ratio of intensities of the 1H NMR spectra, due to partial decomposition during purification by column chromatography, which also led to very low yields. The same reaction behavior was also observed for the nucleophilic substitution of 1,3-dichloro-2-nitrazapropane with potassium 3,4-dinitropyrazolate as reported previously.7c Prior to the presented synthesis route for bipyrazolyl systems, they were solely available by cine substitutions starting from 1,4-dinitropyrazole derivatives.16
:16) and 82 °C (66:34). The isolated yields are not related to the reported ratio of intensities of the 1H NMR spectra, due to partial decomposition during purification by column chromatography, which also led to very low yields. The same reaction behavior was also observed for the nucleophilic substitution of 1,3-dichloro-2-nitrazapropane with potassium 3,4-dinitropyrazolate as reported previously.7c Prior to the presented synthesis route for bipyrazolyl systems, they were solely available by cine substitutions starting from 1,4-dinitropyrazole derivatives.16
The nucleophilic substitution of 1-chloro-2-nitrazapropane with potassium 3,4,5-trinitropyrazolate afforded the 5-chloro-3,4-dinitropyrazolate substituted nitramine 11 due to the specific reactivity of the C5 position towards nucleophiles. This behavior has already been studied by Dalinger et al. for nucleophilic substitution reactions with methylated 3,4,5-trinitropyrazole.17 The reaction of 1-chloro-2-nitrazapropane with potassium 4-amino-3,5-dinitropyrazolate in acetone at ambient conditions afforded 12 in good yield.
Spectroscopy
Vibrational spectroscopic studies of all synthesized compounds were performed with IR and Raman spectroscopy and the frequencies were assigned according to the literature.18 Detailed descriptions are given in the ESI.‡Compounds 1–12 were characterized by 1H, 13C, and 14N NMR spectroscopy in d6-acetone. Selected chemical shifts of 1H, 13C{1H}, and 14N resonances of all presented compounds are summarized in Table 1. The chemical shifts of methylene protons of open-chain nitramines were studied in the 1970s and identified in the range of 5.90–6.10 ppm.19 By introducing electron-withdrawing substituents such as nitro groups in the azoles, the resonances of the methylene protons are shifted downfield. This effect is considerable for the 1,2,4-triazole based nitramines 3 and 5 with resonances at 6.81 ppm and 6.84 ppm, respectively. The resonance signals of methyl protons are in the range of 3.50 ppm to 3.70 ppm. The CH resonances of the imidazolyl and pyrazolyl substituents show the characteristic downfield shifts of aromatic protons of nitrogen-rich heterocycles.
| 1H NMR | 13C{1H} NMR | 14N | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Car–H | CH2 | CH3 | C–NO2 | C–H | Cq | CH2 | CH3 | NO2 | |
| a Due to the low solubility in organic solvents, some carbon signals could not be observed, although the measurements were performed with elongated pulse delays. | |||||||||
| 1 | — | 6.02 | 3.57 | 146.2 | — | 152.8 | 58.0 | 39.6 | −30 |
| −34 | |||||||||
| 2 | — | 6.03 | 3.57 | 145.3 | — | 151.8 | 60.3 | 39.8 | −31 |
| 5.90 | 3.50 | 58.4 | 38.3 | −34 | |||||
| 3 | — | 6.81 | 3.65 | 158.0 | — | — | 67.0 | 39.0 | −32 |
| 151.2 | −37 | ||||||||
| 4 | — | 6.11 | 3.60 | n.o.a | — | 156.9 | 61.6 | 38.5 | −21 |
| −25 | |||||||||
| 5 | — | 6.84 | 3.68 | n.o.a | — | 143.7 | 65.4 | 39.1 | −26 |
| 6 | 8.70 | 6.55 | 3.70 | 143.0 | 125.5 | — | 65.3 | 39.8 | −23 |
| −30 | |||||||||
| −32 | |||||||||
| 7 | 8.28 | 6.43 | 3.66 | 142.3 | 138.0 | — | 62.9 | 39.6 | −24 |
| 129.6 | −30 | ||||||||
| −34 | |||||||||
| 8 | 7.91 | 6.72 | 3.64 | 154.1 | 103.5 | — | 67.9 | 39.9 | −25 |
| 147.7 | −30 | ||||||||
| 9 | 9.03 | 6.33 | 3.63 | 148.4 | 134.0 | — | 66.9 | 38.8 | −28 |
| 126.9 | −31 | ||||||||
| 10 | 9.50 | 6.33 | 3.59 | 150.1 | 138.2 | 132.0 | 63.9 | 39.0 | −20 |
| 8.56 | 129.7 | 137.0 | −25 | ||||||
| 128.2 | −26 | ||||||||
| 11 | — | 6.42 | 3.66 | 148.4 | — | 130.0 | 64.4 | 39.0 | −28 |
| 123.9 | −31 | ||||||||
| 12 | — | 6.64 | 3.59 | 130.5–130.4 | — | 130.5–130.4 | 67.3 | 38.8 | −18 |
| −23 | |||||||||
| −26 | |||||||||
The carbon resonances of the C–NO2 functions are observed as small, broadened signals because of their coupling to the nitrogen cores of the nitro groups. The chemical shifts of the 13C methylene resonances are in the range of 58.0 ppm to 67.0 ppm and the methyl resonances are found in the range from 38.3 ppm to 39.9 ppm.
The resonance signals of the aromatic C–NO2 and the N–NO2 nitro group nitrogens are observed in the 14N NMR from −18 ppm to −37 ppm. An unambiguous assignment of these signals is difficult because of their similar shifts.
Two-dimensional NMR spectra (HMBC) of 10 were recorded for an unambiguous assignment of the protons and carbon atoms of the 3,4,4′-trinitro-1,3′-bipyrazole system (Fig. S1, ESI‡). The assignment is given in Fig. 2. The C–H resonance (no. 3) of the nitramine alkylated pyrazole ring is shifted upfield in comparison to the C–H resonance of 9, whereas C–H resonance no. 6 is shifted downfield, because of the neighboring electron-withdrawing substituted pyrazolyl substituent. The methyl and methylene proton resonances of 10 are in the same range as the ones of 9. Additionally, the assignment of the carbon atoms is given in Fig. 2.
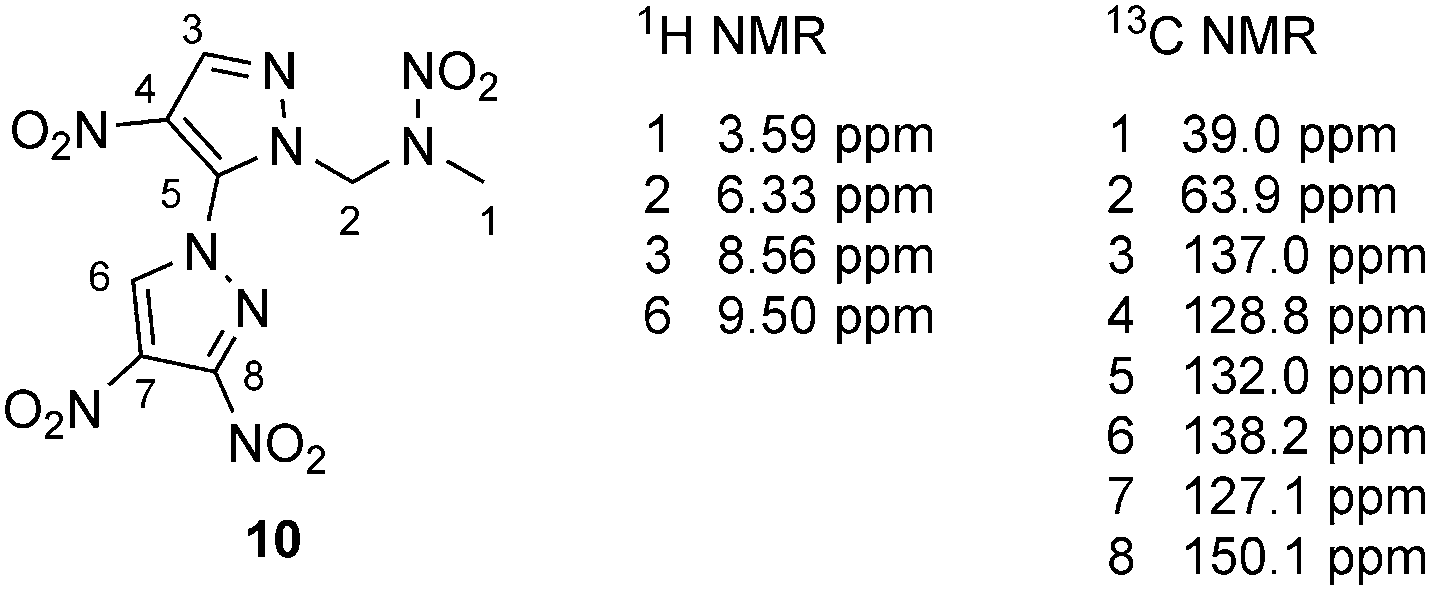 | ||
| Fig. 2 Assigned 1H and 13C{1H} NMR resonances of 10 in d6-acetone. | ||
Crystal structures
All presented compounds were also characterized by low-temperature single-crystal X-ray diffraction. Their molecular structures are depicted in Fig. 3. Selected data and parameters of the measurements and refinements are summarized in Tables S13 to S15 in the ESI.‡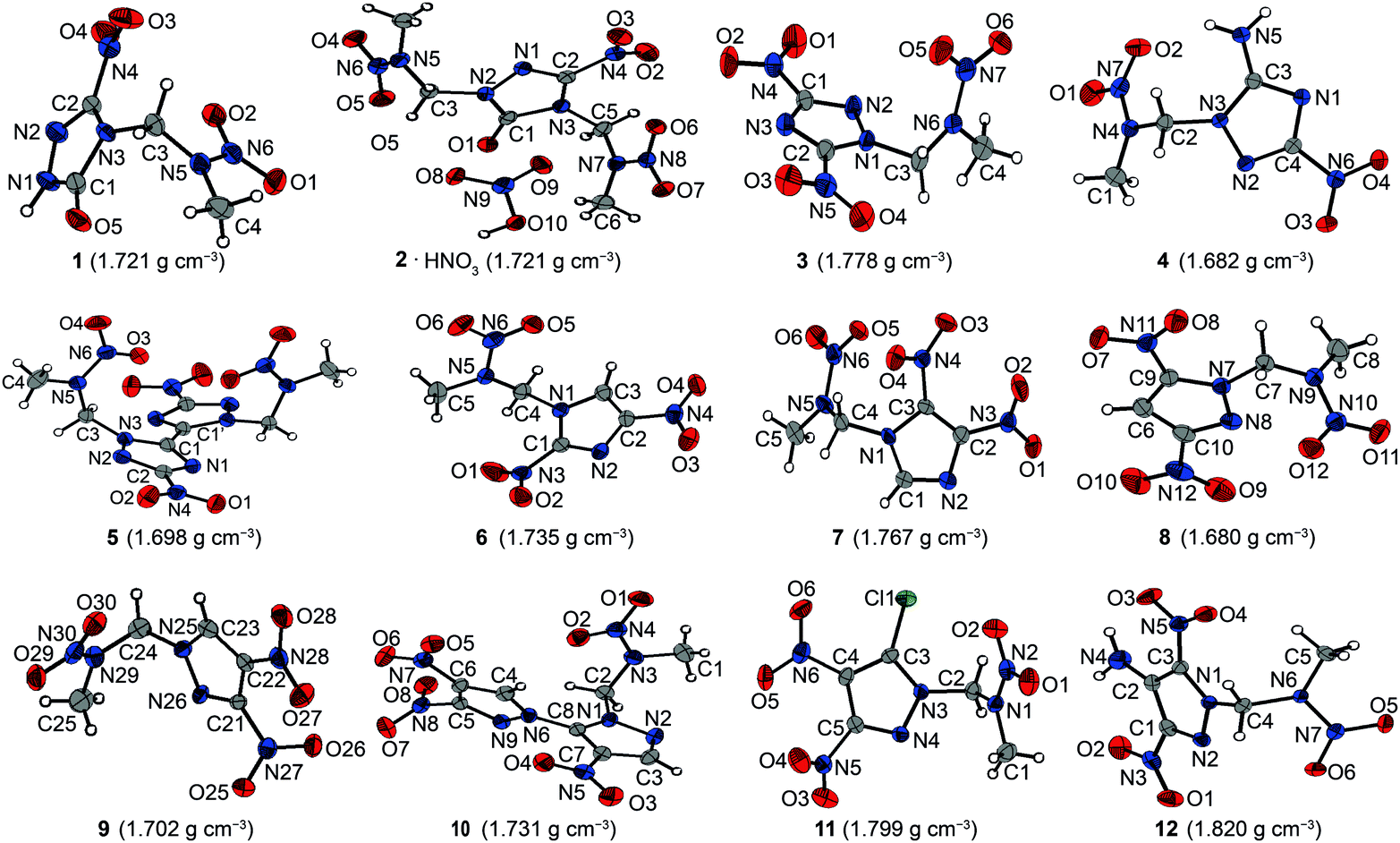 | ||
| Fig. 3 Molecular structures of nitramines 1–12 presented herein. | ||
Nitramine 1 crystallizes from acetone/dichloromethane in the monoclinic space group P21 with two formula units per unit cell and a density of 1.721 g cm−3 at 173 K. Its unit cell is shown in Fig. 4. The twice alkylated nitramine 2 crystallizes as its nitric acid adduct from diluted nitric acid in the monoclinic space group P21. The N–N bond lengths of the nitramine groups of 1 and 2 (1.34 Å) are shorter than the N–N bond length in the triazolone rings (1.37 Å). In comparison to the C–O bond length of γ-lactam systems (1.235 Å),20 the C–O bond length of nitramine 1 (1.214(3) Å) is shorter, whereas the corresponding bond length of 2 is in the same range (1.230(3) Å). The C–N bond length to the nitro group is 1.457(3) Å and, compared to the C–N bond lengths of the triazolone ring (1.293(3)–1.400(3) Å), is considerably elongated. The significant differences in the bond lengths are evidence for the localization of the double bond character. In comparison to β-NTO,21 the C2–N2 bond length (1.293(3) Å) is the shortest one, whereas the alkylation leads to an elongated C1–N3 bond length of 1.400(3) Å instead of 1.378(2) Å.21 The C–NO2 function is twisted out of the triazole plane by 20.4°, in contrast to the almost planar structure in NTO. This twisting enables various intermolecular interactions within the crystal structure of 1. Due to the high acidity of the protons of the 2-nitrazapropyl substituent, non-classical C–H⋯O and C–H⋯N hydrogen bonds are formed. Furthermore, high-grade directed dipolar N⋯O and C⋯O interactions are observed in the range from 2.869–2.970 Å. These dipolar interactions of O2 to the C2–N4 bond are supported by the twisting of the C–NO2 group. The interactions are summarized in Table S1 (ESI‡). The layer-like packing of 1 is formed by an intermolecular hydrogen bond of the acidic N–H function and the oxygen of the γ-lactam system. The layers are connected by weak non-classical hydrogen bonds between the terminal methyl group and the nitro groups, whereas the non-classical hydrogen bond C3–H3A⋯N2 is found within one layer. The dipolar N6⋯O2 interaction of 2.970 Å is observed in between the layers. Similar interactions are observed in the crystal structure of nitramine 2 as listed in Table S2 (ESI‡).
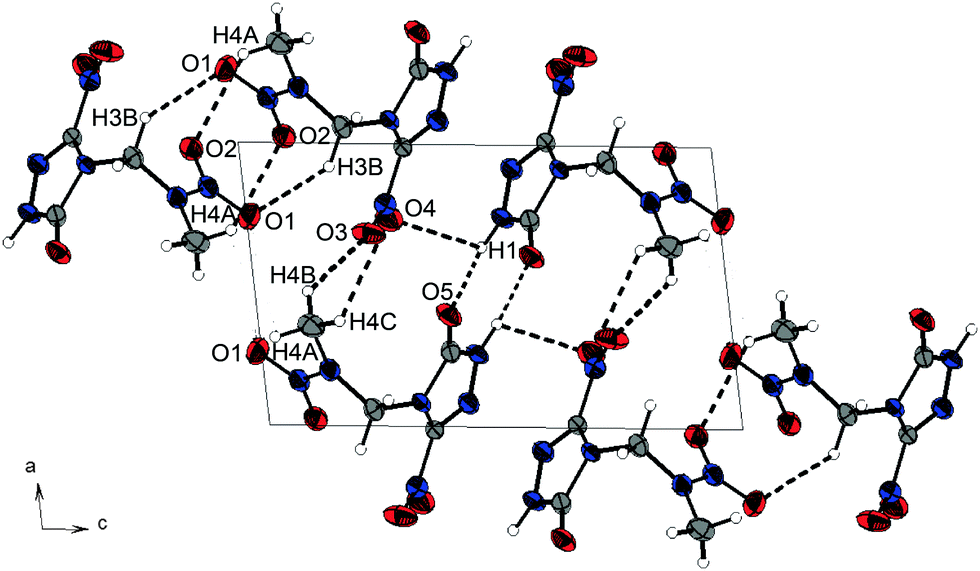 | ||
| Fig. 4 Unit cell of 1-(3-nitro-1H-1,2,4-triazol-5-on-4-yl)-2-nitrazapropane (1) along the b axis. Thermal ellipsoids are drawn at the 50% probability level. Selected hydrogen bonds are shown as black dotted lines. | ||
The crystal structures of the other 1,2,4-triazole based nitramines 3–5 are supported by several non-classical hydrogen bonds and additionally by either classical hydrogen bonds (4) or various dipolar interactions (nitramines 3 and 5) as summarized in Tables S3 to S5 (ESI‡). The structure of nitramine 5 shows C2 symmetry perpendicular to the C1–C1i axis and its triazole rings are twisted by 26.1(1)°. Its crystal structure is depicted in Fig. S8 (ESI‡). The molecules of one layer are connected by a dipolar C⋯O interaction (C3⋯O4: 2.920(2) Å) (Fig. 5).
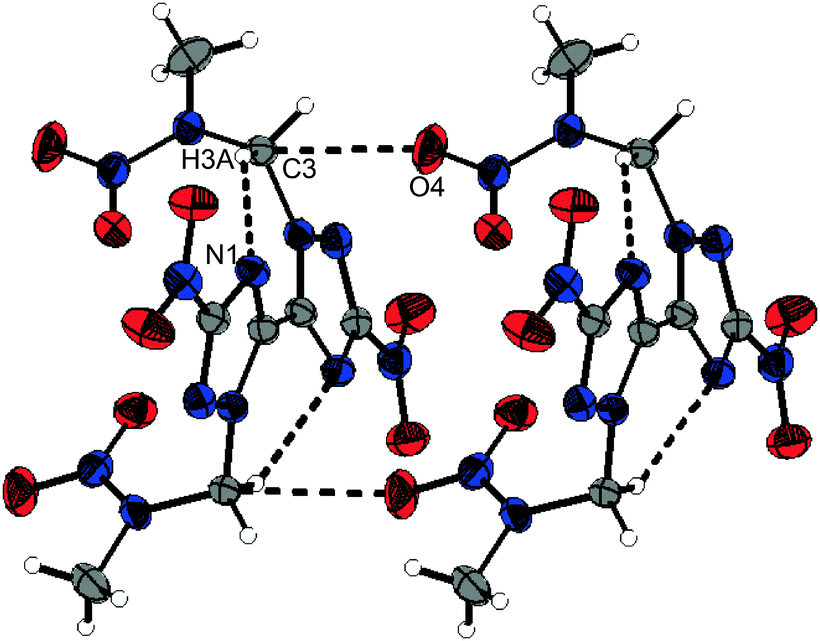 | ||
| Fig. 5 Selected structure showing dipolar intermolecular C⋯O interactions and non-classical hydrogen bonds as black dotted lines within the crystal structure of 5. | ||
The 4,5-dinitroimidazolyl based nitramine 7 crystallizes with a higher crystal density of 1.767 g cm−3 than that of the 2,4-dinitroimidazole based 6 (1.735 g cm−3) at 173 K. Furthermore, a greater extent of non-classical hydrogen bonding and dipolar interactions is observed within the crystal structure of 7 than in that of 6 (Tables S6 and S7, ESI‡). The crystal structures of the two dinitropyrazolyl based nitramines 8 and 9 are supported by various non-classical hydrogen bonds, and dipolar N⋯O and C⋯O interactions with distances below the sum of the corresponding van der Waals radii (Tables S8 and S9, ESI‡).
1-(3,4,4′-Trinitro-1,3′-bipyrazol-2′-yl)-2-nitrazapropane (10) crystallizes from dichloromethane in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two formula units per unit cell and a crystal density of 1.731 g cm−3 at 173 K. The molecular structure of 10 is depicted in Fig. 6. The pyrazole rings are twisted by 55.86(6)°. The almost perpendicular twists of the nitro groups N7 and N8 are caused by the steric repulsion of the neighboring nitro group, which thereby takes part in intra- and intermolecular interactions. The pyrazole N–N bond lengths of 1.367(2) Å and 1.379(2) Å are elongated in comparison to the nitramine N–N bond length of 1.358(2) Å. The alignment of 10 within the crystal structure is formed by non-classical hydrogen bonds. The C–H functions of the bipyrazolyl system and the methylene protons interact with the nitro groups and the nitrogen atoms of the bipyrazole due to their high acidity (Table S10, ESI‡). In addition to the non-classical hydrogen bonds, dipolar high-grade directed N⋯O and C⋯O interactions of oxygen O8 are observed, which are shorter than the corresponding sum of the van der Waals radii.22
with two formula units per unit cell and a crystal density of 1.731 g cm−3 at 173 K. The molecular structure of 10 is depicted in Fig. 6. The pyrazole rings are twisted by 55.86(6)°. The almost perpendicular twists of the nitro groups N7 and N8 are caused by the steric repulsion of the neighboring nitro group, which thereby takes part in intra- and intermolecular interactions. The pyrazole N–N bond lengths of 1.367(2) Å and 1.379(2) Å are elongated in comparison to the nitramine N–N bond length of 1.358(2) Å. The alignment of 10 within the crystal structure is formed by non-classical hydrogen bonds. The C–H functions of the bipyrazolyl system and the methylene protons interact with the nitro groups and the nitrogen atoms of the bipyrazole due to their high acidity (Table S10, ESI‡). In addition to the non-classical hydrogen bonds, dipolar high-grade directed N⋯O and C⋯O interactions of oxygen O8 are observed, which are shorter than the corresponding sum of the van der Waals radii.22
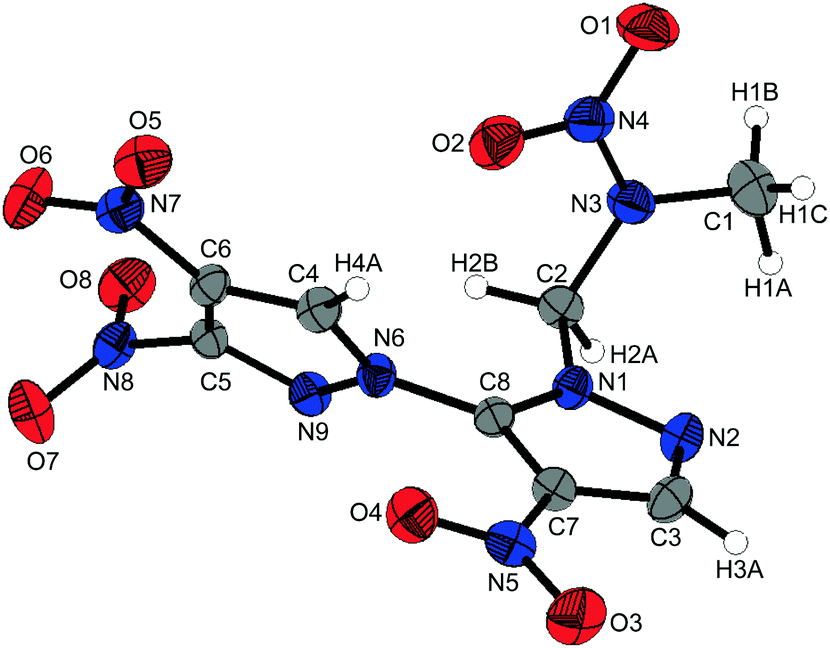 | ||
| Fig. 6 Molecular structure of 1-(3,4,4′-trinitro-1,3′-bipyrazol-2′-yl)-2-nitrazapropane (10). Thermal ellipsoids are drawn at the 50% probability level. | ||
The neighboring nitro groups of nitramine 11 are less twisted than those of 10. The oxygen atoms of the nitro group N6 as well as the nitramine group are involved in non-classical hydrogen bonds and additionally dipolar interactions are observed (Table S11, ESI‡).
1-(4-Amino-3,5-dinitropyrazol-1-yl)-2-nitrazapropane (12) crystallizes from acetonitrile/dichloromethane in the monoclinic space group P21/c with four formula units per unit cell. The bond lengths and angles in the pyrazole ring are similar to the corresponding ones in the nitramine alkylated ring of the bipyrazolyl system 10. In contrast to the crystal structure of 1-chloro-2-nitrazapropane,23 the nitramine nitro group is nonplanar with the C4–C5–N7 plane, but twisted out of the plane by 5.9(1)°. The pyrazolyl ring is turned out of the CNC-plane by 58.11(5)°. Due to the spatial arrangement of the nitramine group, various intramolecular non-classical hydrogen bonds and electrostatic interactions are formed, involving the methyl and methylene groups as donors and the nitramine nitro group as well as the O4 of the C-bonded nitro group as acceptors (Fig. 7). These and further selected interactions are listed in Table 2. The amino group is twisted out of the pyrazole ring by 3.65(3)°, whereas the nitro groups are almost in plane with a twist of less than 0.5°. These twists enable two intramolecular classical hydrogen bonds of 2.1781(16) Å and 2.2521(17) Å. Furthermore, intramolecular dipolar N⋯O and C⋯O interactions considerably below the sum of the van der Waals radii are observed. The unit cell and crystal structure are shown in Fig. S16 (ESI‡). The crystal structure consists of two different layers, which are opposed and staggered to each other. The layers are connected by a large number of non-classical and classical hydrogen bonds with a range from 2.40–2.60 Å. Remarkably, there is a very uncommon four-center bond, which is formed by proton H1 of the amino group interacting intermolecularly with oxygen atoms O3 and O2 and with another O2. In addition to the intermolecular hydrogen bonds, an intermolecular dipolar C⋯O interaction between C3 and O1 of 3.0179(17) Å is observed. The high number of intra- and intermolecular interactions leads to the high density of 1.820 g cm−3 at 173 K, which is the highest of all the herein presented nitramines.
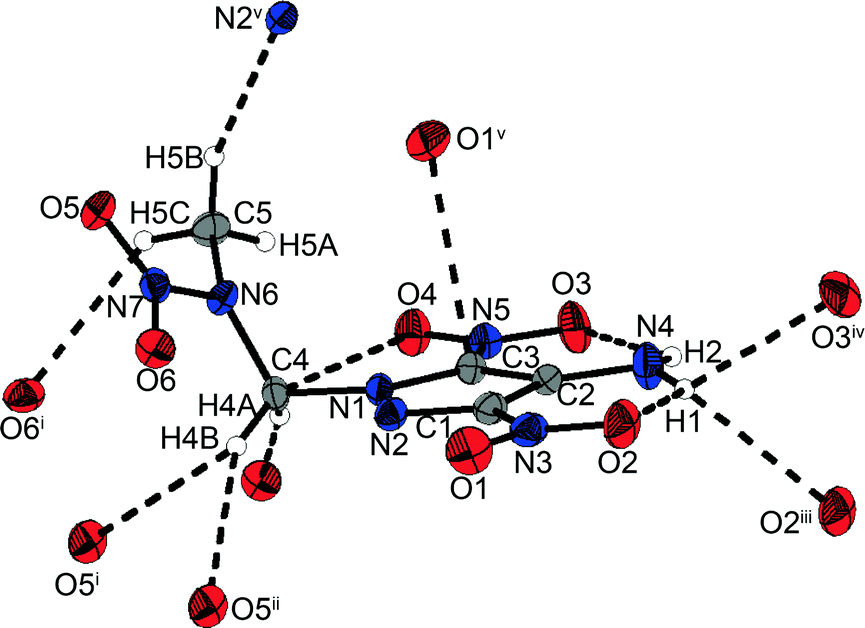 | ||
| Fig. 7 Selected hydrogen bonds within the crystal structure of 12 including a four-center hydrogen bond formed by proton H1 with two different oxygen atoms O2, and oxygen O3. | ||
| D–H⋯A | D–H [Å] | H⋯A [Å] | D⋯A [Å] | ∠DHA [°] |
|---|---|---|---|---|
| Symmetry codes: (i) 1 − x, 0.5 + y, 0.5 − z; (ii) x, −0.5 − y, 0.5 + z; (iii) −x, −y, 1 − z; (iv) −x, −0.5 + y, 0.5 − z; (v) x, −0.5 − y, −0.5 + z. | ||||
| C5–H5A⋯O4 | 0.981(2) | 2.3212(16) | 3.174(3) | 144.94(13) |
| N4–H2⋯O3 | 0.8954(7) | 2.1781(16) | 2.7687(18) | 122.93(6) |
| N4–H1⋯O2 | 0.8763(7) | 2.2521(17) | 2.8219(18) | 122.54(6) |
| C4–H4A⋯O6 | 0.9902(7) | 2.4025(17) | 3.3827(18) | 170.31(6) |
| N4–H1⋯O3iv | 0.8763(7) | 2.4858(14) | 3.0075(16) | 118.79(5) |
| N4–H1⋯O2iii | 0.8763(7) | 2.5768(17) | 3.1444(18) | 123.13(6) |
| C4–H4B⋯O5ii | 0.9904(6) | 2.6017(14) | 3.5332(16) | 156.74(5) |
| C5–H5B⋯N2v | 0.980(2) | 2.5903(16) | 3.479(3) | 150.99(13) |
| C5–H5C⋯O6i | 0.980(2) | 2.5719(14) | 3.293(2) | 130.43(12) |
| C4–H4B⋯O5i | 0.9904(6) | 2.5871(15) | 3.2073(15) | 120.66(5) |
| Dipolar interactions ΣvdW radii (N⋯O) < 3.07 Å22 | ||||
| N4⋯O3 2.7687(18) Å N4⋯O2 2.8219(18) Å | ||||
| Dipolar interactions ΣvdW radii (C⋯O) < 3.22 Å22 | ||||
| C4⋯O4 2.9061(17) Å C3⋯O1v 3.0179(17) Å | ||||
Thermal stabilities, sensitivities and energetic properties
The thermal behavior of all the presented compounds was investigated by differential scanning calorimetry (DSC) (Table 3). With the exception of 1-(3-nitro-1H-1,2,4-triazol-5-on-4-yl)-2-nitrazapropane (1), all nitramines melt before decomposition. For potential applications as energetic ingredients, a thermal stability above 180 °C is desired, which is achieved by the majority of the compounds. They show thermal stabilities from 151 °C to 264 °C.| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|
| a Impact sensitivity (BAM drophammer 1 of 6). b Friction sensitivity (BAM friction tester 1 of 6). c Electrostatic discharge (OZM Research). d Nitrogen content. e Oxygen balance (Ω = (xO − 2yC − 0.5zH)1600/M). f Melting temperature. g Decomposition temperature. h Density at ambient temperature. i Calculated enthalpy of formation. j Calculated energy of formation. k Energy of explosion. l Detonation temperature. m Detonation pressure. n Detonation velocity. o Volume of detonation gases. p Density measured with a He pycnometer. q Values obtained from the EXPLO5 database. | |||||||||
| Formula | C4H6N6O5 | C6H10N8O7 | C4H5N7O6 | C4H7N7O4 | C8H10N12O8 | C5H6N6O6 | C5H6N6O6 | C5H6N6O6 | C5H6N6O6 |
| M (g mol−1) | 218.13 | 306.20 | 247.13 | 217.15 | 402.25 | 246.14 | 246.14 | 246.14 | 246.14 |
| ISa (J) | 10 | >40 | 7 | >40 | >40 | 38 | 25 | 19 | >40 |
| FSb (N) | 160 | 96 | 160 | 252 | 240 | 120 | 144 | 288 | n.d. |
| ESDc (J) | 0.25 | 0.10 | 0.15 | 0.60 | 0.15 | 0.13 | 0.15 | 0.1 | n.d. |
| N (%) | 38.55 | 36.60 | 39.67 | 45.15 | 41.29 | 34.14 | 34.14 | 34.14 | 34.14 |
| Ω CO2 (%) | −44.0 | −52.3 | −29.1 | −55.3 | −51.7 | −45.5 | −45.5 | −45.5 | −45.5 |
| T melt (°C) | — | 117 | 136 | 202 | 225 | 130 | 142 | 126 | 80 |
| T dec (°C) | 151 | 211 | 165 | 224 | 228 | 183 | 190 | 210 | 234 |
| ρ (g cm−3) | 1.69 | 1.66p | 1.74 | 1.65 | 1.66 | 1.70 | 1.73 | 1.65 | 1.67 |
| ΔfHmi (kJ mol−1) | −71 | −51 | 216 | 105 | 311 | 71 | 90 | 111 | 136 |
| ΔfUj (kJ kg−1) | −229 | −65 | 964 | 586 | 866 | 379 | 456 | 542 | 643 |
| EXPLO5 6.02 values | |||||||||
| −ΔExUk (kJ kg−1) | 4598 | 4885 | 5701 | 4756 | 5017 | 5291 | 5374 | 5409 | 5512 |
| T det (K) | 3210 | 3258 | 3942 | 3205 | 3472 | 3610 | 3630 | 3713 | 3747 |
| p CJ (kbar) | 244 | 237 | 305 | 235 | 242 | 263 | 277 | 250 | 259 |
| V det (m s−1) | 7941 | 7925 | 8471 | 7983 | 7919 | 8046 | 8184 | 7899 | 8003 |
| V 0 (L kg−1) | 780 | 798 | 757 | 819 | 764 | 739 | 733 | 749 | 746 |
| 10 | 11 | 12 | TNT | DNAN | PETN | |
|---|---|---|---|---|---|---|
| Formula | C8H7N9O8 | C5H5ClN6O6 | C5H7N7O6 | C7H5N3O6 | C7H6N2O5 | C5H8N4O12 |
| M (g mol−1) | 357.20 | 280.59 | 261.16 | 227.13 | 198.13 | 316.13 |
| ISa (J) | >40 | >40 | >40 | 15 | >40 | 3 |
| FSb (N) | 288 | 288 | >360 | >360 | >360 | 60 |
| ESDc (J) | 0.2 | 0.1 | 0.4 | 0.7 | n.d. | 0.19 |
| N (%) | 38.83 | 29.95 | 37.54 | 18.50 | 14.14 | 17.72 |
| Ω CO2 (%)e | −51.5 | −34.2 | −45.9 | −74.0 | −96.9 | −10.1 |
| T melt (°C) | 133 | 82 | 141 | 81 | 94 | 141 |
| T dec (°C) | 264 | 214 | 165 | 309 | 315 | 165 |
| ρ (g cm−3) | 1.70 | 1.76 | 1.78 | 1.65q | 1.59 | 1.78q |
| ΔfHmi (kJ mol−1) | 331 | 111 | 108 | −59q | −177 | −534 |
| ΔfUj (kJ kg−1) | 1010 | 466 | 508 | −185q | −813 | −1594 |
| EXPLO5 6.02 values | ||||||
| −ΔExUk (kJ kg−1) | 5406 | 4872 | 5284 | 5022 | 4484 | 5994 |
| T det (K) | 3751 | 3761 | 3492 | 3452 | 3004 | 3971 |
| p CJ (kbar) | 258 | 256 | 298 | 207 | 161 | 309 |
| V det (m s−1) | 7986 | 7788 | 8482 | 7241 | 6705 | 8404 |
| V 0 (L kg−1) | 707 | 715 | 753 | 633 | 636 | 743 |
Especially for melt-cast applications the pyrazole based nitramines 8–11 and 2 are of great interest; all of these melt below 140 °C and reach 80–150 °C before decomposition. In general, the pyrazole based nitramines 8–12 show higher thermal stabilities than the imidazole based nitramines 6 and 7. The replacement of one nitro group in 1-(3,5-dinitro-1,2,4-triazol-1-yl)-2-nitrazapropane (3) by an amino group (4) leads to an increase in thermal stability of almost 60 °C. The same effect is observed for the thermal stability of the twice alkylated 3-nitro-1,2,4-triazol-5-one 2 in comparison to the mono alkylated 1.
The impact, friction and electrostatic discharge sensitivity tests were carried out for initial safety testing according to BAM methods.24 All presented nitramines are sensitive towards friction, with the exception of 1-(4-amino-3,5-dinitropyrazol-1-yl)-2-nitrazapropane (12). The impact sensitivities of the 1,2,4-triazolyl based nitramines differ strongly depending on the substituents and vary from sensitive to insensitive. Therefore, the twice alkylated NTO derivative 2 as well as 1-(5-amino-3-nitro-1,2,4-triazol-1-yl)-2-nitrazapropane (4) and the bitriazole 5 are insensitive towards impact, whereas 1 and 3 show sensitivities of 10 J and 7 J. The 2,4-dinitroimidazolyl nitramine 6 is less sensitive than the 4,5-dinitroimidazolyl nitramine 7; they show impact sensitivities of 38 J and 25 J, respectively. The pyrazolyl based derivatives are insensitive towards impact, with the exception of 1-(3,5-dinitropyrazol-1-yl)-2-nitrazapropane (8) (19 J). The electrostatic sensitivities of all presented compounds are within the range of 0.25–0.6 J and so are greater than the values (0.005–0.02 J) that the human body can release.25 In comparison to TNT, the synthesized nitramines mainly show lower impact sensitivities but increased sensitivities towards friction. In the first instance, the performance characteristics of new energetic materials are calculated to evaluate their utility for possible applications. The detonation parameters of all presented nitramines were calculated using the EXPLO5 (version 6.02) computer code.26 The calculations were performed using the maximum densities at 25 °C and the calculated enthalpies of formation. Enthalpies of formation were calculated using the atomization method, with CBS-4M27 based electronic enthalpies computed with the Gaussian 09 A.02 program.28 Gas phase enthalpies were transformed to solid state enthalpies by Trouton's rule, by substracting the corresponding enthalpies of sublimation from the gas-phase enthalpies.29 If the compound had no melting point, the decomposition point was used instead to transform the gas phase enthalpy to the solid state enthalpy by Trouton's rule. The crystal densities at low temperature were corrected to the corresponding crystal densities at 298 K using eqn (1) and the αv coefficient of volume expansion from the related nitramine HMX (αv = 1.6 × 10−4 K30).
| ρ298K = ρT/(1 + αv(298 − T)) | (1) |
Most promising for potential applications as TNT replacements in melt-cast formulations are nitramines 2, 6, 8 and 9 with regard to their thermal behavior, sensitivities and performance.
Furthermore, 1-(3,5-dinitro-1,2,4-triazol-1-yl)-2-nitrazaprop-ane (3) and 1-(4-amino-3,5-dinitropyrazol-1-yl)-2-nitrazaprop-ane (12) could be of potential use for energetic applications, since they show performances comparable to that of pentaerythritol tetranitrate (PETN) while possessing lower sensitivities.
Conclusions
Nucleophilic substitution of 1-chloro-2-nitrazapropane with nitro-substituted azoles afforded various open-chain nitramines, as reported herein. The alkylation of 3,4-dinitro-pyrazole yielded not only the desired nitramine 9 but also the alkylated bipyrazolyl system 10. Prior to this reaction, bipyrazolyl systems were solely obtained by cine substitutions.The crystal structures of all compounds were determined by low-temperature single-crystal X-ray diffraction and deliver insight into structural characteristics. A plurality of hydrogen bonds and dipolar C⋯O and N⋯O interactions is observed within each crystal structure, which leads to higher densities compared to the known crystal structures of the methylated parent compounds. In particular, within the crystal structure of 1-(4-amino-3,5-dinitropyrazol-1-yl)-2-nitrazapropane (12), an abundance of intra- and intermolecular interactions is observed, which leads to the highest density (1.82 g cm−3) of the herein presented nitramines. With the exception of 1, all nitramines melt before decomposition. The decomposition temperatures range from 151 to 264 °C. The thermally most stable compound is the bipyrazolyl nitramine 10. Due to the comparatively low melting points of nitramines 2 and 8–11 (<135 °C) and their considerably higher decomposition points, they are promising as potential melt-cast explosives.
Sensitivities towards impact, friction and electrostatic discharge were investigated by BAM methods. Compounds 1, 3 and 6–8 are found to be sensitive towards impact, while the others are insensitive. All compounds are sensitive towards friction with values between 96 N and 288 N, with the exception of 12 (>360 N, insensitive); these sensitivities are mainly in the range of RDX and PETN or even improved. The nitramines show calculated performances with detonation velocities in the range from 7788–8482 m s−1 and detonation pressures between 235–305 kbar. Nitramines 3 and 12 exhibit performances comparable to PETN, accompanied by lower sensitivities towards impact and friction. However, their thermal stabilities are lower than 180 °C. Therefore, the most promising compounds for potential applications as energetic materials with regard to their sensitivities and performances are the imidazolyl based nitramines 6 and 7. The most promising potential TNT replacements are nitramines 2, 6 and 8, taking into account their syntheses, sensitivities, energetic performances and, especially, their thermal behaviors.
Experimental section
Caution
All materials prepared are energetic compounds with sensitivities to various stimuli. While we encountered no issues in the handling of these materials, proper protective measures (face shield, ear protection, body armor, Kevlar gloves and earthed equipment) should be used at all times.General methods
All reagents and solvents were used as received (Sigma-Aldrich, Fluka, Acros Organics), if not stated otherwise. 5-Nitro-2,4-dihydro-1,2,4-triazol-3-one,12k 3,5-dinitro-1,2,4-tri-azole,10b 5-amino-3-nitro-1,2,4-triazole,12a 3,3′-dinitro-5,5′-bi(1,2,4-triazole),12j 4,5-dinitroimidazole,12c 2,4-dinitroimid-azole,12d 3,5-dinitropyrazole,12e,f 3,4-dinitropyrazole,12f,g 3,4,5-trinitropyrazole,4a and 4-amino-3,5-dinitropyrazole12b were prepared according to literature procedures. Melting and decomposition points were measured with a Linseis PT10 DSC using heating rates of 5 °C min−1. 1H, 13C{1H} and 14N NMR spectra were measured with a JEOL EX 400 or a JEOL Eclipse 400 instrument at ambient temperature. Chemical shifts are quoted in parts per million relative to TMS (1H, 13C) or nitromethane (14N). The solvent used was d6-acetone unless otherwise stated. The solvent residue signal was used as the locking signal for 1H and 13C NMR. Infrared spectra were measured with a PerkinElmer BX FT-IR spectrometer on a Smiths DuraSamplIR II diamond ATR unit at ambient temperature. The following abbreviations are used to characterize the relative signal intensities: vs (very strong), s (strong), m (medium), w (weak), vw (very weak) and br (broad). Raman spectra were recorded on a Bruker BAN II using a Nd:YAG laser (λ = 1064 nm) with a laser output of 300 mW. The vibrational spectroscopy data is given in the ESI.‡ Low-resolution mass spectra were recorded on a JEOL MStation JMS 700. Elemental analyses were performed on a Vario EL and a Vario Micro from Elementar Company. Sensitivity data were determined using a BAM drophammer and a BAM friction tester. The electrostatic sensitivity tests were carried out using an Electric Spark Tester ESD 2010 EN from OZM Research. Single-crystal X-ray diffraction studies were performed on an Oxford Diffraction XCalibur 3 diffractometer with a Kappa CCD detector using monochromatic molybdenum Kα radiation (λ = 0.71073 Å). The data collection was realized by using CrysAlisPro software.31 The structures were solved with SIR-92 implemented in the program package WinGX32 and finally checked using Platon.33The EXPLO5 program is based on the steady-state model of equilibrium and uses the Becker–Kistiakowsky–Wilson equation of state (BKW EOS) for gaseous detonation products and the Cowan-Fickett EOS for solid carbon.34 It is designed to enable the calculation of detonation parameters at the Chapman–Jouguet point.
Syntheses
DSC: Tdec = 151 °C. EA (C4H6N6O5, 218.13 g mol−1): calcd C 22.03, H 2.77, N 38.55%; found C 23.44, H 2.85, N 36.08%. 1H NMR (400 MHz): δ 12.00 (s, 1H, NH), 6.02 (s, 2H, CH2), 3.57 ppm (s, 3H, CH3). 13C{1H} NMR (101 MHz): δ 152.8 (CO), 146.2 (br, CNO2), 58.0 (CH2), 39.6 ppm (CH3). 14N{1H} NMR (29 MHz): δ −30 (NO2), −34 ppm (NO2). MS (DCI+): m/z (%): 219 (16) [M + H+]. Sensitivities (grain size: <100 μm): IS: 10 J; FS: 160 N; ESD: 0.25 J.
:30)). Due to decomposition by chromatography, only a small amount of colorless 9 (36 mg, 0.15 mmol, 3%) was obtained. DSC: Tmelt = 82 °C, Tdec = 234 °C. EA (C5H6N6O6, 246.14 g mol−1): calcd C 24.40, H 2.46, N 34.14%; found C 22.72, H 2.15, N 31.07%. 1H NMR (400 MHz): δ 9.03 (s, 1H, CH), 6.33 (s, 2H, CH2), 3.63 ppm (s, 3H, CH3). 13C{1H} NMR (101 MHz): δ 148.4 (br, CNO2), 134.0 (CH), 126.9 (br, CNO2), 66.9 (CH2), 38.8 ppm (CH3). 14N{1H} NMR (29 MHz): δ −28 (NO2), −31 (NO2), −176 ppm (br). MS (DCI+): m/z (%): 247 (73) [M + H+], 200 (26) [M+ − NO2]. Sensitivities (grain size: <100 μm): IS: >40 J. Rf value (silica gel, ethylacetate/i-hexane (70:30)): 0.72.
:30)) yielded 10 (103 mg, 0.29 mmol, 14%) as a rose solid. DSC: Tmelt = 133 °C, Tdec = 264 °C. EA (C8H7N9O8, 357.20 g mol−1): calcd C 26.90, H 1.98, N 38.83%; found C 26.84, H 1.87, N 34.48%. 1H NMR (400 MHz): δ 9.50 (s, 1H, CH), 8.56 (s, 1H, CH), 6.33 (s, 2H, CH2), 3.59 ppm (s, 3H, CH3). 13C{1H} NMR (101 MHz): δ 150.1 (br, CNO2), 138.2 (CH), 137.0 (CH), 132.0 (Cq), 129.7 (br, CNO2), 128.2 (br, CNO2), 63.9 (CH2), 39.0 ppm (CH3). 14N{1H} NMR (29 MHz): δ −20 (NO2), −25 (NO2), −26 (NO2), −170 ppm (br). MS (DCI+): m/z (%): 358 (4) [M + H+]. Sensitivities (grain size: <100 μm): IS: >40 J; FS: 288 N; ESD: 0.20 J. Rf value (silica gel, ethyl acetate/i-hexane (70:30)): 0.83.
Acknowledgements
The Konrad Adenauer Foundation is gratefully acknowledged for the award of a PhD scholarship to C. P. Financial support for this work by the Ludwig-Maximilian University of Munich (LMU), the U.S. Army Research Laboratory (ARL), the Office of Naval Research (ONR) under grant no. ONR.N00014-16-1-2062, and the Bundeswehr – Wehrtechnische Dienststelle für Waffen und Munition (WTD 91) under grant no. E/E91S/FC015/CF049 is gratefully acknowledged. The authors acknowledge collaborations with Dr Mila Krupka (OZM Research, Czech Republic) in the development of new testing and evaluation methods for energetic materials and with Dr Muhamed Sućeska (Brodarski Institute, Croatia) in the development of new computational codes to predict the detonation and propulsion parameters of novel explosives. We are indebted to and thank Drs Betsy M. Rice, Jesse Sabatini and Brad Forch (ARL, Aberdeen, Proving Ground, MD) for many inspired discussions. The authors would also like to thank Stefan Huber for assistance during the sensitivity measurements.Notes and references
- P. Ravi, D. M. Badgujar, G. M. Gore, S. P. Tewari and A. K. Sikder, Propellants, Explos., Pyrotech., 2011, 36, 393–403 CrossRef CAS.
- D. W. Doll, J. M. Hanks, T. K. Highsmith and G. K. Lund, WO01/46092A1, 2001.
- (a) R. Damavarapu, N. Gelber, R. Surapaneni, M. Zhang, R. Duddu and D. Parithosh, New Trends Res. Energ. Mater., Proc. Semin., 12th, Pardubice (Czech Republic), 2009, pp. 109–114 Search PubMed; (b) J. R. Cho, K. J. Kim, S. G. Cho and J. K. Kim, J. Heterocycl. Chem., 2002, 39, 141–147 CrossRef CAS.
- (a) G. Hervé, C. Roussel and H. Graindorge, Angew. Chem., Int. Ed., 2010, 49, 3177–3181 ( Angew. Chem. , 2010 , 122 , 3245–3249 ) CrossRef PubMed; (b) I. L. Dalinger, I. A. Vatsadze, T. K. Shkineva, G. P. Popova, S. A. Shevelev and Y. V. Nelyubina, J. Heterocycl. Chem., 2013, 50, 911–924 CrossRef CAS.
- S. Ek, K. Dudek, J. Johansson and N. Latypov, New Trends Res. Energ. Mater., Proc. Semin., 17th, Pardubice (Czech Republic), 2014, pp. 180–188 Search PubMed.
- M.-H. V. Huynh, M. A. Hiskey, E. L. Hartline, D. P. Montoya and R. Gilardi, Angew. Chem., Int. Ed., 2004, 43, 4924–4928 ( Angew. Chem. , 2004 , 116 , 5032–5036 ) CrossRef CAS PubMed.
- (a) L. I. Bagal, M. S. Pevzner, A. N. Frolov and N. I. Sheludyakova, Khim. Geterotsikl. Soedin., 1970, 259–264 CAS; (b) P. F. Pagoria, G. S. Lee, A. R. Mitchell and R. D. Schmidt, Thermochim. Acta, 2002, 384, 187–204 CrossRef CAS; (c) T. M. Klapötke, A. Penger, C. Pflüger, J. Stierstorfer and M. Sućeska, Eur. J. Inorg. Chem., 2013, 4667–4678 CrossRef; (d) M. A. Kettner and T. M. Klapötke, Chem. – Eur. J., 2015, 21, 3755–3765 CrossRef CAS PubMed; (e) Y. Tang, C. He, H. Gao and J. M. Shreeve, J. Mater. Chem. A, 2015, 3, 15576–15582 RSC; (f) P. Yin, J. Zhang, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2014, 20, 16529–16536 CrossRef CAS PubMed; (g) P. Yin, D. A. Parrish and J. M. Shreeve, Angew. Chem., Int. Ed., 2014, 53, 12889–12892 ( Angew. Chem. , 2014 , 126 , 13103–13106 ) CrossRef CAS PubMed; (h) O. P. Shitov, V. L. Korolev, V. S. Bogdanov and V. A. Tartakovsky, Russ. Chem. Bull., 2003, 52, 695–699 CrossRef CAS; (i) J. Zhang, C. He, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2013, 19, 8929–8936 CrossRef CAS PubMed.
- (a) G. L. Starova, O. V. Frank-Kamenetskaya and M. S. Pevzner, J. Struct. Chem., 1989, 29, 799–801 CrossRef; (b) Y.-X. Li, X.-J. Wang and J.-L. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o3073 CAS.
- (a) T. M. Klapötke, A. Penger and C. Pflüger, in New Trends Res. Energ. Mater., Proc. Semin., 14th, Pardubice (Czech Republic), 2011, pp. 753–761 Search PubMed; (b) A. Penger, Offenkettige Nitramine als potentielle Ersatzstoffe für Cyclomethylentrinitramin (RDX), PhD thesis, Ludwig Maximilian University of Munich, 2011 Search PubMed.
- (a) J. C. Bottaro, R. J. Schmitt, A. M. Petrie and P. E. Penwell, US62555112, 2001; (b) T. K. Highsmith, J. M. Hanks, S. P. Velarde and J. C. Bottaro, WO2002060881A1, 2002.
- N. Fischer, K. Karaghiosoff, T. M. Klapötke and J. Stierstorfer, Z. Anorg. Allg. Chem., 2010, 636, 735–749 CrossRef CAS.
- (a) K. Y. Lee, C. B. Storm, M. A. Hiskey and M. D. Coburn, J. Energ. Mater., 1991, 9, 415–428 CrossRef CAS; (b) R. D. Schmidt, G. S. Lee, P. F. Pagoria, A. R. Mitchell and R. Gilardi, J. Heterocycl. Chem., 2001, 38, 1227–1230 CrossRef CAS; (c) A. R. Katritzky, S. Singh, K. Kirichenko, M. Smiglak, J. D. Holbrey, W. M. Reichert, S. K. Spear and R. D. Rogers, Chem. – Eur. J., 2006, 12, 4630–4641 CrossRef CAS PubMed; (d) G. Wuellner, F.-W. Herkenrath, A. Juelich, Y. Yamada and S. Kawabe, WO2010021409, 2010; (e) R. Hüttel and F. Büchele, Chem. Ber., 1955, 88, 1586–1590 CrossRef; (f) J. W. A. M. Janssen, H. J. Koeners, C. G. Kruse and C. L. Habraken, J. Org. Chem., 1973, 38, 1777–1782 CrossRef CAS; (g) G. T. Morgan and I. Ackerman, J. Chem. Soc., Trans., 1923, 123, 1308–1318 RSC; (h) Y. V. Serov, M. S. Pevzner, T. P. Kofman and I. V. Tselinskii, Russ. J. Org. Chem., 1990, 26, 773–777 Search PubMed; (i) L. I. Bagal, M. S. Pevzner, A. N. Frolov and N. I. Sheludyakova, Chem. Heterocycl. Compd., 1970, 6, 240–244 CrossRef; (j) A. A. Dippold, T. M. Klapötke and N. Winter, Eur. J. Inorg. Chem., 2012, 3474–3484 CrossRef CAS; (k) K.-Y. Lee, L. B. Chapman and M. D. Cobura, J. Energ. Mater., 1987, 5, 27–33 CrossRef CAS.
- (a) L. I. Bagal, M. S. Pevzner, V. Y. Samarenko and A. P. Egorov, Khim. Geterotsikl. Soedin., 1970, 1701–1703 CAS; (b) M. S. Pevzner, V. Y. Samarenko and L. I. Bagal, Khim. Geterotsikl. Soedin., 1972, 117–119 CAS; (c) T. P. Kofman, G. Y. Kartseva, V. I. Namestnikov and E. A. Paketina, Russ. J. Org. Chem., 1998, 34, 1032–1039 CAS; (d) M. S. Pevzner, T. P. Kofman, E. N. Kibasova, L. F. Sushchenko and T. L. Uspenskaya, Khim. Geterotsikl. Soedin., 1980, 257–261 CAS; (e) T. P. Kofman, Russ. J. Org. Chem., 2001, 37, 1158–1168 CrossRef CAS.
- I. L. Dalinger, I. A. Vatsadze, T. K. Shkineva, I. O. Kortusov, G. P. Popova, V. V. Kachala and S. A. Sheveleva, Russ. Chem. Bull., 2010, 59, 1786–1790 CrossRef CAS.
- I. J. Ferguson, K. Schofield, J. W. Barnett and M. R. Grimmett, J. Chem. Soc., Perkin Trans. 1, 1977, 672–675 RSC.
- P. Cohen-Fernandes, C. Erkelens, C. G. M. Van Eendenburg, J. J. Verhoeven and C. L. Habraken, J. Org. Chem., 1979, 44, 4156–4160 CrossRef CAS.
- I. L. Dalinger, I. A. Vatsadze, T. K. Shkineva, G. P. Popova and S. A. Shevelev, Mendeleev Commun., 2011, 21, 149–150 CrossRef CAS.
- M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der organischen Chemie, Thieme, Stuttgart, 7 edn, 2005 Search PubMed.
- (a) I. J. Solomon, R. K. Momii, F. H. Jarke, A. J. Kacmarek, J. K. Raney and P. C. Adlaf, J. Chem. Eng. Data, 1973, 18, 335–337 CrossRef CAS; (b) A. R. Farminer and G. A. Webb, Tetrahedron, 1975, 31, 1521–1526 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- N. B. Bolotina, E. Zhurova and A. A. Pinkerton, J. Appl. Crystallogr., 2003, 36, 280–285 CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- B. Aas, M. A. Kettner, T. M. Klapötke, M. Sućeska and C. Zoller, Eur. J. Inorg. Chem., 2013, 6028–6036 CrossRef CAS.
- (a) Test Methods According to the UN Manual of Test and Criteria, Recommendations on the Transport of Dangerous Goods, United Nations Publication, New York, Geneva, 4th revised edn, 2003: impact: insensitive >40 J, less sensitive ≥35 J, sensitive ≥4 J, very sensitive ≤3 J; friction: insensitive >360 N, less sensitive = 360 N, sensitive <360 N to >80 N, very sensitive ≤80 N, extremely sensitive ≤10 N; (b) NATO, Standardization Agreement 4489 (STANAG 4489), Explosives, Impact Sensitivity Tests 1999; (c) NATO, Standardization Agreement 4487 (STANAG 4487), Explosives, Friction Sensitivity Tests 2002.
- T. M. Klapötke, Chemistry of High-Energy Materials, De Gruyter, Berlin, Bosten, 2015 Search PubMed.
- M. Sućeska, Explo5 program, Zagreb, Croatia, 6.02 edn, 2014 Search PubMed.
- (a) J. J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532–6542 CrossRef CAS; (b) J. W. Ochterski, G. A. Petersson and J. J. A. Montgomery, J. Chem. Phys., 1996, 104, 2598–2619 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- (a) M. S. Westwell, M. S. Searle, D. J. Wales and D. H. Williams, J. Am. Chem. Soc., 1995, 117, 5013–5015 CrossRef CAS; (b) F. Trouton, Philos. Mag., 1884, 18, 54–57 CrossRef.
- C. Xue, J. Sun, B. Kang, Y. Liu, X. Liu, G. Song and Q. Xue, Propellants, Explos., Pyrotech., 2010, 35, 333–338 CrossRef CAS.
- CrysAlisPro, Version 1.171.35.11 (release 16.05.2011 CrysAlis171.net), Agilent Technologies, 2011.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. L. Spek, Platon, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, NL, 1997 Search PubMed.
- (a) M. Sućeska, Propellants, Explos., Pyrotech., 1991, 16, 197–202 CrossRef; (b) M. Sućeska, Propellants, Explos., Pyrotech., 1999, 24, 280–285 CrossRef.
Footnotes |
| † Parts of this study have been presented at the 14th Seminar on New Trends in Research of Energetic Materials, Pardubice, Czech Republic, April 13–15, 2011 and in the PhD thesis of Alexander Penger. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1439310–1439321. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6nj00202a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |