
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amino acid-derived N-heterocyclic carbene palladium complexes for aqueous phase Suzuki–Miyaura couplings†
Elliot
Steeples
a,
Alexandra
Kelling
b,
Uwe
Schilde
b and
Davide
Esposito
*a
aMax-Planck-Institute of Colloids and Interfaces, D-14424 Potsdam, Germany. E-mail: davide.esposito@mpikg.mpg.de
bUniversity of Potsdam, Institute of Chemistry, D-14476 Potsdam, Germany
First published on 29th March 2016
Abstract
In this work, three ligands produced from amino acids were synthesized and used to produce five bis- and PEPPSI-type palladium–NHC complexes using a novel synthesis route from sustainable starting materials. Three of these complexes were used as precatalysts in the aqueous-phase Suzuki–Miyaura coupling of various substrates displaying high activity. TEM and mercury poisoning experiments provide evidence for Pd-nanoparticle formation stabilized in water.
Introduction
The current dependence of the chemical industry on petrochemically-derived solvents has sparked a recent movement in developing sustainable alternatives.1 Water is one of the leading contenders,2 alongside ionic liquids3,4 and biomass-derived solvents.5,6 The drive to utilize water as a solvent is due to its large heat capacity, non-toxicity, thermal stability, and large abundance on the planet.2,7 The issue of designing catalytic systems suitable for “in-water” and “on-water” organic synthesis still remains and has gained startling momentum within research.8,9 The most popular approach of adapting metal-catalyzed processes for use in water is currently the coordination of water-soluble ligands to the metal centre appropriate for the reaction.9,10 This approach facilitates simple separation of insoluble organic products from the water medium, allowing for the possible recycling of the catalytic water layer.11 Notable examples of homogeneous catalysis in water include Ir in asymmetric hydrogenation,12,13 Ru in olefin metathesis,14,15 and the work on Pd catalyzed C–C coupling reactions.16–18N-Heterocyclic carbenes (NHCs) have played a dominant role in the water-soluble ligand approach.10,19,20 Their strong σ- and π-back donation facilitate their utilization as stable ligands for many catalytic systems.21–23 In addition, the simple functionalization of the N-bound groups allows for easy incorporation of water soluble moieties, thus making them more suitable for water soluble catalyst design as opposed to other competitive ligands such as phosphines,15,16 which can be susceptible to oxidation in aqueous systems.24 The majority of work in this field has been dedicated towards Pd–NHC complexes for Suzuki–Miyaura couplings.9,19 Complexes featuring mono-dentate and multi-dentate chelating ligands have been utilized for homogeneous couplings. Pd–NHC complexes of-bis,25,26 pincer-type,27–29 and PEPPSI-type17,30 (pyridine-enhanced precatalyst, preparation, stabilization and initiation) are among the most active for aqueous cross coupling catalysis.
The most common route towards producing novel NHC precursors involves the functionalization of imidazoles via quaternization with alkyl tails featuring a sulfonate or carboxylate group.31 In this work, we utilize the greener approach of introducing water-soluble R-groups into the imidazolium precursor via a modified one pot Debus-Radziszewski heterocycle synthesis, using amino acids as precursors. This approach was initially developed in our group to obtain building blocks for the preparation of ionic liquids.4,32 Here, we further expand on the use of amino-acid derived imidazoliums using these precursors for the synthesis of novel Palladium precatalysts, which were tested for their activity in the aqueous Suzuki–Miyaura cross-coupling reaction.
Results and discussion
Synthesis and characterization
As established through previous research, imidazolium salts are useful moieties, with applications in various fields of chemistry such as ionic liquids,4 poly-ionic liquids (PILs)33 and NHC precursors.31 The current strategies for synthesizing imidazolium salts fall short in the green and sustainable aspects, as a result of the use of haloalkanes and arenes to introduce novel R-groups. In this research we used the strategy of reacting glyoxal, formaldehyde and two equivalents of amino acid to synthesize imidazolium building blocks as previously described within our group.32 Glyoxal and amino acids can be in principle obtained from sustainable sources, and the heterocycle synthesis is performed rapidly in water as illustrated in Scheme 1. Amino acid-derived NHC precursors have been documented for the synthesis of Au and Ru compounds,34,35 but no examples of similar Pd–NHC complexes from amino-acid precursors have yet been developed to the best of our knowledge.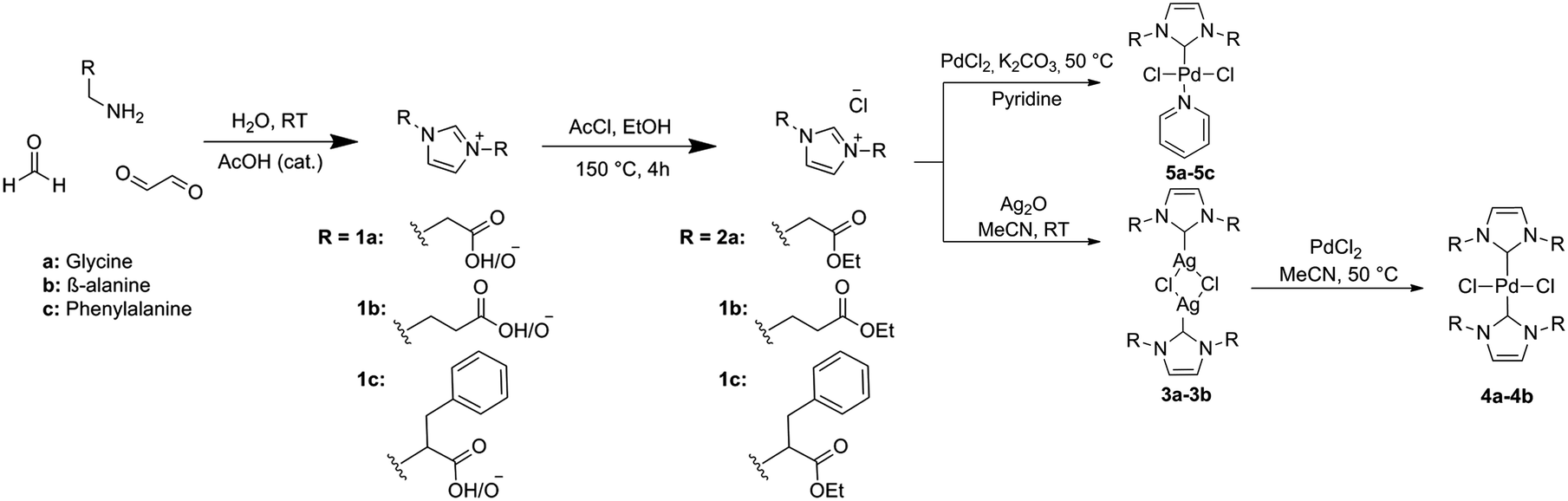 | ||
| Scheme 1 Synthesis of ligand precursors, bis-NHC Pd complexes and PEPPSI NHC Pd complexes. | ||
The building-blocks are isolated as internal carboxylate zwitterions 1a–1c in excellent yields (62–99%). Since initial attempts to directly complex these zwitterions to palladium were unsuccessful, conversion of these building blocks into suitable NHC precursors was performed via a simple esterification of the carboxylate arms in ethanol, using acetyl chloride as the source of both the acidic catalyst and the chloride counterion. In this way, compounds 2a–2c were isolated in very good yields as viscous ionic liquids. Compound 2a was previously synthesized by the Santini group using a different synthesis route.36
Palladium–NHC complexes of bis- and mono-type were then successfully synthesized using two different synthesis methods (Scheme 2). Bis-NHC complexes were prepared using Ag2O to produce the dimeric silver–NHC complexes 3a and 3b, followed by transmetallation with 0.5 equivalents of PdCl2. 3a could be isolated as a white solid, while 3b was unable to be isolated from a mixture of the adduct and unreacted ionic liquid precursor. Both adducts are light and moisture sensitive, however stable under argon atmosphere for up to 5 days. Complexes 4a and 4b were produced as pale yellow solids. Mono-NHC PEPPSI-type complexes are generally more active than bis-NHC complexes due to the presence of the labile pyridine ligand, which can be exchanged allowing the coordination of the reagents to the metal center. Therefore in order to compare the catalytic activity, PEPPSI complexes were also targeted. These were synthesized using the procedure developed by Organ et al.37 from the precursors 2a–2c, using K2CO3 as a base in neat pyridine; producing yellow solids 5a–5c.
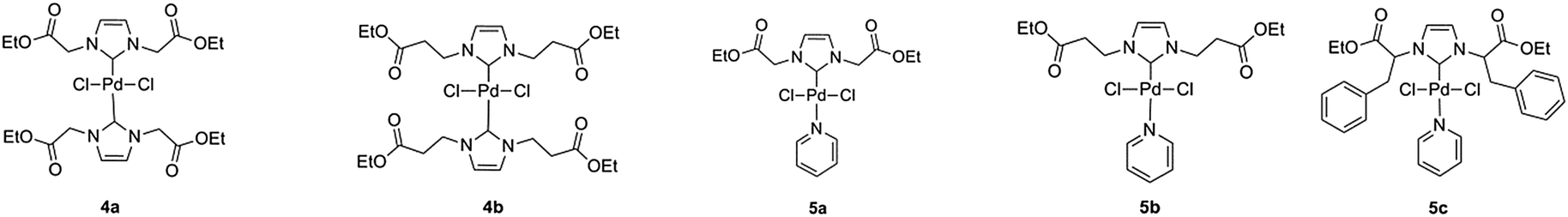 | ||
| Scheme 2 Bis-NHC Pd complexes 4a–4b and PEPPSI NHC Pd complexes 5a–5c. | ||
NHC Precursors 2a–2c incorporate glycine, β-alanine and phenylalanine R-groups into their structure, in order to introduce a variety of steric characteristics into the complexes. These amino acids were selected based solely on the success of their complexation. Attempts were made to synthesize complexes based on NHC precursors featuring alanine and leucine as well as pyruvaldehyde in the imidazolium backbone; however the resulting complexes either gave poor yields or were unable to be isolated. This is likely due to the varying pKa of the proton in the α-position to the N atom, causing non-selective deprotonation of this position over the imidazolium 2-H proton. The attempt to synthesize a bis-complex featuring ligand 2c was also unsuccessful. The presence of a small amount of a silver-2c carbene adduct could be confirmed by MS; however the light-sensitive carbene adduct was unable to be isolated from the unreacted ionic liquid imidazolium precursor.
The structure of the isolated complexes 4a–5b could be confirmed using X-ray structure analysis38–41 with single-crystals grown from slow-evaporation solvent techniques. The molecular structures are presented in Fig. 1–4, bond lengths and angles in Tables 1 and 2. Compound 4a features two complexes within the asymmetric unit, and compound 4b displays a half-molecule in the asymmetric unit; a C2 symmetry point group is revealed upon generating the full molecule. All complexes feature a square-planar Pd(II) center, typical of 16 electron 4-coordinate palladium complexes. Distortions within the Cl–Cl–C–C coordination plane are minimal, the maximum deviations calculated for 4a are 0.027(3) and 0.025(3) Å for the coordinated carbon atoms (C1 and C23) of molecules 1 and 2 respectively. 4b displays no distortion as a result of the symmetry element. Afforded crystal structures confirm the trans-NHC arrangement of the bis-NHC complexes. Dihedral angles between the coordination plane and the imidazolium rings vary between 61–78° (see ESI†), similar torsions to those observed in bis-NHC complexes featuring more bulky ligands such as IPr or IMes.42The Pd–C bond lengths from 2.018(3) to 2.033(3) Å are in the expected range.
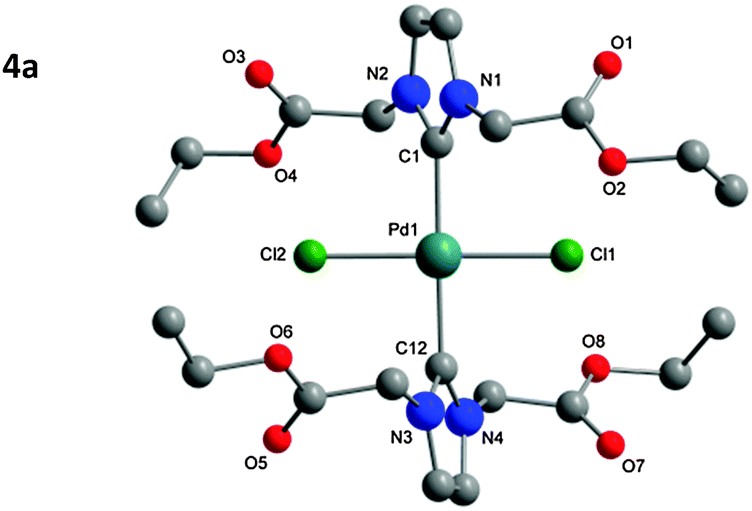 | ||
| Fig. 1 Molecular structure of 4a. Hydrogen atoms omitted for clarity. Molecule 1 is displayed in the figure, for molecule 2 see Table 1 and ESI.† | ||
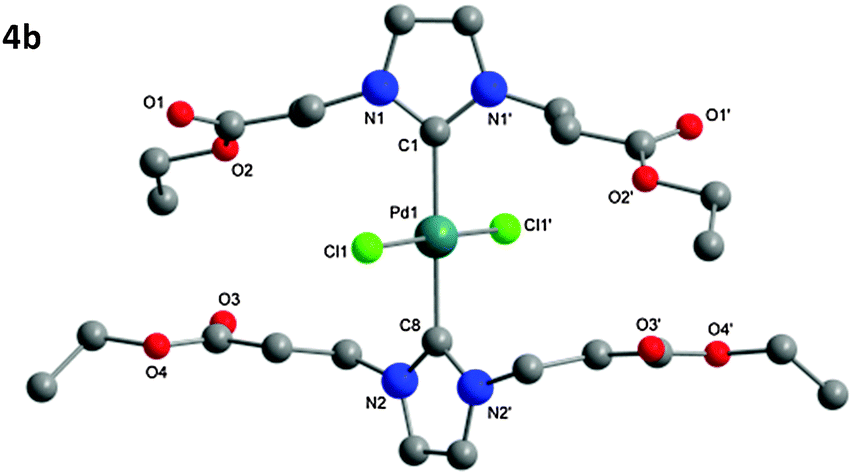 | ||
| Fig. 2 Molecular structure of 4b. Hydrogen atoms omitted for clarity. | ||
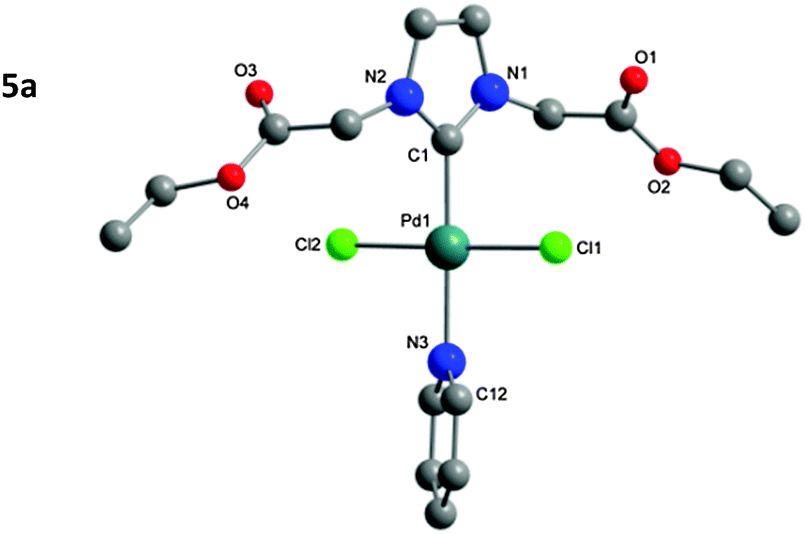 | ||
| Fig. 3 Molecular structure of 5a. Hydrogen atoms omitted for clarity. | ||
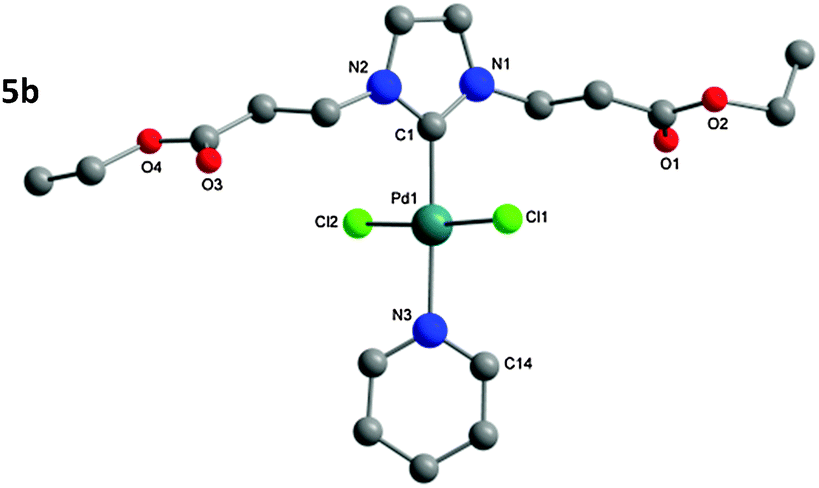 | ||
| Fig. 4 Molecular structure of 5b. Hydrogen atoms omitted for clarity. | ||
| 4a | 4b | |
|---|---|---|
| Bond length [Å] | ||
| Pd–C1 [C34] | 2.033(3) [2.024(3)] | 2.029(3) |
| Pd–C12 [C23]/C8 | 2.029(3) [2.018(3)] | 2.021(3) |
| Pd–Cl1 [Cl3] | 2.3102(7) [2.2991(8)] | 2.3124(5) |
| Pd–Cl2 [Cl4] | 2.3134(7) [2.2982(8)] | 2.3124(5) |
| Bond angle [°] | ||
| N–C1–N [N–C34–N] | 104.0(2) [104.2(2)] | 104.5(2) |
| N–C12/C8–N [N–C23–N] | 104.0(2) [104.4(2)] | 104.7(2) |
| Cl–Pd–Cl | 178.73(3) [178.95(3)] | 178.31(3) |
| C–Pd–C | 177.0(1) [178.4(1)] | 180.0 |
| Cl1–Pd1–C1 [Cl3–Pd2–C34] | 90.81(8) [91.40(8)] | 90.85(1) |
| Cl1–Pd1–C12/C8 | 88.53(8) | 89.15(1) |
| [Cl3–Pd2–C23] | [89.34(8)] | — |
| 5a | 5b | |
|---|---|---|
| Bond length [Å] | ||
| Pd1–C1 | 1.969(2) | 1.958(2) |
| Pd1–N3 | 2.091(2) | 2.110(2) |
| Pd–Cl1 | 2.3046(6) | 2.2988(5) |
| Pd–Cl2 | 2.3014(6) | 2.3167(5) |
| Bond angle [°] | ||
| N1–C1–N2 | 105.0(2) | 105.7(2) |
| Cl1–Pd1–Cl2 | 177.58(3) | 176.21(2) |
| C1–Pd1–N3 | 178.00(9) | 177.50(6) |
| Cl1–Pd1–C1 | 89.12(6) | 87.60(5) |
| Cl1–Pd1–N3 | 90.49(6) | 91.87(5) |
Structures of the PEPPSI complexes confirm the typical trans-arrangement of the NHC ligand to the coordinated pyridine. Once again very little distortion from the Pd(II) square-planar center is observed; the C1 atom of compounds 5a and 5b show plane deviations of 0.100(3) and 0.064(2) Å respectively. Dihedral torsions between the coordination plane and the imidazolium ring are notably larger than those of the bis-NHC compounds; however torsions between the coordination plane and the pyridine ring are smaller, at 47.79(7)° and 39.31(6)° for compounds 5a and 5b respectively. Another notable feature is that the carbonyl oxygen atoms from Glycine-derived complexes 4a and 5a are orientated away from the plane of coordination; whereas in β-alanine-derived complexes 4b and 5b they are aligned in parallel with the coordination plane. This is most likely a result of packing in the unit cell; the distance of the oxygen atoms from the Pd center rules out any coordination influence. The Pd–C bonds (5a: 1.969(2); 5b: 1.958(2) Å) are shorter as those in the bis-NHC complexes. The packing of 5a is completed by a chloroform molecule of crystallization.
Water solubility
All five complexes are not initially soluble in water at pH 7, however PEPPSI-complexes 5a–5c were found to be readily soluble in both acidic and basic aqueous solutions with gentle heating. Within 1 hour at 40 °C, a yellow solution was clearly observed for pH ranges 1–5 and 9–14, suggesting successfully induced water solubility of at least 15 mg mL−1 for all complexes 5a–5c, higher than the required catalyst concentration. However the bis-complexes 4a and 4b remain insoluble even after heating at high temperatures under acidic and basic conditions. As the ester-functionalized NHC complexes are not initially water soluble, we hypothesize that water solubility was induced via hydrolysis of the ester moieties in the presence of base to introduce carboxylate functionalities. This was previously observed by Herrmann et al. as a method of inducing water solubility in Rh and Pd complexes with similar ligands.43 This was probed by dissolving complexes 5a and 5b in D2O under basic conditions and performing a 1H-NMR experiment. Ethanol was clearly observed in the spectrum, suggesting that the N-bound ester groups were hydrolyzed. Ligand peaks were also present at very low intensity. This observation points to the formation of Pd-nanoparticles with surface-bound NHC ligands. Absent or broadened ligand peaks in solution NMR is typical of surface-bound ligand species on nanoparticles, as a result of slow tumbling causing faster T2 spin–spin relaxation times, as observed by Chaudret et al. for Ru–NHC nanoparticle species.44Catalytic screening and optimization of conditions
Although complexes 4a and 4b were observed to produce moderate conversion in the coupling of bromoanisole with phenylboronic acid in ethanolic medium, they were proven to be unsuitable for aqueous-based systems (data not shown). Catalytic activity of the three PEPPSI complexes in pure water was therefore tested. The coupling of bromoanisole with phenylboronic acid using K2CO3 as a base was used as an initial standard for catalyst evaluation (Table 3). Aside from its traditional role in the catalytic cycle, the base is also required to produce the water-soluble complex. The β-alanine-derived precatalyst 5b produced the marginally highest yield over 24 hours, although all precatalysts showed very similar results, despite the large differences in steric bulk, particularly in the phenylalanine-derived 5c (entry 3). A comparison was also made between the PEPPSI precatalysts and palladium acetate, a Pd(II) source typically used in Suzuki couplings (entry 4). Precatalysts 5a–5c all perform substantially better than Pd(OAc)2, highlighting the positive influence of the NHC ligands. The effect of in situ complex dissolution over catalyst pre-dissolution was also tested, comparing the immediate addition of the complex at the start of the reaction, or the pre-dissolution of complexes for 2 hours before addition of substrates. In each case we observed largely no difference in conversion between pre-activated catalysts and in situ dissolution over 24 hours (Table 4, entry 6).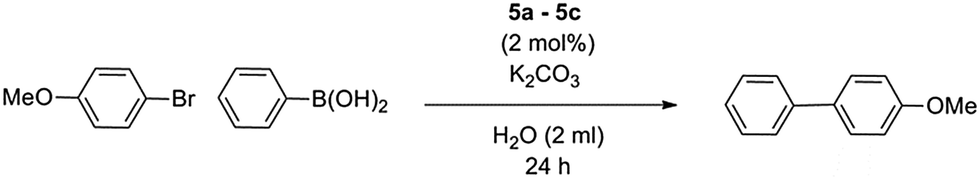
|
|
|||
|---|---|---|---|
| Entry | Base | Temperature [°C] | Yieldd [%] |
| Reaction conditions: bromoanisole (0.5 mmol), phenylboronic acid (0.75 mmol), base (1.0 mmol), 5b (2 mol%), in pure H2O at 60 °C for 24 hours.a Reactants were added after stirring base and precatalyst at temperature for 2 hours.b Catalyst loading was decreased to 1 mol%.c TBAB (1.0 mmol) was added.d Yields determined by NMR using trimethoxybenzene as an internal standard. | |||
| 1 | K2CO3 | 60 | 80 |
| 2 | KOH | 60 | 43 |
| 3 | t BuOK | 60 | 40 |
| 4 | K2CO3 | 80 | 60 |
| 5 | K2CO3 | 40 | 49 |
| 6c | K2CO3 | 60 | 92a, 90b |
| 7c | K2CO3 | 40 | 71 |
| 8c | t BuOK | 60 | 88 |
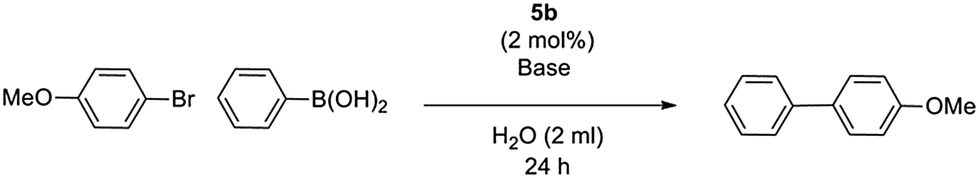
Since 5b exhibited the best activity, it was selected for further condition optimization studies, varying base and temperature (Table 4). After base screenings, K2CO3 still produced the best yield and was selected for further screenings. 60 °C was chosen as the best reaction temperature. Lower temperature screenings were proven to be unsuccessful in producing competitive yields (entry 5), and higher temperatures accelerated the destabilization of catalyst with increased formation of palladium black (entry 4). We also discovered under the optimized conditions that the catalyst loading could be reduced to 1 mol% without loss of activity (entry 6).
Water-soluble catalytic processes are well known to benefit from the addition of a phase-transfer catalyst, which aids the shuttling of base between the catalytic aqueous layer and the immiscible bromoarene.17,18 Addition of two equivalents of tetra-n-butylammonium bromide (TBAB) was seen to dramatically improve yield up to 92% under optimized conditions (entries 6–8).
Further substrate screening
Having determined the optimum conditions for the reaction, screenings of various different substrates with complex 5b were performed in order to comment on the catalytic activity towards different directing groups and aryl ring positions (Table 5). As a general trend, the more electron-withdrawing substituents produce higher yields, as observed in the 4-fluoro and 4-acetyl moieties (entries 4 and 8). The effect of lowering precatalyst concentration was also tested, revealing that catalyst loadings of just 0.01 mol% could activate some of the more active aryl precursors (entries 4 and 8). In a number of cases, lowering the precatalyst loading increases conversion, particularly in the case of bromobenzene (entry 5). 1-Ethylbenzene was also tested, showing the catalyst's ability to also activate benzyl reagents; a moderate 78% yield was observed in this case (entry 7). Good yields were also observed when the boronic acid is exchanged with a pinacol ester (entries 13–15). Entry 13 particularly highlights the increased yield with decreased precatalyst loading. Chloroaryls were also tested but performance was poor.|
|
||||
|---|---|---|---|---|
| Entry | R | R′ | X | Yielda [%] |
| Reaction conditions: aryl/benzyl halide (0.5 mmol), phenylboronic acid/pinacol ester (0.75 mmol), base (1.0 mmol), 5b (2 mol%), TBAB (1.0 mmol) in pure H2O at 60 °C for 24 hours.a Yields determined by NMR using trimethoxybenzene as an internal standard.b Precatalyst loading decreased to 1 mol%.c Precatalyst loading decreased to 0.1 mol%.d Precatalyst loading decreased to 0.01 mol%. | ||||
| 1 | 4-OMe | B(OH)2 | Br | 92, 90b, 85c |
| 2 | 3-OMe | B(OH)2 | Br | 84 |
| 3 | 2-OMe | B(OH)2 | Br | 93 |
| 4 | 4-Ac | B(OH)2 | Br | 95, 94b, 99c, 90d |
| 5 | 4-H | B(OH)2 | Br | 81, 99b, 56c |
| 6 | 1-Napthyl | B(OH)2 | Br | 76 |
| 7 | –CH(Br)CH3 | B(OH)2 | Br | 78 |
| 8 | 4-F | B(OH)2 | Br | 99, 69c, 53d |
| 9 | 4-N(Me)2 | B(OH)2 | Br | 83 |
| 10 | 4-OMe | B(OH)2 | Cl | 8 |
| 11 | 4-Ac | B(OH)2 | Cl | 9 |
| 12 | 4-H | B(OH)2 | Cl | 11 |
| 13 | 4-OMe | B(CH3)4C2O2 | Br | 39, 87b |
| 14 | 4-Ac | B(CH3)4C2O2 | Br | 91 |
| 15 | 4-H | B(CH3)4C2O2 | Br | 99 |
All substrate screenings were performed over 24 hours to ensure full conversion, while a detailed kinetic study was not included in this work. However, preliminary kinetic investigations of the bromoacetophenone coupling indicate that the reaction is finished within 50 minutes for both the pre-dissolved system, and within one hour for the in situ dissolved system. The kinetic plot is available in the ESI.†
Finally, it is worth mentioning that the possibility of catalyst recycling is one of the prominent advantages of using aqueous-phase systems. In a proof-of-principle experiment, the separated aqueous layer was able to be recycled for an additional run. 100% yield was reached in the second cycle within two hours for the bromoacetophenone coupling with 2 mol% catalyst loading, using toluene to extract the product. Since recycling was not of primary interest within this report, detailed investigation into recycling with this family of complexes will be the subject of further study.
Formation of nanoparticles
A number of observations during the initial screenings lead us to believe that Pd(0) nanoparticles were formed during the reaction, a common factor in NHC-complex catalysis in aqueous media.17,18,28,45 In particular, the presence of a black precipitate, the discovery that higher temperatures and catalyst loadings can hinder higher yields, and the very similar yields despite large differences in ligand sterics pointed to the possible formation of Pd(0) nanoparticles. In order to confirm this, TEM analysis was performed on the isolated precipitate from a coupling of bromoanisole with phenylboronic acid using precatalyst 5b (Fig. 5), which clearly shows the formation of nanoparticles taking place. Interestingly, during the screenings using TBAB, we noticed a significantly smaller amount of precipitate being produced in comparison to the initial TBAB-free screenings, suggesting a more effective stabilization of the nanoparticle in solution. To investigate this we performed TEM imaging on the complex 5b after dissolution in alkaline water with TBAB. Nanoparticles were once again observed, gathered inside darker aggregates, with an average size of 8.7 ± 0.1 nm (Fig. 6). This suggests that the nanoparticles are stabilized by TBAB doubling as a surfactant, in line with what was observed in other nanoparticle-based systems.46,47 In order to determine the activity of nanoparticles in the coupling, mercury poisoning experiments were performed for complexes 5a–5c using a molar Hg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd ratio of 300:1 in the presence of TBAB (Table 6). Catalytic activity was significantly reduced, 5b being poisoned entirely, and activity for 5a and 5c reduced to 16% and 14% respectively. This experiment confirms the active role of the nanoparticles as the catalyst, indicating that the reaction is mostly heterogeneously catalyzed. Although the nanoparticles are determined to be responsible for the majority of the catalytic activity, however, the residual activity after Hg addition in the cases of 5a and 5c suggests that a number of catalytic species are likely to be present. This may include different nanoparticle species, metal clusters and molecular complex species with partially-hydrolyzed ester groups. This points to the “catalyst cocktail” model typically observed for in situ generated catalysts within cross-coupling reactions, as described by Ananikov et al.48
:Pd ratio of 300:1 in the presence of TBAB (Table 6). Catalytic activity was significantly reduced, 5b being poisoned entirely, and activity for 5a and 5c reduced to 16% and 14% respectively. This experiment confirms the active role of the nanoparticles as the catalyst, indicating that the reaction is mostly heterogeneously catalyzed. Although the nanoparticles are determined to be responsible for the majority of the catalytic activity, however, the residual activity after Hg addition in the cases of 5a and 5c suggests that a number of catalytic species are likely to be present. This may include different nanoparticle species, metal clusters and molecular complex species with partially-hydrolyzed ester groups. This points to the “catalyst cocktail” model typically observed for in situ generated catalysts within cross-coupling reactions, as described by Ananikov et al.48
 | ||
| Fig. 5 TEM images taken from isolated black precipitate during screening of complex 5b. Precipitate was isolated by centrifugation, and washing with water and ethanol. | ||
 | ||
| Fig. 6 TEM images taken from an aqueous solution of 5b (0.01 mmol) after heating for 2 hours at 60 °C with K2CO3 (1.0 mmol) and TBAB (1.0 mmol). A histogram of the nanoparticle size distribution is included as an inset. | ||
| Complex | Yield (without Hg)a [%] | Yield (with Hg)a [%] |
|---|---|---|
| Reaction conditions: aryl halide (0.5 mmol), phenylboronic acid (0.75 mmol), base (1.0 mmol), 5b (2 mol%), TBAB (1.0 mmol) in pure H2O at 60 °C for 24 hours. Hg was added at the start of the reaction in a Hg:Pd ratio of 300:1.a Yields determined by NMR using trimethoxybenzene as an internal standard. |
||
| 5a | 88 | 16 |
| 5b | 92 | 0 |
| 5c | 88 | 14 |
Conclusions
A simple, green method of synthesizing N-heterocyclic carbene (NHC) precursors with various different functionalities was applied to produce five NHC–palladium complexes. The three pyridine-coordinated complexes were suitable as precatalysts for aqueous phase Suzuki–Miyaura cross-coupling reactions, achieving good catalytic performance with a precatalyst loading as low as 0.01 mol%. It was ascertained that the catalysis proceeds according to a “catalyst cocktail” model, with Pd-nanoparticles responsible for most of the catalytic activity, as evidenced by transmission electron microscopy and mercury poisoning. This research builds on work involving amino-acid derived NHCs and addresses a number of sustainability issues within homogeneous catalysis, which further builds bridges between green chemistry and organometallic catalysis.Materials and methods
Syntheses
All purchased chemicals were used as supplied without further purification. Complex synthesis was carried out under an argon atmosphere using standard Schlenk techniques. The solvents used for complexations were purchased dry from acros organics and stored over 3 Å molecular sieves. Water used as a catalysis medium was purified using an SG Integra UV Plus millipore water purification system. 1H and 13C NMR were performed on a Bruker Ultrashield 400 MHz instrument (1H: 400 MHz, 13C: 101 MHz) at 298 K, using D2O, DMSO-d6, CDCl3 and d3-MeCN as locking solvents. Electrospray mass spectroscopy was performed on a Thermo Scientific Velos Pro MS device coupled with liquid chromatography, using acetonitrile, MeOH and water as solvents, spectra were viewed and analyzed using Xcalibur viewer software. Elemental analysis was performed by the MPIKG microanalytical service on a varioMICRO V 1.4.1 device. Electron micrographs were then recorded on a Zeiss EM 912 Ω microscope operated at an acceleration voltage of 120 kV.Imidazolium carboxylate salts 1a–1c
These compounds were prepared according to the procedure outlined in ref. 32.1a: white solid, yield: 95%. 1H NMR (D2O, 400 MHz): δ 8.87 (s, 1H, CHimi), 7.53 (s, 2H, CHimi), 5.02 (s, 4H). 13C NMR (D2O, 101 MHz) δ 170.88 (Ccarbox), 137.74 (Cimi), 123.34 (Cimi), 50.99. ESI-MS m/z calcd for C7H8N2O4 184.05 [M]+, found 185.1 [M + H]+. Elemental anal: calcd for C7H8N2O4: C, 45.66; H, 4.38; N, 15.21. Found: C, 45.39; H, 4.19; N, 15.03.
1b: white solid, yield: 99%. 1H NMR (D2O, 400 MHz, 298 K): δ 8.83 (s, 1H, CHimi), 7.51 (s, 2H, CHimi), 4.44 (t, J = 6.5 Hz, 4H), 2.87 (t, J = 6.4 Hz, 4H).13C NMR (D2O, 101 MHz) δ 175.92 (Ccarbox), 136.04 (Cimi), 122.33 (Cimi), 45.81, 35.61. ESI-MS m/z calcd for C9H12N2O4 121.08 [M]+, found 213.1 [M + H]+. Elemental anal: calcd for C9H12N2O4: C, 50.94; H, 5.70; N, 13.20. Found: C, 50.76; H, 5.50; N, 12.93.
1c: white solid, yield 62%. 1H NMR (DMSO-d6, 400 MHz) δ 9.34 (s, 1H, CHimi), 7.45 (s, 2H, CHimi), 7.18–7.15 (m, 6H), 6.97–6.95 (m, 4H), 5.20 (dd, J = 11.3, 4.1 Hz, 2H), 3.48 (dd, J = 14.6, 4.0 Hz, 2H), 3.18 (dd, J = 14.5, 11.3 Hz, 2H). 13C NMR (DMSO-d6, 101 MHz) δ 168.99 (Ccarbox), 136.54, 136.42 (Cimi), 128.63, 128.43 (Cimi), 126.66, 121.43, 64.90, 38.34. ESI-MS m/z calcd for C21H21N2O4 365.15 [M]+, found 366.4 [M + H]+. Elemental anal: calcd for C21H21N2O4: C, 69.22; H, 5.53; N, 7.69. Found: C, 68.62; H, 5.52; N, 7.64.
Ligand precursors 2a–2c
A flask with a Soxhlet apparatus containing molecular sieves (3 Å) was charged with the corresponding imidazolium zwitterion (0.01 mol) and a stir bar under argon. Ethanol (150 mL) was then added as a reactant and solvent, followed by acetyl chloride (0.012 mol, 1.2 equiv.). The mixture was refluxed at 150 °C for 4 hours. After cooling, the solution was filtered through a PTFE membrane and the solvent was removed under reduced pressure. The obtained residue was washed with diethyl ether and dried affording the desired ligand precursors, which were stored under an argon atmosphere.2a: white solid, yield: 89%. 1H NMR (CDCl3, 400 MHz) δ 10.21 (s, 1H, CHimi), 7.73 (s, 2H, CHimi), 5.38 (s, 4H), 4.15 (q, J = 7.1 Hz, 4H), 1.21 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 166.15 (Ccarbox), 139.60 (Cimi), 123.41 (Cimi), 63.05 (CH2COOEt), 50.42 (CH2CH3), 14.14 (CH3CH2). ESI-MS: m/z calcd for C11H17ClN2O4 276.09 [M]+, found 241.2 [M − Cl]+. Elemental anal: calcd for C11H17ClN2O4: C, 47.74; H, 6.19; N, 10.12. Found: C, 47.66; H, 5.91; N, 10.05.
2b: brown oil, yield: 84%. 1H NMR (CDCl3, 400 MHz) δ 10.67 (s, 1H, CHimi), 7.51 (s, 2H, CHimi), 4.65 (t, J = 5.8 Hz, 4H), 4.13 (q, J = 7.1 Hz, 4H), 3.05 (t, J = 5.8 Hz, 4H), 1.23 (t, J = 7.1 Hz, 6H). 13C NMR (CDCl3, 101 MHz) δ 170.40 (Ccarbox), 137.80 (Cimi), 122.78 (Cimi), 61.22, 45.24, 34.60, 13.94. ESI-MS: m/z calcd for C13H21ClN2O4 304.12 [M]+, found 269.2 [M − Cl]+. Elemental anal: calcd for C13H21ClN2O4: C, 51.23; H, 6.95; N, 9.19. Found: C, 49.17; H, 6.59; N, 9.00.
2c: brown oil, yield: 71%. 1H NMR (CDCl3, 400 MHz) δ 10.77 (s, 1H, CHimi), 7.23–7.21 (m, 6H), 7.16 (s, 2H, CHimi), 7.09–7.06 (m, 4H), 6.01 (t, J = 7.1 Hz, 2H), 4.22 (q, J = 7.1, 1.0 Hz, 4H), 3.45 (d, J = 7.0 Hz, 4H), 1.24 (t, J = 7.1 Hz, 6H).13C NMR (CDCl3, 101 MHz) δ 167.66 (Ccarbox), 138.89, 133.60 (Cimi), 129.29, 129.15, 128.00, 121.15 (Cimi), 63.12, 62.72, 39.29, 14.05. ESI-MS: m/z calcd for C25H29ClN2O4 456.18 [M]+, found 421.4 [M − Cl]+. Elemental anal: calcd for C25H29ClN2O4: C, 65.71; H, 6.40; N, 6.13. Found: C, 65.69; H, 6.35 N, 5.99.
Silver adducts 3a and 3b
A Schlenk tube was charged with 2a or 2b (0.003 mol) and a stir bar under argon. Dry acetonitrile was added (8 mL) and the mixture was gently heated (45 °C) with stirring for 10 minutes. Silver oxide (0.32 g, 0.0015 mol) was then added under flow of argon and the mixture stirred at room temperature for 12 hours. The mixture was then gently heated again (45 °C) for 5 minutes to aid dissolution of the adduct, and filtered under argon by use of a cannula. The solvent was removed from the filtrate in vacuo to give the desired product.3a: white solid, yield 83%. 1H NMR (CDCl3, 400 MHz) δ 7.13 (s, 2H, CHimi), 4.93 (s, 4H), 4.26 (q, J = 7.1 Hz, 4H), 1.32 (t, J = 7.2 Hz, 6H). 13C NMR (CDCl3, 101 MHz) δ 183.54 (Ccarbene), 167.24 (Ccarbox), 122.75, 62.61, 52.83, 14.21. ESI-MS: m/z calcd for C11H17AgClN2O4 384.99 [M]+, found 587.3 [2L + Ag − Cl]+. Elemental anal: calcd for C11H17AgClN2O4: C, 34.35; H, 4.46; N, 7.28. Found: C, 34.57; H, 4.28; N, 7.33.
Bis-NHC Pd complexes 4a and 4b
4a: a Schlenk tube was charged with 3a (0.79 g, 0.002 mol) and a stir bar under argon. Dry acetonitrile was added (8 mL) and the mixture was stirred until dissolution. Palladium(II) chloride (0.18 g, 0.001 mol) was then added under flow of argon and the mixture was further stirred at 50 °C for 12 hours. The suspension was filtered and the filtrate dried in vacuo. The obtained crude solid was recrystallized from CHCl3 to afford the desired product. Crystals suitable for X-ray analysis were prepared from a slow evaporation chamber of CH2Cl2/THF. Yellow solid, yield 36%.1H NMR (CDCl3, 400 MHz) δ 7.03 (s, 2H, CHimi), 5.32 (s, 4H), 4.26 (q, J = 7.1 Hz, 4H), 1.29 (t, J = 7.1 Hz, 6H).13C NMR (101 MHz, CDCl3) δ 171.88 (Ccarbene), 167.83 (Ccarbox), 122.50, 62.03, 51.99, 14.22. ESI-MS: m/z calcd for C22H32Cl2N4O8Pd 658.08 [M]+, found 623.3 [M − Cl]+. Elemental anal. calcd for C22H32Cl2N4O8Pd: C, 40.17; H, 4.90; N, 8.52. Found: C, 40.29; H, 4.75; N, 7.84.
4b: a Schlenk tube was charged with 3b (0.94 g, 0.0023 mol) and a stir bar under argon. Dry acetonitrile was added (8 mL) and the mixture was stirred until dissolution. Palladium(II) chloride (0.2 g, 0.0011 mol) was then added under flow of argon and the mixture was stirred at room temperature for 12 hours. The mixture was filtered and the filtrate was transferred to a freezer at −20 °C overnight to afford the desired product. Crystals suitable for X-ray analysis were prepared from a slow evaporation chamber of CH2Cl2/THF. Yellow solid, yield 33%.
1H NMR (CDCl3, 400 MHz) δ 6.96 (s, 2H, CHimi), 4.79 (t, J = 6.7 Hz, 4H), 4.16 (q, J = 7.1 Hz, 4H), 3.23 (t, J = 6.7 Hz, 4H), 1.26 (t, J = 7.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 171.63 (Ccarbene), 170.50 (Ccarbox), 121.71, 60.98, 46.20, 35.96, 14.27. ESI-MS: m/z calcd for C26H40Cl2N4O8Pd 714.13 [M]+, found 679.3 [M − Cl]+. Elemental anal: calcd for C26H40Cl2N4O8Pd: C, 43.74; H, 5.65; N, 7.85. Found: C, 43.74; H, 5.44; N, 7.84.
PEPPSI–NHC complexes 5a–5c
A Schlenk tube was charged with 2a–2c (0.0033 mol), K2CO3 (0.004 mol), Palladium(II) chloride (0.69 g, 0.004 mol), freshly powdered 3 Å molecular sieves (80 mg) and a stir bar under argon. Dry pyridine (7 mL) was added and the mixture stirred at 50 °C for 36 hours. The mixture was then filtered and the filtrate was evaporated to dryness. The crude solid was recrystallized from hot ethanol to afford the desired product.5a: yellow solid, yield 36%. Crystals suitable for X-ray analysis were prepared from a slow evaporation chamber of CHCl3/petroleum ether 60°–80°.
1H NMR (CDCl3, 400 MHz) δ 8.96–8.94 (m, 2H), 7.79–7.74 (m, 1H), 7.35 (dd, J = 7.7, 6.5 Hz, 2H), 7.15 (s, 2H, CHimi), 5.45 (s, 4H), 4.29 (q, J = 7.2 Hz, 4H), 1.32 (t, J = 7.1 Hz, 6H). 13C NMR (CDCl3, 101 MHz,) δ 167.33, (Ccarbox) 153.19 (Ccarbene), 151.44, 138.27, 124.62, 123.46, 62.27, 52.07, 14.23. ESI-MS: m/z calcd for C16H21Cl2N3O4Pd 496.99 [M]+, found 462.1 [M − Cl]+. Elemental anal: calcd for C16H21Cl2N3O4Pd: C, 38.69; H, 4.26; N, 8.46. Found: C, 38.71; H, 4.11; N, 8.40.
5b: yellow solid, yield 30%. Crystals suitable for X-ray analysis were prepared from a slow evaporation chamber of CHCl3/petroleum ether 60°–80°.
1H NMR (CDCl3, 400 MHz) δ 8.99–8.97 (m, 2H), 7.79–7.76 (m, 1H), 7.36 (dd, J = 7.7, 6.5 Hz, 2H), 7.06 (s, 2H, CHimi), 4.84 (t, J = 6.5 Hz, 4H), 4.15 (q, J = 7.1 Hz, 4H), 3.22 (t, J = 6.4 Hz, 4H), 1.24 (t, J = 7.1 Hz, 6H). 13C NMR (CDCl3, 101 MHz) δ 171.67 (Ccarbox), 151.37, 149.76 (Ccarbene), 138.28, 124.65, 122.97, 61.13, 46.32, 35.50, 14.28. ESI-MS: m/z calcd for C18H25Cl2N3O4Pd 525.02 [M]+, found 489.2 [M − Cl]+. Elemental anal: calcd for C18H25Cl2N3O4Pd: C, 41.20; H, 4.80; N, 8.01. Found: C, 41.26; H, 4.72; N, 7.88.
5c: yellow solid, yield 26%. 1H NMR (Acetonitrile-d3, 400 MHz) δ 8.90 (dd, J = 6.5, 1.6 Hz, 2H), 7.94 (tt, J = 7.7, 1.7 Hz, 1H), 7.51 (dd, J = 7.7, 6.5 Hz, 2H), 7.33–7.26 (m, 10H), 7.22 (s, 2H, CHimi), 6.51 (dd, J = 8.1, 6.9 Hz, 2H), 4.14 (q, J = 7.1 Hz, 4H), 3.52 (dd, J = 7.5, 2.7 Hz, 4H), 1.13 (t, J = 7.1 Hz, 6H). 13C NMR (CD3CN, 101 MHz) δ 170.23 (Ccarbox), 151.91(Ccarbene), 139.87, 136.50, 130.31, 129.52, 128.12, 125.88, 121.91, 64.83, 62.75, 39.25, 14.23. ESI-MS: m/z calcd for C30H33Cl2N3O4Pd 677.09 [M]+, found 642.3 [M − Cl]+. Elemental anal: calcd for C30H33Cl2N3O4Pd: C, 53.23; H, 4.91; N, 6.21. Found: C, 53.39; H, 4.75; N, 6.27.
Catalytic screening
Microscopy
Acknowledgements
The authors would like to thank the Max-Planck Society for financial support of this work.Notes and references
- P. Pollet, E. A. Davey, E. E. Urena-Benavides, C. A. Eckert and C. L. Liotta, Green Chem., 2014, 16, 1034–1055 RSC.
- M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- S. Kirchhecker, M. Antonietti and D. Esposito, Green Chem., 2014, 16, 3705–3709 RSC.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- V. Molinari, M. Antonietti and D. Esposito, Catal. Sci. Technol., 2014, 4, 3626–3630 CAS.
- P. Tundo, P. Anastas, D. Black, J. Breen, T. Collins, S. Memoli, J. Miyamoto, M. Polyakoff and W. Tumas, Pure Appl. Chem., 2000, 72, 1207–1228 CAS.
- M. B. Gawande, V. D. B. Bonifacio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522–5551 RSC.
- K. H. Shaughnessy, Chem. Rev., 2009, 109, 643–710 CrossRef CAS PubMed.
- L.-A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270–289 CrossRef CAS PubMed.
- A. Azua, S. Sanz and E. Peris, Organometallics, 2010, 29, 3661–3664 CrossRef CAS.
- H. Horváth, Á. Kathó, A. Udvardy, G. Papp, D. Szikszai and F. Joó, Organometallics, 2014, 33, 6330–6340 CrossRef.
- V. Pénicaud, C. Maillet, P. Janvier, M. Pipelier and B. Bujoli, Eur. J. Org. Chem., 1999, 1745–1748 CrossRef.
- J. Tomasek and J. Schatz, Green Chem., 2013, 15, 2317–2338 RSC.
- B. Mohr, D. M. Lynn and R. H. Grubbs, Organometallics, 1996, 15, 4317–4325 CrossRef CAS.
- A. L. Casalnuovo and J. C. Calabrese, J. Am. Chem. Soc., 1990, 112, 4324–4330 CrossRef CAS.
- R. Zhong, A. Pothig, Y. Feng, K. Riener, W. A. Herrmann and F. E. Kühn, Green Chem., 2014, 16, 4955–4962 RSC.
- L. Li, J. Wang, C. Zhou, R. Wang and M. Hong, Green Chem., 2011, 13, 2071–2077 RSC.
- E. Levin, E. Ivry, C. E. Diesendruck and N. G. Lemcoff, Chem. Rev., 2015, 115, 4607–4692 CrossRef CAS PubMed.
- H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032–7060 RSC.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 1999, 100, 39–92 CrossRef.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS.
- A. Cervilla, F. Pérez-Pla, E. Llopis and M. Piles, Inorg. Chem., 2006, 45, 7357–7366 CrossRef CAS PubMed.
- F. Godoy, C. Segarra, M. Poyatos and E. Peris, Organometallics, 2011, 30, 684–688 CrossRef CAS.
- C.-C. Yang, P.-S. Lin, F.-C. Liu, I. J. B. Lin, G.-H. Lee and S.-M. Peng, Organometallics, 2010, 29, 5959–5971 CrossRef CAS.
- F. Churruca, R. SanMartin, B. Inés, I. Tellitu and E. Domínguez, Adv. Synth. Catal., 2006, 348, 1836–1840 CrossRef CAS.
- B. Inés, R. SanMartin, M. J. Moure and E. Domínguez, Adv. Synth. Catal., 2009, 351, 2124–2132 CrossRef.
- T. Tu, X. Feng, Z. Wang and X. Liu, Dalton Trans., 2010, 39, 10598–10600 RSC.
- H. Turkmen, R. Can and B. Cetinkaya, Dalton Trans., 2009, 7039–7044 RSC.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed.
- D. Esposito, S. Kirchhecker and M. Antonietti, Chem. – Eur. J., 2013, 19, 15097–15100 CrossRef CAS PubMed.
- K. Täuber, Q. Zhao, M. Antonietti and J. Yuan, ACS Macro Lett., 2015, 4, 39–42 CrossRef.
- J. DePasquale, N. J. White, E. J. Ennis, M. Zeller, J. P. Foley and E. T. Papish, Polyhedron, 2013, 58, 162–170 CrossRef CAS.
- M. A. Reynoso-Esparza, I. I. Rangel-Salas, A. A. Peregrina-Lucano, J. G. Alvarado-Rodríguez, F. A. López-Dellamary-Toral, R. Manríquez-González, M. L. Espinosa-Macías and S. A. Cortes-Llamas, Polyhedron, 2014, 81, 564–571 CrossRef CAS.
- M. Pellei, V. Gandin, M. Marinelli, C. Marzano, M. Yousufuddin, H. V. R. Dias and C. Santini, Inorg. Chem., 2012, 51, 9873–9882 CrossRef CAS PubMed.
- C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem. – Eur. J., 2006, 12, 4743–4748 CrossRef PubMed.
- Crystal data of 4a. C22H32Cl2N4O8Pd. M = 657.81 g mol−1, monoclinic, a = 12.2261(3) Å, b = 27.7257(6) Å, c = 17.0282(4) Å, β = 99.793(2)°, V = 5688.1(2) Å3, T = 210 K, space group P21/n, (no. 14), Z = 8, 69663 reflections measured, 9541 unique, (Rint = 0.0616), which were used in all calculations. The final wR(F2) was 0.0978 (all data). CCDC 1438609.
- Crystal data of 4b. C26H40Cl2N4O8Pd. M = 713.29 g mol−1, monoclinic, a = 10.3920(5) Å, b = 12.0677(7) Å, c = 25.5618(10) Å, β = 98.377(3)°, V = 3171.4(3) Å3, T = 210 K, space group C2/c, (no. 15), Z = 4, 10088 reflections measured, 2804 unique, (Rint = 0.0178), which were used in all calculations. The final wR(F2) was 0.0610 (all data). CCDC 1438610.
- Crystal data of 5a CHCl3. C17H22Cl5N3O4Pd. M = 616.02 g mol−1, monoclinic, a = 11.7256(4) Å, b = 23.0938(8) Å, c = 9.0944(6) Å, β = 99.520(4)°, V = 2428.7(2) Å3, T = 210 K, space group P21/c, (no. 14), Z = 4, 15567 reflections measured, 4269 unique, (Rint = 0.0364), which were used in all calculations. The final wR(F2) was 0.0613 (all data). CCDC 1438611.
- Crystal data of 5b. C18H25Cl2N3O4Pd. M = 524.71 g mol−1, triclinic, a = 9.4985(8) Å, b = 9.7104(7) Å, c = 12.2951(10) Å, α = 93.265(6)°, β = 95.848(6)°, γ = 98.828(6)°, V = 1111.72(15) Å3, T = 210 K, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , (no. 2), Z = 2, 14413 reflections measured, 3924 unique, (Rint = 0.0243), which were used in all calculations. The final wR(F2) was 0.0533 (all data). CCDC 1438612.
, (no. 2), Z = 2, 14413 reflections measured, 3924 unique, (Rint = 0.0243), which were used in all calculations. The final wR(F2) was 0.0533 (all data). CCDC 1438612. - H. Lebel, M. K. Janes, A. B. Charette and S. P. Nolan, J. Am. Chem. Soc., 2004, 126, 5046–5047 CrossRef CAS PubMed.
- W. A. Herrmann, L. J. Gooβen and M. Spiegler, J. Organomet. Chem., 1997, 547, 357–366 CrossRef CAS.
- P. Lara, O. Rivada-Wheelaghan, S. Conejero, R. Poteau, K. Philippot and B. Chaudret, Angew. Chem., Int. Ed., 2011, 50, 12080–12084 CrossRef CAS PubMed.
- A. Ferry, K. Schaepe, P. Tegeder, C. Richter, K. M. Chepiga, B. J. Ravoo and F. Glorius, ACS Catal., 2015, 5414–5420 CrossRef CAS.
- S. Singh and J. Datta, J. Mater. Sci., 2010, 45, 3030–3040 CrossRef CAS.
- L. Adak, K. Chattopadhyay and B. C. Ranu, J. Org. Chem., 2009, 74, 3982–3985 CrossRef CAS PubMed.
- A. S. Kashin and V. P. Ananikov, J. Org. Chem., 2013, 78, 11117–11125 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Featuring detailed NMR spectra and crystallographic information. CCDC 1438609–1438612. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj03337c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |