
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Growth of strontium hydrogen phosphate/gelatin composites: a biomimetic approach
Mazeyar Parvinzadeh
Gashti
 *a,
Manuela
Stir
b and
Jürg
Hulliger
*b
*a,
Manuela
Stir
b and
Jürg
Hulliger
*b
aYoung Researchers and Elites Club, Yadegar-e-Imam Khomeini (RAH) Branch, Islamic Azad University, P.O. Box 18155/144, Tehran, Iran. E-mail: mparvinzadeh@gmail.com; Fax: +98(021)22593135; Tel: +98(0)9123137115
bDepartment of Chemistry & Biochemistry, University of Berne, Freiestrasse 3, CH-3012 Berne, Switzerland. E-mail: juerg.hulliger@dcb.unibe.ch
First published on 26th April 2016
Abstract
Recent research has focused on the crystal growth of strontium phosphates via different methods due to the bioactivity and biocompatibility of these materials with bone tissue. Here, we use a biomimetic method to synthesize strontium hydrogen phosphate/gelatin composites via single diffusion in gelatin. We compare the composite crystals with analytical-grade strontium hydrogen phosphate using infrared spectroscopy (IR), scanning electron microscopy (SEM), thermal gravimetric analysis, and X-ray diffraction. The IR spectra reveal that gelatin is incorporated into strontium hydrogen phosphate crystals due to CH2 and amide I vibrations. The SEM data exhibit arrow-like crystals and microspheres of strontium hydrogen phosphate with bundled nano-flakes and flower-like surface arrangements of crystals. Furthermore, we find that the thermal stability of the strontium hydrogen phosphate/gelatin composite crystals is higher than that of the analytical-grade strontium hydrogen phosphate. The ion concentration and the preparation method had only a small influence on the broadening of the X-ray Bragg reflections. Our study confirms that gelatin is a useful hydrogel for studying the crystallization of strontium hydrogen phosphate.
1. Introduction
Strontium has been demonstrated as a trace element in the human body that effectively stimulates bone formation and remodeling.1–4 SrHPO4 (SrHP) as one of its main compounds has been considered to be an ion exchanger biomaterial for holding both hydrogen phosphate (HPO42−) and strontium (Sr2+) ions.5–9 A significant deal of attention has been paid to strontium containing hydroxyapatite cements to increase their bioactivity, biocompatibility and compressive strength.10–14 The majority of previous studies devoted to the synthesis of SrHP have been focused on the hydrothermal method,15,16 cathodic reduction17 and solution growing.18–25 Taher et al. studied the hydrothermal synthesis of SrHP from a mixture of Li2CO3, SrCO3 and H3PO4 in water after conducting a thermal procedure at 180 °C for 6 h.15 More recently, Zhang et al. developed SrHP nano-belts without any surfactant as the template at 160 °C.16 It has been shown that the SrHP coating can be controlled on the cathode via the electrochemical reduction of alkaline earth metal-containing electrolytes.17 First introduced by Mooney et al.,18,19,21 growing solutions of SrHP has been the most popular approach in recent decades. This process consists of the slow evaporation of a solution containing Sr(NO3)2 and NH4H2PO4, which leads to the precipitation of SrHP.24 Furthermore, Roming and Feldman prepared nanoscale SrHP by rapidly mixing Sr(NO3)2, diethylene glycol and H3PO4 in water. These authors found that the rapid cooling of the process temperature from 170 to 15 °C resulted in the formation of α-SrHP and a continuous slow-heating process resulted in the appearance of β-SrHP.25 The only study about SrHP crystallization in gelatin was conducted by Banks et al. who focused on double diffusion in a gel. These authors, who found that polycrystalline spherulites of SrHP were formed in gelatin, did not discuss the morphology of the crystals or the influence of the gel on the crystallization.26 The question, therefore, arises of how the polar domains of gelatin influence the SrHP nucleation and morphology. In 2013, we aimed to study the effect of glutamic acid on the biomineralization of brushite in gelatin hydrogel. Interestingly, we found that glutamic acid capable of changing the morphology of brushite from spherulitic growth to mushroom-like crystals. Such structures were shown to be very similar to brushite kidney stones which play an important role in the study of crystal deposition diseases. Our further studies demonstrated that gelatin, starch and iota-carrageenan gels can modulate the diffusion of barium and calcium ions.27–30 In this regard, we reported the novel morphologies of barium hydrogen phosphate in gelatin for different metal phosphate crystals. As a result, the gel pH, the gel type, the concentration of ions as well as the method of crystallization strongly contributed to morphological changes of different inorganic–organic composite crystals. Our results strongly revealed that the morphology of crystals is altered and controlled using the gel growth method.The mineralization of inorganic biocompounds in living organisms typically occurs in collagenous scaffolds.31 Here, we use gelatin as a model system for the gel growth of SrHP because it is an irreversibly hydrolyzed form of collagen, the primary organic compound found in bone. Gelatin contains peptides and proteins that are excellent biocompatible and biodegradable materials for bone tissue engineering applications. Furthermore, the gel diffusion method has been shown to be a straightforward, inexpensive and controllable model for the reproduction of biomineralization conditions that occur in nature.32,33 This method has been previously employed by several groups to gain new insights into the formation mechanisms of fluorapatite,34 brushite,27 octacalcium phosphate,30 calcium carbonate and calcite.35 Hydrogels provide an effective medium with high cross-linking density for controlling the morphology of a crystal by adjusting the ratio of ion diffusion.36 On the other hand, SrHP is known as a transient in vivo phase to produce apatite. Strontium ions bear a chemical resemblance to calcium ions due to their charge-to-size ratio, and strontium ions play a special role in bone remodeling via apatite lattice distortion, osteoblast proliferation, osteoclast differentiation, bone strength promotion and alkaline phosphatase activation.10,11 These benefits arise because strontium has a larger ionic radius than calcium, which strongly suppresses the biological performance of strontium-containing cements.12,13
Here, we developed new types of SrHP crystals as key compounds in bone tissue engineering based on our previously stated approaches to calcium hydrogen phosphate and barium hydrogen phosphate. This supports the further understanding of how different parameters in biomineralization via the gel diffusion method affect the morphology of biomimetic composite materials. Therefore, this study may be useful for explaining the mechanisms of biomineralization of phosphate and strontium ions in vivo.
2. Experimental section
2.1. Synthesis of composites
We dissolved 1 g of pig skin gelatin (300 bloom, Sigma-Aldrich, USA) in 10 mL deionized water, and 7.5 cc of 0.5 M SrCl2 solution (Sigma-Aldrich) was added to the dissolved gelatin. The solution was then stirred for 2 h at 50 °C. The pH of the mixture was changed using tris(hydroxymethyl)aminoethane/HCl (Sigma-Aldrich) to adjust it to a value of 7.4. The mixture was then poured into the tube and left at room temperature for 24 h until gelation occurred. Next, we added 7.5 cc of 0.5 M Na2HPO4 solution (Sigma-Aldrich) buffered at pH 7.4 to the top of the gelatin tube. The diffusion was allowed to take place at 25 °C for 1 month. At the same time, gelatin was mixed with a Na2HPO4 solution to allow the gelation to come into contact with a buffered SrCl2 solution. In the third single tube, a mixture of gelatin and 0.8 M Na2HPO4 was placed in contact with buffered 1 M SrCl2. SrHP/gelatin composite crystals were produced inside the gel near the interface between the gel and the top liquid in all of the tubes. The samples were washed several times with hot water to remove the excess gel, and the experiments were repeated 10 times for each set of samples.2.2. Characterization
We carried out the chemical characterization of the gelatin and SrHP crystals using a Perkin Elmer Spectrum One IR spectrometer integrated with an IBM personal computer.We analyzed the surface morphology of the composite crystals by means of a Hitachi scanning electron microscope S-3000 N. The samples were covered with a layer of gold under vacuum conditions prior to the measurement.
We performed thermogravimetric analysis on a Mettler-Toledo TG/SDTA 851e analyser. In each case, a 5 mg sample was examined under an N2 atmosphere at a heating rate of 5 °C min−1 from 30 to 600 °C.
We collected powder X-ray diffraction patterns at ambient temperature using a STOE StadiP diffractometer in the reflection Bragg–Brentano geometry (monochromatic Cu Kα1 radiation, wavelength λ = 0.15406 nm). The scattered intensity was detected using a linear position sensitive detector.
3. Results and discussion
3.1. Chemical characterization
The infrared (IR) spectra of pure gelatin, SrHP (Sigma-Aldrich) and SrHP/gelatin composite crystals are shown in Fig. 1. In gelatin, evidence for amide I and amide A consists of two strong peaks at 1644 and 3423 cm−1 for a random coil and the α-helix conformation of gelatin, respectively.37,38 These bands are obvious based on C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching coupled with COO− vibrations and NH stretching coupled with hydrogen bonding. The signature of amide II in gelatin is seen in a band at 1534 cm−1 for an out-of-phase combination of CN stretching and in-plane NH deformation modes of the peptide groups.39 On the other hand, the NH bending of amide III is evidenced by the peak at 1233 cm−1. Moreover, all of the bands at 1384, 1400, 1437 and 2933 cm−1 appeared for CH2 wagging and the asymmetrical CH2 stretching of proline. We assign the bands at 1163, 1079 and 1030 cm−1 to the carbonyl and the skeletal vibrations of gelatin.40,41
O stretching coupled with COO− vibrations and NH stretching coupled with hydrogen bonding. The signature of amide II in gelatin is seen in a band at 1534 cm−1 for an out-of-phase combination of CN stretching and in-plane NH deformation modes of the peptide groups.39 On the other hand, the NH bending of amide III is evidenced by the peak at 1233 cm−1. Moreover, all of the bands at 1384, 1400, 1437 and 2933 cm−1 appeared for CH2 wagging and the asymmetrical CH2 stretching of proline. We assign the bands at 1163, 1079 and 1030 cm−1 to the carbonyl and the skeletal vibrations of gelatin.40,41
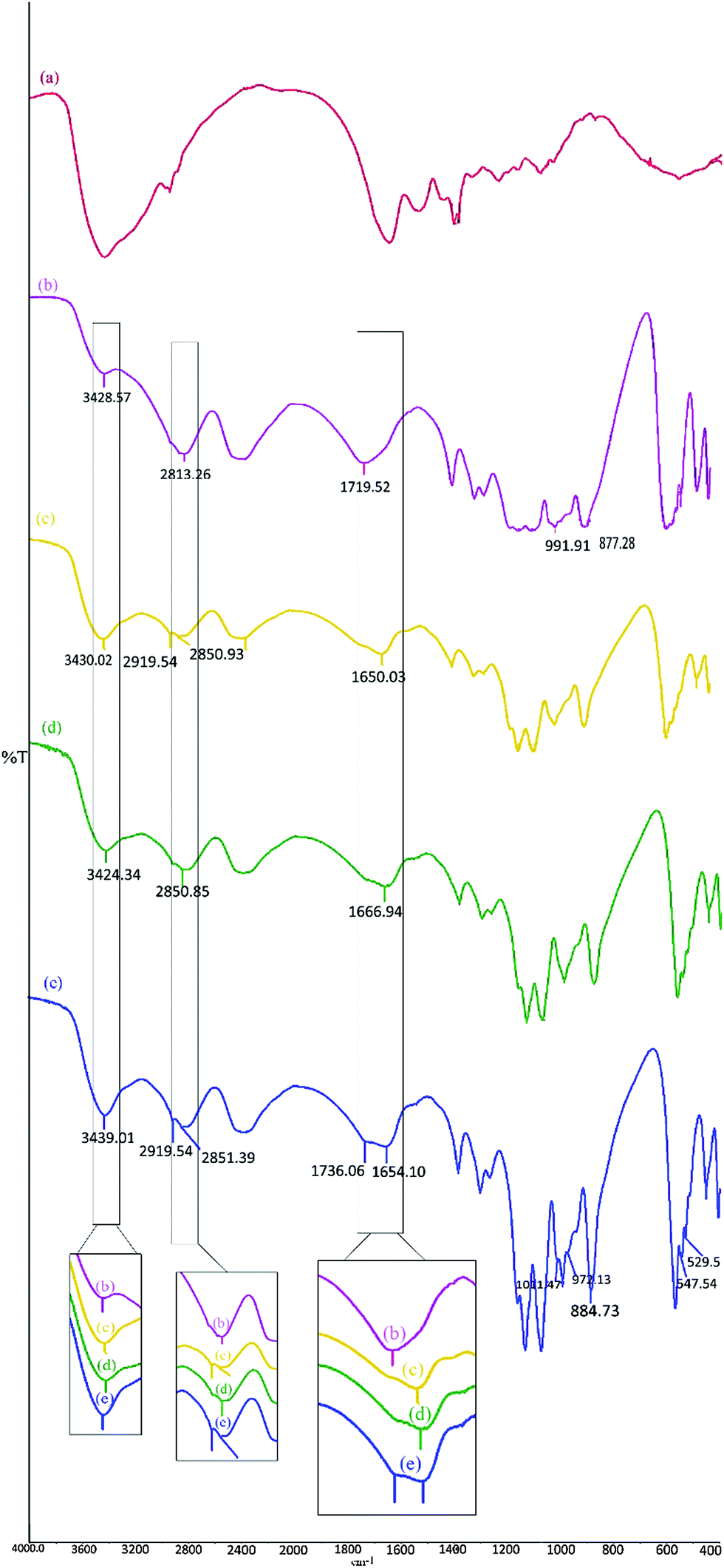 | ||
| Fig. 1 The IR spectra of (a) gelatin, (b) analytical grade SrHP (Sigma-Aldrich), (c) SrHP crystals prepared in tube 1, (d) SrHP crystals prepared in tube 2, and (e) SrHP crystals prepared in tube 3. | ||
Analytical-grade SrHP provided by Sigma-Aldrich (Fig. 1b) was used as a reference for comparison with different SrHP/gelatin composite crystals obtained in this study (Fig. 1, panels c–e). Four intense bands at 3428, 2813, 2390 and 1719 cm−1 are related to H2O bending, hydrogen bonding, O–H stretching of the hydrogen phosphate anions and vibration of the hydrogen ions in the H+–PO43− contacts, respectively.28,30,42,43 The bands located in the range 1400–1200 cm−1 are attributed to the in-plane P–O–H bending modes. The PO asymmetric and symmetric stretching modes in the PO43− fragment of SrHP are associated with the three strong bands at 1131, 1077 and 991 cm−1. The symmetric stretching of P–O(H) is represented by a strong band at 877 cm−1. The bands in the range 600–400 cm−1 arise from the P–O anti-symmetric and symmetric bending modes in the PO43− fragment.25
The IR spectrum for the crystals collected from tube 1 (Fig. 1c) revealed the appearance of two new peaks at 2919 and 2850 cm−1 after the crystallization of SrHP in gelatin and the intensity of the band at 3430 cm−1 increased. Furthermore, the peak at 1719 cm−1 (in Fig. 1b) was shifted to 1650 cm−1 (in Fig. 1c) and the band at 511 cm−1 disappeared compared with analytical grade SrHP. For the crystals produced in tube 2 (Fig. 1d), we observed several changes including the appearance of a new peak at 2850 cm−1, shifting of a band from 1719 to 1666 cm−1 and disappearance of a peak at 511 cm−1. On the other hand, the collected crystals from tube 3 (Fig. 1e) showed an increase in the intensity of the band at 3439 cm−1; the appearance of several new peaks at 2919, 2851, 1654, 1011, 972, 547, 542 and 529 cm−1; and the disappearance of a peak at 511 cm−1 as well as shifting of bands at 1719 and 877 cm−1 to 1736 and 844 cm−1, respectively.
As previously stated, polypeptide chains generally produce several characteristic bands as amide A (about 3500 cm−1), amide B (about 3100 cm−1), amide I (1600–1700 cm−1), amide II (1550 cm−1) and amide III (1300–1350 cm−1). In other words, CH bonds can be vibrated at 2850–3100 cm−1 depending on the vibration mode. According to all the FTIR results, all the changes in the FTIR spectra of crystals associated with the above-mentioned regions are due to the fact that gelatin is entrapped within the crystal sheets. In this regard, we previously measured the gelatin content in barium hydrogen phosphate and calcium hydrogen phosphate crystals to be about 2–3 wt%. We also stated that the changes occurred in the FTIR spectra of composite crystals produced in biopolymeric gels depend on the ion concentration, gel pH and ion diffusion method. This observation is confirmed in the FTIR spectra of SrHP/gelatin composites where the type of ion firstly diffused and the ion concentration was different.
Note that we have previously demonstrated similar peak changes in amide I after the incorporation of gelatin into calcium hydrogen phosphate and barium hydrogen phosphate minerals. Such intermolecular interactions were due to hydrogen bonds and the coupling of positive ions with the gelatin carboxylate anions.27,28 The ionic strength of proteinous gels can be further enhanced via salts, which may lead to more rigid conformations of peptides and changes in the ordering of triple helices, as was found by Feng et al.44 Furthermore, the intensity of the amide A bands and the CH2 stretching of gelatin were not the same for the SrHP samples in different tubes. This result clearly demonstrates that the interaction between gelatin and ions is dependent on the type of ion diffusing in the gel and the ion concentration.
3.2. Microscopic observation of composite crystals
Fig. 2 illustrates the morphology of SrHP/gelatin composites obtained from the gel diffusion. We observed two different morphologies for the SrHP/gelatin composites: arrow-like and spherulitic crystals. In the first tube, arrow-like crystals approximately 60 μm in width and 300 μm in length were obtained. These crystals were composed of aligned bundles of single crystals, which functioned as the building blocks of a three-dimensional supercomposite crystal. We also observed highly porous microspheres in all of the tubes. The surface of the spheres in tube 1 (Fig. 2c and d) included bundled nano-flakes originating from a common center with a thickness of approximately 140 nm. It was obvious from tubes 2 and 3 that the crystalline structure of SrHP was similar to that of the spheres in tube 1. However, their surface morphology was different depending on the variation of the molarity of the ions and the type of ion diffused in the gelatin. It should be noted that the molarities of HPO42− and Sr2+ were similar for tubes 1 and 2 but different for the diffusion procedure in the gel. This situation resulted in changes in the crystallization and was responsible for the absence of arrow-like crystals in tube 2. Moreover, the molarity of ions in tube 3 was higher than that in tube 2, which possibly changed the surface arrangement, roughness and porosity of the spheres. The spheres in tube 2 were more uniform compared with those in tube 3, and two-dimensional bundled sheets were arranged on the surface of spheres via self-assembly. As a result, spheres with flower-like surface arrangements were generated (Fig. 2, panels e–h). | ||
| Fig. 2 SEM images of SrHP/gelatin composites: (a) arrow-like composites obtained in tube 1, (b) spherulitic composites obtained in tube 1, (c and d) the surface of spherulitic crystals obtained in tube 1 with higher magnification, (e) spherulitic composites obtained in tube 2 at low magnification, (f) spherulitic composites obtained in tube 2 at high magnification, (g) spherulitic composites obtained in tube 3 at low magnification, and (h) spherulitic composites obtained in tube 3 at high magnification. | ||
We previously demonstrated that the crystal growth habits of barium hydrogen phosphate and calcium hydrogen phosphate crystals in gels depended on several factors including the electrostatic charge density, the topology of the binding surface, van der Waals forces, the surface distribution of biopolymers and the type and sequence of the functional groups.27,28,45,46 Researchers have found that gelatin macromolecular chains have randomly coiled and untangled single conformations in water at about 40–50 °C (i.e., their melting point region). These chains have a reduced energy level at lower temperatures. In this regard, three individual helices are organized into a clockwise triple-helix rich in glycine–proline–hydroxyproline amino acids. Hydrogen bonds among the peptide groups play a pivotal role in the stabilization of this structure. It has been shown that the nucleation and growth of crystals in gelatin are influenced by the impregnation of calcium, barium and phosphate ions. In this regard, pre-ordering of the gelatin network is related to the type of the first ion impregnated in the gel prior to the diffusion of a second ion. Since the HPO42− ion was mixed with gelatin in tubes 2 and 3, it interacted with the outer surface of the protein helices, which resulted in the bending of the triple-helices and the formation of hydrogen and electrostatic bondings.47 The Sr2+ cations, which are diffused later, were able to react with the aggregated HPO42− ions in gelatin to induce the crystallization of SrHP. This mechanism differs from the as-prepared mixture of gelatin and Sr2+, which possibly resulted in changes in the crystallization mechanism and morphology of the SrHP/gelatin composites. We conclude that the experimental conditions included the ion concentrations and the type of ion impregnated in the gel control, the reactant diffusion and the morphology of the SrHP crystals.
3.3. Thermal studies
Fig. 3 shows the thermal degradation of SrHP/gelatin composites and analytical-grade SrHP crystals. We observed three zones or regions of decomposition – initial, main and char – for all of the samples. In the first zone, the weight loss is the result of the removal of surface-adsorbed water molecules.48 The SrHP/gelatin composites lost about 3% of their weight in the first region, which was negligible for the analytical-grade SrHP crystals.25 Significant weight loss for the analytical-grade SrHP crystals occurred at earlier temperatures of the second region between 310 and 350 °C due to the generation of Sr–O–P2O5, as stated by Sales et al. However, a higher thermal stability of the SrHP/gelatin composites was observed by shifting the second zone to temperatures between 410 and 470 °C.23 This finding may be due to strong interactions between the inorganic and organic compounds. We observed that the degradation of composites started at 470 °C, which was most definitely higher than the degradation level of 450 °C observed for analytical-grade SrHP pure crystals. We have previously shown the pyrolytic split between the polar domain of amine and carboxylic acids in gelatin on the one hand and the strontium and phosphate ions of SrHP on the other hand in composites of barium hydrogen phosphate/gelatin and calcium hydrogen phosphate/gelatin.28 Furthermore, we observed that the char content was dissimilar for the SrHP/gelatin composites produced in different tubes at temperatures higher than 470 °C. This result further confirms the interaction between gelatin and SrHP, which is in agreement with the findings of the IR spectra.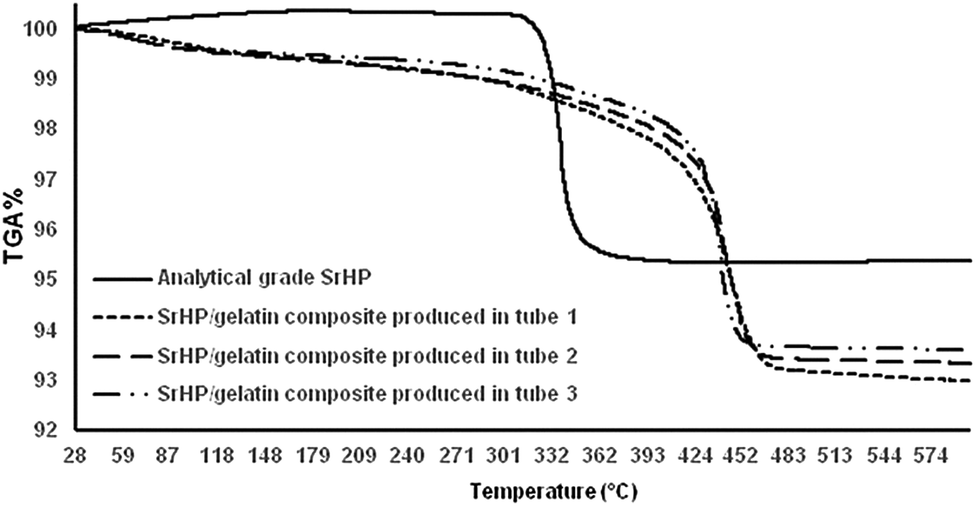 | ||
| Fig. 3 TGA curves for analytical grade SrHP and SrHP/gelatin composites. | ||
3.4. X-ray diffraction analysis
The X-ray powder diffraction patterns revealed a single crystalline phase, α – SrHPO4 (PDF 01-070-1215), for all the samples (S1, S2 and S3) synthesized in the presence of gelatin (Fig. 4).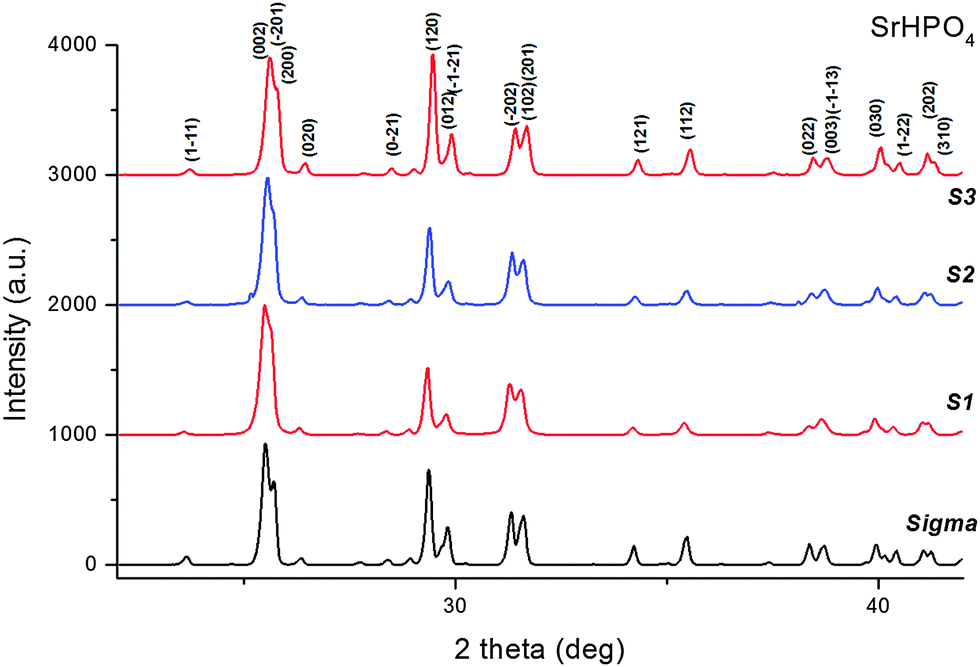 | ||
| Fig. 4 X-ray diffraction patterns collected on specimen tubes 1–3 and on the reference sample (Sigma-Aldrich). | ||
We used pattern decomposition to investigate the influence of the amount of gelatin and the preparation method on the crystals’ structure and the line broadening of the SrHP Bragg reflections. The monotonic increase in the intensity of the (120) reflection as a function of the amount of gelatin (Fig. 4) may well suggest a gelatin-induced preferential (textured) growth of the SrHP crystals with respect to the (120) planes.
In terms of the hypothesis of negligible micro-strain, we applied the Scherrer eqn (1) to quantitatively estimate the size of the coherent scattering domains (i.e., the crystallite size):
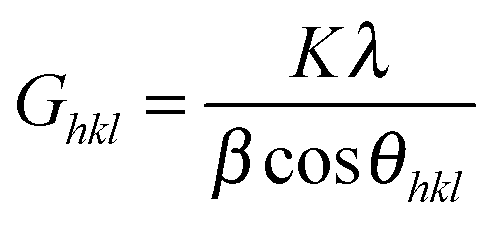 | (1) |
In Table 1, we relate G002 to the mean crystallite size normal to the (002) plane; G120 refers to the mean crystal size along a direction perpendicular to the (120) planes.
| hkl | G S1 (nm) | G S2 (nm) | G S3 (nm) | G Sigma (nm) |
|---|---|---|---|---|
| (002) | 45.90 | 45.75 | 46.02 | 50.11 |
| (120) | 59.74 | 66.66 | 66.72 | 67.34 |
The difference in the crystallite size values in different directions suggests that the crystallites possess a needle or plate-like shape, which is in agreement with the scanning electron microscopy (SEM) results (see, for example, Fig. 2d, f and h). The values of the lattice parameters did not change significantly as a function of the preparation method or the ion concentration (Table 2).
| Samples | a (Å) | b (Å) | c (Å) | α (deg) | β (deg) | γ (deg) |
|---|---|---|---|---|---|---|
| S1 (tube 1) | 7.1870(12) | 6.7921(9) | 7.2606(10) | 94.651(14) | 104.954(16) | 88.778(16) |
| S2 (tube 2) | 7.1721(25) | 6.7829(16) | 7.2462(19) | 94.660(26) | 104.967(28) | 88.733(33) |
| S3 (tube 3) | 7.1476(35) | 6.7582(27) | 7.2403(23) | 94.520(42) | 105.021(36) | 88.691(50) |
| Sigma | 7.1774(20) | 6.7878(15) | 7.2532(17) | 94.613(24) | 105.070(28) | 88.759(27) |
We refined the lattice parameters using the powder diffraction file of triclinic α – SrHPO4 (PDF 01-070-1215, space group P1 (point group 1, centrosymmetric)).
4. Summary and conclusion
In this work, we have investigated the influence of gelatin and the concentration of ions and their diffusion on the preparation of SrHP/gelatin biocomposite crystals using various analytical techniques. IR spectra revealed strong interactions among the gelatin functional groups and the strontium and phosphate ions, which promoted the formation of composites. The IR results also confirmed that the carboxylate, amine and hydroxyl groups of gelatin provided the crystal backbone precursors for the nucleation and growth of SrHP. As a result, various morphologies were produced such as micro-crystals, spheres and nano-flakes. The macromolecular conformation of double helices in gelatin in the presence of strontium cations or phosphate anions induced pre-structural disorder for SrHP deposition via the production of highly porous spheres. Our findings clearly show that gelatin can produce novel SrHP crystal morphologies depending on the first ion diffused and the ion concentrations. Such morphologies have not yet been reported elsewhere. Irrespective of the ion concentrations, the single-diffusion method leads to the formation of single-phase α-SrHPO4. Our SrHP–gelatin composites contribute to a better understanding of the mechanisms of mineralization of strontium and phosphate ions in bone tissue. However, additional studies should focus on different factors such as the gel type and pH, the type of diffusion method, the duration of the diffusion process and the gel concentration.Acknowledgements
This project was supported by SNF project no. 200020_146413. We thank Mrs Beatrice Frey for providing SEM images and Dr Matthias Burgener for assistance and support.References
- G. Romieu, X. Garric, S. Munier, M. Vert and P. Boudeville, Acta Biomater., 2010, 6, 3208–3215 CrossRef CAS PubMed.
- H. B. Pan, Z. Y. Li, W. M. Lam, J. C. Wong, B. W. Darvell, K. D. K. Luk and W. W. Lu, Acta Biomater., 2009, 5, 1678–1685 CrossRef CAS PubMed.
- P. E. Kazin, A. S. Karpov, M. Jansen, J. Nuss and Y. D. Tretyakov, Z. Anorg. Allg. Chem., 2003, 629, 344–352 CrossRef CAS.
- G. M. Kuang, W. P. Yau, W. M. Lam, J. Wu, K. Y. Chiu, W. W. Lu and H. Pan, J. Biomed. Mater. Res., Part B, 2012, 100, 778–787 CrossRef PubMed.
- F. Q. Zhuang, R. Q. Tan, W. F. Shen, X. P. Zhang, W. Xu and W. J. Song, Acta Metall. Sin., 2015, 28, 438–443 CrossRef CAS.
- D. Guo, K. Xu, X. Zhao and Y. Han, Biomaterials, 2005, 26, 4073–4083 CrossRef CAS PubMed.
- W. Zhang, Y. Shen, H. Pan, K. Lin, X. Liu, B. W. Darvell, W. W. Lu, J. Chang, L. Deng, D. Wang and W. Huang, Acta Biomater., 2011, 7, 800–808 CrossRef CAS PubMed.
- W. M. Lam, H. B. Pan, Z. Y. Li, C. Yang, W. K. Chan, C. T. Wong, K. D. K. Luk and W. W. Lu, Ceram. Int., 2010, 36, 683–688 CrossRef CAS.
- E. A. Abdel-Aal, Int. J. Nanopart., 2011, 4, 77–92 CrossRef CAS.
- M. Schumacher, A. Henß, M. Rohnke and M. Gelinsky, Acta Biomater., 2013, 9, 7536–7544 CrossRef CAS PubMed.
- T. G. Kim and B. Park, Inorg. Chem., 2005, 26, 9895–9901 CrossRef PubMed.
- G. Bechkoff, J. Radisson, L. Bessueille, K. Bouchekioua-Bouzaghou and R. Buchet, Biochem. Biophys. Res. Commun., 2008, 29, 378–381 CrossRef PubMed.
- D. Guo, M. Mao, W. Qi, H. Li, P. Ni, G. Gao and K. Xu, J. Mater. Sci.: Mater. Med., 2011, 22, 2631–2640 CrossRef CAS PubMed.
- H.-B. Pan, Z.-Y. Li, T. Wang, W. M. Lam, C. T. Wong, B. W. Darvell, K. D. K. Luk, Y. Hu and W. W. Lu, Cryst. Growth Des., 2009, 9, 3342–3345 CAS.
- L. B. Taher, L. Smiri, Y. Laligant and V. Maisonneuve, J. Solid State Chem., 2000, 152, 428–434 CrossRef.
- Y. Zheng, Y. Cheng, Y. Wang, Y. Yu, D. Chen and F. Bao, J. Cryst. Growth, 2005, 280, 569–574 CrossRef CAS.
- M. Dinamani and P. Vishnu Kamath, Mater. Res. Bull., 2001, 36, 2043–2050 CrossRef CAS.
- R. W. Mooney, M. A. Aia, C. W. W. Hoffman and R. C. Ropp, J. Am. Chem. Soc., 1959, 81, 826–829 CrossRef CAS.
- R. C. Ropp, M. A. Aia, C. W. W. Hoffman, T. J. Veleker and R. W. Mooney, Anal. Chem., 1959, 31, 1163–1166 CrossRef CAS.
- M. H. Frere, Soil Sci. Soc. Am. J., 1962, 26, 48–51 CrossRef CAS.
- M. A. Aia, J. E. Mathers and R. W. Mooney, J. Chem. Eng. Data, 1964, 9, 335–338 CrossRef CAS.
- T. P. Feenstra, H. A. Van Straten and P. L. De Bruyn, J. Colloid Interface Sci., 1981, 80, 255–270 CrossRef CAS.
- B. C. Sales, B. C. Chakoumakos, L. A. Boatner and J. O. Ramey, J. Non-Cryst. Solids, 1993, 159, 121–139 CrossRef CAS.
- B. Louati, K. Guidara, M. Gargouri and M. Fourati, Z. Naturforsch., 2005, 60, 121–126 CAS.
- M. Roming and C. Feldmann, J. Mater. Sci., 2008, 43, 5504–5507 CrossRef CAS.
- E. Banks, R. Chianelli and F. Pintchovsky, J. Cryst. Growth, 1973, 18, 185–190 CrossRef CAS.
- M. Parvinzadeh Gashti, M. Bourquin, M. Stir and J. Hulliger, J. Mater. Chem. B, 2013, 1, 1501–1508 RSC.
- M. Parvinzadeh Gashti, M. Burgener, M. Stir and J. Hulliger, J. Colloid Interface Sci., 2014, 431, 149–156 CrossRef PubMed.
- M. Parvinzadeh Gashti, M. Bourquin, M. Stir and J. Hulliger, Cryst. Growth Des., 2013, 13, 2166–2173 CAS.
- M. Parvinzadeh Gashti, M. Stir and J. Hulliger, Colloids Surf., B, 2013, 110, 426–433 CrossRef PubMed.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef PubMed.
- S. V. Dorozhkin, Biomaterials, 2010, 31, 1465–1485 CrossRef CAS PubMed.
- J. L. Gornall and E. M. Terentjev, Soft Matter, 2008, 4, 544–549 RSC.
- H. Tlatlik, P. Simon, A. Kawska, D. Zahn and R. Kniep, Angew. Chem., Int. Ed., 2006, 45, 1905–1910 CrossRef CAS PubMed.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed.
- M. Burgener, T. Putzeys, M. Parvinzadeh Gashti, S. Busch, H. Oulevey Aboulfadl, M. Wübbenhorst, R. Kniep and J. Hulliger, Biomacromolecules, 2015, 16, 2814–2819 CrossRef CAS PubMed.
- M. Parvinzadeh Gashti, D. Hegemann, M. Stir and J. Hulliger, Plasma Processes Polym., 2014, 11, 37–43 CrossRef CAS.
- M. C. Chang, C. C. Ko and W. H. Douglas, Biomaterials, 2003, 24, 2853–2862 CrossRef CAS PubMed.
- K. J. Payne and A. Veis, Biopolymers, 1988, 27, 1749–1760 CrossRef CAS PubMed.
- M. Parvinzadeh Gashti and A. Almasian, Composites, Part B, 2013, 52, 340–349 CrossRef CAS.
- C. Pouchert, The Aldrich Library FTIR Spectra, Aldrich Chemical Company, Milwaukee, 1985 Search PubMed.
- J. H. Muyonga, C. G. B. Cole and K. G. Duodu, Food Chem., 2004, 86, 325–332 CrossRef CAS.
- M. Parvinzadeh, S. Moradian, A. Rashidi and M. E. Yazdanshenas, Appl. Surf. Sci., 2010, 256, 2792–2802 CrossRef CAS.
- Y. Feng, M. Taraban and Y. Bruce Yu, Soft Matter, 2012, 1, 11723–11731 RSC.
- S. Schweizer and A. Taubert, Macromol. Biosci., 2007, 7, 1085–1099 CrossRef CAS PubMed.
- K. Gorna, R. Muñoz-Espí, F. Gröhn and G. Wegner, Macromol. Biosci., 2007, 7, 163–173 CrossRef CAS PubMed.
- D. Zahn, O. Hochrein, A. Kawska, J. Brickmann and R. Kniep, J. Mater. Sci., 2007, 42, 8966–8973 CrossRef CAS.
- M. Parvinzadeh Gashti, M. Yousefpour Navid and M. H. Rahimi, Pigm. Resin Technol., 2013, 42, 34–44 CrossRef.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |