
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon-rich cyclopentadienyl ruthenium allenylidene complexes†
Susanne
Spörler
a,
Frank
Strinitz
a,
Philipp
Rodehutskors
a,
Lisa
Müller
a,
Andreas R.
Waterloo
b,
Maximilian
Dürr
a,
Eike
Hübner
c,
Ivana
Ivanović-Burmazović
a,
Rik R.
Tykwinski
b and
Nicolai
Burzlaff
*a
aInorganic Chemistry, Department of Chemistry and Pharmacy, Friedrich-Alexander University Erlangen Nürnberg (FAU), Egerlandstraße 1, 91058 Erlangen, Germany. E-mail: nicolai.burzlaff@fau.de; Fax: +49 9131 85 27387; Tel: +49 9131 85 28976
bOrganic Chemistry, Department of Chemistry and Pharmacy, Friedrich-Alexander University Erlangen Nürnberg (FAU), Henkestraße 42, 91054 Erlangen, Germany. E-mail: rik.tykwinski@fau.de; Fax: +49 9131 85 26865; Tel: +49 9131 85 22540
cOrganic Chemistry, Technical University Clausthal, Leibnizstraße 6, 38678 Clausthal Zellerfeld, Germany
First published on 6th May 2016
Abstract
Ruthenium allenylidene complexes with carbon-rich polyaromatic moieties have been synthesized by using [RuCl(η5-C5H5)(PPh3)2] (η5-C5H5 = cyclopentadienyl) as a precursor and the propargyl alcohols 10-ethynyl-10-hydroxyanthracen-9-one (ACO), 13-ethynyl-13-hydroxypentacen-6-one (PCO), 1-phenyl-1-(pyren-1-yl)prop-2-yn-1-ol (PyrPh), 9-ethynyl-9H-fluoren-9-ol (FN) and 6-ethynyl-6H-benzo[cd]pyren-6-ol (BPyr) as ligands. The resulting cationic allenylidene complexes, [Ru(η5-C5H5)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CC(AO))(PPh3)2]PF6 (1), [Ru(η5-C5H5)(CC(PCO))(PPh3)2]PF6 (2), [Ru(η5-C5H5)(CC(PyrPh))(PPh3)2]PF6 (3), [Ru(η5-C5H5)(CC(FN))(PPh3)2]PF6 (4), and [Ru(η5-C5H5)(CC(BPyr))(PPh3)2]PF6 (5) show interesting intermolecular π-interactions in the solid-state structure as well as solution state complexation with pyrene (documented by Job's plots experiments). CV data indicate possible Ru(II)/Ru(III) oxidation, as well as the potential reduction of the carbon-rich allenylidene moiety.
CC(AO))(PPh3)2]PF6 (1), [Ru(η5-C5H5)(CC(PCO))(PPh3)2]PF6 (2), [Ru(η5-C5H5)(CC(PyrPh))(PPh3)2]PF6 (3), [Ru(η5-C5H5)(CC(FN))(PPh3)2]PF6 (4), and [Ru(η5-C5H5)(CC(BPyr))(PPh3)2]PF6 (5) show interesting intermolecular π-interactions in the solid-state structure as well as solution state complexation with pyrene (documented by Job's plots experiments). CV data indicate possible Ru(II)/Ru(III) oxidation, as well as the potential reduction of the carbon-rich allenylidene moiety.
Introduction
In recent years, there has been great interest in bringing metal complexes in close contact with single-wall carbon nanotubes (SWCNTs). Apart from covalent attachment, π-interactions of SWCNTs with polyaromatic moieties, such as pyrene or diazaacenes, could lead to applications in catalysis, photoinduced electron transfer or information storage.1–3Moreover, various acene derivatives and their precursors, the acenequinones, are promising candidates for small-molecule semiconductor applications and have been studied regarding their potential applications in thin film transistors.4–6 Even anthraquinones and pentacenequinones are, like the anthracenes and pentacenes, potentially useful organic semiconductors, although their applications as organic semiconductors remain largely unexplored.4–8 The derivatization or functionalization of acenes is mostly achieved by attaching alkyl, aryl, and alkyne residues at the middle of the acene framework in order to tune the HOMO–LUMO gap. Furthermore, these residues can have an influence on the acene arrangement in the solid state, e.g. regarding the manner of the π-stacking.4,5,9–11
Ruthenium carbon-rich allenylidene complexes with poly-aromatic substituents have the ability to exchange electrons via the allenylidene unit, which might lead to a variety of applications like magnetic and optical devices.12–17 Since its first examples reported independently in 1976 by Fischer18,19 as well as by Berke,20 the chemistry of transition metal allenylidene complexes increased significantly due to their promising chemical and physical properties and has been reviewed extensively by various authors.14,21–26 Transition metal allenylidene complexes turned out to be interesting and useful complexes and intermediates for a huge range of chemistry and catalytic processes.23,27–29 Especially, the building block properties of allenylidene complexes in organic syntheses as well as their catalytic properties, for example as catalysts or pre-catalysts for the olefin metathesis,30–38 arouse interest. Other reports focus on the esterification of propargyl alcohols39 or the ring opening polymerization.40–43 Usually, electrophiles add to C-β of the allenylidene moieties, affording cationic carbyne complexes,44 whereas nucleophiles attack at C-α or C-γ.27,45–48
Recently, we reported on ruthenium allenylidene complexes [Ru(bdmpza)Cl(PPh3)(CCCR2)] (bdmpza = bis(3,5-dimethylpyrazol-1-yl)acetate; CR2 = polyaromatic residue) bearing polyaromatic moieties.49–51 Most of these complexes show strong π-stacking interactions in solid state, which could be, therefore, promising candidates for metal-tuned FETs or “organic” metal-semiconductor field-effect transistors (OMESFETs). The synthesis followed the procedure first reported by Selegue52 by using the corresponding propargyl alcohols. It is worthwhile mentioning that Fürstner et al. reported on a series of cationic ruthenium allenylidene complexes of the type [(η6-(p-cymene))(R3P)RuCl(CCCR2′)]+ X− gained by a Selegue type reaction.23 Moreover, other cationic Cp-ruthenium allenylidene complexes such as [Ru(η5-C5H5)(PPh3)2(CCCR2)]PF6 are also well-known for almost two decades53,54 and therefore, spark our interest. Thus, here we report on cationic cyclopentadienyl allenylidene complexes [RuCl(η5-C5H5)(PPh3)(CCCR2)]+ with various carbon-rich polyaromatic moieties (CR2).
Results and discussion
The synthesis of the allenylidene complexes followed the procedure first reported by Selegue52 by using the corresponding propargyl alcohols as building blocks. 10-Ethynyl-10-hydroxyanthracen-9-one (AO), 13-ethynyl-13-hydroxypentacen-6-one (PCO), 1-phenyl-1-(pyren-1-yl)prop-2-yn-1-ol (PyrPh), 9-ethynyl-9H-fluoren-9-ol (FN) and 6-ethynyl-6H-benzo[cd]pyren-6-ol (BPyr) have been chosen as starting materials. These precursors were reacted with [RuCl(η5-C5H5)(PPh3)2] and NH4[PF6] in MeOH for one to two days (Scheme 1) to obtain the deep colorful allenylidene complexes after removal of the solvent, recrystallisation from CH2Cl2/Et2O and subsequent purification via column chromatography (silica; CH2Cl2/acetone). | ||
| Scheme 1 Synthesis of mononuclear ruthenium allenylidene complexes 1–5. | ||
The resulting mono-cationic complexes are rather stable towards oxygen and were characterized by 1H, 13C and 31P NMR spectroscopy, IR spectroscopy, ESI mass spectrometry and elemental analysis. Crystals suitable for X-ray structure determinations were obtained of 1, 2 and 5 (Fig. 1).
 | ||
| Fig. 1 Molecular structures of 1, 2 and 5. π-Stacking (3.38 Å) resulting in (a) formation of a dimer for 1, (b) a staircase for 2 packing (3.37 and 3.39 Å) along the pentacenequinone axis (three molecules are shown) while (c) no π-stacking is observed for 5. The hexafluorophosphate anions, solvent molecules and hydrogen atoms have been omitted for clarity. | ||
The structures of the complexes 1, 2 and 5 exhibit similar bond figure lengths and angles referring to the allenylidene unit and the anthraquinone, the pentacenequinone and the benzopyrene moieties (Table S2, ESI†). The angle between Ru–Cα–Cβ = 168.7° of complex 2 is more bent in comparison to ∠Cα–Cβ–Cγ = 172.1° for 1 and 176.2° for 5, which could be the consequence of the staircase π-stacking (see below). However, these angles are closer to the ideal 180° compared to the recently reported complex [Ru(bdmpza)Cl(PPh3)(CC(PCO))].49–51,55 The Cα–Cβ bond lengths (1.251 Å (1); 1.256 Å (2) and 1.250 Å (5)) and the Cβ–Cγ distances (1.356 Å (1), 1.360 Å (2) and 1.378 Å (5)) are typical for the allenylidene unit when compared to other published allenylidene complexes.14,21,49–51,55 The Ru–Cα bond distances of 1.895 Å (1), 1.907 Å (2) and 1.925 Å (5) are in good agreement with the distances of Ru–C cumulenylidene bonds.14,21,49–51,55
The experimental distances of the molecular structure of 2 are in good accordance to the theoretical values gained by DFT calculations (Table S2, ESI†).
While the packing analysis of complex 1 exhibits the formation of dimeric units in the solid state due to π-stacking, the π-stacking of 2 causes the formation of a staircase arrangement with short interplanar distances of 3.39 and 3.37 Å (Fig. 1). The pentacenone units are arranged by this face-to-face π-stacking, which typically comes along with some displacement along the molecular axis, to form this staircase packing. Thus, one of the common packing motifs adopted by semiconducting π-molecules is reflected in this crystal structure. As a result, complex 2 could represent a good design for n-type semiconductors in thin film transistors. It was also possible to investigate the aggregation of 1–5 with pyrene in solution, by UV/Vis titration experiments. The stoichiometry of the aggregation was determined by recording Job's plots.56 The plots of the complexes 3, 4 and 5 show maxima at χ = 0.5, which indicate the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts in solution (Job's plots of 1, 4 and 5 with pyrene: Fig. S1–S3, ESI†). Moreover, also the graphs of the anthraquinone and pentacenequinone based complexes 1 and 2 seem to show maxima at χ = 0.5 at a first glance. But at a closer look, these graphs might point out local maxima at χ = 0.4 and χ = 0.6 (Fig. 2), which could indicate an equilibrium of 1:2 and 2:1 pyrene:complex adducts at the concentrations used for the titration experiments, besides the 1:1 adducts. A shoulder in the Job's plot of 5 at χ = 0.8 might also indicate the presence of 2:1 pyrene:complex adduct species.
:1 adducts in solution (Job's plots of 1, 4 and 5 with pyrene: Fig. S1–S3, ESI†). Moreover, also the graphs of the anthraquinone and pentacenequinone based complexes 1 and 2 seem to show maxima at χ = 0.5 at a first glance. But at a closer look, these graphs might point out local maxima at χ = 0.4 and χ = 0.6 (Fig. 2), which could indicate an equilibrium of 1:2 and 2:1 pyrene:complex adducts at the concentrations used for the titration experiments, besides the 1:1 adducts. A shoulder in the Job's plot of 5 at χ = 0.8 might also indicate the presence of 2:1 pyrene:complex adduct species.
 | ||
| Fig. 2 Job's plots of the aggregation of 2 (top) and 3 (bottom) with pyrene. | ||
To back these titration results, we then investigated the aggregation of the complexes 2 and 5 with pyrene via cryospray-ionisation mass spectrometry (CSI-MS). The data is in accordance with the conclusions drawn from the Job's plots measurements and indicate an 1:1 adduct in case of complex 2 and a 2:1 pyrene:complex adduct in case of 5, respectively (CSI-MS, see Fig. S4 and S5, ESI†).
The cyclic voltammetric (CV) analysis of the complexes 1–5 reveals several ligand based reductions and one ruthenium-based oxidation for some of the complexes. For complexes 1, 3 and 4, the Ru2+/Ru3+ couple shows either a reversible process or an irreversible event at about 0.50 to 0.60 V in the cyclo-voltammogram. We ascribe this oxidation event to the Ru2+/Ru3+ redox couples due to previous observations for related carbon-rich allenylidene complexes reported by our group.49,50 Moreover, this assignment agrees well with those of other authors.57–59 In contrast, Skelton and Koutsantonis considered it unlikely that such oxidation processes may correspond to the Ru2+/Ru3+ oxidation couple but preferred a ligand-centered oxidation instead.60 Furthermore, they reported on irreversible ligand-centered oxidation processes around 1.4 V. It is worthwhile mentioning that complexes 2–5 exhibit similar irreversible oxidations in the range of 1.25 V to 1.48 V which thus might also be ligand-centered.
However, for complex 2 and 5, two reversible processes at −0.64 V and −1.28 V and −0.73 V and −1.65 V, respectively, correspond to the reduction of the organic moiety of the complex, i.e. the allenylidene unit bearing the pentacene or the benzopyrene moiety. Exemplary scans for complexes 2 and 5 are depicted in Fig. 3 (for full CV data of the complexes see Fig. S6–S10, ESI†). Obviously, the complexes possess a significant electron-acceptor potential compared to PCBM ([6,6]-phenyl-C61-butyric acid methyl ester), which is known as an excellent electron-acceptor (pristine PCBM exhibits three reduction peaks at −1.11 V, −1.33 V, and −1.92 V vs. Ag/Ag+).61 Thus, according to the observed reduction processes future application of complexes 2 and 5 as electron-acceptors might be possible. On the other hand suggest the observed (metal centered) oxidation processes electron donor properties for some of the complexes.
 | ||
| Fig. 3 Reductive part of the cyclic voltammogram of 2 and 5 (1.00 mM) in n-Bu4NPF6 (0.1 M) solution of MeCN at scan rate of 500 mV s−1. | ||
UV/Vis spectra of the complexes were recorded in CH2Cl2 (Fig. 4 and Fig. S11–S15, ESI†). All allenylidene complexes 1–5 show characteristic absorptions corresponding to MLCT transitions in the range of 400 nm to 700 nm. Complexes 2, 3, and 5 exhibit rather intense absorption bands with extinction coefficients in a range of 20000 to 45000 L mol−1 cm−1 (Fig. 4 and 5).
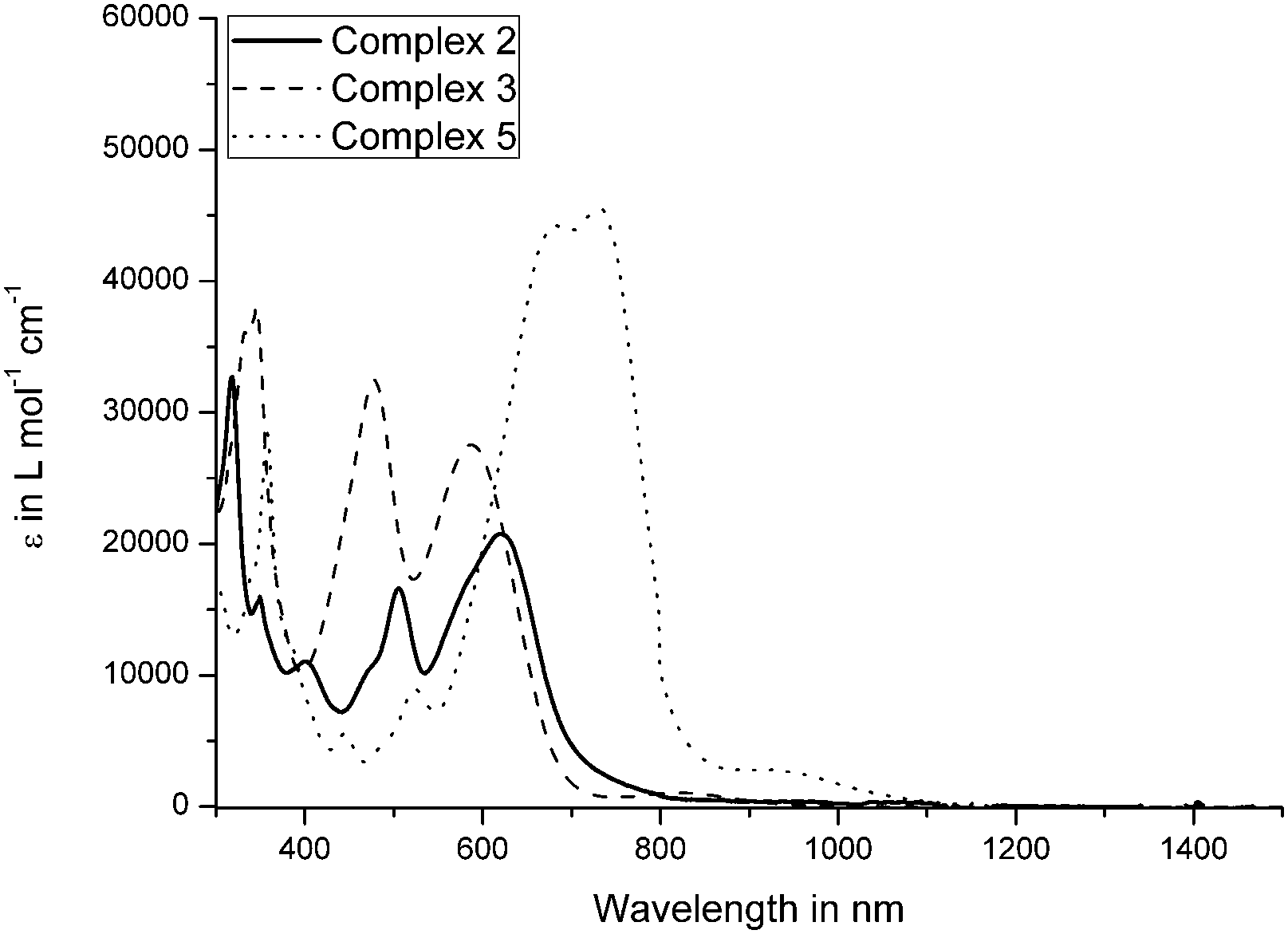 | ||
| Fig. 4 UV/Vis absorption spectra of 2, 3 and 5 in CH2Cl2 (300–1500 nm). | ||
 | ||
| Fig. 5 UV/Vis-NIR absorption spectra of 3 and 5 in CH2Cl2 in the range of 800–1200 nm. | ||
Due to the nearly panchromic absorption and high extinction coefficients, the pentacenequinone, pyrenophenone and benzopyrenone-based complexes 2, 3 and 5 could represent promising dyes for dye-sensitized solar cells (DSSC). Furthermore, the allenylidene complexes show broad transitions at the edge of the NIR region, at about 900–1000 nm, which can be attributed to HOMO−1 → LUMO and HOMO → LUMO excitations. Surprisingly, UV/Vis measurements regarding complex 2 revealed no absorptions located in this region, which is in accordance to the TD-DFT calculations of the excited state of 2. The transitions at lower wavelength of 3 and 5 are in good agreement with former TD-DFT calculations.49,50
Experimental
All air sensitive compounds were prepared under dry argon or nitrogen atmosphere using conventional Schlenk techniques. The yields refer to analytically pure substances. The starting compounds were used as purchased without further purification. The 1H, 13C and 31P NMR were measured with either a Bruker AVANCE DPX300, a Bruker AVANCE DRX400 WB, a Bruker AVANCE III HD 400 MHz spectrometer and a Bruker AVANCE III HD 600 MHz spectrometer. The δ values are given in ppm relative to tetramethylsilane and were calibrated by the solvent peaks of the deuterated solvent. Electrospray-ionization MS (ESI-MS) measurements were performed on an UHR-TOF Bruker Daltonik (Bremen, Germany) maXis plus 5G, an ESI-ToF MS capable of resolution of at least 60000 FWHM. Detection was in positive ion mode, the source voltage was 2.8 kV. The flow rates were 180 μL per hour. The drying gas (N2), to aid solvent removal, was held at 180 °C and the spray gas was held at 20 °C. Cryospray-ionization MS (CSI-MS) measurements were performed on an UHR-TOF Bruker Daltonik (Bremen, Germany) maXis plus 5G, an ESI-TOF MS capable of resolution of at least 60000 FWHM, which was coupled to a Bruker Daltonik Cryospray unit. Detection was in positive ion mode, the source voltage was 4.5 kV. The flow rates were 280 μL per hour. The drying gas (N2), to aid solvent removal, was held at −35 °C and the spray gas was held at −40 °C. The machine was calibrated prior to every experiment via direct infusion of the Agilent ESI-TOF low concentration tuning mixture, which provided a m/z range of singly charged peaks up to 2700 Da in both ion modes. IR spectra were recorded with a Varian EXCALIBUR FTS-3500 FT-IR spectrometer in CaF2 cuvettes (0.2 mm) or in a KBr matrix. For elemental analysis, an Euro EA 3000 (Euro Vector) and EA 1108 (Carlo Erba) instrument were used. Thermogravimetric analyses (TGA) were achieved on a Mettler Toledo DSC 821e/Sensor FRS5-Ceramic. All thermal analyses were carried out under a flow of nitrogen with a heating rate of 10 °C min−1. Thermal decomposition temperature as measured by TGA (as sample weight loss) is reported as Td in which the temperature listed corresponds to the intersection of the tangent lines of the baseline and the edge of the peak corresponding to the first significant weight loss, typically >5%. UV/Vis spectroscopy was performed with a Shimadzu UV-2401PC or a Varian Cary 5000 spectrometer. [Ru(η5-C5H5)Cl(PPh3)2],62 10-ethynyl-10-hydroxyanthracen-9-one,49 13-ethynyl-13-hydroxy-pentacen-6-one,50 1-phenyl-1-(pyren-1-yl)prop-2-yn-1-ol55 and 6H-benzo[cd]pyren-6-one63 were prepared according to the literature.
Synthesis of 6-((trimethylsilyl)ethynyl)-6H-benzo[cd]pyren-6-ol
n-Butyllithium (1.95 mL, 1.6 M in hexanes, 3.12 mmol, 8 equiv.) was added dropwise to a solution of TMS-acetylene (0.55 mL, 3.90 mmol, 10 equiv.) in THF (20 mL) at −50 °C under argon atmosphere. The mixture was warmed to room temperature and stirred for 45 minutes. 6H-Benzo[cd]pyren-6-one (100 mg, 0.394 mmol) was added as a solid in one portion and the resulting solution was stirred at room temperature for 14 hours. The reaction mixture was quenched by the addition of water (5 mL) and the solvent was removed in vacuo. The crude product was loaded on a column (silica, ∅ 4 cm, length 10 cm) and eluted with acetone/n-pentane (1:1, v/v) to obtain the brown product (Rf = 0.6) in yield of 128 mg (0.241 mmol, 93%). 1H NMR (300 MHz, CDCl3): δ = 8.50 (d, 3JH,H = 7.7 Hz, 2H, H3), 8.07 (d, 3JH,H = 7.7 Hz, 2H, H5), 8.03 (d, 3JH,H = 8.7 Hz, 2H, H7/H8), 7.95 (d, 3JH,H = 8.7 Hz, 2H, H7/H8), 7.82 (t, 3JH,H = 7.7 Hz, 2H, H4), 0.26 (s, 9 H, SiMe3) ppm; 13C NMR (75 MHz, CDCl3): δ = 137.2 (C), 136.8 (C), 131.6 (C), 129.4 (C), 128.8 (CH), 127.9 (CH), 126.3 (CH), 125.7 (CH), 124.2 (C), 106.6 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 93.0 (CC), 68.9 (C–OH), −0.1 (Me) ppm; ESI-MS (MeOH) m/z (%) = 531.13 (30) [M − H]−; IR (CH2Cl2):
C), 93.0 (CC), 68.9 (C–OH), −0.1 (Me) ppm; ESI-MS (MeOH) m/z (%) = 531.13 (30) [M − H]−; IR (CH2Cl2): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3258 (s, OH), 2948 (m, CH), 2114 (w, CC), 1617 (CC), 1583 (CC) cm−1; EA C24H20OSi (352.51 g mol−1): calc.: C 81.78, H 5.72; found: C 81.43, H 5.77%.
= 3258 (s, OH), 2948 (m, CH), 2114 (w, CC), 1617 (CC), 1583 (CC) cm−1; EA C24H20OSi (352.51 g mol−1): calc.: C 81.78, H 5.72; found: C 81.43, H 5.77%.
Synthesis of 6-ethynyl-6H-benzo[cd]pyren-6-ol
6-((Trimethylsilyl)ethynyl)-6H-benzo[cd]pyren-6-ol (200 mg, 0.567 mmol) was dissolved in a mixture of methanol:THF (1:1) (10 mL). KOH (47.7 mg in 5 mL H2O, 0.850 mmol, 1.5 equiv.) was added dropwise by syringe. The mixture was stirred at 23 °C for 3 hours. The solvent was removed in vacuo and the crude product was loaded on a column (silica, length 10 cm, ∅ 3 cm) with chloroform as eluent. The product could be isolated at Rf = 0.8 in yield of 128 mg (0.457 mmol, 93%). 1H NMR (300 MHz, CDCl3): δ = 7.91 (t, 3JH,H = 7.7 Hz, 2H, H4), 8.03 (d, 3JH,H = 8.7 Hz, 2H, H7/H8), 8.14 (d, 3JH,H = 8.7 Hz, 2H, H7/H8), 8.35 (dd, 3JH,H = 7.8 Hz, 3JH,H = 0.9 Hz, 2H, H5), 8.90 (dd, 3JH,H = 7.5 Hz, 3JH,H = 0.9 Hz, 2H, H3) ppm; 13C NMR (75 MHz, CDCl3): δ = 138.2 (C), 136.4 (C), 131.2 (C), 129.4 (C), 128.8 (CH), 127.6 (CH), 126.9 (CH), 126.7 (CH), 124.2 (C), 88.5 (CC), 93.0 (CCH), 68.9 (C–OH) ppm; IR (CH2Cl2): = 3258 (s, OH), 2115 (w, CC) cm−1; ESI-MS (MeOH) m/z (%) = 279.09 (100) [M − H]−; EA C21H12O (280.33 g mol−1): calc.: C 89.98, H 4.31; found: C 89.43, H 4.54%.
Synthesis of [Ru(η5-C5H5)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CC(AO))(PPh3)2]PF6 (1)
CC(AO))(PPh3)2]PF6 (1)
A mixture of [RuCl(η5-C5H5)(PPh3)2] (290 mg, 0.400 mmol), 10-ethynyl-10-hydroxyanthracen-9-one (100 mg, 0.427 mmol) and NH4[PF6] (65.2 mg, 0.400 mmol) was stirred for 1 d. The deep-purple solution was filtered and the solvent removed under vacuum. The residue was dissolved in CH2Cl2 (10 mL) and filtered into an excess of rapidly stirred Et2O. The suspension was filtered and the purple precipitate was washed with cold Et2O (3 × 5 mL) and dried in vacuo. The filtrate was worked up as before to give a second crop of the product, yield 235 mg (0.223 mmol, 56%). 1H NMR (CD2Cl2, 300.13 MHz): δ = 8.31 (d, 3JH,H = 7.8 Hz, 2H, AO–H), 8.00 (t, 3JH,H = 7.8 Hz, 2H, AO–H), 7.92 (d, 3JH,H = 7.6 Hz, 2H, AO–H), 7.67 (t, 3JH,H = 7.6 Hz, 2H, AO–H), 7.40 (t, 3JH,H = 7.0 Hz, 6H, p-PPh3), 7.22–7.12 (m, 24H, o-PPh3, m-PPh3), 5.27 (s, 5H, Cp) ppm. 13C NMR (CD2Cl2, 75.48 MHz): δ = 293.5 (d, 2JC,P = 27.2 Hz, C-α), 234.3 (C-β), 181.9 (CO), 146.0 (C-γ), 134.5 (AO–C), 133.5 (d, 1JC,P = 50.0 Hz, i-PPh3), 132.3 (t, 2JC,P = 10.5 Hz, o-PPh3), 131.7 (AO–CH), 131.1 (AO–CH), 130.1 (p-PPh3), 129.7 (AO–CH), 128.4 (AO–CH), 128.0 (t, 2JC,P = 10.5 Hz, m-PPh3), 127.4 (AO–C), 94.6 (Cp) ppm. 31P NMR (CD2Cl2, 121.50 MHz): δ = 47.12 (s, PPh3), −144.48 (sept, PF6) ppm. ESI-MS (CH2Cl2): m/z (%) = 907.1827 (100) [M]+. IR (CH2Cl2): = 1919 (CCC), 1662 (CO), 1593 (CC) cm−1. EA C57H43F6OP3Ru (1051.95 g mol−1): calc.: C 65.08, H 4.12; found: C 65.22, H 4.14%. TGA: Td ≈ 158 °C. UV/Vis (CH2Cl2): λmax in nm (ε/dm3 mol−1 cm−1) = 933 (261), 574 (16846), 372 (12746).
Synthesis of [Ru(η5-C5H5)(CC(PCO))(PPh3)2]PF6 (2)
A mixture of [RuCl(η5-C5H5)(PPh3)2] (290 mg, 0.400 mmol), 13-ethynyl-13-hydroxypentacen-6-one (140 mg, 0.420 mmol) and NH4[PF6] (65.2 mg, 0.400 mmol) was stirred at reflux for 2 d. The deep-blue solution was filtered and the solvent removed under vacuum. The residue was dissolved in CH2Cl2 (10 mL) and filtered into an excess of rapidly stirred Et2O. The suspension was filtered and the dark blue precipitate was washed with Et2O (3 × 5 mL) and dried in vacuo. The filtrate was worked up as before to give a second crop of the product, yield 308 mg (0.267 mmol, 67%). 1H NMR (CD2Cl2, 300.13 MHz): δ = 8.96 (s, 2H, PCO–H), 8.59 (s, 2H, PCO–H), 8.17 (d, 3JH,H = 8.1 Hz, 2H, PCO–H), 7.90–7.83 (m, 4H, PCO–H), 7.70 (t, 2H, 3JH,H = 9.0 Hz, PCO–H), 7.37–7.32 (m, 6H, p-PPh3), 7.17–7.07 (m, 24H, o-PPh3, m-PPh3), 5.26 (s, 5H, Cp) ppm. 13C NMR (CD2Cl2, 75.48 MHz): δ = 281.2 (d, 2JC,P = 23.7 Hz, C-α), 208.8 (C-β), 183.8 (CO), 144.8 (C-γ), 136.8 (PCO–C), 136.2 (PCO–C), 135.2 (d, 1JCP = 49.1 Hz, i-PPh3), 134.8 (PCO–C), 133.4 (t, 2JCP = 10.5 Hz, o-PPh3), 132.2 (PCO–CH), 131.2 (PCO–CH), 131.1 (p-PPh3), 130.8 (PCO–CH), 130.7 (PCO–CH), 130.3 (PCO–CH), 130.2 (PCO–CH), 129.0 (t, 2JCP = 10.5 Hz, m-PPh3), 128.2 (PCO–C), 93.6 (Cp) ppm. 31P NMR (CD2Cl2, 121.50 MHz): δ = 46.93 (s, PPh3), −144.53 (qi, PF6) ppm. ESI-MS (CH2Cl2): m/z (%) = 1007.22 (100) [M]+. IR (CH2Cl2): = 1929 (CCC), 1679 (CO), 1617 (CC), 1583 (CC) cm−1. EA C65H47F6OP3Ru (1152.07 g mol−1): calc.: C 67.77, H 4.11; found: C 67.40, H 4.38%. TGA: Td ≈ 152 °C. UV/Vis (CH2Cl2): λmax in nm (ε/dm3 mol−1 cm−1) = 940 (262), 619 (20759), 506 (5225), 402 (16650).
Synthesis of [Ru(η5-C5H5)(CC(PyrPh))(PPh3)2]PF6 (3)
A mixture of [RuCl(η5-C5H5)(PPh3)2] (580 mg, 0.800 mmol), 1-phenyl-1-(pyren-1-yl)prop-2-yn-1-ol (300 mg, 0.886 mmol) and NH4[PF6] (130 mg, 0.800 mmol) was stirred for 2 d. The deep-red solution was filtered and the solvent removed under vacuum. The residue was dissolved in CH2Cl2 (20 mL) and filtered into an excess of rapidly stirred Et2O. The suspension was filtered and the black precipitate was washed with cold Et2O (3 × 5 mL) and dried in vacuo. The filtrate was worked up as before to give a second crop of the product, yield 258 mg (0.224 mmol, 28%). 1H NMR (CD2Cl2, 300.13 MHz): δ = 8.38 (d, 3JH,H = 9.0 Hz, 1H, Pyr–H), 8.33 (d, 3JH,H = 9.0 Hz, 1H, Pyr–H), 8.23–8.10 (m, 5H, Pyr–H), 7.83–7.75 (m, 5H, Pyr–H, o-Ph–H, p-Ph–H), 7.46 (t, 3JH,H = 6.0 Hz, 2H, m-Ph–H), 7.26 (t, 3JH,H = 6.0 Hz, 6H, p-PPh3), 7.09–6.89 (m, 24H, o-PPh3, m-PPh3), 5.08 (s, 5H, Cp) ppm. 13C NMR (CD2Cl2, 75.48 MHz): δ = 302.0 (d, 2JC,P = 26.3 Hz, C-α), 213.1 (C-β), 145.5 (C-γ), 142.2 (Pyr–C1), 139.1 (Pyr–C), 135.9 (Pyr–C), 135.2 (d, 1JC,P = 50.0 Hz, i-PPh3), 135.1 (d, 1JC,P = 50.2 Hz, i-PPh3), 133.5 (t, 2JC,P = 9.8 Hz, o-PPh3), 133.2 (o-Ph–C), 133.4 (Pyr–C), 132.0 (Pyr–C), 131.2 (p-Ph–C), 131.2 (p-PPh3), 130.0 (Pyr–C), 128.7 (t, 2JC,P = 9.8 Hz, m-PPh3), 125.6 (Pyr–C), 93.9 (Cp) ppm. 31P NMR (CD2Cl2, 121.50 MHz): δ = 45.95 (s, PPh3), −144.52 (sept, PF6) ppm. ESI-MS (CH2Cl2): m/z (%) = 1005.23 (100) [M]+. IR (CH2Cl2): = 1931 (CCC), 1653 (CC), 1593 (CC) cm−1. EA C66H49F6P3Ru (1150.10 g mol−1): calc.: C 68.93, H 4.29; found: C 68.49, H 4.20%. TGA: Td ≈ 238 °C. UV/Vis (CH2Cl2): λmax in nm (ε/dm3 mol−1 cm−1) = 821 (924), 587 (27563), 479 (32469).
Synthesis of [Ru(η5-C5H5)(CC(FN))(PPh3)2]PF6 (4)
A mixture of [RuCl(η5-C5H5)(PPh3)2] (363 mg, 0.500 mmol), 9-ethynyl-9-fluorenyl (107 mg, 0.520 mmol) and NH4[PF6] (81.5 mg, 0.500 mmol) was stirred for 1 d. The deep-pink solution was filtered and the solvent removed under vacuum. The residue was dissolved in CH2Cl2 (10 mL) and filtered into an excess of rapidly stirred Et2O. The suspension was filtered and the purple precipitate was washed with cold Et2O (3 × 5 mL) and dried in vacuo. The filtrate was worked up as before to give a second crop of the product, yield 343 mg (0.335 mmol, 67%). 1H NMR (CD2Cl2, 300.13 MHz): δ = 7.64 (t, 3JH,H = 7.5 Hz, 2H, FN–H), 7.46 (d, 3JH,H = 7.5 Hz, 2H, FN–H), 7.42–7.37 (m, 4H, FN–H), 7.26–7.13 (m, 24H, o-PPh3, m-PPh3, p-PPh3), 5.16 (s, 5H, Cp) ppm. 13C NMR (CD2Cl2, 75.48 MHz): δ = 294.7 (d, 2JC,P = 17.5 Hz, C-α), 213.4 (C-β), 145.9 (C-γ), 142.0 (FN–C), 135.4 (FN–C), 134.2 (FN–CH), 134.9 (d, 1JC,P = 49.8 Hz, i-PPh3), 133.7 (t, 2JC,P = 10.5 Hz, o-PPh3), 131.2 (p-PPh3), 130.0 (FN–CH), 129.2 (t, 2JC,P = 10.5 Hz, m-PPh3), 124.4 (FN–CH), 122.1 (FN–CH), 94.8 (Cp) ppm. 31P NMR (CD2Cl2, 121.50 MHz): δ = 47.60 (s, PPh3), −144.48 (sept, PF6) ppm. ESI-MS (CH2Cl2): m/z (%) = 879.13 (100) [M]+. IR (CH2Cl2): = 1939 (CCC), 1600 (CC) cm−1. EA C56H43F6P3Ru (1023.94 g mol−1): calc.: C 65.69, H 4.23; found: C 66.02, H 4.32%. TGA: Td ≈ 219 °C. UV/Vis (CH2Cl2): λmax in nm (ε/dm3 mol−1 cm−1) = 920 (183), 535 (26705), 397 (11343).
Synthesis of [Ru(η5-C5H5)(CC(BPyr))(PPh3)2]PF6 (5)
A mixture of [RuCl(η5-C5H5)(PPh3)2] (109 mg, 0.150 mmol), 6-ethynyl-6H-benzo[cd]pyren-6-ol (50.0 mg, 0.178 mmol) and NH4[PF6] (24.4 mg, 0.150 mmol) was stirred for 2 d. The deep-turquoise solution was filtered and the solvent removed under vacuum. The residue was dissolved in CH2Cl2 (10 mL) and filtered into an excess of rapidly stirred Et2O. The suspension was filtered and the turquoise precipitate was washed with cold Et2O (3 × 5 mL) and dried in vacuo. The filtrate was worked up as before to give a second crop of the product, yield 97.0 mg (0.088 mmol, 59%). 1H NMR (CD2Cl2, 300.13 MHz): δ = 8.69 (d, 3JH,H = 6.0 Hz, 2H, BPyr–H), 8.64 (d, 3JH,H = 9.0 Hz, 2H, BPyr–H), 8.38 (d, 3JH,H = 6.0 Hz, 2H, BPyr–H), 8.14 (d, 3JH,H = 9.0 Hz, 2H, BPyr–H), 7.83 (t, 3JH,H = 6.0 Hz, 2H, BPyr–H), 7.36 (t, 3JH,H = 9.0 Hz, 6H, p-PPh3), 7.36 (t, 3JH,H = 9.0 Hz, 6H, p-PPh3), 7.31–7.25 (m, 12H, m-PPh3), 7.18 (t, 3JH,H = 9.0 Hz, 12H, o-PPh3), 5.07 (s, 5H, Cp) ppm. 13C NMR (CD2Cl2, 75.48 MHz): δ = 286.5 (d, 2JC,P = 22.7 Hz, C-α), 207.0 (C-β), 143.3 (C-γ), 138.8 (BPyr–C), 136.3 (BPyr–C), 136.2 (BPyr–CH), 135.9 (d, 1JC,P = 40.0 Hz, i-PPh3), 134.0 (BPyr–C), 133.7 (t, 2JCP = 10.5 Hz, o-PPh3), 132.5 (BPyr–C), 130.7 (p-PPh3), 130.3 (BPyr–CH), 129.3 (BPyr–CH), 128.8 (t, 2JC,P = 10.5 Hz, m-PPh3), 125.8 (BPyr–C), 91.8 (Cp) ppm. 31P NMR (CD2Cl2, 121.50 MHz): δ = 50.27 (s, PPh3), −143.27 (sept, PF6) ppm. ESI-MS (CH2Cl2): m/z (%) = 953.20 (100) [M]+. IR (CH2Cl2): = 1942 (CCC), 1605 (CC), 1570 (CC) cm−1. EA C62H45F6P3Ru (1098.02 g mol−1): calc.: C 67.82, H 4.13; found: C 67.44, H 4.52%. TGA: Td ≈ 185 °C. UV/Vis (CH2Cl2): λmax in nm (ε/dm3 mol−1 cm−1) = 928 (235), 733 (45592), 681 (44334), 523 (8962), 445 (5576).
X-ray structure determination
The structure determination for 1 and 2 were carried out on a Bruker-Nonius Kappa-CCD diffractometer. For the structure of 5 X-ray intensity data was collected on an Agilent Supernova Dual Source diffractometer equipped with an Atlas S2 CCD detector. In all cases Mo-Kα radiation (λ = 0.71073 Å) was used. Single crystals of 1, 2 and 5 were coated with perfluoropolyether, picked with a glass fiber or loop, and immediately mounted in the nitrogen cold gas stream of the diffractometer. The structures were solved by using direct methods and refined with full-matrix least squares against F2 (SHELX-97).64 A weighting scheme was applied in the last steps of the refinement with w = 1/[σ2(Fo2) + (aP)2 + bP] and P = [2Fc2 + max(Fo2,0)]/3. The hydrogen atoms were included in their calculated positions and refined in a riding model. The asymmetric unit of 1 contains one disordered diethyl ether molecule that was refined in a 1:1 ratio on two positions. The asymmetric unit of 2 contains one acetone and one diethyl ether molecule and in case of complex 5 one dichloromethane and half a molecule of disordered n-hexane were found. Since it was not possible to resolve the disorder of this n-hexane, SQUEEZE was applied in case of 5.65,66 The structure pictures were prepared with the program Mercury.67
Calculations
All density-functional theory (DFT)-calculations were carried out by using the Jaguar 7.7.107 software running on Linux 2.6.18-238.el5 SMP (×86_64) on two AMD Phenom II X6 1090T processor workstations (Beowulf-cluster) parallelized with OpenMPI.68 The X-ray crystal structure of 2 was used as starting geometry. Complete geometry optimization was carried out on the implemented LACVP* (Hay–Wadt effective core potential (ECP) basis on heavy atoms, N31G6* for all other atoms) basis set and with the B3LYP density functional. The calculated structure was proven to be a true minima by the absence of large imaginary frequencies. Orbital plots were obtained using Maestro 9.1.207, the graphical interface of Jaguar. UV/Vis transitions were obtained by time dependent (TD) DFT-calculations on the geometry of the minimized structure.Conclusions
Carbon-rich, cationic Cp-ruthenium allenylidene complexes bearing a range of polyaromatic moieties have been synthesized. These complexes show significant π-stacking in the solid-state structures, especially 1 and 2, and in solution π-stacking interactions were established by UV/Vis experiments with pyrene and the associated Job's plots. High extinction coefficients in the visible spectrum, as well as absorptions in the near-IR, demonstrated by the complexes suggest potential applications in DSSCs, particularly since these complexes are very stable according to TGA analyses. The CV data implies significant electron-acceptor potential of the complexes. Especially, complex 2 shows a staircase π-stacking in solid state that might be a good design for future metal-tuned FETs or “organic” metal-semiconductor field-effect transistors (OMESFETs). Future work will focus on the question whether SWCNTs can be decorated with such complexes by π-interactions.Acknowledgements
This work was generously supported by ‘Solar Technologies go Hybrid’, an initiative of the Bavarian State Ministry for Science, Research and Art. Funding of a Bruker AVANCE III HD 400 MHz spectrometer and a Bruker AVANCE III HD 600 MHz spectrometer as well as a Super Nova A S2 (Dual) diffractometer with Atlas S2 CCD detector and Nova (Cu) and Mova (Mo) X-ray sources by the German Research Council (DFG) is gratefully acknowledged.Notes and references
- D. Jain, A. Saha and A. A. Marti, Chem. Commun., 2011, 47, 2246–2248 RSC.
- J. Bartelmess, B. Ballesteros, G. de la Torre, D. Kiessling, S. Campidelli, M. Prato, T. Torres and D. M. Guldi, J. Am. Chem. Soc., 2010, 132, 16202–16211 CrossRef CAS PubMed.
- J. Yu, S. Mathew, B. S. Flavel, M. R. Johnston and J. G. Shapter, J. Am. Chem. Soc., 2008, 130, 8788–8796 CrossRef CAS PubMed.
- Q. Miao, Synlett, 2012, 326–336 CrossRef CAS.
- Z. Liang, Q. Tang, J. Liu, J. Li, F. Yan and Q. Miao, Chem. Mater., 2010, 22, 6438–6443 CrossRef CAS.
- A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- Q. Tang, J. Liu, H. S. Chan and Q. Miao, Chem. – Eur. J., 2009, 15, 3965–3969 CrossRef CAS PubMed.
- S. H. Etschel, A. R. Waterloo, J. T. Margraf, A. Y. Amin, F. Hampel, C. M. Jager, T. Clark, M. Halik and R. R. Tykwinski, Chem. Commun., 2013, 49, 6725–6727 RSC.
- D. Lehnherr, A. H. Murray, R. McDonald and R. R. Tykwinski, Angew. Chem., Int. Ed., 2010, 49, 6190–6194 CrossRef CAS PubMed.
- S. Guesmi, D. Touchard and P. H. Dixneuf, Chem. Commun., 1996, 2773–2774 RSC.
- S. K. Costuas, D. Touchard, J.-Y. Saillard, S. Golhen and P. H. Dixneuf, J. Am. Chem. Soc., 2004, 126, 4072–4073 CrossRef PubMed.
- S. Rigaut, D. Touchard and P. H. Dixneuf, Coord. Chem. Rev., 2004, 248, 1585–1601 CrossRef CAS.
- S. Rigaut, O. Maury, D. Touchard and P. H. Dixneuf, Chem. Commun., 2001, 373–374 RSC.
- N. Mantovani, M. Brugnati, L. Gonsalvi, E. Grigiotti, F. Laschi, L. Marvelli, M. Peruzzini, G. Reginato, R. Rossi and P. Zanello, Organometallics, 2005, 24, 405–418 CrossRef CAS.
- F. Paul and C. Lapinte, Coord. Chem. Rev., 1998, 178–180(part 1), 431–509 CrossRef CAS.
- E. O. Fischer, H. J. Kalder, A. Frank, F. H. Köhler and G. Huttner, Angew. Chem., 1976, 88, 683–684 CrossRef CAS.
- E. O. Fischer and A. Maasböl, Angew. Chem., 1964, 76, 645 CrossRef CAS.
- H. Berke, Angew. Chem., 1976, 88, 684–685 CrossRef CAS.
- M. I. Bruce, Chem. Rev., 1998, 98, 2797–2858 CrossRef CAS PubMed.
- R. Castarlenas, C. Vovard, C. Fischmeister and P. H. Dixneuf, J. Am. Chem. Soc., 2006, 128, 4079–4089 CrossRef CAS PubMed.
- A. Fürstner, M. Liebl, C. W. Lehmann, M. Picquet, R. Kunz, C. Bruneau, D. Touchard and P. H. Dixneuf, Chem. – Eur. J., 2000, 6, 1847–1857 CrossRef.
- D. Touchard and P. H. Dixneuf, Coord. Chem. Rev., 1998, 178–180(part 1), 409–429 CrossRef CAS.
- V. Cadierno and J. Gimeno, Chem. Rev., 2009, 109, 3512–3560 CrossRef CAS PubMed.
- J. W. Herndon, Coord. Chem. Rev., 2013, 257, 2899–3003 CrossRef CAS.
- I. García de la Arada, J. Díez, M. P. Gamasa and E. Lastra, Organometallics, 2015, 34, 1345–1353 CrossRef.
- E. Bustelo, M. Jiménez-Tenorio, M. C. Puerta and P. Valerga, Organometallics, 2007, 26, 4300–4309 CrossRef CAS.
- S. Conejero, J. Díez, M. P. Gamasa and J. Gimeno, Organometallics, 2004, 23, 6299–6310 CrossRef CAS.
- M. Lichtenheldt, S. Kress and S. Blechert, Molecules, 2012, 17, 5177 CrossRef CAS PubMed.
- A. Antonucci, M. Bassetti, C. Bruneau, P. H. Dixneuf and C. Pasquini, Organometallics, 2010, 29, 4524–4531 CrossRef CAS.
- M. J. Queensen, N. P. Rath and E. B. Bauer, Organometallics, 2014, 33, 5052–5065 CrossRef CAS.
- C. E. Diesendruck, E. Tzur and N. G. Lemcoff, Eur. J. Inorg. Chem., 2009, 4185–4203 CrossRef CAS.
- X. Sauvage, Y. Borguet, G. Zaragoza, A. Demonceau and L. Delaude, Adv. Synth. Catal., 2009, 351, 441–455 CrossRef CAS.
- H. Kopf, B. Holzberger, C. Pietraszuk, E. Hübner and N. Burzlaff, Organometallics, 2008, 27, 5894–5905 CrossRef CAS.
- R. Castarlenas, C. Fischmeister, C. Bruneau and P. H. Dixneuf, J. Mol. Catal. A: Chem., 2004, 213, 31–37 CrossRef CAS.
- B. Cetinkaya, S. Demir, I. Ozdemir, L. Toupet, D. Semeril, C. Bruneau and P. H. Dixneuf, New J. Chem., 2001, 25, 519–521 RSC.
- A. Fürstner, A. Furstner, M. Picquet, C. Bruneau and P. H. Dixneuf, Chem. Commun., 1998, 1315–1316 RSC.
- D. F. Alkhaleeli, K. J. Baum, J. M. Rabus and E. B. Bauer, Catal. Commun., 2014, 47, 45–48 CrossRef CAS.
- S. Csihony, C. Fischmeister, C. Bruneau, I. T. Horvath and P. H. Dixneuf, New J. Chem., 2002, 26, 1667–1670 RSC.
- M. Saoud, A. Romerosa and M. Peruzzini, Organometallics, 2000, 19, 4005–4007 CrossRef CAS.
- S. M. Maddock and M. G. Finn, Angew. Chem., Int. Ed., 2001, 40, 2138–2141 CrossRef CAS.
- D. A. Valyaev, M. G. Peterleitner, O. V. Semeikin, K. I. Utegenov, N. A. Ustynyuk, A. Sournia-Saquet, N. Lugan and G. Lavigne, J. Organomet. Chem., 2007, 692, 3207–3211 CrossRef CAS.
- N. E. Kolobova, L. L. Ivanov, O. S. Zhvanko, O. M. Khitrova, A. S. Batsanov and Y. T. Struchkov, J. Organomet. Chem., 1984, 262, 39–47 CrossRef CAS.
- D. Touchard, N. Pirio and P. H. Dixneuf, Organometallics, 1995, 14, 4920–4928 CrossRef CAS.
- C. Cosset, I. Del Rio and H. Le Bozec, Organometallics, 1995, 14, 1938–1944 CrossRef CAS.
- R. Le Lagadec, E. Roman, L. Toupet, U. Mueller and P. H. Dixneuf, Organometallics, 1994, 13, 5030–5039 CrossRef CAS.
- R. Dussel, D. Pilette, P. H. Dixneuf and W. P. Fehlhammer, Organometallics, 1991, 10, 3287–3291 CrossRef CAS.
- F. Strinitz, J. Tucher, J. A. Januszewski, A. R. Waterloo, P. Stegner, S. Förtsch, E. Hübner, R. R. Tykwinski and N. Burzlaff, Organometallics, 2014, 33, 5129–5144 CrossRef CAS.
- F. Strinitz, A. Waterloo, J. Tucher, E. Hübner, R. R. Tykwinski and N. Burzlaff, Eur. J. Inorg. Chem., 2013, 5181–5186 CrossRef CAS.
- H. Kopf, C. Pietraszuk, E. Hübner and N. Burzlaff, Organometallics, 2006, 25, 2533–2546 CrossRef CAS.
- J. P. Selegue, Organometallics, 1982, 1, 217–218 CrossRef CAS.
- M. Tamm, T. Jentsch and W. Werncke, Organometallics, 1997, 16, 1418–1424 CrossRef CAS.
- M. I. Bruce, P. J. Low and E. R. T. Tiekink, J. Organomet. Chem., 1999, 572, 3–10 CrossRef CAS.
- F. Strinitz, PhD thesis, University of Erlangen-Nürnberg, 2014.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- R. F. Winter, Eur. J. Inorg. Chem., 1999, 2121–2126 CrossRef CAS.
- S. Hartmann, R. F. Winter, B. Sarkar and F. Lissner, Dalton Trans., 2004, 3273–3282 RSC.
- S. Rigaut, J. Perruchon, S. Guesmi, C. Fave, D. Touchard and P. H. Dixneuf, Eur. J. Inorg. Chem., 2005, 447–460 CrossRef CAS.
- G. A. Koutsantonis, P. A. Schauer and B. W. Skelton, Organometallics, 2011, 30, 2680–2689 CrossRef CAS.
- S. H. Yoo, J. M. Kum and S. O. Cho, Nanoscale Res. Lett., 2011, 6, 1–7 CrossRef PubMed.
- M. I. Bruce, C. Hameister, A. G. Swincer, R. C. Wallis and S. D. Ittel, Inorganic Syntheses, John Wiley & Sons, Inc., 2007, pp. 270–272 Search PubMed.
- J. Morgenthaler and C. Rüchardt, Eur. J. Org. Chem., 1999, 2219–2230 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., 1990, A46, 194–201 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- Jaguar, version 7.7, Schrodinger, LLC, New York, NY, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Job's plots, CSI-MS, CV, UV/Vis and X-ray data of 1, 2 and 5. CCDC 1406943, 1406944 and 1417389. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj03556b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |