
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSparingly fluorinated maltoside-based surfactants for membrane-protein stabilization†
Ange
Polidori
a,
Simon
Raynal
a,
Laurie-Anne
Barret
ab,
Mohamed
Dahani
a,
Cherone
Barrot-Ivolot
a,
Colette
Jungas
b,
Erik
Frotscher
c,
Sandro
Keller
c,
Christine
Ebel
def,
Cécile
Breyton
def and
Françoise
Bonneté
*a
aInstitut des Biomolécules Max Mousseron (IBMM) UMR 5247 CNRS-UM-ENSCM, Chimie BioOrganique et Systèmes Amphiphiles, Université d'Avignon, 301, rue Baruch de Spinoza, F84000 Avignon, France. E-mail: francoise.bonnete@univ-avignon.fr
bLaboratoire de Génétique et de Biophysique des Plantes (L.G.B.P) UMR 7265 CNRS-CEA-AMU, Faculté des Sciences de Luminy, 163 avenue de Luminy, F-13009 Marseille, France
cMolecular Biophysics, University of Kaiserslautern, Erwin-Schrödinger-Str. 13, 67663 Kaiserslautern, Germany
dUniversité Grenoble Alpes, IBS, F-38044 Grenoble, France
eCNRS, IBS, F-38044 Grenoble, France
fCEA, IBS, F-38044 Grenoble, France
First published on 7th April 2016
Abstract
Membrane proteins pose formidable challenges during in vitro investigations, as they require amphiphilic molecules for their solubilization, stabilization, and crystallization for structural characterization. Therefore, numerous, chemically diverse new amphiphiles have been developed for membrane-protein applications. Among these, both perfluorinated and hemifluorinated surfactants have long been known to stabilize membrane proteins, but the contribution of the fluorine content in the aliphatic chain has not yet been examined in detail. We have synthesized two new maltose-based fluorosurfactants bearing either a perfluoroethyl (F2H9) or a perfluorobutyl (F4H5) tip at the end of the chain and compared them with the common detergent dodecyl maltoside and a commercial highly fluorinated octyl maltoside derivative. We describe the physicochemical properties, aggregate morphologies, and micellization thermodynamics of these sparingly fluorinated surfactants as a function of the length of the fluorinated segment and evaluate their biochemical use for membrane-protein stabilization. Intriguingly, the surfactant carrying a perfluorobutyl (F4H5) tip trumps both nonfluorinated dodecyl maltoside and a more extensively fluorinated octyl maltoside derivative in conferring extraordinary long-term functional and colloidal stability to the model membrane protein bacteriorhodopsin.
Introduction
Membrane proteins (MPs) play key roles in signal and mass transfer across lipid bilayers, which form dynamic interfaces between cellular compartments or between a cell and its environment. Although the genes encoding MPs represent on average 30% of genomes and the corresponding proteins 60% of all therapeutic targets,1 there is a lack of structural information compared with that available for soluble proteins. During the last four decades, the characterization of MPs has been a major challenge in structural biology, one of the main bottlenecks consisting in finding detergents suitable for their extraction and handling in a native and active state. The stability of protein–detergent complexes (PDCs) depends both on the type and on the concentration of the detergent. Detergent concentrations below the critical micellar concentration (CMC) can cause MPs to aggregate and precipitate; conversely, high detergent concentrations may inactivate them. The amount of detergent molecules bound to transmembrane regions is highly variable and can be very large, thus modifying considerably the size and the mass of PDCs. For example, Bamber et al. have shown that, as the length of the aliphatic chain in n-alkyl-β-D-maltosides decreases, PDCs become smaller and thus expose more MP surface area.2It is thus clear that detergent packing around MPs will significantly contribute to understand how complexes interact and self-assemble during phase transition and crystallization and how PDCs pack into the crystal.3,4 For example, detergents forming large micelles, such as n-dodecyl-β-D-maltoside (H12βM) and polyoxyethylene dodecyl ether (C12E9), are more likely to keep a MP in solution by efficiently shielding its apolar surface; however, the large size of the micelle means that less protein surface is available to form protein–protein interactions essential for crystal-lattice formation.5 By contrast, small-micelle detergents, such as n-octyl-β-D-glucoside (βOG) and n-octyl-β-D-maltoside (βOM), leave more of the protein exposed to form protein–protein interactions necessary for strong crystal contacts but may also inactivate MPs by the intrusion of the detergent alkyl chain into the interior of the protein or by stripping away stabilizing lipids, cofactors, or subunits. At this stage, one might wonder if a detergent suitable for stabilization is also good for crystallization.
Different strategies have been developed over two decades to synthesize new surfactants that allow in vitro synthesis,6,7 solubilization and purification,8,9 or trapping and stabilization10 of MPs. Most surfactants developed to date have been valuable for solubilizing and stabilizing MPs, but the number of surfactants amenable to crystallization and X-ray crystallography is rather limited. It seems reasonable that a hydrophobic chain that is less lipophilic towards residual lipids and cofactors and less intrusive towards transmembrane regions of the protein should be more stabilizing and thus more favorable for crystallization. Furthermore, new powerful amphiphiles for crystallization would cover less protein surface to favor protein–protein contacts in crystals while keeping MPs stable. This could be achieved by tighter control of the micellar assembly and the surfactant interactions with MPs.
Recently, we have characterized the physicochemical and structural properties of a new polycyclic maltoside surfactant, propyl(bi)cyclohexyl-α-maltoside (PCCαM).11 The stability of the cytochrome b6f complex was higher in PCCαM than in H12βM, rendering PCCαM suitable for crystallization. We further showed by small-angle X-ray scattering (SAXS) that micelles of H12βM and PCCαM possess similar ellipsoidal sizes and shapes. However, aggregation numbers obtained from molar masses revealed that PCCαM micelles (Nagg ≈ 165) contain more surfactant monomers than H12βM micelles (Nagg ≈ 125).12 Moreover, second virial coefficients determined by SAXS revealed that intermicellar interactions are more attractive for PCCαM than H12βM, thus highlighting that the hydrophobic chain can greatly impact this kind of interaction. This resulted in a lower cloud point boundary for PCCαM than for H12βM, which favored the crystallization of the MP target RC-LH1-pufX in this new surfactant; however, diffraction quality was not sufficiently improved to allow structure determination. In order to increase X-ray diffraction by a better packing of MP–surfactant complexes in crystals, new surfactants need to be designed with a degree of hydrophobicity that stabilizes MPs in a native and active form and, at the same time, promotes suitable attraction for crystallization.
Several series of fluorinated surfactants have been synthesized over the past 20 years and tested for handling MPs in aqueous solutions.13–18 These surfactants have the same general structure as classical detergents (i.e. a hydrophilic headgroup and a hydrophobic tail), but their hydrophobic tail is fully or partially fluorinated (e.g. F6, F8). Since fluoroalkanes are both hydrophobic and poorly miscible with alkanes (i.e. lipophobic),19 this renders perfluorinated surfactants poor solvents for lipids and hydrophobic cofactors, although exceptions have recently been reported.20 As a result, most fluorinated surfactants are unable to efficiently extract MPs from native membranes. Furthermore, fluorinated surfactants present bulkier and more rigid hydrophobic chains than their hydrogenated counterparts and, thus, are less likely to intrude into the protein structure itself, which makes them less denaturing for MPs. While the stabilization of MPs has been demonstrated in fluorinated surfactants (reviewed in ref. 21), no successful in surfo crystallization has yet been reported. For the crystallization of MPs in surfo, crucial parameters include the micelle morphology and the belt size around the protein to allow polar contacts in the crystal. Previous results obtained with fluorinated and hemifluorinated derivatives of H12βM22 have shown that the structural heterogeneity observed with the (H)F6-maltoside series was because the fluorinated alkyl chain being bulkier than the hydrogenated one. The length and the volume of the F6 segment influence the packing parameter23 of fluorinated surfactants and, therefore, the morphology of their aggregates formed in water. In order to form small globular micelles with maltose-based fluorinated surfactants, we have developed new fluorinated surfactants with a perfluoroethyl (F2H9) or a perfluorobutyl (F4H5) tip at the end of the aliphatic chain, whose overall hydrophilic/lipophobic balance is expected to be equivalent to that of H12βM (Scheme 1).
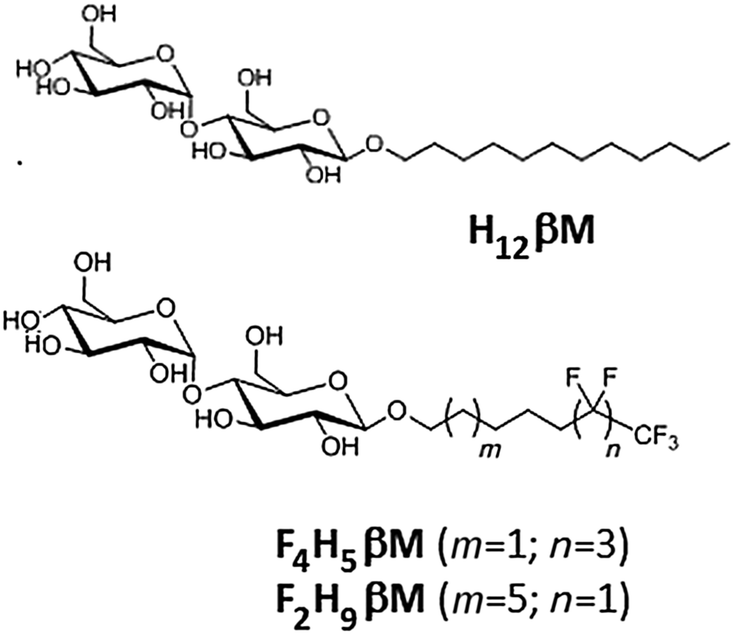 | ||
| Scheme 1 Chemical structures of surfactants: H12βM, F2H9βM and F4H5βM. | ||
In this paper, we describe the synthesis of these two new sparingly fluorinated surfactants. We compare their physicochemical and micellar properties with those of commercial n-dodecyl-β-D-maltoside (H12βM) and fluorinated octyl maltoside (F6H2βM) and perform a biochemical evaluation of MP complex stability and homogeneity using bacteriorhodopsin (bR) from the purple membrane as a model MP.
Experimental section
Materials
All starting materials were commercially available and used without further purification. All solvents were of reagent grade and used as received unless noted otherwise. THF and MeOH were dried over Na, and CH2Cl2 was dried over CaH2 under an argon atmosphere. Commercially available anhydrous DMF was stored over activated molecular sieves (3 Å). The progress of reactions was monitored by thin-layer chromatography (TLC, Merck 254, silica plates), and compounds were detected either by exposure to ultraviolet light (254 nm), iodine, or by spraying with a 0.05% sulfuric acid solution in ethanol followed by heating to 150 °C. Flash chromatography purifications were carried out on silica gel (40–63 mm granulometry). Size exclusion chromatography purifications were carried out on Sephadex LH-20 resin. 1H and 13C NMR spectra were recorded on a Bruker Ascend 9.4-T spectrometer at 400 MHz for 1H, 376 MHz for 19F, and 100 MHz for 13C. Chemical shifts (δ values) are reported in ppm downfield from the internal residual solvent as a heteronuclear reference. High-resolution mass spectrometry by electrospray ionization (HRMS-ESI) was carried out on a QStar Elite mass spectrometer. Peracetylated β-maltoside was prepared according to the study of Vill et al.24Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate 6:4). A mixture of water/acetic acid/12 N HCl (30/15/5: v/v/v) was added which dissolved zinc. The mixture was filtered through a pad of celite and washed with saturated NaHCO3 solution (25 mL). After drying over anhydrous sodium sulfate and the evaporation of the solvent, the residue was purified by column chromatography on silica gel (cyclohexane/ethyl acetate: 6:4) to give 2.2 g of liquid 1 (70%).
:ethyl acetate 6:4). A mixture of water/acetic acid/12 N HCl (30/15/5: v/v/v) was added which dissolved zinc. The mixture was filtered through a pad of celite and washed with saturated NaHCO3 solution (25 mL). After drying over anhydrous sodium sulfate and the evaporation of the solvent, the residue was purified by column chromatography on silica gel (cyclohexane/ethyl acetate: 6:4) to give 2.2 g of liquid 1 (70%).
1H NMR δ (CDCl3): 1.45–1.51 (m, CH2–CH2–CH2–, 2H), 1.58–1.68 (m, CF2–CH2–CH2–, CH2–CH2–OH, 4H), 2.07 (m, 2H, CH2–CF2, 2H), 3.67 (t, CH2OH, 2H). 19F NMR δ (CDCl3): −126.08 (t, 2F, CF2–CF2–CF2–CF3), −124.52 (t, 2F, CF2–CF2–CF2–CF3), −114.63 (t, 2F, CH2–CF2–CF2–CF2–CF3), −81.06 (s, 3F, –CF3). 13C NMR δ (CDCl3): 19.96, 19.99 (CF2–CH2–CH2–), 25.33 (CH2–CH2–CH2–), 30.75 (t, CH2–CF2), 32.24 (CH2–CH2–OH), 62.48 (CH2OH).
1H-NMR δ (CDCl3): 1.3–1.65 (m, 8H, CH2–(CH2)3–CH2–CF2–), 2.0–2.14 (m, 23H, acetyl groups, CH2–CF2), 3.5 (m, 1H, O–CH2), 3.65 (m, 1H, H5), 3.87 (m, 1H, O–CH2), 3.9–4.1 (m, 3H, H4, H5′, H6′a), 4.15–4.35 (m, 2H, H6b, H6′b), 4.45 (d, 1H, H6a), 4.5–4.55 (d, 1H, H1, 3J1,2 = 7.5 Hz), 4.75–4.95 (m, 2H, H2, H2′), 5–5.15 (d, 1H, H4′), 5.25 (t, 1H, H3), 5.36 (t, 1H, H3′), 5.42 (d, 1H, H1′, 3J1,2 = 4 Hz). 19F NMR δ (ppm, CDCl3): −126.07 (t, 2F, CH2–CF2–CF2–CF2–CF3), −124.52 (t, 2F, CF2–CF2–CF2–CF3), −114.6 (t, 2F, CH2–CF2–CF2–CF2–CF3), −81.06 (t, 3F, CF3). 13C NMR δ (CDCl3): 19.80 (CH2–CH2–CF2), 20.55, 20.58, 20.65, 20.78, 20.89 (CH3, OAc), 25.41 (CH2), 29.05 (CH2–CF2), 30.65 (t, CF2CH2), 61.50 (C6′), 62.80 (C6), 68.02 (C5′), 68.48 (C4), 69.33 (C3′), 69.47 (CH2O), 69.99 (C2′) 72.13 (C5), 72.18 (C2), 72.74 (C4), 75.40 (C3), 95.52 (C1′), 100.25 (C1), 170.52, 170.43, 170.24, 169.96, 169.56, 169.40 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, OAc). ESI-TOF MS: [M + H]1+ 925.2.
O, OAc). ESI-TOF MS: [M + H]1+ 925.2.
1H NMR δ (CD3OD): 1.4 (m, 2 H, O–(CH2)2–CH2–(CH2)2–CF2), 1.54 (m, 4H, CH2–CH2–CF2, CH2–CH2–O), 2.1 (2H, m, CH2–CF2), 3.11–3.81 (complex signal, 17H, H glycosyl and CH2O), 4.17–4.19 (d, 1H, H1, 3J1,2 = 8 Hz), 5.06–5.07 (d, 1H, H1′, 3J1′,2′ = 4 Hz). 19F NMR δ (CD3OD): −127.3 (t, 2F, CF2–CF3), −125.55 (t, 2F, CF2–CF2–CF2–CF3), −115.67 (t, 2F, CH2–CF2–CF2–CF2–CF3), −81.70 (t, 3F, CF3). 13C NMR δ (CD3OD): 21.06 (CH2CH2CF2), 26.58 (CH2–CH2–CH2–), 30.38 (t, CH2–CF2), 31.60 (CH2CF2), 62.18 (C6), 62.75 (C6′), 70.42 (CH2O), 71.50 (C4′), 74.17 (C5′), 74.70 (C2), 74.79 (C2′), 75.08 (C3′), 76.59 (C5), 77.87 (C3), 81.35 (C4), 102.93 (C1′), 104.33 (C1). ESI-TOF HRMS: calcd for C21H32F9O11 [M + H]1+ 631.1800, found 631.1774.
1H-NMR δ (CDCl3): 1.21–1.55 (m, 12H, (CH2)6), 1.93–2.07 (m, 23H, CH3CO), 3.45 (m, 1H, OCH2), 3.7 (m, 1H, H5), 3.8 (m, 1H, OCH2), 3.9–4.2 (m, 4H), 4.25 (m, 2H), 4.5 (m, 2H, H1, J = 7,5 Hz), 4.75–4.9 (m, 2 H), 5.0–5.1 (m, 1H), 5.2–5.3 (m, 1H), 5.31–5.45 (m, 2H, H′1), 5.58 (m, 3H, vinyl function), 5.7–5.8 (m, 1H, CH2CH). 13C NMR δ (CDCl3): 20.56, 20.59, 20.61, 20.67, 20.83, 20.91 (CH3CO), 25.73, 28.80, 29.01, 29.10, 29.34 (OCH2–(CH2)7–CH2–CF2), 33.72 (CH2–CF2), 61.51 (C6′), 62.90 (C6), 68.03 (C5′), 68.46 (C4), 69.35 (C3′), 69.98 (CH2O), 70.16 (C2′) 72.05 (C5), 72.22 (C2), 72.78 (C4), 75.46 (C3), 95.49 (C1′), 100.28 (C1), 114.21 (CH2CH), 139.05 (CH2CH), 169.41, 169.58, 169.95, 170.26, 170.48, 170.52 (CO, OAc).
1H-NMR δ (CDCl3): 1.32–1.59 (m, 10H, (CH2)5–CH2–CHI–), 1.7–1.85 (CH2–CHI), 2.01–2.15 (m, 23H, acetyl groups, CH2–CF2), 2.6–2.95 (m, 1H, CHI), 3.45 (m, 1H, O–CH2), 3.65 (m, 1H, H5), 3.84 (m, 1H, O–CH2), 3.9–4.06 (m, 3H, H4, H5′, H6′a), 4.20–4.27 (m, 2H, H6b, H6b′), 4.46 (d, 1H, H6a), 4.5–4.51–4.53 (m, 1H, H1, 3J1,2 = 8 Hz), 4.80–4.88 (m, 2H, H2, H2′), 5.06 (t, 1H, H4′), 5.37 (t, 1H, H3′, H3), 5.42 (d, 1H, H1′, 3J1,2 = 4 Hz). 19F NMR δ (ppm, CDCl3): −85.82 (CF3), −115.3 to −119.2 (dd, CF2). 13C NMR δ (CDCl3): 20.57, 20.60, 20.65, 20.68, 20.84, 20.91 (CH3CO), 25.71, 28.43, 29.04, 29.31, 29.47 (OCH2–(CH2)7–CH2–CF2), 40.16 (CHI), 41.48 (t, CH2–CF2), 61.50 (C6′), 62.88 (C6), 68.03 (C5′), 68.47 (C4), 69.35 (C3′), 69.99 (CH2O), 70.09 (C2′); 72.22 (C2, C5), 72.76 (C4), 75.45 (C3), 95.50 (C1′), 100.30 (C1), 169.41, 169.57, 169.95, 170.26, 170.46, 170.53 (CO, OAc). ESI-TOF HRMS: calcd for C37H53O18F5 I[M + H]1+ 1007.2197 found 1007.2214.
1H-NMR δ (CDCl3): 1.2–1.9 (m, 12H, (CH2)6), 1.9–2.1 (m, 21H, CH3CO), 3.45 (m, 1H, OCH2), 3.7 (m, 1H, H5), 3.85 (m, 1H, OCH2), 3.9–4.3 (m, 5H, 3H, H4, H5′, H6), 4.5 (m, 2H, H6, H1, J = 7.5 Hz), 4.75–4.9 (m, 2 H, H2, H2′), 5.1 (t, 1H, H4′), 5.25 (t, 1H, H3′), 5.35 (t, 1H, H3), 5.45 (d, 1H, H1′), 5.5–5.65 (m, 1H, –CHCH–CF2), 6.35–6.45 (m, 1H, –CHCH–CF2). 19F NMR δ (ppm, CDCl3): −85.42 (CF3), −115.06 (CF2). 13C NMR δ (CDCl3): 20.54, 20.56, 20.64, 20.79, 20.88 (CH3, OAc), 25.67, 25.84, 28.85, 28.97, 29.22, 29.30, 31.89, 33.64 (CH2), 61.52, 62.00 (C6′), 62.88, (C6), 68.05 (C5′), 68.47 (C4), 69.35 (C3′), 70.00, 70.04 (CH2O), 71.35, 71.75 (C2′), 72.09 (C5), 72.23 (C2), 72.81, 72.85 (C4), 75.43 (C3), 95.51 (C1′), 100.28, 100.81 (C1), 114.41, 116.60 (CF2–CH), 138.74, 142.98 (CH), 170.50, 170.43, 170.29, 170.23, 169.93, 169.54, 169.38, 169.23 (CO, OAc). ESI-TOF HRMS: calcd for C37H52O18F5 [M + H]1+ 879.3074 found 879.3074.
:6) to afford compound 6 (1.25 g) with 83% yield. The product was recrystallized in hot methanol to give compound 6 as a pure white powder.
1H-NMR δ (CDCl3): 1.23–1.6 (m, 8H, CH2–(CH2)3–CH2–CF2–), 1.96–2.14 (m, 23H, acetyl groups, CH2–CF2), 3.45 (m, 1H, O–CH2), 3.65 (m, 1H, H5), 3.86 (m, 1H, O–CH2), 3.98–4.06 (m, 3H, H4′, H5′, H6a), 4.24 (m, 2H, H6b, H6′a), 4.45–4.49 (dd, 1H, H6′b), 4.50–4.52 (m, 1H, H1, 3J1,2 = 8 Hz), 4.82–4.88 (m, 2H, H2, H2′), 5.08 (t, 1H, H4), 5.25 (t, 1H, H3′), 5.36 (t, 1H, H3, H3), 5.41–5.42 (d, 1H, H1′, 3J1,2 = 4 Hz). 19F NMR δ (ppm, CDCl3): −118.24 (t, 2F, –CF2–CF3), −85.44 (s, 3F, CF2–CF3). 13C NMR δ (CDCl3): 19.80 (CH2–CH2–CF2), 20.55, 20.58, 20.65, 20.78, 20.89 (CH3, OAc), 25.41 (CH2), 29.05 (CH2–CH2O), 30.65 (t, CF2CH2), 61.50 (C6′), 62.80 (C6), 68.02 (C5′), 68.48 (C4), 69.33 (C3′), 69.47 (CH2O), 69.99 (C2′) 72.13 (C5), 72.18 (C2), 72.74 (C4), 75.40 (C3), 95.52 (C1′), 100.25 (C1), 170.52, 170.43, 170.24, 169.96, 169.56, 169.40 (CO, OAc). ESI-TOF HRMS: calcd for for C37H54O18F5 [M + H]1+ 881.3230, found 881.3245.
1H NMR δ (CD3OD): 1.3–1.5 (m, 10 H, O–(CH2)2–(CH2)5–(CH2)2–CF2–), 1.55–1.63 (m, 4H, CH2–CH2–CF2, CH2–CH2–O), 2.08–2.12 (2H, m, CH2–CF2), 3.21–3.91 (complex signal, 17H, H glycosyl and CH2O), 4.27–4,29 (d, 1H, H1, 3J1,2 = 8 Hz), 5.16–5.17 (d, 1H, H1′, 3J1′,2′ = 4 Hz). 19F NMR δ (CD3OD): −119.39 (2F, CF2–CF3), −87.00 (3F, CF3). 13C NMR δ (CD3OD): 21.43 (CH2CH2CF2), 27.07, 30.11, 30.29, 30.46, 30.48, 30.78 ((CH2)n), 31.49 (t, CH2CF2), 62.22 (C6), 62.79 (C6′), 70.94 (CH2O), 71.56 (C4′), 74.19 (C5′), 74.73 (C2), 74.79 (C2′), 75.11 (C3′), 76.62 (C5), 77.90 (C3), 81.36 (C4), 102.93 (C1′), 104.34 (C1). ESI-TOF HRMS: calcd for C23H40F5O11 [M + H]1+ 587.2491, found 587.2489.
Techniques
Surface tensiometry
After dissolving the surfactants in Milli-Q water, surface tensiometry (ST) was performed with the aid of a K100 tensiometer (Kruss, Hamburg, Germany) using the Wilhelmy plate technique. Surfactant solutions at 5–10 times the expected CMC were prepared 24 h prior to measurements, and 20 mL of the sample was transferred to a 50 mL vessel supplied with a stir bar. Surface tensions were determined by the automatic dilution of the stock solutions using a Metrohm 700 Dosino. In a typical experiment, 100 concentration steps were used with ∼20 min between consecutive concentration steps. The platinum plate was cleaned by flaming before each experiment. The glassware was cleaned with sulfochromic solution and rinsed with Milli-Q water. All measurements were performed at 20 °C. Sets of measurements to obtain equilibrium surface tension were taken until the change in surface tension was less than 0.01 mN m−1. The CMC and the surface tension at the CMC (γCMC) were determined from, respectively, the abscissa and the ordinate at the break point of the γ − logC curve. The maximum surface excess Γmax was calculated using the following Gibbs adsorption isotherm equation:25| Γmax = −(1/RT) (dγ/dlnC) |
lnC) is the slope below the CMC in the surface tension plots. The minimum area occupied per surfactant molecule at the air/water interface (Amin, in Å2) is related to Γmax by the relation Amin = 1016/NaΓmax, where Na is Avogadro's number.
The standard Gibbs free energy of micellization is given by the following equation  , where XCMC is the mole fraction of the surfactant in the liquid phase at the CMC, that is, XCMC = CMC/55.5 M for surfactants in water. The standard Gibbs free energy of adsorption
, where XCMC is the mole fraction of the surfactant in the liquid phase at the CMC, that is, XCMC = CMC/55.5 M for surfactants in water. The standard Gibbs free energy of adsorption 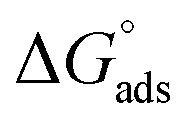 is then given as follows
is then given as follows  with the surface pressure ΠCMC denoting the difference between the surface tension of pure water (γo) and that of the solution at the CMC (γCMC).
with the surface pressure ΠCMC denoting the difference between the surface tension of pure water (γo) and that of the solution at the CMC (γCMC).
19F NMR for CMC determination
13 samples of each surfactant (F2H9βM, F4H5βM, and F6H2βM) at different concentrations were prepared from stock solutions (2.5 g L−1 for F2H9βM and F6H2βM and 4 g L−1 for F4H5βM). The solutions were diluted by taking 4 mL of previous solutions to which 1 mL of a solution of water containing 1 mg of CF3COONa was added. 19F NMR spectra were recorded on a Bruker Avance 400 spectrometer equipped with a pulse-field gradient module (Z-axis) using a 5 mm BBO probe. The instrument was operated at a frequency of 376.72 MHz at 25 °C. The observed chemical (“obs”) shifts of the CF3 of the surfactants were examined as a function of concentration below and above the CMC. All samples were dissolved in H2O, and chemical shifts were referenced to the center of the CF3COONa signal (−73.53 ppm).Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) demicellization experiments were carried out on a VP-ITC (Malvern Instruments, Worcestershire, UK) at 21 °C. A surfactant solution at a concentration of 10–15 times the CMC was loaded in the syringe and titrated into triple-distilled water. Experimental settings included an injection volume of 5 μL, a reference power of 42–84 μJ s−1, and a filter period of 2 s. Time spacings between injections were set long enough to allow the power signal to reach the baseline before the next injection. Baseline adjustment and peak integration were accomplished using NITPIC.26 The first injection was always excluded from further analysis. For F2H9βM, demicellization isotherms were fitted using a generic sigmoid function to determine the CMC and the molar enthalpy of micellization, as explained elsewhere.27 Demicellization isotherms of F4H5βM reproducibly revealed a local maximum within the transition region, which precluded such a detailed fit. Hence, the CMC was taken as the maximum in the first derivative of the heat of reaction with respect to surfactant concentration, and the molar enthalpy of micellization was determined from the difference between linear pre- and post-transition baselines at the CMC.Analytical ultracentrifugation (AUC) sedimentation velocity
Stock solutions at 15 g L−1 in water were prepared, and diluted samples of F2H9βM at 8, 6, 4, 2, 1, and 0.5 g L−1, of F4H5βM at 8, 6, 4, and 2 g L−1, and of F6H2βM at 15, 10, 7.5, 5, 2, and 1 g L−1 were investigated by sedimentation velocity (SV) experiments, conducted in an XLI analytical ultracentrifuge (Beckman, Palo Alto, CA) using an ANTi-50 rotor and detected at 280 nm and with interference optics, at 42000 revolutions per min (rpm) and 20 °C overnight, using double-channel center pieces (Nanolytics, Germany) of 3 or 12 mm optical path length (the reference channel being filled with water) equipped with sapphire windows. Continuous distribution of sedimentation coefficients, c(s), was derived using the SEDFIT software28 version 14.1 (freely available at: http://www.analyticalultracentrifugation.com), correcting for errors in the time acquisition of the acquisition software Proteomelab XLI V6.0.29 and analyzed as described in ref. 30, to obtain the CMC and refractive index increment, ∂n/∂c, the sedimentation coefficient at infinite dilution (s0), and the concentration dependency factor (ks′).31 The value of s0 is interpreted through the Svedberg equation, in terms of the molar mass, M, the hydrodynamic radius, RH, and the frictional ratio, f/fmin. We considered the partial specific volumes, ![[v with combining macron]](https://www.rsc.org/images/entities/i_char_0076_0304.gif) , from ref. 32 reported in Table S1 (ESI†). Values calculated from chemical composition33 differ somewhat but are in the same range, that is, 0.718, 0.629, and 0.564 mL g−1 for F2H9βM, F4H5βM, and F6H2βM, respectively.
, from ref. 32 reported in Table S1 (ESI†). Values calculated from chemical composition33 differ somewhat but are in the same range, that is, 0.718, 0.629, and 0.564 mL g−1 for F2H9βM, F4H5βM, and F6H2βM, respectively.
Dynamic light scattering
Dynamic light scattering (DLS) experiments were performed on a Zetasizer Nano-S model 1600 (Malvern Instruments, UK) equipped with a He–Ne laser (λ = 633 nm) at an angle of 173° (backscattering detection). Surfactant solutions were prepared 24 h prior to measurements using filtered Milli-Q water. Surfactant solutions were centrifuged at 15000 rpm for 2 h before being transferred into a 45 μL low-volume quartz batch cuvette (Hellma). Time-dependent correlation functions were measured for different concentrations of surfactants. The hydrodynamic radius (RH) of micelles was calculated from extrapolation to zero concentration of diffusion coefficients, D = D0(1 + kD(c − CMC)), using the Stokes–Einstein equation D0 = kBT/6πηRH, where kB is Boltzmann's constant, T is the absolute temperature, and η is the viscosity of the solvent. kD refers to an interaction parameter between micelles in solution.34 If kD is positive (negative), the interactions between micelles are repulsive (attractive). All measurements were carried out at 20 °C.
Small-angle X-ray scattering
Micellar assemblies in water were characterized by small-angle X-ray scattering (SAXS) on a bioSAXS beamline ID14-eh3 and BM2935 at the European Synchrotron Radiation Facility in Grenoble, France. To prevent radiation damage during the scattering experiments, data were collected in 10 successive 2s frames, and the solution was moved into the capillary during exposure. All measurements were carried out at 20 °C. Averaged scattered intensities were subtracted from water. Forward scattering values (i.e., q → 0; I(c,0)) and radii of gyration (RG) were evaluated using the Guinier approximation I(c,q) = I(0)·exp(−q2RG2/3) assuming that qRG < 1 at very small angles. CMCs of surfactants were determined from the plot of I(c,0) as a function of surfactant concentration. Molar masses and aggregation numbers of micelles were then calculated from the absolute forward intensity, normalized to a reference of pure water.36 The aggregation number Nagg was determined by dividing the micelle molar mass by that of the surfactant monomer:with Na denoting Avogadro's number, r0 the classical electron radius (r0 = 0.28179 × 10−12 cm e−1),
 the surfactant partial specific volume (cm3 g−1), Msurf the surfactant molar mass, ρsurf and ρ0 the scattering length densities of the surfactant and water (e cm−3), respectively, and dΣ/dΩ|water the absolute scattering intensity of water equal to 0.01632 cm−1 at 20 °C.
the surfactant partial specific volume (cm3 g−1), Msurf the surfactant molar mass, ρsurf and ρ0 the scattering length densities of the surfactant and water (e cm−3), respectively, and dΣ/dΩ|water the absolute scattering intensity of water equal to 0.01632 cm−1 at 20 °C.
Biochemistry
The purified purple membrane was solubilized for 36 h at 4 °C with 52 mM n-octyl-β-D-thioglucoside (OTG; CMC = 9 mM) at a membrane concentration of 1.3 g L−1 in 20 mM sodium phosphate buffer, pH 6.8. Samples were diluted to reach a final OTG concentration of 15 mM, supplemented with 3 mM of the surfactant to be tested, and incubated for 15 min prior to being loaded onto a 10–30% (w/w) sucrose gradient containing 20 mM sodium phosphate buffer pH 6.8 and 6 mM of the same surfactant. A control experiment was performed in a gradient containing 6 mM H12βM. Gradients were centrifuged for 5 h at 55000 rpm (200000g) using a TLS55 rotor of a TL100 ultracentrifuge (Beckman). Bands containing the colored protein were collected with a syringe, and protein samples were kept at 4 °C in the dark for UV-visible spectrophotometry.
Results and discussion
Synthesis of novel fluorinated maltosides
Two novel fluorosurfactants were synthesized with the aim of emulating the structure and the overall hydrophilic/lipophobic balance of the commonly used detergent H12βM. To this end, we modulated the length of hydrogenated and fluorinated segments, taking into account that the hydrophobic contribution of CF2 is about 1.5–1.7 times greater than that of CH2, depending on the nature of the polar head.37,38 In order to avoid the formation of large and polydisperse aggregates as previously observed with F6H3βM and H2F6H3βM,22 we combined shorter fluorinated segments, namely, either a fluorinated segment of four carbons with a hydrogenated segment made of five carbons (F4H5βM) or a fluorinated segment of two carbons with an aliphatic segment comprising nine hydrogenated carbons (F2H9βM). Two chemical pathways were used to prepare these two compounds.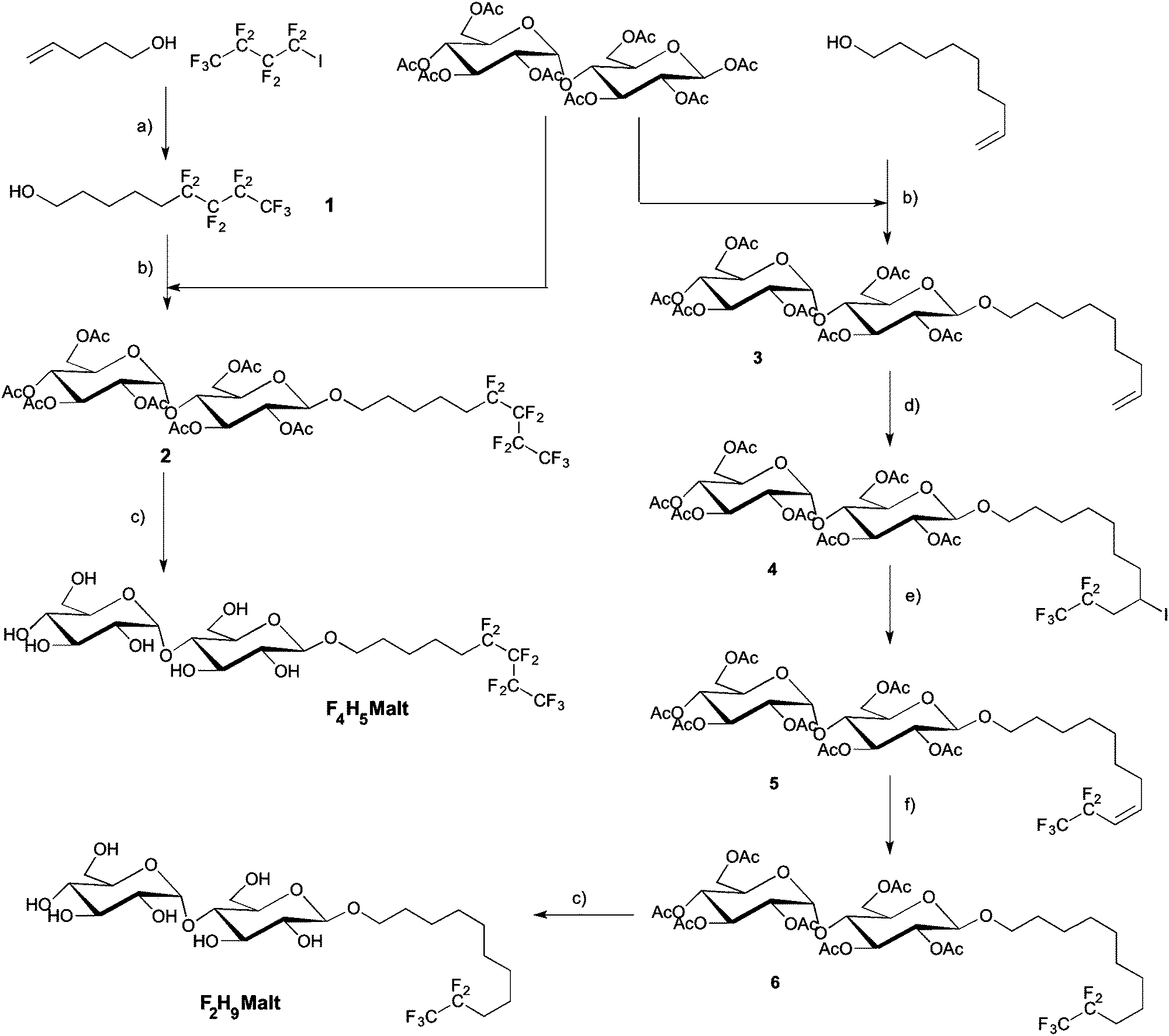 | ||
| Scheme 2 Synthetic route leading to the poorly fluorinated H12βM derivatives F2H9βM and F4H5βM: (a) activated zinc, CH2Cl2, Argon, for 4 h then H2O/AcOH/12 N HCl: 30/15/5, 70%; (b) BF3–Et2O, 0 °C, 24 h, then Ac2O for 2 h, 32–34%; (c) MeONa, MeOH, rt, 4 h, 70–94%; (d) CF3CF2I, THF, −80 °C Et3B, 24 h, 94%; (e) DBU, toluene, 70 °C, 3 h, 92%; (f) H2, Pd–C, MeOH, 83%. | ||
The iodinated addition product was dehalogenated in a one-pot reaction by the addition of acetic acid and 2 N HCl. The progress of the reaction was monitored by 1H NMR spectroscopy. The iodinated and the dehalogenated products have the same ratio front in TLC. In a first step, we observed the complete disappearance of the vinylic signal and the formation of a multiplet corresponding to the CHI signal at 2.75 ppm. This signal disappeared after acidification of the reaction. After chromatography, the hemifluorinated alcohol 1 was obtained pure with a 70% overall yield. An effective method to graft maltose onto the alcohol 1 is Lewis acid-catalyzed glycosylation using maltose peracetate. Maltosidation of alcohol was carried out by the treatment of β-D-maltose peracetate with equimolar amounts of alcohols and BF3–Et2O in dry dichloromethane.40–42 Such Lewis acid glycosylation afforded the β-D-glucoside 2 with 32% yield after chromatography and crystallization with a high degree of stereoselectivity. β-Anomeric stereochemistry was expected for compound 2 and was confirmed by its 1H NMR spectrum (J = 7.75 Hz). However, the α isomer always contaminates the samples after glycosylation (5 to 10%). Indeed it is impossible during glycosylation to have a 100% stereoselective reaction. Crystallization in ethanol is the most efficient methodology to completely remove the α anomer and obtain the β anomer with a chiral purity higher than 99%. We observed the formation of deacetylated compounds by transesterification. This side reaction uses large quantities of alcohol and contributes to greatly reduced yields of glycosylation. The addition of an acetic anhydride and pyridine mixture allowed reacetylation of partially deacetylated compounds and improved reaction yields after crystallization in ethanol. The remaining acetyl protecting groups in 2 were removed with MeONa in MeOH to provide F4H5βM in good yield.
The β-anomeric configuration of 3 was confirmed by 1H and 13C NMR spectra. The addition of perfluoroethyl iodide in THF to the olefin 3 catalyzed by Et3B was performed at low temperature in order to liquefy the perfluoroalkyl iodide at room temperature. The reaction was completed after 24 h and gave the adduct 4 in 94% yield. Dehalogenation had to be quantitative, as the deiodinated product and compound 4 had the same ratio front in TLC and were very difficult to separate. Reductive radical dehalogenation reactions with Bu3SnH/AIBN or Zn/HCl/CH3COOH were not efficient and produced mixtures with low amounts of compound 4 difficult to separate by chromatography. Elimination of HI with DBU in toluene at 70 °C for 3 h gave the olefin 5 in 92% yield.
Compound 4 was entirely transformed, as confirmed by the disappearance of the CHI signal in 1H NMR at 2.75 ppm. The reduction of olefin 5 was performed in methanol with hydrogen in the presence of Pd–C and gave product 6 in 83% yield after chromatography and crystallization in methanol. NMR spectra showed the high chiral purity of the sample. No α anomer was observed after the crystallization process. So we can confirm the high chiral purity of the sample. In a last step, Zemplen deprotection of 6 gave the fully deprotected F2H9βM in good yield after chromatography.
Physicochemical characterization of surfactants
The synthesis and the use of new surfactants for MP stabilization and crystallization should be accompanied by the characterization of micelle behavior in solution (in particular CMC, micelle size and shape, and homogeneity) and MP stability studies in the presence of these novel surfactants.The two novel fluorosurfactants, F2H9βM and F4H5βM, were fully soluble in water (up to 10% w/v) for further biophysical and biostructural studies in solution.
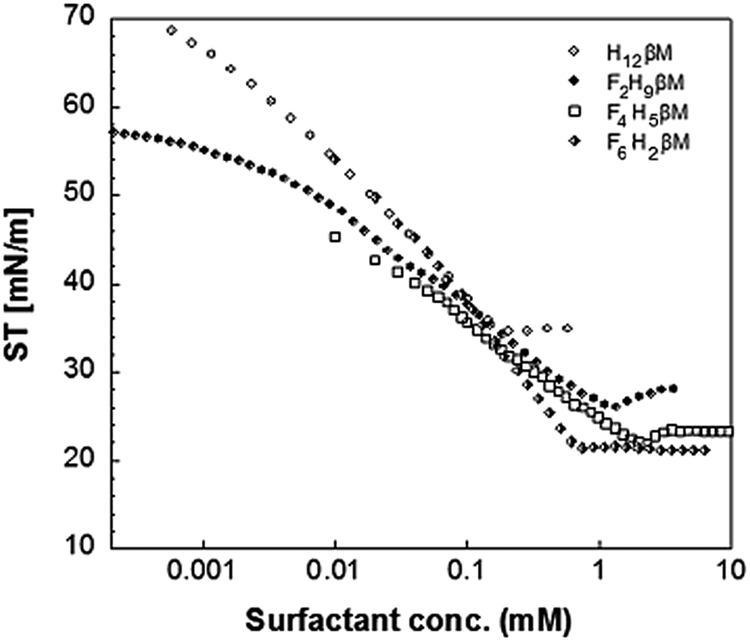 | ||
| Fig. 1 Surface tension of F2H9βM, F4H5βM, F6H2βM, and H12βM as a function of their concentration. | ||
The values of the CMC, surface tension at the CMC (γCMC), maximum surface excess concentration (Γmax), minimum occupied area per molecule (Amin), free energy of micellization 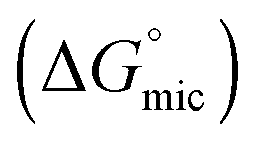 , and free energy of adsorption
, and free energy of adsorption 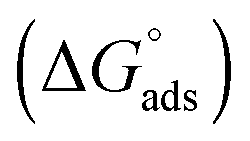 of the fluorinated surfactants extracted from this graph are listed in Table 1, along with the data of highly fluorinated maltosides and different alkyl maltosides. The CMCs corresponding to the break point of the surface tension versus surfactant concentration are presented in Table 2.
of the fluorinated surfactants extracted from this graph are listed in Table 1, along with the data of highly fluorinated maltosides and different alkyl maltosides. The CMCs corresponding to the break point of the surface tension versus surfactant concentration are presented in Table 2.
values from the Anatrace catalog; n.a. not available in the literature)
| Surfactant | Calculated hydrophobic contribution if CH2 ≈ 1.5CF2 | CMC (mM) | N agg/RH | γ CMC (mN m−1) | Γ max (μmol m−2) | πCMC (mN m−1) |
|
|
|
pC 20 | A min (Å2) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H8βM | 8 C | 19.5* | 55/2.15 | n.a. | n.a. | n.a. | −19.6 | n.a | n.a. | n.a. | n.a. |
| H9βM | 9 C | 6* | 66/2.55 | n.a. | n.a. | n.a. | −22.6 | n.a. | n.a. | n.a. | n.a. |
| H10βM | 10 C | 1.8* | 81/2.75 | n.a. | n.a. | n.a. | −25.6 | n.a. | n.a. | n.a. | n.a. |
| H11βM | 11 C | 0.59* | 105/3.15 | n.a. | n.a. | n.a. | −28.4 | n.a. | n.a. | n.a. | n.a. |
| H12βM | 12 C | 0.17 | 125/3.45 | 34.7 | 2.72 | 37.3 | −30.7 | −44.4 | −13.7 | 1.87 | 61 |
| F2H9βM | 12 C | 1.14 | 65/2.7 | 26 | 1.93 | 46 | −26 | −49.8 | −23.8 | 2.42 | 86 |
| F4H5βM | 11 C | 2.16 | 50/4.6 | 22 | 1.87 | 50 | −24.5 | −51.3 | −26.8 | 2.81 | 89 |
| F6H2βM | 11 C | 0.71 | 800/15.6 | 21.2 | 3.52 | 50.8 | −27.2 | −41.6 | −14.4 | 1.84 | 47 |
| F6H3βM | 12 C | 0.2 | n.a. | 17.5 | 4.37 | 54.5 | −30.3 | −42.8 | −12.5 | n.a. | 38 |

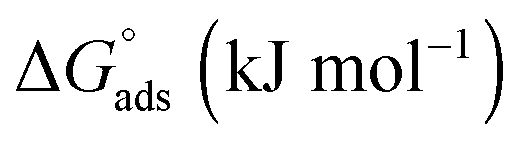

As expected, the value of the surface tension at the CMC (γCMC) decreases when the length of the fluorinated segment increases. In agreement with the literature,38,44 the fluorinated maltoside-derived surfactants FnHmβM have a higher tendency to form aggregates than their analogues Hm′βM (m′ = n + m) with the same number of carbon atoms. In particular, the eight carbons of the partly fluorinated chain of F6H2βM (or the nine carbons of F6H3βM) have the same hydrophobic contribution as the eleven carbons of the aliphatic chain of H11βM (or the twelve carbons of H12βM). This contribution for the highly fluorinated surfactants is in agreement with the 1CF2 ≈ 1.5CH2 rule. By contrast, for the fluorinated surfactants F2H9βM and F4H5βM, this rule is not reflected in the measured values. While the CMC of a highly fluorinated chain (F6H2βM) is close to the CMC of an H11 chain, the CMCs of the partly fluorinated chains are far from the values expected by this rule (i.e., for H12 and H11 chains, respectively). The CMCs of F4H5βM (2.16 mM) and F2H9βM (1.14 mM) correspond approximately to that of H10βM (1.8 mM). Hence, it appears that the shorter the length of the fluorinated segment is, the less will be the ‘hydrophobic’ contribution of the fluorinated carbons. Thus, if we consider that the hydrophobic contribution of CH2 is constant for H10βM, F4H5βM, and F2H9βM, we deduce that for F4H5βM, 1CF2 ≈ 1.25CH2 and for F2H9βM, 1CF2 ≈ 0.5CH2. Therefore, for a short fluorinated tip at the end of the aliphatic chain, the hydrophobic contribution of a CF2 becomes lower than that of a CH2.
This reduced, context-dependent hydrophobic contribution of a CF2 group residing in a short fluorinated tip becomes even more obvious upon comparison with nonfluorinated detergents in terms of the thermodynamics of micellization. In agreement with literature reports,38 we observe that 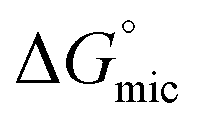 of F6H3βM (−30.3 kJ mol−1) is more exergonic than that of H9βM (−22.6 kJ mol−1). By contrast, however, F2H9βM (−26.0 kJ mol−1) has a reduced affinity for micellization compared with its hydrocarbon counterpart H11βM (−28.4 kJ mol−1). This is not the case for F4H5βM (−24.5 kJ mol−1), which has a greater tendency to micellize than its hydrocarbon counterpart H9βM (−22.6 kJ mol−1). Thus, the usual increase in hydrophobic contribution conferred by fluorine atoms becomes marginal for very low fluorinated derivatives, and a small perfluoroethyl tip even seems to disturb the formation of micelles, presumably because of packing constraints in the micelle core. A good measure of the adsorption efficiency of a surfactant is its concentration required to produce a 20 mN m−1 reduction in surface tension,45 as expressed by the negative logarithm of this concentration, pC20. The larger the pC20 value is, the more efficiently the surfactant will reduce the surface tension. As shown in Table 1, pC20 values for the fluorinated surfactants were observed to increase for low fluorinated surfactants compared with fluorinated F6H2βM. This result indicates that low fluorinated surfactants have a higher tendency to adsorb at the interface.
of F6H3βM (−30.3 kJ mol−1) is more exergonic than that of H9βM (−22.6 kJ mol−1). By contrast, however, F2H9βM (−26.0 kJ mol−1) has a reduced affinity for micellization compared with its hydrocarbon counterpart H11βM (−28.4 kJ mol−1). This is not the case for F4H5βM (−24.5 kJ mol−1), which has a greater tendency to micellize than its hydrocarbon counterpart H9βM (−22.6 kJ mol−1). Thus, the usual increase in hydrophobic contribution conferred by fluorine atoms becomes marginal for very low fluorinated derivatives, and a small perfluoroethyl tip even seems to disturb the formation of micelles, presumably because of packing constraints in the micelle core. A good measure of the adsorption efficiency of a surfactant is its concentration required to produce a 20 mN m−1 reduction in surface tension,45 as expressed by the negative logarithm of this concentration, pC20. The larger the pC20 value is, the more efficiently the surfactant will reduce the surface tension. As shown in Table 1, pC20 values for the fluorinated surfactants were observed to increase for low fluorinated surfactants compared with fluorinated F6H2βM. This result indicates that low fluorinated surfactants have a higher tendency to adsorb at the interface.
The Gibbs free energy change for adsorption 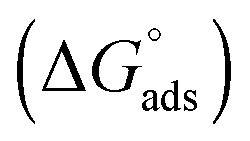 was calculated using the following equation:
was calculated using the following equation:

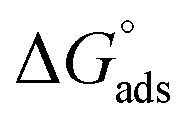 values are more negative than their corresponding
values are more negative than their corresponding 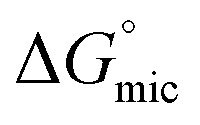 values, indicating that the transfer of the surfactant from the air/water interface to the micelle is unfavorable. The difference between the
values, indicating that the transfer of the surfactant from the air/water interface to the micelle is unfavorable. The difference between the 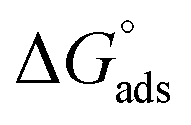 and
and 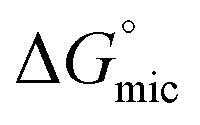 values is called the effective Gibbs free energy change
values is called the effective Gibbs free energy change 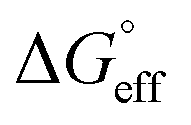 .46 It was observed (Table 1) that
.46 It was observed (Table 1) that 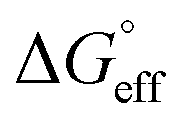 is significantly more negative when the degree of fluorination in the hemifluorinated chain is low, indicating that micellization is less favorable as compared with surface adsorption.
is significantly more negative when the degree of fluorination in the hemifluorinated chain is low, indicating that micellization is less favorable as compared with surface adsorption.
The surface excess concentration at surface saturation (Γmax) is another useful measure of the effectiveness of surfactant adsorption at the air/water interface, as it is the maximum value that adsorption can attain.47 For instance, straight chains favor close, effective packing, whereas branched chains experience steric hindrance at the interface. The larger the Γmax is, the more tightly is the surfactant packed at the interface. We found that the sparingly fluorinated surfactants have a lower surface excess than the highly fluorinated surfactants F6H2βM and F6H3βM and even than the non-fluorinated H12βM. However, fluorinated surfactants have a very strong ability to form tightly packed and organized films at the air/water interface38 and, thus, are excellent emulsifying and foaming agents. This general rule is confirmed by comparing the excess surface of F6H2βM and F6H3βM with that of H12βM. Therefore, the air/water interface behavior of the novel fluorinated surfactants presents an anomaly, as failure to tightly pack at the air/water interface is usually related to the presence of bulky, branched, or multiple aliphatic chains.
The area occupied per surfactant molecule at the air/water interface can be obtained from the value of the excess surface since Amin = 1016/NaΓmax. A greater efficacy of adsorption at the interface means that the area occupied by the surfactant molecule is smaller.
In the case of F4H5βM and F2H9βM, we observed a strong, that is, two-fold increase in surface area compared with the highly fluorinated surfactants F6H2βM and F6H3βM. Sadtler et al.48 have found similar behavior in a series of partially fluorinated surfactants with a dimorpholinophosphate head group. They observed an unexplained break when the number of fluorinated carbon atoms is lower than eight. We believe that the increase in the area occupied by the low fluorinated chain is related to a kink in the hydrophobic tail, that is bending between the fluorinated tip and the hydrogenated segment of the aliphatic chain. This bending can also affect their adsorption efficiency at the air/water interface and the mode of micellar aggregation.
| δobs = pδmon + (1 − p)δmic | (1) |
To obtain the CMC, the concentration of monomers above the CMC is assumed to equal the CMC value irrespective of the total surfactant concentration (c).52
| p = CMC/c | (2) |
| δobs = (CMC/c)δmon + (1 − CMC/c)δmic |
| δobs = δmic + (δmon − δmic)CMC/c | (3) |
| Δδ = δobs − δmon | (4) |
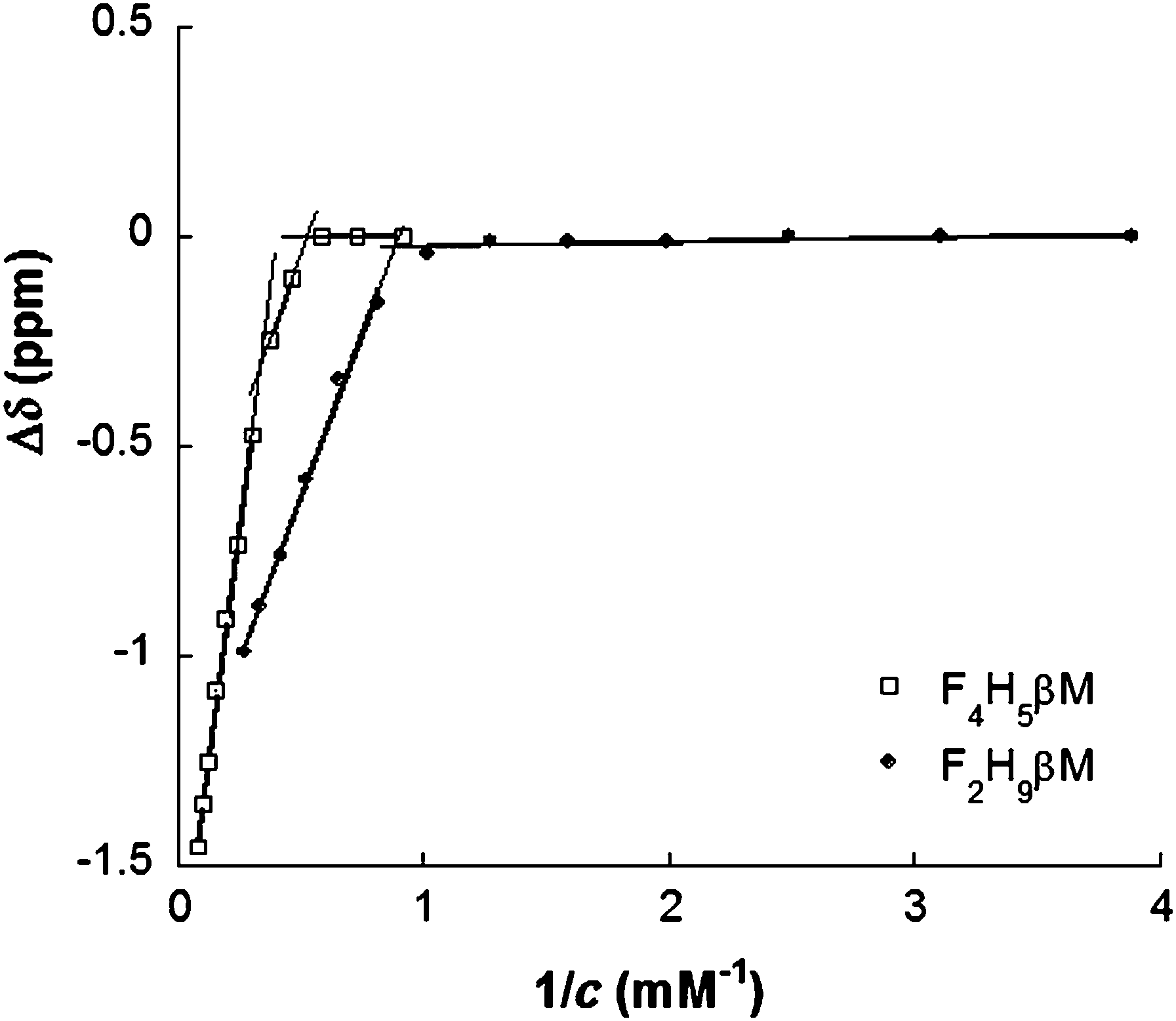 | ||
| Fig. 2 19F NMR chemical shift changes (Δδ) of the CF3 group of F2H9βM and F4H5βMversus the reciprocal value of their concentration in water. | ||
By contrast, 19F chemical shifts of CF3 in the fluorocarbon chain of F4H5Malt as a function of surfactant concentration reveal two break points at 1.9 mM and 3.0 mM, indicating that a premicellization process takes place before the formation of “mature” micelles. This particular behavior has already been described for fluorinated surfactants dissolved in ionic liquids.53,54 Consistent with NMR titrations, demicellization isotherms derived from high-sensitivity ITC revealed a well-defined micellization behavior for F2H9βM (Fig. 3A and C).
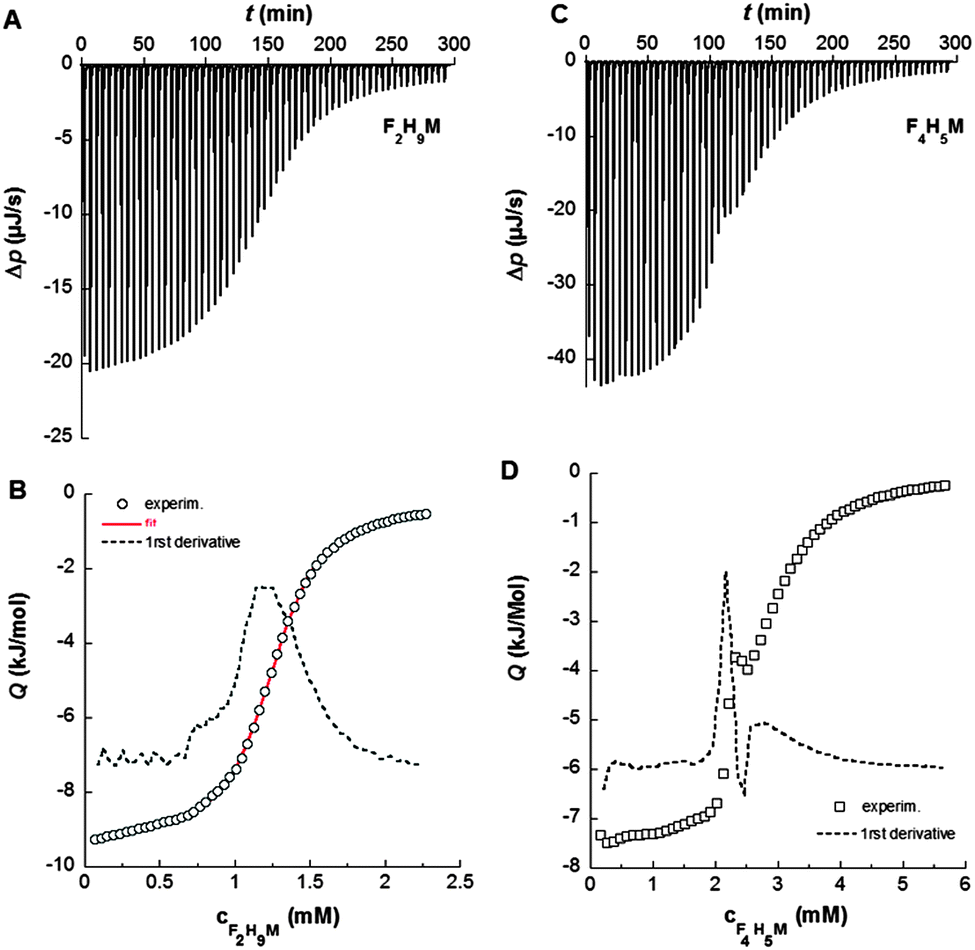 | ||
| Fig. 3 ITC demicellization experiments at 21 °C. (A) Thermogram displaying differential heating power, Δp, against time, t, for a titration of 12 mM F2H9βM into water. (B) Corresponding isotherm (circles) showing molar reaction heats, Q, against F2H9βM concentration with the first derivative (dashed line) and a sigmoidal fit (red solid line). (C) Thermogram for a titration of 30 mM F4H5βM into water. (D) Corresponding isotherm (squares) with the first derivative (dashed line). | ||
These isotherms displayed a single sigmoid transition from the submicellar to the micellar concentration range at 1.19 mM and, hence, were straightforward to analyze, yielding essentially the same CMC value as obtained from NMR and ST.
By contrast, for F4H5βM, there was a pronounced, reproducible dip within the transition region of the isotherm (Fig. 3B and D). A first maximum appears at a surfactant concentration of 2.15 mM and a second one at 2.77 mM, suggesting that there is some structural rearrangement with increasing surfactant concentration. Again, these concentration values agreed with those found using ST and NMR (Table 2).
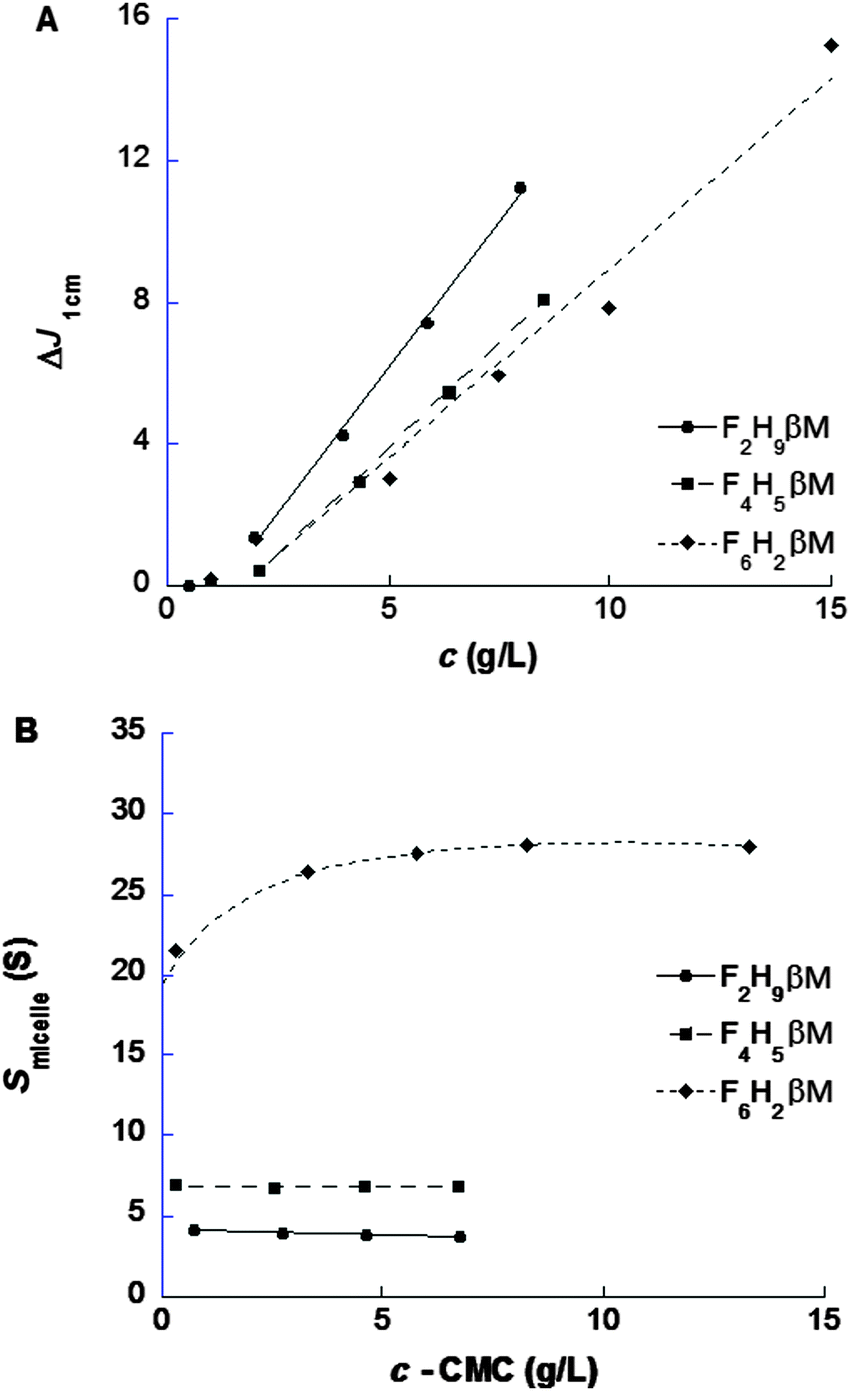 | ||
| Fig. 4 Sedimentation velocity experiments. Plots of the micelle signal, in fringe shifts, versus surfactant concentration (A), and of the mean s for the micelle versus micelle concentration (B). The lines in (A) and (B) are linear fits for concentrations ≥2 g L−1, except for F6H2βM in (B), where it gives a guide to the eye. | ||
Light and X-ray scattering are suitable techniques to characterize molecular and structural parameters of particles in solution such as the size and the molar mass and to derive low-resolution models. These techniques also allow the characterization of weak interactions between particles in solution (i.e., repulsion or attraction) to control the phase diagrams for crystallization.55–58 DLS experiments were performed on both fluorinated surfactants prior to SAXS in order to evaluate their dispersity (i.e., the presence of different kinds of aggregates) and micelle size. Concentration series for the three fluorosurfactants above their respective CMC (from ST) were measured to determine size distributions weighted by intensity or volume (Fig. S2, ESI†) and micelle diffusion coefficients (Dt) as functions of surfactant concentration.32 Although large aggregates (>100 nm) appeared in intensity-weighted size distributions for F2H9βM (Fig. S2B, ESI†) and F4H5βM (Fig. S2E, ESI†), their contribution was negligible in volume-weighted distributions (<1%). The hydrodynamic radius (RH) of fluorosurfactant micelles obtained via the Stoke–Einstein equation was determined by extrapolation to zero concentration of Dt = f(c − CMC) (Table 3) of the major distribution peak to account for intermicellar interaction effects. RH increases as the length of the fluorinated segment increases, although the total number of carbons (i.e., the chain length) decreases, in contrast to what is observed with alkyl chains.59,60 The micelle size of hemifluorinated surfactants is thus influenced by the length of the fluorinated segment. With a short fluorinated segment, micelles of F2H9βM are smaller than H12βM micelles and behave like micelles of a hydrogenated surfactant with an alkyl chain having ∼10 carbons, as it was observed above for CMC and micellization thermodynamics. In contrast to a long fluorinated chain and a linear maltoside headgroup, F6H2βM forms elongated micelles like F6H3βM,22 as expected from the packing parameter concept23 and recently described.20,32 For F4H5βM, which is composed of half-hydrogenated and half-fluorinated segments, RH is larger than for H12βM micelles, although the hydrophobic chain is shorter. We expected peculiar behavior because of the poor miscibility of fluorinated and hydrogenated segments, which may confer packing defects to micellar assemblies.
Complementary structural information on fluorosurfactant micelle assemblies in solution was obtained from SAXS experiments. Fig. 5A depicts SAXS curves for each fluorosurfactant at 10 g L−1 (c > CMC). SAXS curves at different concentrations between 2 and 40 g L−1, pair distribution functions, and forward scattering intensity for CMC determination are shown in Fig. S3 and S4 (ESI†). For F2H9βM and F6H2βM, the variation of forward intensity was linear with surfactant concentration; for F4H5βM, by contrast, we observed a break in the slope, in line with the chemical shifts observed by NMR. CMC values obtained from the extrapolation of forward intensity as a function of total concentration for F2H9βM and F6H2βM were found to be consistent with other techniques (ST, ITC, and 19F NMR). For F4H5βM, two CMC values were apparent, as observed by 19F NMR and ITC, which we attributed to premicellar rearrangement due to the particular structure of the hydrophobic chain. F2H9βM and F6H2βM present a stable shape from SAXS patterns at concentrations 6–8 times the CMC, as seen in the large q-range (q > 0.5 nm−1) and repulsive interactions at high concentrations (>20 g L−1) (Fig. S3A and C, ESI†). While F2H9βM forms small globular micelles with Nagg ≈ 65 (i.e., M ≈ (38000 ± 1000) g mol−1), as derived from the forward scattering intensity, F6H2βM forms elongated micelles with Nagg > 800 (i.e., M ≈ (550000 ± 5000) g mol−1), as obvious in the log–log representation. By contrast, F4H5βM micelles changed their size as a function of concentration (Fig. S3E, ESI†) and experienced attractive interactions at high concentrations (>20 g L−1), as the forward intensity increased with concentration without a change in the form factor (q > 1 nm−1). The molar mass and the aggregation number of F4H5βM micelles were determined from forward intensities to amount to ∼32000 g mol−1 and 50 molecules per micelle, respectively; this is less than the corresponding values of F2H9βM but as expected for this less hydrophobic chain. A thorough determination of micelle dimensions for the three fluorosurfactants was described in Dahani et al.32 The maximum distances in micelle assemblies (Dmax) obtained from the pair distribution function P(r) for the three fluorosurfactants (Fig. 5B) are in agreement with the hydrodynamic diameters obtained from DLS.
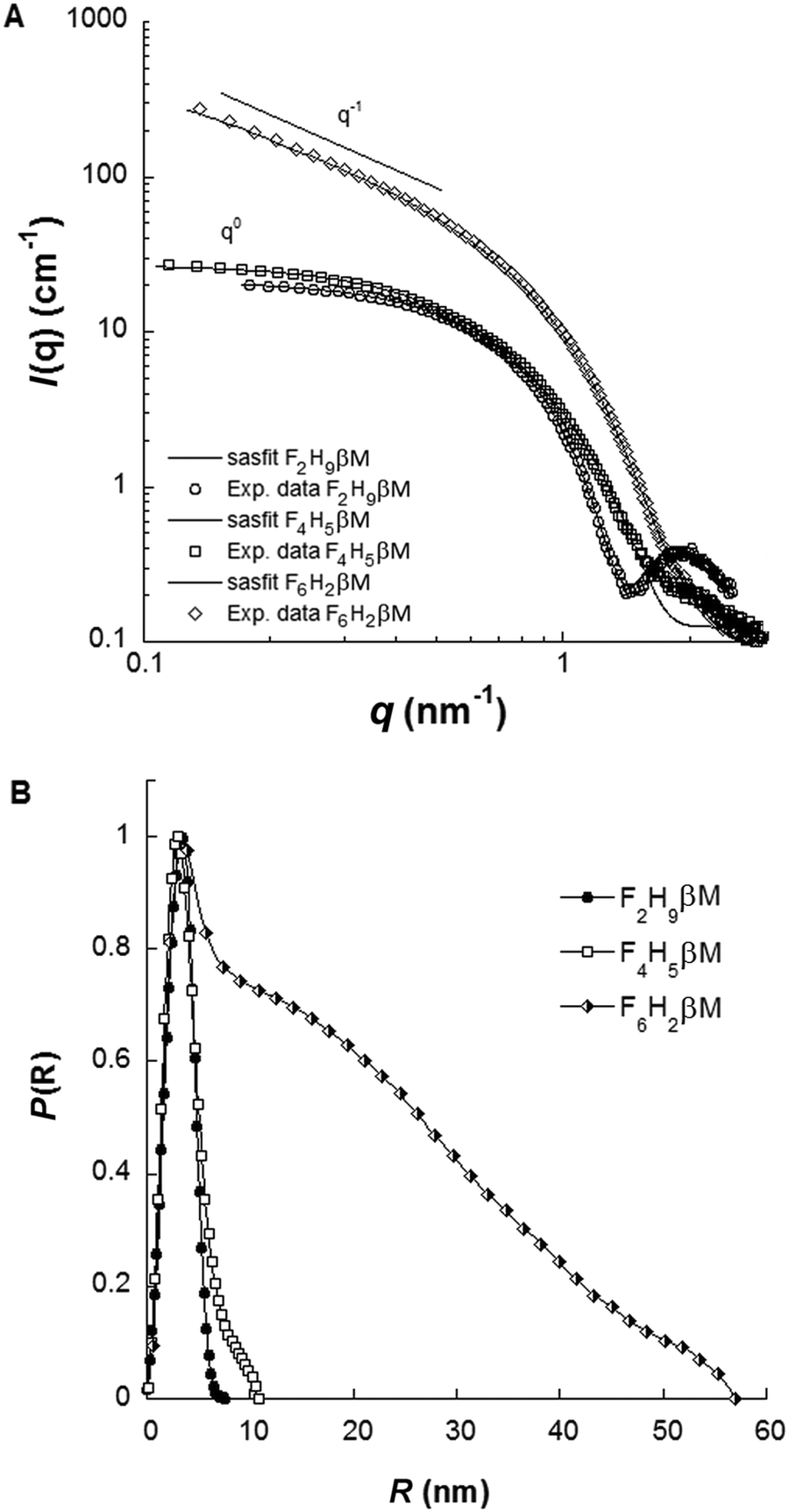 | ||
| Fig. 5 (A) SAXS patterns in log–log representation and (B) pair distribution functions of F2H9βM, F4H5βM, and F6H2βM at 10 g L−1 in water. | ||
Finally, the SAXS form factors at 10 g L−1 were analyzed using least-squares models using the SASfit software package.61 Different models were used, either an ellipsoidal core–shell model for F2H9βM and F4H5βM or a cylindrical core–shell model with an elliptical cross-section for F6H2βM. ρcore, ρshell, and ρsolv representing the scattering length densities of the core, the shell, and the solvent, respectively, are listed in Table S1 (ESI†). F2H9βM is well fitted by an oblate ellipsoid with short and long axes estimated to be 1.3 and 2.5 nm and a shell thickness of 0.6 nm. Our best model for F4H5βM was found to be a triaxial prolate ellipsoid with parameters a, b, c, and t equal to 4.4, 1.7, 8.8 and 0.8 nm, respectively. For F6H2βM, the parameters for the cylindrical model are a core cross-section radius of 1.2 nm, a shell thickness of 0.5 nm, and a total length of 56 nm. Aggregation numbers could be evaluated from these models by dividing the core volume by the hydrophobic chain volume. For F2H9βM, F4H5βM, and F6H2βM, we thus estimated aggregation numbers of ∼90, 86, and 511, respectively, which are in good agreement with values from forward intensities, given that molar masses are typically subjected to 10–15% error from sample preparation and concentration estimation.
Biochemical evaluation
To test the biochemical usefulness of F2H9βM and F4H5βM and compare them to commercial F6H2βM and H12βM, their ability to keep MPs soluble and in their native form was tested by using bacteriorhodopsin (bR) as a model protein. bR is a light-driven proton pump purified from the archaea Halobacterium.62 It is composed of seven transmembrane α-helices and binds a covalent cofactor, a retinal molecule, whose visible absorption spectrum is very sensitive to its local environment.63 This makes it a convenient reporter of the native state of the protein: the trimeric protein in its native membranes reveals a visible absorption spectrum with a maximum at λmax = 570 nm. When solubilized in detergent, the monomeric protein displays λmax = 554 nm; in both cases, the protein appears purple. When the protein denatures, the retinal is released, and λmax shifts to 400–380 nm, that is, the protein solution turns yellow. We have also reported that when the solubilized monomeric protein is transferred into a fluorinated surfactant, λmax shifts to 615 nm, giving a blue color to the protein–surfactant complex.30 bR dimers and trimers, however, still appear purple in the same surfactants (i.e., λmax ≈ 570 nm). Fig. 6 shows the results of sucrose density gradient experiments. This technique allows both surfactant exchange and evaluation of the colloidal homogeneity of the protein–surfactant complex. As expected from the small globular micelles formed by both surfactants, the sharp colored bands indicate homogeneous protein–surfactant complexes. Interestingly, we note that bR in F2H9βM migrated as a blue band, nearly at the same position as that in H12βM. | ||
| Fig. 6 Migration of bR in 10–30% sucrose gradients in the presence of 6 mM of each surfactant (from left to right: H12βM, F2H9βM, F4H5βM, and F6H2βM). Gradients were centrifuged for 5 h at 200000g. | ||
This is in agreement with the fact that F2H9βM, being less fluorinated, has a density closer to that of H12βM. This blue band implied that bR was present as a monomer in this fluorinated surfactant, even though it migrated deeper into the gradient than monomeric bR in conventional detergents (see, e.g., ref. 30). When transferred into F4H5βM, bR still gave rise to a blue band but migrated a bit deeper into the gradient, as expected for a protein–surfactant complex of higher density (note that the F2H9βM and F4H5βM gradients are not filled to the same extent). In stark contrast to our novel fluorosurfactants, F6H2βM led to the accumulation of bR from the pellet of the gradient, suggesting large protein–surfactant complexes. In another experiment, where the gradients were centrifuged more gently, the band appeared very broad and purple. This is in agreement with the fact that this surfactant forms large and heterogeneous micelles. Even though F6H2βM is a fluorinated surfactant, which normally yields blue bR,13,21 the pellet appeared purple. When resuspended, the spectrum of the protein–surfactant complex displayed λmax ≈ 568 nm, which suggests that the protein was not monomeric (Fig. 7C); it also diffused slightly, confirming the presence of large particles in the sample. As already observed for bR in F6-Monoglu, which also forms large protein–surfactant particles, the protein was very stable over a year and a half, albeit with an increase in diffusion suggesting further oligomerization or even aggregation of the protein. When transferred into F2H9βM, bR displayed λmax = 610 nm (Fig. 7A). This species was stable over nearly a month but was slowly converted into a species with λmax = 400 nm, suggesting the denaturation of the protein. This was accompanied by the aggregation of the protein, as witnessed by the diffusion of the spectrum and by the conversion of the remaining 610 nm species into a species absorbing at ∼590 nm, which probably resulted in the formation of higher-molecular-weight species such as dimers, trimers, or higher. This species was, in turn, slowly converted into a 400 nm species, indicating that the protein continued to denature. When collected from the gradient, bR in F4H5βM displayed λmax = 615 nm (Fig. 7B). After ∼5 months (163 days) of incubation at 4 °C in the dark, the spectrum remained largely unchanged; hence the protein appeared very stable in this environment, showing no signs of denaturation or aggregation. Slight diffusion of the spectrum was observed after one year of incubation, but the same was observed for the control in the H12βM sample (Fig. 7D). After a year, the 615 nm peak looses intensity, without the appearance of a peak at 400 nm and thus suggests either some bleaching of the 615 nm F4H5βM-bR species, or its denaturation or bleaching of the 400 nm species.
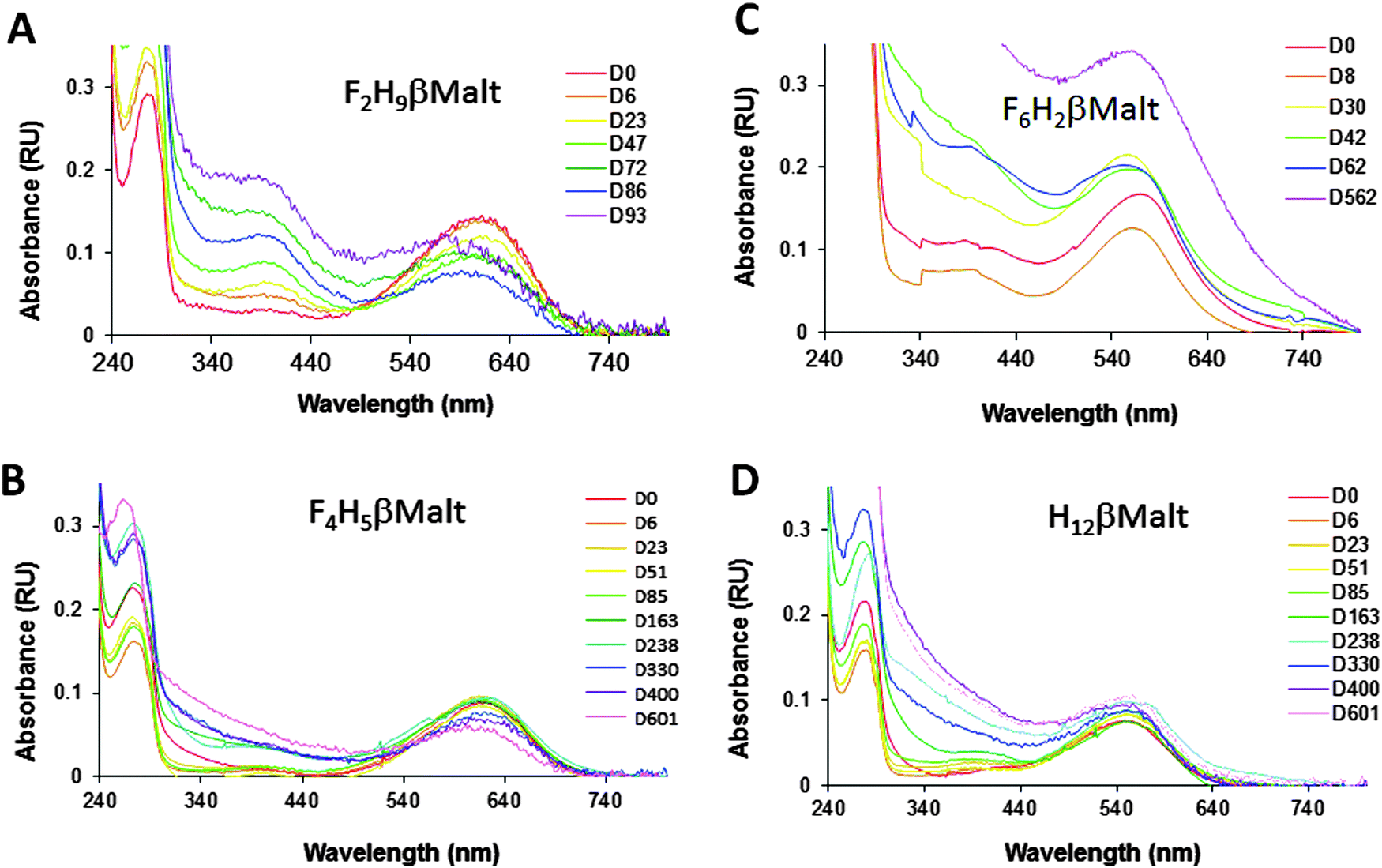 | ||
| Fig. 7 Spectral time course of bR collected from the gradients in F2H9βM (A), F4H5βM (B), F6H2βM (C), and H12βM (D). Samples were incubated at 4 °C in the dark and UV-visible spectra were recorded at the indicated time (given in days, D). | ||
In summary, we conclude that bR is stable as a homogeneous monomer over approximately a month in F2H9βM but for more than a year in F4H5βM. By contrast, when transferred into F6H2βM, bR forms large protein–surfactant complexes, as might be expected from the large micelles formed by that surfactant,20 and as already observed for F6H3βM.22
Conclusions
We have synthesized and characterized two novel fluorinated maltose-based surfactants having the same theoretical hydrophobicity as the common detergent dodecyl maltoside but bearing a sparingly fluorinated chain. Physicochemical analysis revealed the atypical properties of these fluorinated compounds as compared with their more extensively fluorinated counterparts. Most notably, the hydrophobicity of a fluorinated carbon in a short segment appears to be much lower than predicted by the 1CF2 ≈ 1.5CH2 rule, being even negative in the case of a very short fluorinated segment (i.e., perfluoroethyl tip). Biochemical analysis underlined the great potential of tailoring sparingly fluorinated surfactants for membrane-protein applications, as indicated by their differential ability to stabilize the model protein bacteriorhodopsin over extended periods of time. Bacteriorhodopsin is stable in the monomer state over more than a year in F4H5βM. Future work will focus on expanding such systematic correlations between physicochemical surfactant properties and their suitability for biochemical applications to enable a more rational design of fluorinated surfactants for membrane-protein research.Acknowledgements
This work used the platforms of the Grenoble Instruct centre (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). This study was supported by a PhD grant from the region PACA (Provence Alpes Côte d'Azur-France) via European Regional Development funds (FEDER). We are grateful to the European Synchrotron Radiation Facility for the provision of synchrotron radiation facilities Petra Pernot and Adam Round for assistance in using beamlines ID14-eh3 and BM29.References
- M. A. Yildirim, K. I. Goh, M. E. Cusick, A. L. Barabasi and M. Vidal, Nat. Biotechnol., 2007, 25, 1119 CrossRef CAS PubMed.
- L. Bamber, M. Harding, P. J. G. Butler and E. R. S. Kunji, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16224 CrossRef CAS PubMed.
- E. Pebay-Peyroula, R. Garavito, J. Rosenbusch, M. Zulauf and P. Timmins, Structure, 1995, 3, 1051 CrossRef CAS PubMed.
- S. Penel, E. Pebay-Peyroula, J. Rosenbusch, G. Rummel, T. Schirmer and P. A. Timmins, Biochimie, 1998, 80, 543 CrossRef CAS PubMed.
- E. R. Kunji, M. Harding, P. J. Butler and P. Akamine, Methods, 2008, 46, 62 CrossRef CAS PubMed.
- K. H. Park, C. Berrier, F. Lebaupain, B. Pucci, J. L. Popot, A. Ghazi and F. Zito, Biochem. J., 2007, 403, 183 CrossRef CAS PubMed.
- K.-H. Park, E. Billon-Denis, T. Dahmane, F. Lebaupain, B. Pucci, C. Breyton and F. Zito, New Biotechnol., 2007, 28, 255 CrossRef PubMed.
- C. Duval-Terrié, P. Cosette, G. Molle, G. Muller and E. Dé, Protein Sci., 2003, 12, 681 CrossRef PubMed.
- S. M. Yu, D. T. McQuade, M. A. Quinn, C. P. R. Hackenberger, M. P. Krebs, A. S. Polans and S. H. Gellman, Protein Sci., 2000, 9, 2518 CrossRef CAS PubMed.
- J. M. Dörr, M. C. Koorengevel, M. Schäfer, A. V. Prokofyev, S. Scheidelaar, E. A. W. van der Cruijsen, T. R. Dafforn, M. Baldus and J. A. Killian, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18607 CrossRef PubMed.
- J. Hovers, M. Potschies, A. Polidori, B. Pucci, S. Raynal, F. Bonneté, M. Serrano-Vega, C. Tate, D. Picot, Y. Pierre, J.-L. Popot, R. Nehmé, M. Bidet, I. Mus-Veteau, H. Bußkamp, K.-H. Jung, A. Marx, P. A. Timmins and W. Welte, Mol. Membr. Biol., 2011, 28, 171 CrossRef PubMed.
- L.-A. Barret, C. Barrot-Ivolot, S. Raynal, C. Jungas, A. Polidori and F. Bonneté, J. Phys. Chem., 2013, B117, 8770 CrossRef PubMed.
- M. Abla, S. Unger, S. Keller, F. Bonnete, C. Ebel, B. Pucci, C. Breyton and G. Durand, J. Colloid Interface Sci., 2015, 445, 127 CrossRef CAS PubMed.
- P. Bazzacco, K. S. Sharma, G. Durand, F. Giusti, C. Ebel, J.-L. Popot and B. Pucci, Biomacromolecules, 2009, 10, 3317 CrossRef CAS PubMed.
- C. Breyton, E. Chabaud, Y. Chaudier, B. Pucci and J.-L. Popot, FEBS Lett., 2004, 564, 312 CrossRef CAS PubMed.
- O. Joubert, R. Nehmé, D. Fleury, M. De Rivoyre, M. Bidet, A. Polidori, M. Ruat, B. Pucci, P. Mollat and I. Mus-Veteau, Biochim. Biophys. Acta, 2009, 1788, 1813 CrossRef CAS PubMed.
- K. H. Park, C. Berrier, F. Lebaupain, B. Pucci, J. L. Popot, A. Ghazi and F. Zito, Biochem. J., 2007, 403, 183 CrossRef CAS PubMed.
- J.-C. Talbot, A. Dautant, A. Polidori, B. Pucci, T. Cohen-Bouhacina, A. Maali, B. n. d. Salin, D. Brèthes, J. Velours and M.-F. Giraud, J. Bioenerg. Biomembr., 2009, 41, 349 CrossRef CAS PubMed.
- P. Barthélémy, V. Tomao, J. Selb, Y. Chaudier and B. Pucci, Langmuir, 2002, 18, 2557 CrossRef.
- E. Frotscher, B. Danielczak, C. Vargas, A. Meister, G. Durand and S. Keller, Angew. Chem., Int. Ed., 2015, 54, 5069 CrossRef CAS PubMed.
- C. Breyton, B. Pucci and J.-L. Popot, Methods Mol. Biol., 2010, 601, 219 CAS.
- A. Polidori, M. Presset, F. Lebaupain, B. Ameduri, J. L. Popot, C. Breyton and B. Pucci, Bioorg. Med. Chem. Lett., 2006, 16, 5827 CrossRef CAS PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, Biochim. Biophys. Acta, Biomembr., 1977, 470, 185 CrossRef CAS.
- V. Vill, T. Böcker, J. Thiem and F. Fischer, Liq. Cryst., 1989, 6, 349 CrossRef CAS.
- M. J. Rosen, A. W. Cohen, M. Dahanayake and X. Y. Hua, J. Phys. Chem., 1982, 86, 541 CrossRef CAS.
- S. Keller, C. Vargas, H. Zhao, G. Piszczek, C. A. Brautigam and P. Schuck, Anal. Chem., 2012, 84, 5066 CrossRef CAS PubMed.
- J. Broecker and S. Keller, Langmuir, 2013, 29, 8502 CrossRef CAS PubMed.
- P. Schuck, Biophys. J., 2000, 78, 1606 CrossRef CAS PubMed.
- H. Zhao, R. Ghirlando, G. Piszczek, U. Curth, C. A. Brautigam and P. Schuck, Anal. Biochem., 2013, 437, 104 CrossRef CAS PubMed.
- C. Breyton, F. Gabel, M. Abla, Y. Pierre, F. Lebaupain, G. Durand, J. L. Popot, C. Ebel and B. Pucci, Biophys. J., 2009, 97, 1077 CrossRef CAS PubMed.
- A. Solovyova, P. Schuck, L. Costenaro and C. Ebel, Biophys. J., 2001, 81, 1868 CrossRef CAS PubMed.
- M. Dahani, L.-A. Barret, P. Loll, C. Jungas, A. Polidori and F. Bonneté, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 838 CrossRef CAS PubMed.
- H. Durchschlag and P. Zipper, Jorn. Com. Esp. Deterg., Comun., 1995, 29, 275 Search PubMed.
- S. Li, D. Xing and J. Li, J. Biol. Phys., 2004, 30, 313 CrossRef CAS PubMed.
- P. Pernot, A. Round, R. Barrett, A. De Maria Antolinos, A. Gobbo, E. Gordon, J. Huet, J. Kieffer, M. Lentini, M. Mattenet, C. Morawe, C. Mueller-Dieckmann, S. Ohlsson, W. Schmid, J. Surr, P. Theveneau, L. Zerrad and S. McSweeney, J. Synchrotron Radiat., 2013, 20, 660 CrossRef CAS PubMed.
- D. Orthaber, A. Bergmann and O. Glatter, J. Appl. Crystallogr., 2000, 33, 218 CrossRef CAS.
- K. Shinoda, M. Hato and T. Hayashi, J. Phys. Chem., 1972, 76, 909 CrossRef CAS.
- J. C. Ravey, A. Gherbi and M. J. Stébé, in Trends in Colloid and Interface Science, ed. V. Degiorgio, Steinkopff, 1988, ch. 40, vol. 76, p. 234 Search PubMed.
- N. Requirand, H. Blancou and A. Commeyras, Bull. Soc. Chim. Fr., 1993, 130, 798 CAS.
- S. Hanessian and J. Banoub, Carbohydr. Res., 1977, 59, 261 CrossRef CAS.
- J. Dahmén, T. Frejd, G. Grönberg, T. Lave, G. Magnusson and G. Noori, Carbohydr. Res., 1983, 116, 303 CrossRef.
- G. Milkereit, S. Gerber, K. Brandenburg, M. Morr and V. Vill, Chem. Phys. Lipids, 2005, 135, 1 CrossRef CAS PubMed.
- T. Ogawa, K. Beppu and S. Nakabayashi, Carbohydr. Res., 1981, 93, C6 CrossRef CAS.
- P. Mukerjee, J. Am. Oil Chem. Soc., 1982, 59, 573 CrossRef CAS.
- M. J. Rosen, Surfactants and Interfacial Phenomena, New York, 2nd edn, 1989 Search PubMed.
- M. S. Borse and S. Devi, Adv. Colloid Interface Sci., 2006, 123–126, 387 CrossRef CAS PubMed.
- M. J. Rosen and J. T. Kunjappu, Surfactants and interfacial phenomena, New Jersey, 2012 Search PubMed.
- V. M. Sadtler, F. Giulieri, M. P. Krafft and J. G. Riess, Chem. – Eur. J., 1998, 4, 1952 CrossRef CAS.
- N. Muller and R. H. Birkhahn, J. Phys. Chem., 1967, 71, 957 CrossRef CAS.
- W. Guo, T. A. Brown and B. M. Fung, J. Phys. Chem., 1991, 95, 1829 CrossRef CAS.
- S. Dong, X. Li, G. Xu and H. Hoffmann, J. Phys. Chem. B, 2007, 111, 5903 CrossRef CAS PubMed.
- D. P. Bossev, M. Matsumoto and M. Nakahara, J. Phys. Chem. B, 1999, 103, 8251 CrossRef CAS.
- N. Li, S. Zhang, L. Zheng and T. Inoue, Langmuir, 2009, 25, 10473 CrossRef CAS PubMed.
- P. Long, J. Chen, D. Wang, Z. Hu, X. Gao, Z. Li and J. Hao, J. Phys. Chem. B, 2012, 116, 7669 CrossRef CAS PubMed.
- B. W. Berger, C. M. Gendron, C. R. Rlobinson, E. W. Kaler and A. M. Lenhoff, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 724 CrossRef PubMed.
- S. Finet, D. Vivares, F. Bonneté and A. Tardieu, Macromolecular Crystallography, Pt C, 2003, vol. 368, p. 105 Search PubMed.
- P. Loll, C. Hitscherich, V. Aseyev, M. Allaman and J. Wiencek, Cryst. Growth Des., 2002, 2, 533 CAS.
- O. D. Velev, E. W. Kaler and A. M. Lenhoff, Biophys. J., 1998, 75, 2682 CrossRef CAS PubMed.
- A. Meyer, K. Dierks, R. Hussein, K. Brillet, H. Brognaro and C. Betzel, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 75 CAS.
- R. C. Oliver, J. Lipfert, D. A. Fox, R. H. Lo, S. Doniach and L. Columbus, PLoS One, 2013, 8, e62488 CAS.
- J. Kohlbrecher, SASfit ver. 0.93.3, available at http://kur.web.psi.ch/sans1/SANSSoft/sasfit.html.
- E. Pebay-Peyroula, G. Rummel, J. P. Rosenbusch and E. M. Landau, Science, 1997, 277, 1676 CrossRef CAS PubMed.
- J. Wang, S. Link, C. D. Heyes and M. A. El-Sayed, Biophys. J., 2002, 83, 1557 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) is available: Additional characterization. See DOI: 10.1039/c5nj03502c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |