DOI:
10.1039/C5NJ03211C
(Paper)
New J. Chem., 2016,
40, 4981-5001
Experimental and computational analysis of supramolecular motifs involving Csp2(aromatic)–F and CF3 groups in organic solids†
Received
(in Montpellier, France)
14th November 2015
, Accepted 14th March 2016
First published on 16th March 2016
Abstract
A detailed experimental (SCXRD) and theoretical (PIXEL and QTAIM) investigation of the evolution of different supramolecular motifs formed via the presence of both C(sp2)/(sp3)–F groups in the crystal packing has been performed in a series of newly synthesized substituted benzanilides (containing “both” the fluorine and the trifluoromethyl group in the same molecule) along with previously reported similarly related crystal structures [CrystEngComm, 2008, 10, 54–67; CrystEngComm, 2012, 14, 1972–1989, CrystEngComm, 2013, 15, 3711–3733]. It was observed that the highest stabilized molecular motifs primarily consist of C(sp2)–H⋯F–C(sp2) H-bonds in preference to C(sp2)–H⋯F–C(sp3) H-bonds in the crystal. The motifs involving C(sp2)–H⋯F–C(sp2)/(sp3) H bonds were observed to be present over the entire distance range between 2.2 and 2.7 Å, albeit the difference in energies of stabilization involving fluorine atoms attached to sp2 and sp3 carbon is not significant in molecular crystals. From QTAIM analysis, the C(sp2)/(sp3)–F⋯F–C(sp2)/(sp3) interactions were observed to be a closed shell in nature and provide local stabilization, indicating the formation of bonds, similar to weak hydrogen bonds observed in crystals.
Introduction
The introduction of a fluorine atom to the carbon atom (termed as “Organic Fluorine”) can lead to the formation of many weak interactions like C–H⋯F–C hydrogen bonds,1,2 C–F⋯F–C3,4 and C–F⋯π5–7 interactions and the study of these interactions involving organic fluorine is still an expanding area of research amongst the scientific community.8 However, there has been an enduring discussion regarding the ability of organic fluorine to act as a hydrogen bond acceptor9–18 because of its low polarizability. However, the crystal structure analysis of fluorobenzenes12 and ribonucleic acids13 unfasten the area of research regarding the study of interactions involving organic fluorine. Further, such interactions with the protein active site through C–F⋯C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–F⋯H–Cα interactions have been reviewed by Diederich et al.19 Since then many studies via inputs from crystallography, spectroscopy and theoretical calculations have established the fact that the interactions involving fluorine are ubiquitous and can play an important role in the stabilization of the crystal packing and influence the phenomenon and properties in the solid state, one such example is polymorphism. The extensive literature on compounds related to the presence of organic fluorine has been excellently combined in reviews8,20–22 and a book chapter.23 It was initially postulated that the weak interactions involving fluorine like C–H⋯F–C hydrogen bonds are only significant in the absence of any other strong intermolecular forces.24 The analysis of the nature of fluorine interactions on many molecules has been performed, wherein the possibility of the formation of strong hydrogen bonds was eliminated. Amongst these, benzene,12,18 naphthalene, anthracene and phenanthrene,16 isoquinolines,24,25 trifluoroacetophenones,26N-phenylmaleimides and corresponding phthalimides,27 benzonitriles,28 pyridines,29 azobenzenes,30N-benzylideneanilines,31,32 toluene,33 and N-methyl-N-phenylbenzamides34 are a few examples to be mentioned. There is no sufficient literature on the study of these intermolecular interactions in molecules wherein strong intermolecular forces are present.20 However, fluorinated N-(2-chloropyridin-4-yl)-N′-phenylureas,35 fluoro-N-(pyridyl)benzamides,36 fluorine-substituted benzoic acid,37 and fluorine-substituted benzimidazoles38 are a few examples wherein the evaluation of interactions involving fluorine was realized in the presence of a strong hydrogen bond. Keeping in mind the above-mentioned points, improvements aimed towards the understanding of the nature, capability and energetics of interactions involving organic fluorine (in particular C–H⋯F and C–F⋯F–C interactions) in the presence of a strong hydrogen bond have been undertaken by our research in the past few years.17,39–41 Our main goal was (i) a systematic exploration of the participation of the fluorine atom in different intermolecular interactions, (ii) robustness of the interaction (i.e. how often they are present) in the formation of different supramolecular motifs, (iii) the role of hybridization of the C-atom to which fluorine is attached and finally (iv) the calculation of stabilization energy and topological parameters such as the electron densities (ρ), Laplacian (∇2ρ), local potential energy (Vb), and kinetic potential energy (Gb) at the bond critical point by quantum theory of atoms in molecules (QTAIM).42 In this regard, a large library of molecules containing the organic fluorine group have been synthesized, crystallized and investigated for different intermolecular interactions involving organic fluorine. The systems were designed such that they have only one strong donor and acceptor (the amide group –NH–CO– in our studies) atom, which connects at least one phenyl ring wherein the position of the fluorine (connected to the C-atom) of different hybridization, [C(sp2)–F and –C(sp3)–F3 groups] may vary, keeping the main molecular connectivity invariant. Hence, different molecules, namely fluorine substituted benzanilides,17 fluorine and –CF3 substituted N-phenylacetamides and N-methylbenzamides,39 and –CF3 substituted benzanilides have been studied.40 The purpose of selecting the CF3 group is as follows: (i) the –CF3 group being strongly electron-withdrawing in nature increases the acidity of hydrogen atoms in its vicinity, (ii) better H-bond acceptor abilities of C(sp3)–F can be exploited, (iii) in addition to C–H⋯F–C hydrogen bonds, the propensity of the formation of other interactions namely C–F⋯F–C and C–F⋯π is now increased and (iv) in addition to all the above-mentioned points, a comparative study of the role of hybridization of the C-atom to which fluorine is attached can be achieved in the crystal.
O and C–F⋯H–Cα interactions have been reviewed by Diederich et al.19 Since then many studies via inputs from crystallography, spectroscopy and theoretical calculations have established the fact that the interactions involving fluorine are ubiquitous and can play an important role in the stabilization of the crystal packing and influence the phenomenon and properties in the solid state, one such example is polymorphism. The extensive literature on compounds related to the presence of organic fluorine has been excellently combined in reviews8,20–22 and a book chapter.23 It was initially postulated that the weak interactions involving fluorine like C–H⋯F–C hydrogen bonds are only significant in the absence of any other strong intermolecular forces.24 The analysis of the nature of fluorine interactions on many molecules has been performed, wherein the possibility of the formation of strong hydrogen bonds was eliminated. Amongst these, benzene,12,18 naphthalene, anthracene and phenanthrene,16 isoquinolines,24,25 trifluoroacetophenones,26N-phenylmaleimides and corresponding phthalimides,27 benzonitriles,28 pyridines,29 azobenzenes,30N-benzylideneanilines,31,32 toluene,33 and N-methyl-N-phenylbenzamides34 are a few examples to be mentioned. There is no sufficient literature on the study of these intermolecular interactions in molecules wherein strong intermolecular forces are present.20 However, fluorinated N-(2-chloropyridin-4-yl)-N′-phenylureas,35 fluoro-N-(pyridyl)benzamides,36 fluorine-substituted benzoic acid,37 and fluorine-substituted benzimidazoles38 are a few examples wherein the evaluation of interactions involving fluorine was realized in the presence of a strong hydrogen bond. Keeping in mind the above-mentioned points, improvements aimed towards the understanding of the nature, capability and energetics of interactions involving organic fluorine (in particular C–H⋯F and C–F⋯F–C interactions) in the presence of a strong hydrogen bond have been undertaken by our research in the past few years.17,39–41 Our main goal was (i) a systematic exploration of the participation of the fluorine atom in different intermolecular interactions, (ii) robustness of the interaction (i.e. how often they are present) in the formation of different supramolecular motifs, (iii) the role of hybridization of the C-atom to which fluorine is attached and finally (iv) the calculation of stabilization energy and topological parameters such as the electron densities (ρ), Laplacian (∇2ρ), local potential energy (Vb), and kinetic potential energy (Gb) at the bond critical point by quantum theory of atoms in molecules (QTAIM).42 In this regard, a large library of molecules containing the organic fluorine group have been synthesized, crystallized and investigated for different intermolecular interactions involving organic fluorine. The systems were designed such that they have only one strong donor and acceptor (the amide group –NH–CO– in our studies) atom, which connects at least one phenyl ring wherein the position of the fluorine (connected to the C-atom) of different hybridization, [C(sp2)–F and –C(sp3)–F3 groups] may vary, keeping the main molecular connectivity invariant. Hence, different molecules, namely fluorine substituted benzanilides,17 fluorine and –CF3 substituted N-phenylacetamides and N-methylbenzamides,39 and –CF3 substituted benzanilides have been studied.40 The purpose of selecting the CF3 group is as follows: (i) the –CF3 group being strongly electron-withdrawing in nature increases the acidity of hydrogen atoms in its vicinity, (ii) better H-bond acceptor abilities of C(sp3)–F can be exploited, (iii) in addition to C–H⋯F–C hydrogen bonds, the propensity of the formation of other interactions namely C–F⋯F–C and C–F⋯π is now increased and (iv) in addition to all the above-mentioned points, a comparative study of the role of hybridization of the C-atom to which fluorine is attached can be achieved in the crystal.
Therefore, we have synthesized a library of substituted benzanilides (eighteen in number) containing both the fluorine and trifluoromethyl group on the same molecule (Scheme 1). Initial investigations into two molecules in this series, namely N-(4-fluorophenyl)-3-(trifluoromethyl)benzamide and 4-fluoro-N-[3-(trifluoromethyl)phenyl]benzamide, demonstrated the existence of short H-bonds with organic fluorine in the presence of strong N–H⋯OC H-bonds.41 The nature of these interactions was tested with criteria for H-bonds proposed recently by IUPAC43 and has also been analyzed using the PIXEL method44–47 and the QTAIM approach.42 It is now well established that the C–H⋯F interactions at short distances are indeed a “true H-bond” and these are not the consequence of crystal packing. In this current work, along with the detailed crystal packing analysis of the remaining newly synthesized compound in this series, our main focus will be on the (i) identification of different robust or reoccurring motifs formed by the interactions involving organic fluorine in the crystal, (ii) investigations of these in terms of their nature, energetics and topological properties using the PIXEL method and the QTAIM approach and (iii) the comparative study on the role of hybridization48 of the C-atom to which fluorine is attached, based on the inputs obtained from current and previous series of molecules17,39,40 having a similar molecular framework.
 |
| | Scheme 1 Synthetic scheme of all the compounds along with their nomenclature plan used in this manuscript are presented. | |
Experimental section
All the compounds were synthesized by the procedure that is already reported in the literature.40Scheme 1 describes the general route for the synthesis of all the 18 compounds and their corresponding nomenclature code used in this paper. All the synthesized compounds were characterized by FTIR [Fig. S1(a–p)], and 1H NMR [Fig. S2(a–p)] (Section S1, ESI†). Melting points were recorded using a DSC [Fig. S3(a–p), ESI†] on the pure powder compounds. Powder X-ray diffraction (PXRD) data were recorded for all the solid compounds and then compared with their calculated PXRD patterns [Fig. S4(a–p), ESI†]. In order to ensure the phase purity, a profile fitting refinement (Section S2, ESI†) was performed using a JANA2000.49 In the case of 1F2T, 2F3T, 2T1F and 3T1F, high values of the profile fitting parameters (Rp, Rwp) were observed, which may indicate the possibility of the presence of more than one phase in the bulk powder.
The details of all the crystallization experiments of all the solid compounds from different solvents and solvent mixtures are presented in the ESI† (Table S1).
Data collection, structure solution and refinement
Single crystal X-ray diffraction data were collected on a Bruker AXS SMART APEX II CCD diffractometer at 100 K. All the data were collected at 100(2) K. All the crystal structures were solved by direct methods using SIR 9250 and refined using the full matrix least-squares method using SHELXL201351 present in the program suite WinGX.52 The non-hydrogen atoms were refined anisotropically and the hydrogen atoms bonded to C and N atoms were positioned geometrically and refined using a riding model with Uiso(H) = 1.2Ueq [C(sp2)]. The disorder associated with the CF3 group (in the case of compounds 2F2T and 2T1F_w) and the positional disorder of the F-atom (in the case of compounds 1F1T, 2F1T, 2F2T, 2T1F, 2T1F_w and 2T2F) were modeled with PART command in SHELXL 2013 at two independent orientations (the major component was labeled ‘A’) (Fig. S5, ESI†). Molecular and packing diagrams were generated using Mercury software.53 Table S2 (ESI†) lists all the crystallographic and refinement data. ORTEPs of all compounds are presented in Fig. S5(a–m) in the ESI.†
Computational tools and theoretical calculations
The PIXEL method [in the CLP computer program package (version 10.2.2012)] has been used for the interaction energy of the selected molecular pairs, extracted from the crystal packing and related by the corresponding symmetry element as mentioned in our previous work.41 In the method, the total interaction energy is partitioned into their Coulombic (Ecoub), polarization (Epol), dispersion (Edisp) and repulsion (Erep) contributions. In the case of disordered molecules, the molecular conformation with the maximum population was considered for the calculations. The PIXEL method was observed to provide better and useful insights into the nature of different types of intermolecular interactions present in the different molecular pairs/motifs.54–57 The PIXEL interaction energy was further compared with the interaction energies obtained from theoretical calculations at the DFT+Disp/B97D58,59 level at the higher aug-cc-pVTZ basis set using TURBOMOLE.60 The hydrogen atoms were moved to neutron values (1.083 Å for C–H) before the calculations. The basis set superposition error (BSSE) for the interaction energies was corrected by using the counterpoise method.61 Table S3 (ESI†) (divided into two parts: S3a and b, separately for C–F⋯F–C in Table S3b, ESI†) lists the selected intermolecular interactions (in the decreasing order of their stabilization energy) in different motifs along with their interaction energies (I.E.) of the motifs. In the case of highly disordered compounds 2T1F_w [having the rotational disorder associated with the CF3 group and the positional disorder of the fluorine atom at the phenyl ring along with the presence of half molecule of water in the asymmetric unit, Fig. S5(h), ESI†], PIXEL calculations have not been performed. Instead, BSSE corrected interaction energies for the selected dimers were calculated at the DFT+Disp/B97D level using the aug-cc-pVTZ basis set (Table S5, ESI†).
Analysis of topological parameters (QTAIM calculations)
Topological calculations on the selected dimers at the crystal geometry were performed using the same procedure as mentioned previously41 using AIMALL (version 13.05.06).62 The selected topological parameters like electron densities (ρc), Laplacian (∇2ρc), local potential energy (Vb), and kinetic energy density (Gb) at the bond critical points (BCPs) were calculated. The dissociation energies for different intermolecular interactions were also estimated through the following two empirical approaches: (i) D.E.V(int) = −0.5Vb (in atomic units)63 and (ii) D.E.G(int) = 0.429Gb (in atomic units),64,65 where D.E.(int) is the dissociation energy of the interaction. Interaction energy (I.E.) = −D.E. Vb and Gb are the local potential and kinetic energy density at the bond critical points (BCPs), respectively. The results of the topological analysis on different molecular pairs are presented in Section S4 (Tables S4 and S5) in the ESI.† Topological parameters of the selected C–H⋯F and C–F⋯F–C interactions in different motifs along with their dissociation energy are presented in Table S4b (ESI†). Compounds having disorder associated with the CF3 group or fluorine (positional disorder) were not considered for the calculations except for 1F1T and 2T2F wherein the positional disorder of fluorine with an occupancy ratio at the two positions was observed to be 0.875(2):0.125(2) and 0.944(3):0.056(3) respectively. In these cases, the molecular conformation with the maximum population was considered for the calculations.
Results and discussion
(1)
N-(2-Fluorophenyl)-2-(trifluoromethyl)benzamide (1F1T)
Compound 1F1T crystallizes in the orthorhombic non-centrosymmetric space group Pna21 with Z = 4. A strong N–H⋯OC H-bond along with a weak C–H⋯π hydrogen bond (motif I, −12.7 kcal mol−1, Table S3a, ESI†) was observed to connect the molecules along the crystallographic a-axis in the formation of the molecular chain [Fig. 1(a)]. Such chains are interconnected via a weak C–H⋯F–Csp3 hydrogen bond (motif V, −1.5 kcal mol−1). Furthermore, there exists the formation of the herringbone pattern observed down the bc plane in the crystal packing, stabilized via the presence of motif II (involving C–H⋯O and C–H⋯F–Csp2 hydrogen bonds, −6.2 kcal mol−1) and weak C–H⋯F–Csp3 hydrogen bonds in motif IV (−1.9 kcal mol−1). Weak C–H⋯OC and C–H⋯F–Csp3 hydrogen bonds were found to stabilize the crystal packing (motif III, −4.8 kcal mol−1) in the generation of a molecular chain [Fig. 1(c)]. It is to be noted that the stabilization energy for a C–H⋯F hydrogen bond was reported to be ∼−0.40 kcal mol−1 (−1.6 kJ mol−1) by ab initio theoretical calculation for the neutral molecule by D'Oria and Novoa.66
 |
| | Fig. 1 (a) Packing of molecules in 1F1Tvia the network of strong N–H⋯OC, weak C–H⋯π and C–H⋯F–Csp3 hydrogen bonds. The Roman numbers in red (in this and also subsequent diagrams) indicate the molecular motifs presented in Table S3 (ESI†). (b) Formation of the herringbone layer of molecules down the bc plane with the utilization of weak C–H⋯O, C–H⋯F–Csp2 and C–H⋯F–Csp3 hydrogen bonds in 1F1T. (c) Formation of the molecular chain in 1F1Tvia weak C–H⋯O and C–H⋯F–Csp3 hydrogen bonds. (d) Selected molecular motifs (denoted with Roman numbers from Table S4, ESI†) in 1F1T, showing different intermolecular interactions. The brown small spheres represent bond critical points (BCPs) on the bond path. | |
The presence of weak C–H⋯F–C hydrogen bonds in the motifs III, IV and V was characterized topologically by using the QTAIM approach. There was the presence of (3, −1) bond critical points (BCPs) recognized for these interactions (Table S4b, ESI†) along with other related interactions present in the respective motifs [Fig. 1(d)].
(2)
N-(3-Fluorophenyl)-2-(trifluoromethyl)benzamide (2F1T)
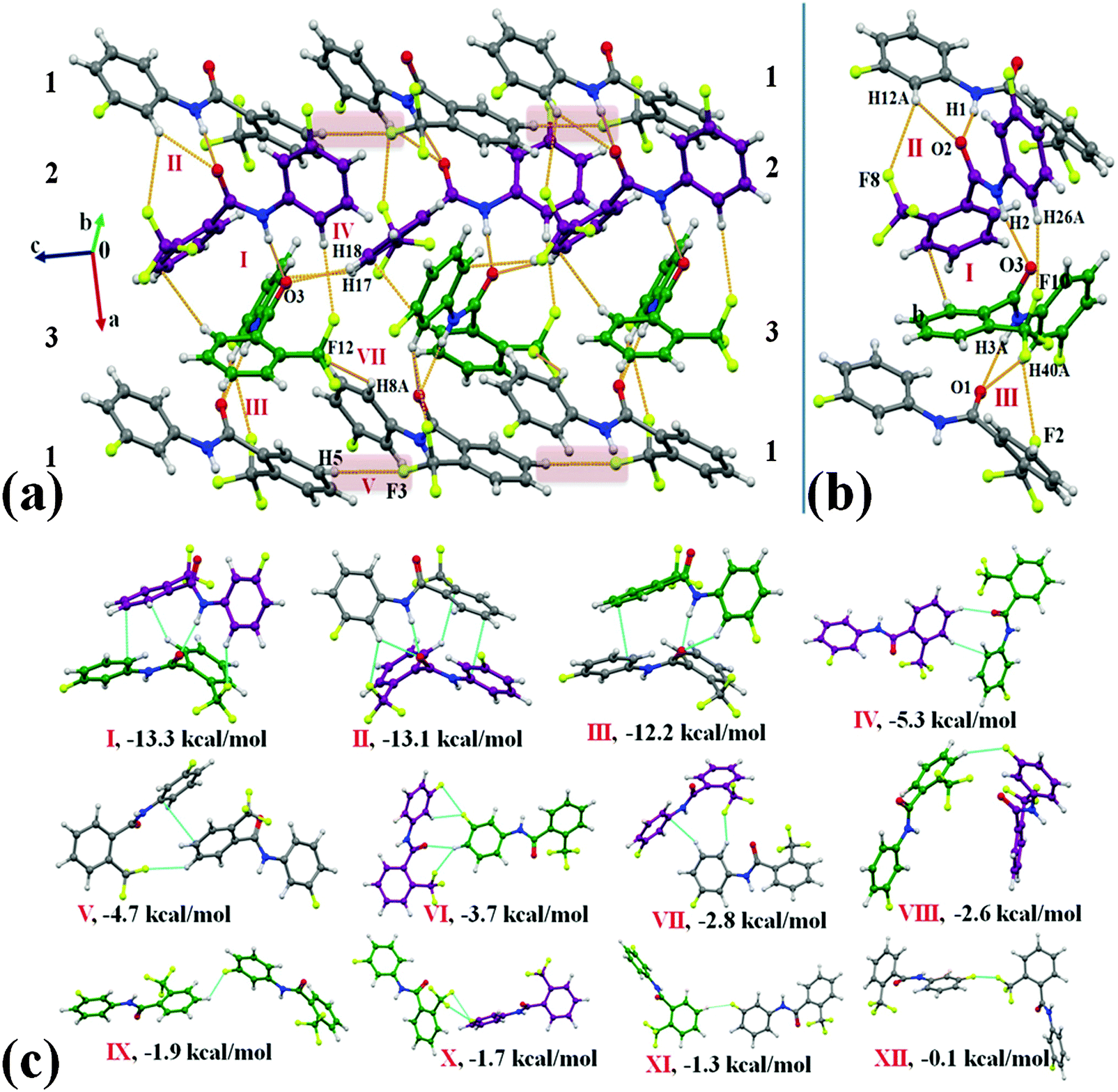
Compound 2F1T crystallizes in the monoclinic centrosymmetric space group P21/c with three molecules in the asymmetric unit (Z′ = 3) (Fig. 2). Two (molecule 2 and 3) out of three molecules in the asymmetric unit observed in the formation of the most stabilized molecular motif I (−13.3 kcal mol−1) involved strong N–H⋯OC, weak C–H⋯π and C–H⋯F–Csp3 hydrogen bonds along with π⋯π interactions. Selected molecular pairs, which contribute towards the stabilization of the crystal packing in 2F1T, are presented in Fig. 2(c) along with their interaction energies (I.E.). The three most stabilized molecule motifs I, II (−13.1 kcal mol−1) and III (−12.2 kcal mol−1), consisting of strong N–H⋯OC hydrogen bonds along with other weak interactions (Table S3a, ESI†), were observed to be involved in the formation of a molecular chain along the crystallographic a-axis in the crystal packing [Fig. 2(a) and (b)]. Such chains were observed to be connected with the utilization of motifs IV, V and VII in the crystal packing [Fig. 2(a)]. The motif IV (I.E. = −5.3 kcal mol−1) consists of short C–H⋯OC (2.39 Å/149°) and C–H⋯π (2.68 Å/141°) hydrogen bonds while the motif V (−4.7 kcal mol−1) involves the presence of weak C–H⋯F–Csp3 hydrogen bonds [Fig. 2(a)] along with π⋯π interactions. A short C–H⋯F–Csp3 (2.39 Å/127°) and a weak C–H⋯π (2.86 Å/155°) hydrogen bond were recognized to connect the molecules in motif VII. Moreover, the packing of molecules in 2F1T are also stabilized by the presence of weak C–H⋯F–Csp2 hydrogen bonds in the motifs VIII, IX and XI with the stabilization energy ranging from −1.3 to −2.6 kcal mol−1 [Table S3, ESI† and Fig. 2(c)]. It is to be noted that the motif X, involves the presence of bifurcated Csp2–F⋯F–Csp3 interactions [type I (2.883 Å, 110°, 94°) and other a “near” type II (3.133 Å, 152°, 83°) contact], provides stabilization towards the crystal packing with an interaction energy of 1.7 kcal mol−1, the nature being primarily of dispersive origin. A very recent charge density analysis has revealed the polarization of the electron density on the fluorine atoms on the trifluoromethyl group, which facilitate the formation of type II C–F⋯F–C contacts in the crystal.67 Furthermore, a very short type I Csp2–F⋯F–Csp3 interaction [2.736 Å, 146°, 149°, motif XII] was observed in the crystal packing, providing almost negligible stabilization [0.1 kcal mol−1] [Fig. 2(c)]. This stabilization energy is similar to the value reported in a recent analysis by ab initio calculations on all the unique dimers, extracted from the crystal structure of CF4, C2F4 and C6F6 by Osuna et al.68
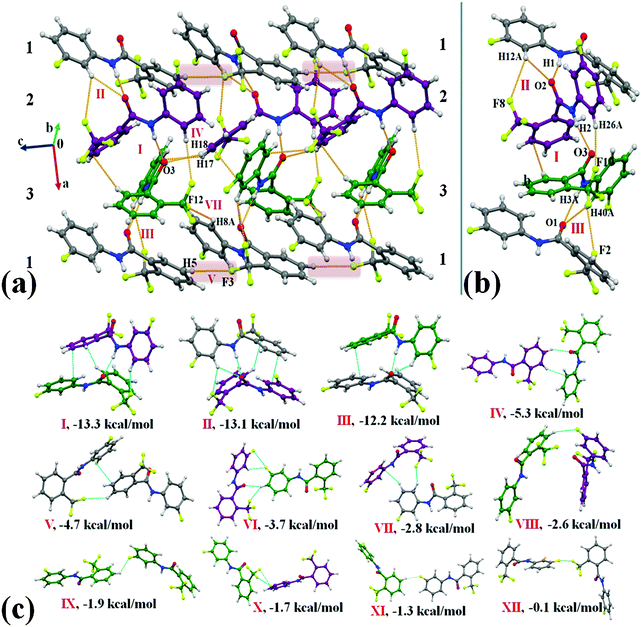 |
| | Fig. 2 (a) Packing of molecules in 2F1Tvia the network of strong N–H⋯OC, weak C–H⋯π and C–H⋯F–Csp3 hydrogen bonds. (b) Clear depiction of the …123… molecular chain along the crystallographic a-axis. Different color codes of carbon atoms indicate the presence of three molecules in the asymmetric unit. Grey: molecule 1, purple: molecule 2 and green: molecule 3. (c) Selected molecular pairs extracted from the crystal packing of 2F1T along with their interaction energies (Table S3, ESI†). | |
(3)
N-(2-Fluorophenyl)-3-(trifluoromethyl)benzamide (1F2T)
Compound 1F2T crystallizes in the monoclinic centrosymmetric space group with Z = 4. Fig. 3(a) depicts the packing of molecules in 1F2T in all the three directions with the utilization of different molecular motifs I to VIII (Table S2, ESI†). The highest stabilized molecular motif I (I.E. = −11.8 kcal mol−1) consists of strong N–H⋯OC, a short and directional C–H⋯F–Csp2 (2.47 Å, 168°) and a short C–H⋯π (2.66 Å, 158°) hydrogen bond along with C–F⋯π interactions. The motif I along with motif III [I.E. = −4.8 kcal mol−1; consists of a short C–H⋯π (2.61 Å, 159°) and C–H⋯OC hydrogen bond] and motif V (I.E. = −1.9 kcal mol−1; involves a bifurcated C–H⋯F–Csp3 along with the presence of C–F⋯π interactions) were observed to pack the molecules along the c-axis with the utilization of c-glide perpendicular to b-axis. A short and directional C–H⋯F–Csp2 (2.45 Å, 168°) hydrogen bond along with the two π⋯π interactions in the motif II (−6.4 kcal mol−1) was observed to connect the molecules along the crystallographic a-axis. The molecular chains, formed along the a-axis, with the utilization of motif II [Fig. 3(b)], were observed to be connected via motifs IV, VI to VIII in the generation of a molecular layer down the ab plane. Amongst these, motif VI (−1.5 kcal mol−1) and VII (−1.1 kcal mol−1) were found to consist of a short C–H⋯F–Csp3 (2.47 Å, 161°; 2.57 Å, 139°) hydrogen bond while the motif IV (I.E. = −2.0 kcal mol−1) involves dimeric Csp3–F⋯F–Csp3 interaction and a weakly stabilized (−0.2 kcal mol−1) motif VIII consists of type I Csp3–F⋯F–Csp3 (2.942 Å, 158°, 158°) interactions. Further, QTAIM calculations reveal the presence of a (3, −1) BCP for all C–H⋯F and C–F⋯F–C interactions [Fig. 3(c)].
 |
| | Fig. 3 (a) Packing of the molecules down the bc crystallographic plane in 1F2Tvia the network of strong N–H⋯OC, weak C–H⋯π, C–H⋯F–Csp2 and C–H⋯F–Csp3 hydrogen bonds along with π⋯π, Csp3–F⋯π and Csp3–F⋯F–Csp3 interactions. (b) Part of the crystal packing in 1F2T down the ab plane, displaying the presence of weak C–H⋯F–Csp2 and C–H⋯F–Csp3 hydrogen bonds along with π⋯π (off set) and Csp3–F⋯F–Csp3 interactions. (c) Selected molecular motifs (denoted with Roman numbers from Table S4, ESI†) in 1F2T, showing different intermolecular interactions. The small brown spheres represent bond critical points (BCPs) on the bond path. Only the interacting part of the motifs is shown in case of IV, VI–VIII. | |
(4)
N-(3-Fluorophenyl)-3-(trifluoromethyl)benzamide (2F2T)
Compound 2F2T crystallizes in the orthorhombic centrosymmetric space group Pbcn with Z = 8. The most stabilized molecular motif I (I.E. = −12.1 kcal mol−1) consists of strong N–H⋯OC hydrogen bonds supported by weak C–H⋯O and C–H⋯π hydrogen bonds. A molecular chain, formed via motif I utilizing c-glide, was observed to connect via the weak C–H⋯π hydrogen bonds recognized in the motif II (−4.2 kcal mol−1), VI (−2.1 kcal mol−1), VIII (−1.6 kcal mol−1) [Fig. 4(a)]. Furthermore, the packing of molecules in 2F2T was stabilized by the formation of motifs III (−3.0 kcal mol−1) and IV (−2.4 kcal mol−1), comprising of dimeric weak C–H⋯F–Csp3 hydrogen bonds. It is of interest to note that a short and directional Csp3–F⋯π [3.134 Å, 150°, Table S3, ESI†] interaction, utilizing 2-fold rotation parallel to the b-axis, provided (motif V) stabilization to the crystal packing [2.3 kcal mol−1] with dispersion being a major contributor. Moreover, bifurcated weak C–H⋯F–Csp2 hydrogen bonds (motif VII, −1.7 kcal mol−1) were realized in the formation of a molecular chain, utilizing a c-glide plane perpendicular to the b-axis. Such chains were connected with the presence of motif IX (–0.2 kcal mol−1), possessing a pair of weak type I Csp3–F⋯F–Csp3 interactions including the one at a short distance (2.873 Å, 122°, 122°) [Fig. 4(b)]. It was also noticed that the two sides of the phenyl ring, substituted with the fluorine and –CF3 group, were observed in the formation of different supramolecular motifs in the crystal packing [Fig. 4(c)].
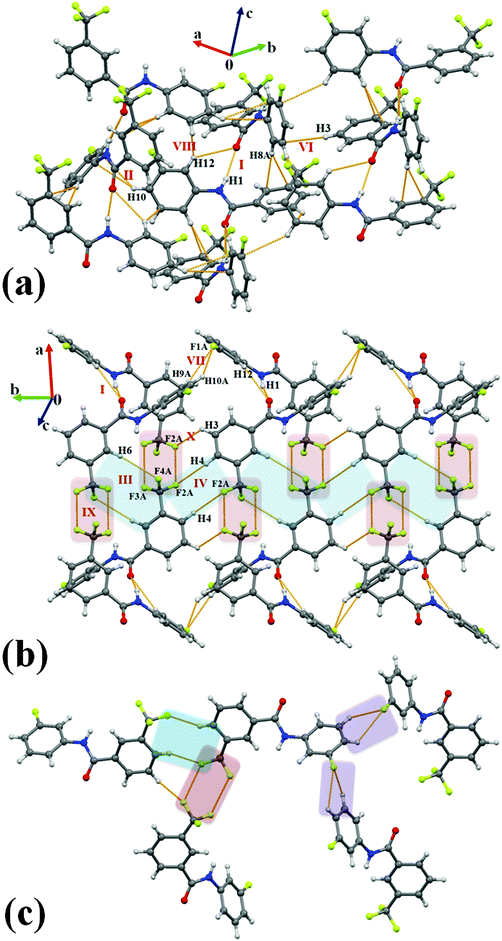 |
| | Fig. 4 (a) Packing of molecules in 2F2Tvia the network of strong N–H⋯OC, and weak C–H⋯O, C–H⋯π hydrogen bonds. (b) Packing of molecules in 2F2T, displaying the network of strong N–H⋯OC, weak C–H⋯O, C–H⋯F–Csp2 and C–H⋯F–Csp3 hydrogen bonds along with Csp3–F⋯F–Csp3 interactions. (c) Part of crystal packing in 2F2T, depicting the presence of different motifs involving fluorine interactions. | |
(5)
N-(3-Fluorophenyl)-4-(trifluoromethyl)benzamide (2F3T)
Compound 2F3T was found to crystallize in the monoclinic centrosymmetric space group C2/c with Z = 8. The strong N–H⋯OC hydrogen bond along with the weak C–H⋯OC hydrogen bond and π⋯π interactions (motif I, −12.3 kcal mol−1) were observed to engage in the formation of molecular chains along the crystallographic b-axis. Such a chain is connected by the utilization of weak C–H⋯π hydrogen bonds and π⋯π interactions in the formation of a molecular layer down the bc plane [Fig. 5(a)], involved in the next three stabilized motifs II (−7.6 kcal mol−1), III (−7.4 kcal mol−1) and IV (−6.1 kcal mol−1). The packing of molecules down the bc plane [Fig. 5(b)] was detected as the formation of a molecular sheet with the utilization of bifurcated weak and short C–H⋯F–Csp3 hydrogen bonds [2.55 Å, 145° (motif VIII, −1.3 kcal mol−1); 2.55 Å, 125° (motif IX, −1.1 kcal mol−1)], along the b-axis. Furthermore, the crystal packing of 2F3T was also observed to be stabilized by the formation of similarly stabilized molecular motifs V (−2.2 kcal mol−1) and VI (−2.1 kcal mol−1) [Fig. 5(c)]. The motif V consists of bifurcated weak C–H⋯F–Csp2 hydrogen bonds while the motif VI involves in the formation of dimeric Csp3–F⋯F–Csp3 interactions. In addition, a type II Csp3–F⋯F–Csp3 interaction (motif VII, −1.9 kcal mol−1) was also observed to form a chain, utilizing the 21 screw axis along the crystallographic b-axis in the crystal packing [Fig. 5(d)]. The selected motifs, containing weak C–H⋯F and Csp3–F⋯F–Csp3 interactions, were characterized by the QTAIM theory and the presence of (3, −1) BCPs was observed on the bond path of these interactions [Fig. 5(e) and Table S5, ESI†].
 |
| | Fig. 5 (a) Formation of a molecular layer down the bc crystallographic plane with the utilization of strong N–H⋯OC, weak C–H⋯π hydrogen bonds and π⋯π interactions in 2F3T. (b) Packing of molecules down the ab plane via the networks of strong N–H⋯OC and weak C–H⋯F–Csp3 hydrogen bonds in 2F3T. (c) Part of the crystal packing in 2F3T, displaying the formation of bifurcated weak C–H⋯F–Csp2 hydrogen bonds and dimeric Csp3–F⋯F–Csp3 interactions. (d) Part of the crystal packing 2F3T showing the presence of Csp3–F⋯F–Csp3 interactions forming a chain motif along with a strong N–H⋯OC hydrogen bond. (e) Selected molecular motifs (Table S4, ESI†) in 2F3T, showing different intermolecular interactions. The small brown spheres represent bond critical points (BCPs) on the bond path. Only the interacting part of the motif was shown in the case of VI–IX. | |
(6)
N-(4-Fluorophenyl)-4-(trifluoromethyl)benzamide (3F3T)
Compound 3F3T crystallizes in the triclinic centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with Z = 2. As expected, the most stabilized molecular motif I (−11.7 kcal mol−1) was noticed to involve strong N–H⋯OC hydrogen bonds along with weak C–H⋯OC hydrogen bonds and π⋯π interactions. The motif I was found to be involved in the formation of a molecular chain along the crystallographic a-axis. Such chains were interlinked via dimeric weak C–H⋯F–Csp2 hydrogen bonds [motif VII (−1.4 kcal mol−1) and VIII (−1.1 kcal mol−1)] and dimeric weak Csp3–F⋯F–Csp3 interactions [motif IX (−0.7 kcal mol−1)] in the formation of a molecular layer down the ab plane. Further, in the molecular packing of 3F3T, the molecular chains formed with the utilization of motifs VIII and IX were observed to be connected by the molecular motifs II to VI which consist of other weak interactions in the crystal packing [Fig. 6(b)]. The motif II involves the presence of a weak C–H⋯π hydrogen bond and a short Csp3–F⋯OC (3.118 Å, 120°) interaction, the interaction energy being −6.1 kcal mol−1. The motifs III and IV were noticed to provide similar stabilization towards the crystal packing (−4.7 kcal mol−1 and −4.0 kcal mol−1 respectively) but observed to involve different interactions. The motif III consists of weak C–H⋯F–Csp3 and C–H⋯π hydrogen bonds while the weak π⋯π interaction was observed in motif IV. Moreover, a dimeric short C–H⋯F–Csp3 (2.45 Å, 157°) contact was observed to stabilize (motif V, −2.8 kcal mol−1) the crystal packing in 3F3T. In addition, dimeric weak Csp3–F⋯π interactions (motif VI, −2.2 kcal mol−1) were also observed to provide stabilization to the crystal packing in 3F3T. The weak interactions involving fluorine in 3F3T were studied with QTAIM theory and the presence of (3, −1) BCP was observed for these interactions in their corresponding molecular motifs [Fig. 6(c)].
with Z = 2. As expected, the most stabilized molecular motif I (−11.7 kcal mol−1) was noticed to involve strong N–H⋯OC hydrogen bonds along with weak C–H⋯OC hydrogen bonds and π⋯π interactions. The motif I was found to be involved in the formation of a molecular chain along the crystallographic a-axis. Such chains were interlinked via dimeric weak C–H⋯F–Csp2 hydrogen bonds [motif VII (−1.4 kcal mol−1) and VIII (−1.1 kcal mol−1)] and dimeric weak Csp3–F⋯F–Csp3 interactions [motif IX (−0.7 kcal mol−1)] in the formation of a molecular layer down the ab plane. Further, in the molecular packing of 3F3T, the molecular chains formed with the utilization of motifs VIII and IX were observed to be connected by the molecular motifs II to VI which consist of other weak interactions in the crystal packing [Fig. 6(b)]. The motif II involves the presence of a weak C–H⋯π hydrogen bond and a short Csp3–F⋯OC (3.118 Å, 120°) interaction, the interaction energy being −6.1 kcal mol−1. The motifs III and IV were noticed to provide similar stabilization towards the crystal packing (−4.7 kcal mol−1 and −4.0 kcal mol−1 respectively) but observed to involve different interactions. The motif III consists of weak C–H⋯F–Csp3 and C–H⋯π hydrogen bonds while the weak π⋯π interaction was observed in motif IV. Moreover, a dimeric short C–H⋯F–Csp3 (2.45 Å, 157°) contact was observed to stabilize (motif V, −2.8 kcal mol−1) the crystal packing in 3F3T. In addition, dimeric weak Csp3–F⋯π interactions (motif VI, −2.2 kcal mol−1) were also observed to provide stabilization to the crystal packing in 3F3T. The weak interactions involving fluorine in 3F3T were studied with QTAIM theory and the presence of (3, −1) BCP was observed for these interactions in their corresponding molecular motifs [Fig. 6(c)].
 |
| | Fig. 6 (a) Packing of molecules in 3F3T down the ab crystallographic plane with the utilization of strong N–H⋯OC, weak C–H⋯OC, C–H⋯F–Csp2 hydrogen bonds along with Csp2–F⋯F–Csp2, Csp3–F⋯F–Csp3 and π⋯π interactions. (b) Packing of molecules in 3F3T down the bc crystallographic plane via weak C–H⋯F–Csp2, C–H⋯F–Csp3 and C–H⋯π hydrogen bonds along with Csp2–F⋯F–Csp2, Csp3–F⋯F–Csp3, Csp3–F⋯OC, Csp3–F⋯π and π⋯π interactions. (c) Selected molecular motifs (Table S4, ESI†) in 3F3T, showing different intermolecular interactions. The small brown spheres represent bond critical points (BCPs) on the bond path. Only the interacting part of the motif is shown in the case of III, V–VII, IX and X. | |
(7) 2-Fluoro-N-(3-(trifluoromethyl)phenyl)benzamide (2T1F)
Compound 2T1F crystallizes in the centrosymmetric orthorhombic space group Pbca with Z = 8. A strong N–H⋯OC hydrogen bond along with π⋯π interactions (motif I, −11.6 kcal mol−1) was observed in the generation of a zig-zag chain with the utilization of b-glide perpendicular to the a-axis. The chains are interconnected utilizing motif VII which consists of the weak near type II Csp3–F⋯F–Csp3 (2.990 Å, 108°, 154°) interactions (I.E. being −1.0 kcal mol−1). The next most stabilized motif II (−4.5 kcal mol−1), consisting of weak π⋯π interaction, was found to connect the molecular chain [Fig. 7(b)] generated via a 21-screw along the b-axis utilizing weak C–H⋯π hydrogen bonds (motif V, −2.8 kcal mol−1). Furthermore, there was the formation of a herringbone pattern down the ac plane [Fig. 7(c)] observed in the crystal packing of 2T1F, exploiting motifs III, IV and VI. The motif III (−3.0 kcal mol−1) was noticed to involve weak C–H⋯O and C–H⋯F–Csp3 hydrogen bonds while it was short bifurcated C–H⋯F–Csp2 hydrogen bonds which were observed in the motif IV, the stabilization energy being 2.8 kcal mol−1. Moreover, a weak C–H⋯F–Csp3 hydrogen bond was recognized in the motif VI (−1.4 kcal mol−1).
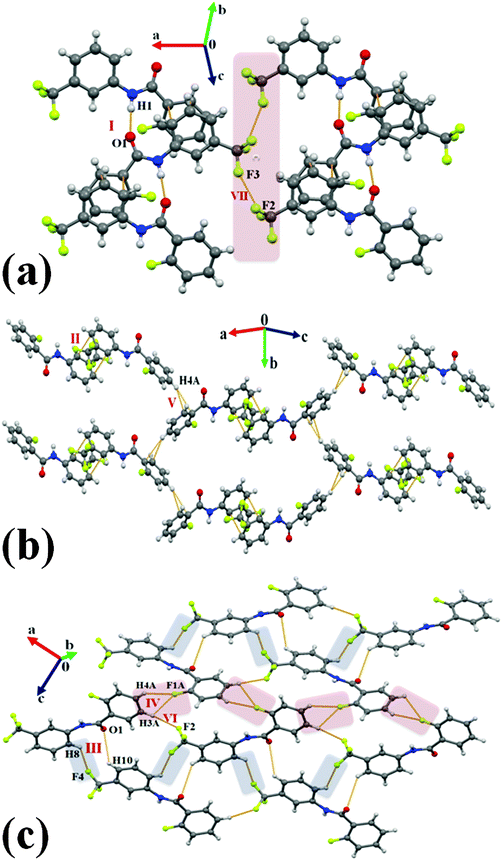 |
| | Fig. 7 (a) Packing of molecules in 2T1Fvia strong N–H⋯OC hydrogen bonds along with Csp3–F⋯F–Csp3 and π⋯π interactions. (b) Packing of molecules in 2T1Fvia the network of weak C–H⋯π hydrogen bonds and π⋯π interactions. (c) Formation of a molecular layer in 2T1F down the ac crystallographic plane with the utilization of weak C–H⋯O, C–H⋯F–Csp2 and C–H⋯F–Csp3 hydrogen bonds. | |
(8) 2-Fluoro-N-(3-(trifluoromethyl)phenyl)benzamide hydrate (2T1F_w)
Hydrate of the compound 2-fluoro-N-(3-(trifluoromethyl)phenyl)benzamide hydrate (2T1F_w) crystallizes in the orthorhombic centrosymmetric space group Pbcn with half molecule of water in the asymmetric unit (Table S2, ESI†). It has been characterized using differential scanning calorimetry (DSC), thermal gravimetry analysis (TGA) and hot stage microscopy (HSM) (Section S3 in the ESI†). The packing of the molecules in 2T1F_w was observed to be stabilized by the solvent molecule with the formation of strong hydrogen bonds like N–H⋯Owater and Owater–H⋯OC, noticed in motifs I and II with I.E. being −5.04 kcal mol−1 and −5.63 kcal mol−1 respectively [Table S3c, ESI,† and Fig. 8(a)]. A stacked motif III along the a-axis (utilizing a 21-screw), consisting of two weak C–H⋯F–Csp3 hydrogen bonds along with π⋯π interactions, is a highly stabilized (−14.2 kcal mol−1) molecular pair in the crystal packing [Fig. 8(a) and Table S3c, ESI†]. The packing of the molecules down the bc plane shows the formation of a molecular layer utilizing motifs I, II, IV and V [Fig. 8(b)]. The molecular chain with alternate interactions of the compound and solvent was observed to be connected with weak C–H⋯F–Csp3 hydrogen bonds (motif V, −1.64 kcal mol−1) utilizing c-glide perpendicular to the b-axis [Fig. 8(b)]. Moreover, the presence of highly short type I Csp3–F⋯F–Csp3 interactions was recognized in the weakly stabilized molecular motif VI (−0.42 kcal mol−1) and relatively destabilized motif VII (+1.29 kcal mol−1) (Table S3c, ESI†). It is to be noted here that the molecules in the destabilized motif VII, consisting of highly short type I Csp3–F⋯F–Csp3 interactions (2.663 Å, 132°, 132°), were observed to be stabilized by the presence of water molecules via the formation of two strong Owater–H⋯OC hydrogen bonds (motif II) in the crystal packing [Fig. 8(a)].
 |
| | Fig. 8 (a) Part of the crystal packing in 2T1F_w displaying the formation of strong N–H⋯Owater and Owater–H⋯OC, and weak C–H⋯F–Csp3 hydrogen bonds along with Csp3–F⋯F–Csp3 and π⋯π interactions. (b) Packing view down the bc plane in 2T1F_w displaying the formation of the molecular layer utilizing interactions involving the solvent along with C–H⋯F–Csp3 hydrogen bonds. | |
(9) 2-Fluoro-N-(4-(trifluoromethyl)phenyl)benzamide (3T1F)
Compound 3T1F crystallizes in the non-centrosymmetric orthorhombic space group P21cn with Z = 4. The packing of molecules in 3T1F involves the formation of a molecular chain along the b-axis with the utilization of strong N–H⋯OC along with weak C–H⋯OC hydrogen bonds and π⋯π interactions (motif I, −9.7 kcal mol−1, Table S3, ESI†). Such chains are interconnected by the utilization of next two similarly stabilized motifs II (−6.5 kcal mol−1) and III (−6.3 kcal mol−1) in the formation of a molecular layer down the (101) plane [Fig. 9(a)]. The motif II involved a pair of weak C–H⋯π hydrogen bonds along with short Csp3–F⋯F–Csp3 (2.840 Å, 127°, 171°) and short Csp2–F⋯CO (3.134 Å, 146°) interactions (Table S3, ESI†) whereas a short and directional C–H⋯π (2.65 Å, 156°) and C–H⋯F–Csp3 (2.56 Å, 160°) hydrogen bonds were present in motif III. Furthermore, a molecular chain formed by the utilization of bifurcated C–H⋯F–Csp3 hydrogen bonds (motif IV, −1.9 kcal mol−1, the 21 screw parallel to the a-axis) was observed to be interconnected via motifs II, III and V in the packing of molecules down the ac plane in 3T1F. The motif V (−1.6 kcal mol−1) was found to consist of weak C–H⋯F–Csp3 hydrogen bonds. These were the presence of (3, −1) BCPs identified in the case of weak C–H⋯F and C–F⋯F–C interactions [Fig. 9(c) and Table S4, ESI†].
 |
| | Fig. 9 (a) Packing of molecules in 3T1Fvia the network of strong N–H⋯OC, weak C–H⋯π, and C–H⋯F–Csp3 hydrogen bonds along with Csp3–F⋯F–Csp3, Csp2–F⋯CO and π⋯π interactions. (b) Packing view down the ac plane in 3T1Fvia the network of weak C–H⋯π and C–H⋯F–Csp3 hydrogen bonds along with Csp3–F⋯F–Csp3, Csp2–F⋯CO interactions. (c) Selected molecular motifs (Table S4, ESI†) in 3T1F, showing different intermolecular interactions. Only the interacting part of the motif is shown in the case of IV–V. | |
(10) 3-Fluoro-N-(2-(trifluoromethyl)phenyl)benzamide (1T2F)
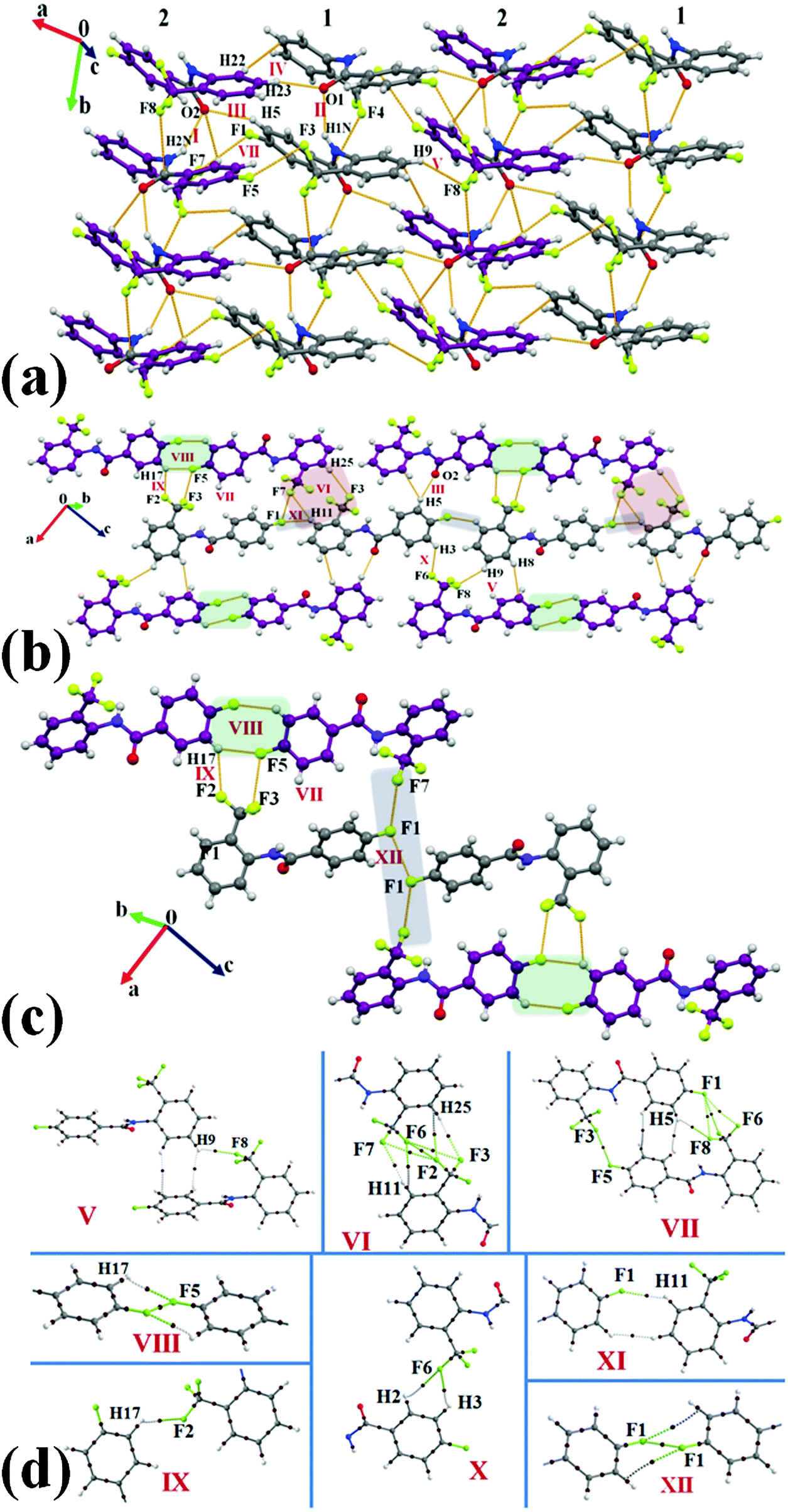
Compound 1T2F crystallizes in the centrosymmetric monoclinic space group P21/c with two molecules in the asymmetric unit (Z = 8). The two most and similarly stabilized molecular motifs I (−12.9 kcal mol−1) and II (−12.4 kcal mol−1) were observed to consist of strong N–H⋯OC hydrogen bonds along with short Csp3–F⋯CO (3.090 Å, 141°; 3.011 Å, 141° respectively, Table S3, ESI†) and π⋯π interactions. These motifs would appear to be involved in the formation of a …1212… type molecular chain along the crystallographic a-axis. Such chains are recognized to interlink with the exploitation of next four stabilized molecular motifs III, IV, V and VI. In the similarly stabilized motifs III (−4.6 kcal mol−1) and IV (−4.4 kcal mol−1), there were short and directional C–H⋯OC (2.33 Å, 149°; 2.23 Å, 168°) hydrogen bonds recognized with the contribution from electrostatic (Coulombic + polarization) being significant (46% and 54% respectively). Further, motifs V (involving a bifurcated weak C–H⋯F–Csp3 and C–H⋯π hydrogen bonds) and VI (involving a bifurcated weak C–H⋯F–Csp3 hydrogen bond) were observed to provide similar stabilization (−3.5 and −3.2 kcal mol−1 respectively) to the crystal packing. Moreover, intermolecular interactions involving organic fluorine of type C–H⋯F, C–F⋯F–C were noticed to stabilize the crystal packing in 1T2F and participate in the generation of different supramolecular motifs VII to XII (Table S3, ESI†) in the crystal packing [Fig. 10(b) and (c)]. The motif VII (−2.8 kcal mol−1) was observed to be composed of dimeric weak C–H⋯F–Csp3 hydrogen bonds along with dimeric Csp3–F⋯F–Csp3 interactions while a dimeric weak C–H⋯F–Csp2 hydrogen bond, interacting side wise, was recognized in the equally stabilized motif VIII. Further, a bifurcated weak C–H⋯F–Csp3 hydrogen bond was observed in motif IX (−1.8 kcal mol−1) whereas in the similarly stabilized motif X (−1.7 kcal mol−1), a short C–H⋯F–Csp2 hydrogen bond (2.48 Å, 137°) was recognized. In the last two weakly stabilized motifs XI (−1.1 kcal mol−1) and XII (−0.6 kcal mol−1), a weak C–H⋯F–Csp2 hydrogen bond and weak type I Csp3–F⋯F–Csp2 interactions were observed respectively. All the molecular pairs consisting of weak C–H⋯F and C–F⋯F–C interactions were analyzed by the theory of QTAIM and the topological parameters were obtained (Table S4, ESI†). There was presence of (3, −1) BCPs noticed for these interactions along with other related contacts in the respective motifs [Fig. 10(d)].
 |
| | Fig. 10 (a) Formation of the …1212… type molecular layer in 1T2Fvia strong N–H⋯OC, weak C–H⋯O, C–H⋯π, and C–H⋯F–Csp3 hydrogen bonds along with Csp3–F⋯CO and π⋯π interactions. C-atoms in purple are shown for the second molecule in the asymmetric unit. (b) Packing view down the bc plane, displaying the network of weak C–H⋯π, C–H⋯F–Csp2, C–H⋯F–Csp3 hydrogen bonds along with Csp3–F⋯F–Csp3 interactions in 1T2F. (c) Packing of molecules in 1T2Fvia the network of weak C–H⋯O, C–H⋯F–Csp2, C–H⋯F–Csp3 hydrogen bonds along with Csp3–F⋯F–Csp3 interactions. C-atoms in purple are shown for the second molecule in the asymmetric unit. (d) Selected molecular motifs (Table S4, ESI†) in 1T2F showing different intermolecular interactions. Only the interacting part of the motif is shown in the case of VII–XII. | |
(11) 3-Fluoro-N-(3-(trifluoromethyl)phenyl)benzamide (2T2F)
Compound 2T2F crystallizes in the non-centrosymmetric monoclinic space group Cc with four molecules in the unit cell. A strong N–H⋯OC hydrogen bond along with π⋯π interactions (motif I, −11.3 kcal mol−1) steer the molecules along [110] in the formation of a molecular chain. Such chains are interlinked with the involvement of next two stabilized motifs II (−5.5 kcal mol−1) and III (–4.8 kcal mol−1) in the crystal packing. The motif II was recognized to involve weak π⋯π interactions with substantial contribution from dispersion energy towards the total stabilization whereas a weak C–H⋯OC along with two short C–H⋯F–Csp2/Csp3 (2.45 Å, 133°; 2.53 Å, 136° respectively) hydrogen bonds were observed in motif III. Further the molecular ladder formed via the motif III was observed to be connected by the utilization of motifs IV, V and VI in the crystal packing [Fig. 11(b) and (c)]. The motif IV (−2.2 kcal mol−1) consists of bifurcated weak C–H⋯F–Csp3 hydrogen bonds while a short C–H⋯F–Csp3 (2.50 Å, 146°) hydrogen bond was observed in motif V (–1.5 kcal mol−1). Moreover, a weak type I Csp3–F⋯F–Csp3 interaction (motif VI, −1.2 kcal mol−1) was also recognized to stabilize the packing of the molecules in 2T2F (Table S3, ESI†). The presence of (3, −1) BCPs were observed for the weak C–H⋯F and C–F⋯F–C interactions in the respective motifs [Fig. 11(d)].
 |
| | Fig. 11 (a) Packing of molecules in 2T2F with the utilization of strong N–H⋯OC, weak C–H⋯O, C–H⋯F–Csp2, and C–H⋯F–Csp3 hydrogen bonds along with π⋯π interactions. (b) Formation of a molecular layer down the bc plane by the utilization of weak C–H⋯O, C–H⋯F–Csp2, and C–H⋯F–Csp3 hydrogen bonds and Csp3–F⋯F–Csp3 interactions in 2T2F. (c) Packing of molecules in 2T2Fvia weak C–H⋯OC, C–H⋯F–Csp2, and C–H⋯F–Csp3 hydrogen bonds. (d) Selected molecular motifs (Table S4, ESI†) in 2T2F showing different intermolecular interactions. Only the interacting part of the motif is shown in the case of IV–VI. | |
(12) 4-Fluoro-N-(2-(trifluoromethyl)phenyl)benzamide (1T3F)
Compound 1T3F crystallizes in the centrosymmetric monoclinic space group P21/n with two molecules in the asymmetric unit (Z = 8). The two most stabilized motifs I (−12.8 kcal mol−1) and II (−12.7 kcal mol−1), involving similar interactions (strong N–H⋯OC hydrogen bonds along with π⋯π interactions), were observed in the formation of a molecular chain of molecules 1 and 2 in the asymmetric unit, respectively, utilizing n-glide along the b-axis [Fig. 12(a)]. Such chains are interconnected alternatively with the utilizations of motifs III, IV, V and VII. The motifs III and IV provide similar stabilization (−4.4 kcal mol−1 and −4.1 kcal mol−1 respectively) and both are involved in the formation of highly short and directional C–H⋯OC (2.29 Å, 152°; 2.22 Å, 167° respectively, Table S3, ESI†) hydrogen bonds. A short C–H⋯F–Csp3 (2.56 Å, 132°) along with a weak C–H⋯π hydrogen bond were noticed to connect the molecules in motif V whereas a near type II along with a near type I Csp3–F⋯F–Csp2 interactions (2.983 Å, 110°, 165°; 3.128 Å, 90°, 117°) were recognized to stabilize (I.E. being −2.7 kcal mol−1) the crystal packing, existing as motif VII (Table S2, ESI†). Further, the packing of molecules in 1T3F stabilized the formation of two dimeric motifs VI (−3.3 kcal mol−1) and VIII (−2.1 kcal mol−1) [Fig. 12(b)]. The motif VI involved two C–H⋯F–Csp3 hydrogen bonds along with weak Csp3–F⋯F–Csp3 interactions (Table S3, ESI†). And the dimeric motif VIII was observed to possess short C–H⋯F–Csp2 (2.48 Å, 131°) hydrogen bonds in the crystal packing in 1T3F. Moreover, there were three equally stabilized (1.5 kcal mol−1) molecular motifs IX, X and XI observed in the crystal packing and all were noticed to involve weak C–H⋯F–C hydrogen bonds (Table S2, ESI†). Further a motif (XII, −1.3 kcal mol−1), consisting of weak Csp2–F⋯F–Csp2 interactions and connecting molecules ‘1’ of the asymmetric unit, was observed to stabilize the crystal packing in 1T3F [Fig. 12(c)]. The weak C–H⋯F–C hydrogen bonds and C–F⋯F–C interactions present in the motifs V to XII in 1T3F were characterized by the theory of AIM. The presence of (3, −1) BCPs was observed for these interactions [Fig. 12(d)].
 |
| | Fig. 12 (a) Packing of molecules in 1T3Fvia the network of strong N–H⋯OC, weak C–H⋯O, C–H⋯π, and C–H⋯F–Csp3/sp2 hydrogen bonds along with Csp3–F⋯CO and Csp3–F⋯F–Csp2 interactions. C-atoms in purple are shown for the second molecule in the asymmetric unit. (b) Formation of the molecular layer down the ac plane with the utilization of weak C–H⋯OC, C–H⋯π, C–H⋯F–Csp3/sp2 hydrogen bonds along with Csp3–F⋯F–Csp2 interactions in 1T3F. (c) Part of crystal packing in 1T3F showing the presence of weak C–H⋯F–Csp3/sp2 hydrogen bonds along with C–F⋯F–C interactions. (d) Selected molecular motifs (Table S4, ESI†) in 1T3F showing different intermolecular interactions. Only the interacting part of the motif is shown in the case of V–XII. | |
(13) 4-Fluoro-N-(4-(trifluoromethyl)phenyl)benzamide (3T3F)
Compound 3T3F crystallizes in the centrosymmetric monoclinic space group P21/c with Z = 4. A strong N–H⋯OC hydrogen bond, supported by weak C–H⋯OC and C–H⋯π hydrogen bonds (motif I, 11.6 kcal mol−1), was observed in the formation of a molecular chain utilizing c-glide perpendicular to the b-axis in 3T3F [Fig. 13(a) and (b)]. Such chains are interconnected via the motifs II, III, V and VIII. The weak π⋯π interactions were observed to connect molecules in the dimeric motifs II (−5.9 kcal mol−1) and III (−4.6 kcal mol−1). In the motif V (−2.6 kcal mol−1), weak C–H⋯F–Csp2 hydrogen bonds along with C–H⋯CO interactions were recognized whereas a short C–H⋯F–Csp3 hydrogen bond was observed in motif VIII (−1.4 kcal mol−1). The packing of the molecules in 3T3F involved the formation of a herringbone pattern down the crystallographic ab plane with the utilization of motifs IV [−2.7 kcal mol−1; involving short C–H⋯π hydrogen bonds (2.79 Å, 133°; Table S2, ESI†)], V and VIII [Fig. 13(c)]. Moreover, a bifurcated C–H⋯F–Csp2 hydrogen bond (motif VII, −2.2 kcal mol−1), utilizing c-glide, was noticed in the formation of a molecular chain [Fig. 13(d)]. The chain was interlinked via the presence of motifs VI, VIII and IX. The motif VI (−2.6 kcal mol−1) was found to involve the weak C–H⋯F–Csp3 hydrogen bond in the formation of a molecular chain utilizing c-glide [Fig. 13(d)]. Dimeric and weakly stabilized (−0.4 kcal mol−1) short Csp3–F⋯F–Csp3 interactions were identified in the motif IX. The molecular motifs V–VIII and IX, consisting of weak interactions involving fluorine, were studied topologically using the approach of QTAIM. Bond critical points at the bond path for these interactions were observed [Fig. 13(e)].
 |
| | Fig. 13 (a) Packing of molecules in 3T3Fvia the network of strong N–H⋯OC, weak C–H⋯O, C–H⋯π, C–H⋯F–C hydrogen bonds along with π⋯π interactions. (b) Depiction of a molecular chain via the motif I in 3T3F. (c) Formation of a layer down the ab plane, utilizing weak C–H⋯F–C hydrogen bonds and C–H⋯CO interactions in 3T3F. (d) Formation of a weak bifurcated C–H⋯F–C hydrogen bonds along with Csp3–F⋯F–Csp3 interactions down the bc plane in 3T3F. (e) Selected molecular motifs (Table S4, ESI†) in 3T3F, showing different intermolecular interactions. Only the interacting part of the motif is shown in the case of V–VII and IX. | |
Therefore, the detailed analysis of all crystal structures revealed that the C(sp2)/(sp3)–F group was observed in the formation of different robust structural motifs in the presence of a strong N–H⋯OC hydrogen bond and other related weak interactions like C–H⋯OC, C–H⋯π, and π⋯π. Moreover, in many cases, different supramolecular motifs (Table S3, ESI†) by C(sp2)/(sp3)–F from both sides of the molecules can combine co-operatively in the formation of bigger structural motifs in the crystal structures.
Insights from atoms in molecules calculations
It is of interest to conduct topological characterization of H⋯F and F⋯F interactions, observed in this class of compounds (including the previously reported crystal structures17,38–40) and the analysis of the nature of these interactions along with the relationship of different topological parameters at BCP with bond path length. For this purpose, the selected dimers involving H⋯F or F⋯F interactions were identified and QTAIM calculations have been performed at their crystal geometry in accordance with the procedure reported in our earlier work.41 The results of the calculations are presented in Tables S4 and S5 of Section S4 in the ESI.† Topological parameters of the selected C–H⋯F and C–F⋯F–C interactions are given in Table S4b (ESI†).
It is to be noted here that some of the important criteria for an interaction to be called a hydrogen bond or a closed shell type are:69,70 (i) ρ value at BCP lies within the range [0.013, 0.270] e Å−3, (ii) positive value of the Laplacian of the electron density [∇2ρ > 0] indicates closed shell interactions, (iii) the range of Laplacian values [0.578 < ∇2ρ (e Å−5) < 3.350] indicates the presence of a “H-bond” and (iv) the value of |Vb|/Gb < 1 for hydrogen bonds71,72 and closed shell interactions; Vb and Gb are the potential and kinetic energy density at BCP. It is of interest to validate the nature of H⋯F and F⋯F interactions with these criteria.
Analysis of H⋯F bonding interactions
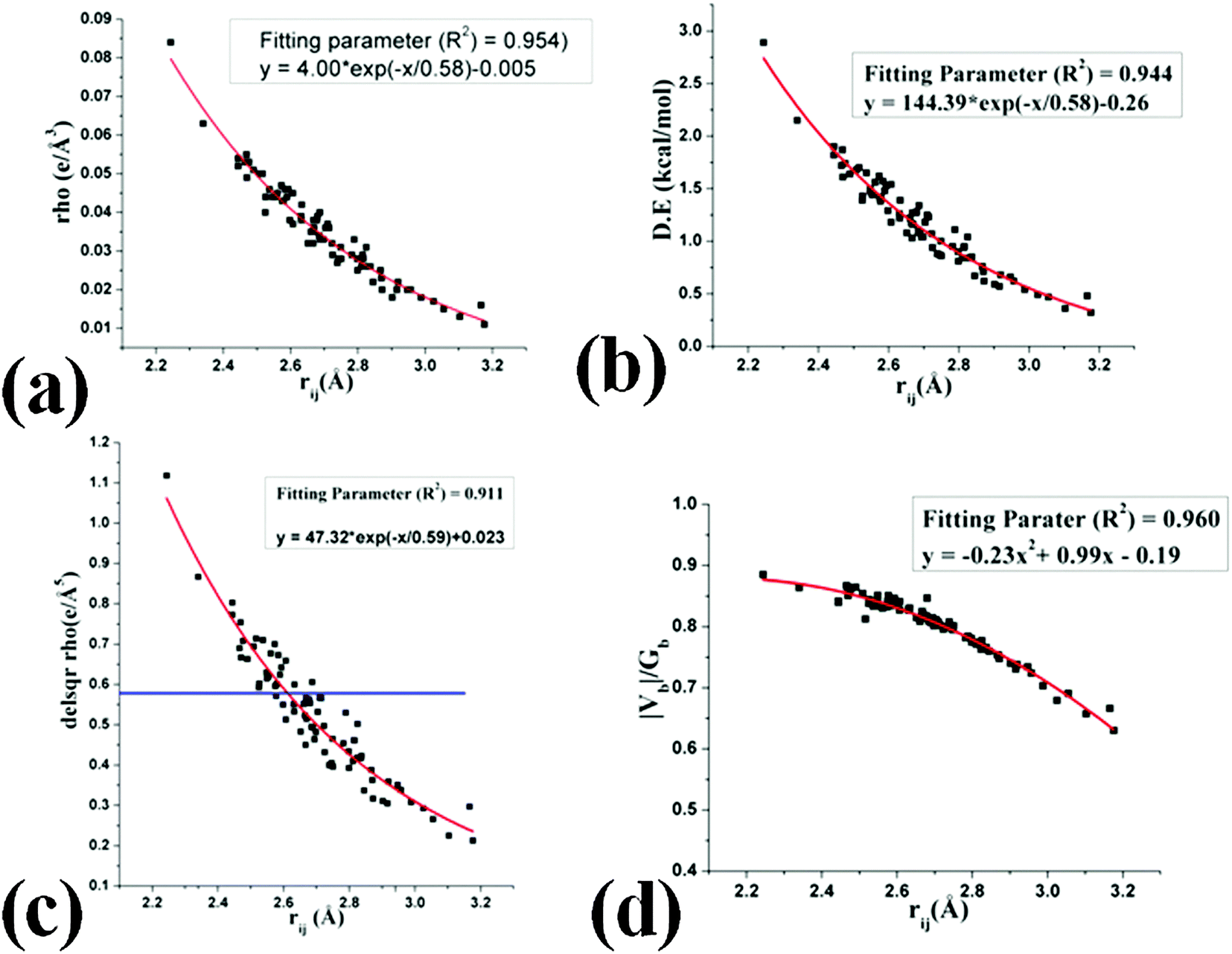
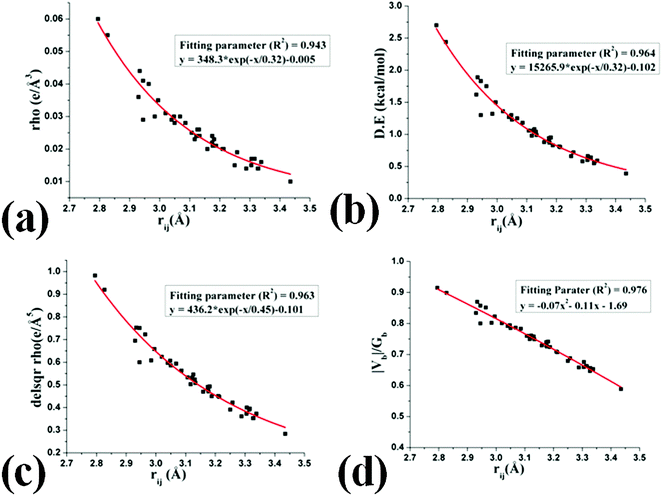
H⋯F interactions were observed to lie in the range of 2.2–3.0 Å for all structures in the current work. As observed earlier,41 the dependency of electron density (ρb) shows exponential dependence with the bond path length (rij) [Fig. 14(a)] with the values in the range of 0.085 > ρb > 0.015 e Å−3. Hence these fulfill the criterion for the interactions to be called a hydrogen bond. The dissociation energy of these interactions (vary exponentially with rij, [Fig. 14(b)]) were observed between ∼3.0 to ∼0.4 kcal mol−1. The value of Laplacian observed to be positive [(∇2ρ > 0), Fig. 14(c)] for the entire bond path length (rij) suggesting the closed shell nature of these H⋯F interactions. It is to be noted that for a short C–H⋯F bond path length (rij) at less than the sum of van der Waals radii73 of H and F, 2.67 Å, the values of the Laplacian are in accordance with the Koch and Popelier criteria for the existence of hydrogen bonds.74 Moreover, the values of |Vb|/Gb were also observed to be less than one (criteria for hydrogen bonds71,72) for the entire range of the bond path [Fig. 14(d)].
 |
| | Fig. 14 Variation of (a) electron density (e Å−3) at the BCP, (b) dissociation energy (D.E) (kcal mol−1) with the H⋯F bond path length (Å), (c) Laplacian (∇2ρ) at the BCP and (d) |Vb|/Gb with the H⋯F bond path length (Å). | |
Furthermore, it was of interest to study the role of hybridization of the C-atoms to which fluorine is attached in the C–H⋯F interactions. Hence the variation of dissociation energy (D.E.) (kcal mol−1) and electron density (e Å−3) at the BCP and with H⋯F bond path length (Å) for C–H⋯F–C(sp2) and C–H⋯F–C(sp3) has been compared [Fig. 15(a) and (b), Tables S4 and S5, ESI†]. It was observed that the highest stabilized molecular motifs primarily consist of C(sp2)–H⋯F–C(sp2) hydrogen bonds in preference to C(sp2)–H⋯F–C(sp3) hydrogen bonds in the crystal with the difference in energies of stabilization involving fluorine atoms attached to sp2 and sp3 carbon is not significant in the molecular crystal [circled area in Fig. 15(a) and (b)].
 |
| | Fig. 15 Comparison of variation of (a) dissociation energy (D.E) (kcal mol−1) and (b) electron density (e Å−3) at the BCP and with the H⋯F bond path length (Å) for C–H⋯F–C(sp2) [red curve] and C–H⋯F–C(sp3) [blue curve] [total 153 data points (98 for the red curve and 55 for the blue curve)]. | |
Analysis of F⋯F bonding interactions
There are numerous C–F⋯F–C interactions observed in the crystal packing of this series of compounds (Tables S4 and S5, ESI†). Hence, it was also of interest to do the topological characterization of F⋯F interactions. The nature and role of C–F⋯F–C interactions were recently studied both experimentally and theoretically by many researchers.75,76 The previous report by the QTAIM analysis of intramolecular C–F⋯F–C interactions on a rigid isolated molecular system showed that these are closed shell interactions and can impart as much as 14 kcal mol−1 of local stabilization.77 In the present work, QTAIM analysis of intermolecular C–F⋯F–C interactions has been performed and the results show the exponential dependence of electron density at the BCP (ρb), dissociation energy (D.E.) and Laplacian (∇2ρ) of the electron density at the BCP with the F⋯F bond path length (rij) (Fig. 16). Values of electron density and dissociation energy (local stabilization) were observed between ∼0.060 > ρb (e Å−3) > ∼0.010 and ∼2.8 < D.E. (kcal mol−1) < ∼0.5 for the ∼2.80 > rij (Å) > 3.45. These values are slightly less or comparable with weak C–H⋯F–C interactions in the present work and observed to be similar to the previously observed values.67,77–80 Furthermore, for the entire range of the F⋯F bond path length, Laplacian values (∇2ρ) > 1 and |Vb|/Gb < 1 were observed. Hence it can be concluded that the intermolecular F⋯F interactions in the present work are of closed shell in nature. Furthermore, the nature of C(sp3)/(sp2)–F⋯F–C(sp3)/(sp2) interactions from current and previous studies along with reports from experimental charge density analysis (Table 1) have been compared in Fig. 17. The results show the small differences in the ρb (e Å−3) at the BCP for different types of C–F⋯F–C interactions involving fluorine atoms attached to sp2 and sp3 carbon atoms. The shorter C–F⋯F–C interactions were observed to be associated with the C(sp2)–F bond in the molecule in the crystal. It is also of interest to notice variations in ρb, dissociation energy (D.E.) and Laplacian (∇2ρ) of the electron density at the BCP for a given value of rij. Different values of electron density at the BCP for C(sp3)/(sp2)–F⋯F–C(sp3)/(sp2) interactions for the nearly same rij (A) of the F⋯F bond path (near to the sum of van der Waals radii of the F-atom, 2.94 A) in the case of 3, 5, 6, 7, 8, 10 have been observed [Fig. 18]. This feature may be explained due to the cooperative interplay amongst the different possible intermolecular bond paths present in the motifs 3, 5, 6, 7 and 8 whereas in the case of 10 (having the least value of ρb among these), only one C(sp3)–F⋯F–C(sp3) is present.
 |
| | Fig. 16 Variation of (a) electron density (e Å−3), (b) dissociation energy (D.E) (kcal mol−1) with the F⋯F bond path length (Å) [42 data points], (c) Laplacian (∇2ρ) at the BCP and (d) |Vb|/Gb with the F⋯F bond path length (Å). | |
Table 1 Topological parameters from previous reports on C–F⋯F–C interactions by charge density analysis of high resolution X-ray data
| Nature of interactions |
R
ij
(Å) |
ρ (e Å−3) |
∇2ρ (e Å−5) |
Ref. |
| C(sp2)–F⋯F–C(sp2); type I |
2.8091 |
0.049 |
1.030 |
78
|
| C(sp2)–F⋯F–C(sp2); type I |
2.8187 |
0.040 |
0.820 |
79
|
| C(sp2)–F⋯F–C(sp2); type II |
2.8240 |
0.040 |
0.900 |
80
|
| C(sp3)–F⋯F–C(sp3); type II |
2.9255 |
0.030 |
0.633 |
67
|
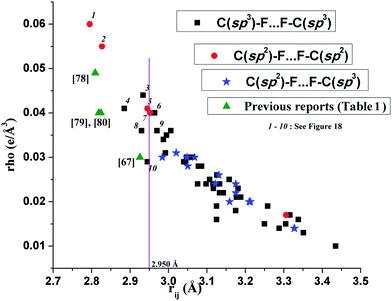 |
| | Fig. 17 Comparison of the variation of the electron density (e Å−3) at the BCP and with the F⋯F bond path length (Å) for C(sp3)/(sp2)–F⋯F–C(sp3)/(sp2) interactions from current and previous studies [total 64 data points]. | |
 |
| | Fig. 18 Showing first ten more stabilized motifs in Fig. 17, having C(sp3)/(sp2)–F⋯F–C(sp3)/(sp2) interactions near or below the sum of van der Waals radii of two F-atoms, 2.94 Å. | |
Conclusion
Following the analysis of the 15 crystal structures (including one hydrate) from the 18 newly synthesized compounds along with the inputs from previously reported structures, the role of organic fluorine in the crystal packing has been analyzed in the presence of strong hydrogen bonds. The formation of many “reoccurring” structural motifs by the Csp2–F and CF3 group has been identified and investigated in terms of their nature, energetics and topological properties, which were quantified by the PIXEL method and the QTAIM approach. It was observed that the highest stabilized molecular motifs primarily consist of C(sp2)–H⋯F–C(sp2) hydrogen bonds in preference to C(sp2)–H⋯F–C(sp3) H bonds in the crystal. Moreover, the formation of hydrogen bonds by the C(sp2)/(sp3)–F group was observed to be present over the entire distance range between 2.2 and 2.7 Å (from the QTAIM approach), albeit the difference in energies of stabilization involving fluorine atoms attached to sp2 and sp3 carbon is not of significance in molecular crystals. Following the analysis of C(sp2)/(sp3)–F⋯F–C(sp2)/(sp3) interactions from the QTAIM approach, it was observed that these fulfill the criteria of these being of the closed shell type for the entire F⋯F bond path length and provide local stabilization (indicates the formation of bonds) similar to the case of weak hydrogen bonds in crystals. For future study, it would be of interest to extend this study to donor atoms in different hybridization environments with the F–C(sp2)/(sp3) group in different chemical environments. Furthermore, it is also of interest to investigate the effect of increasing fluorination on the molecule and the impact on the stabilization energies for the different supramolecular motifs involving organic fluorine present in the crystal.
Acknowledgements
PP thanks UGC-India for research scholarship. D. C. thanks IISER Bhopal for research facilities and infrastructure and DST-SERB for research funding.
References
- H.-J. Schneider, Chem. Sci., 2012, 3, 1381–1394 RSC.
- G. K. S. Prakash, F. Wang, M. Rahm, J. Shen, C. Ni, R. Haiges and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 11761–11764 CrossRef CAS PubMed.
- F. F. Awwadi, R. D. Willett, K. A. Peterson and B. Twamley, Chem. – Eur. J., 2006, 12, 8952 CrossRef CAS PubMed.
- M. Perez-Torralba, M. A. Garcia, C. Lopez, M. C. Torralba, M. R. Torres, R. M. Claramunt and J. Elguer, Cryst. Growth Des., 2014, 14, 3499–3509 CAS.
- I. Saraogi, V. G. Vijay, S. Das, K. Sekar and T. N. G. Row, Cryst. Eng., 2003, 6, 69–77 CrossRef CAS.
- M. T. Scerba, S. Bloom, N. Haselton, M. Siegler, J. Jaffe and T. Lectka, J. Org. Chem., 2012, 77, 1605–1609 CrossRef CAS PubMed.
- X. Xu, B. Pooi, H. Hirao and S. H. Hong, Angew. Chem., Int. Ed., 2014, 53, 1283–1287 CrossRef CAS PubMed.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC and references therein.
- L. Shimoni and J. P. Glusker, Struct. Chem., 1994, 5, 383–397 CrossRef CAS.
- J. A. K. Howard, V. J. Hoy, D. O'Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613–12622 CrossRef CAS.
- J. D. Dunitz and R. Taylor, Chem. – Eur. J., 1997, 3, 89–98 CrossRef CAS.
- V. R. Thalladi, H.-C. Weiss, D. Blaser, R. Boese, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1998, 120, 8702–8710 CrossRef CAS.
- J. Parsch and J. W. Engels, J. Am. Chem. Soc., 2002, 124, 5564–5572 CrossRef.
- J. D. Dunitz, ChemBioChem, 2004, 5, 614–621 CrossRef CAS PubMed.
- J. D. Dunitz and W. B. Schweizer, Chem. – Eur. J., 2006, 12, 6804–6815 CrossRef CAS PubMed.
- F. Cozzi, S. Bacchi, G. Filippini, T. Pilati and A. Gavezzotti, Chem. – Eur. J., 2007, 13, 7177–7184 CrossRef CAS PubMed.
- D. Chopra and T. N. G. Row, CrystEngComm, 2008, 10, 54–67 RSC.
- T. S. Thakur, M. T. Kirchner, D. Blaser, R. Boese and G. R. Desiraju, CrystEngComm, 2010, 12, 2079–2085 RSC.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- D. Chopra and T. N. G. Row, CrystEngComm, 2011, 13, 2175–2186 RSC and references therein.
- D. Chopra, Cryst. Growth Des., 2012, 12, 541–546 CAS and references therein.
- P. A. Champagne, J. Desroches and J.-F. Paquin, Synthesis, 2015, 306–322 CAS and references therein.
-
P. Panini and D. Chopra, in Hydrogen Bonded Supramolecular Structures, ed. Z. Li and L. Wu, Lecture Notes in Chemistry, Springer-Verlag, Berlin, Heidelberg, 2015, vol. 87, pp. 37–67, ISBN: 978-3-662-45755-9 Search PubMed.
- A. R. Choudhury and T. N. G. Row, Cryst. Growth Des., 2004, 4, 47–52 CAS.
- A. R. Choudhury and T. N. G. Row, CrystEngComm, 2006, 8, 265–274 RSC.
- D. Chopra, V. Thiruvenkatam, S. G. Manjunath and T. N. G. Row, Cryst. Growth Des., 2007, 7, 868–874 CAS.
- A. Schwarzer and E. Weber, Cryst. Growth Des., 2008, 8, 2862–2874 CAS.
- V. Vasylyeva and K. Merz, Cryst. Growth Des., 2010, 10, 4250–4255 CAS.
- V. Vasylyeva, O. V. Shishkin, A. V. Maleev and K. Merz, Cryst. Growth Des., 2012, 12, 1032–1039 CAS.
- M. Karanam and A. R. Choudhury, Cryst. Growth Des., 2012, 13, 4803–4814 Search PubMed.
- G. Kaur, P. Panini, D. Chopra and A. R. Choudhury, Cryst. Growth Des., 2012, 12, 5096–5110 CAS.
- G. Kaur and A. R. Choudhury, Cryst. Growth Des., 2014, 14, 1600–1616 CAS.
- K. Merz, M. V. Evers, F. Uhl, R. I. Zubatyuk and O. V. Shishkin, Cryst. Growth Des., 2014, 14, 3124–3130 CAS.
- P. Panini and D. Chopra, New J. Chem., 2015, 39, 8720–8738 RSC.
- A. Abad, C. Agullo, A. C. Cunat, C. Vilanova and M. C. R. de Arellano, Cryst. Growth Des., 2006, 6, 46–57 CAS.
- P. Mocilac, K. Donnelly and J. F. Gallagher, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 189–203 CAS.
- R. Dubey, M. S. Pavan and G. R. Desiraju, Chem. Commun., 2012, 48, 9020–9022 RSC.
- M. Perez-Torralba, M. A. Garcia, C. Lopez, M. C. Torralba, M. R. Torres, R. M. Claramunt and J. Elguer, Cryst. Growth Des., 2014, 14, 3499–3509 CAS.
- P. Panini and D. Chopra, CrystEngComm, 2013, 15, 3711–3733 RSC.
- P. Panini and D. Chopra, CrystEngComm, 2012, 14, 1972–1989 RSC.
- P. Panini and D. Chopra, Cryst. Growth Des., 2014, 14, 3155–3168 CAS.
-
R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, UK, 1990 Search PubMed.
-
(a) E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaeergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CAS;
(b) E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaeergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- A. Gavezzotti, New J. Chem., 2011, 35, 1360–1368 RSC.
- A. Gavezzotti, Mol. Phys., 2008, 106, 1473–1485 CrossRef CAS.
- L. Maschio, B. Civalleri, P. Ugliengo and A. Gavezzotti, J. Phys. Chem. A, 2011, 115, 11179–11186 CrossRef CAS PubMed.
- J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2012, 12, 5873–5877 CAS.
- R. Shukla and D. Chopra, CrystEngComm, 2015, 17, 3596–3609 RSC.
-
V. Petricek, M. Dusek and L. Palatinus, JANA 2000, 18/12/2007, Institute of Physics, Czech Republic, 2007, http://jana.fzu.cz.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, WinGX, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS
http://www.ccdc.cam.ac.uk/mercury
.
- P. Panini, K. N. Venugopala, B. Odhav and D. Chopra, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 681–696 CAS.
- R. Shukla, T. P. Mohan, B. Vishalakshi and D. Chopra, CrystEngComm, 2014, 16, 1702–1713 RSC.
- D. Dey, T. P. Mohan, B. Vishalakshi and D. Chopra, Cryst. Growth Des., 2014, 14, 5881–5896 CAS.
- P. Panini, R. Shukla, T. P. Mohan, B. Vishalakshi and D. Chopra, J. Chem. Sci., 2014, 126, 1337–1345 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- W. Hujo and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 13942–13950 RSC.
- R. Ahlrichs, M. Baer, M. Haeser, H. Horn and C. Koelmel, Electronic structure calculations on work-station computers: the program system TURBOMOLE, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- F. Bernardi and S. F. Boys, Mol. Phys., 1970, 19, 553 CrossRef.
-
T. A. Keith, AIMALL, version 13.05.06, TK Gristmill Software, Overland Park KS, USA, 2013, http://aim.tkgristmill.com Search PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- I. Mata, I. Alkorta, E. Espinosa and E. Molins, Chem. Phys. Lett., 2011, 507, 185–189 CrossRef CAS.
- M. V. Vener, A. N. Egorova, A. V. Churakov and V. G. Tsirelson, J. Comput. Chem., 2012, 33, 2303–2309 CrossRef CAS PubMed.
- E. D'Oria and J. J. Novoa, CrystEngComm, 2008, 10, 423–436 RSC.
- V. R. Hathwar, D. Chopra, P. Panini and T. N. G. Row, Cryst. Growth Des., 2014, 14, 5366–5369 CAS.
- R. M. Osuna, V. Hernàndez, J. T. L. Navarrete, E. D'Oria and J. J. Novoa, Theor. Chem. Acc., 2011, 128, 541–553 CrossRef CAS.
-
P. L. Popelier, Atoms in Molecules: An Introduction, Prentice Hall, London, 2000 Search PubMed.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef CAS.
- I. Mata, I. Alkorta, E. Molins and E. Espinosa, Chem. – Eur. J., 2010, 16, 2442–2452 CrossRef CAS PubMed.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- G. Zhang, W. He and D. Chen, Mol. Phys., 2014, 112, 1736–1744 CrossRef CAS.
- R. J. Baker, P. E. Colavita, D. M. Murphy, J. A. Platts and J. D. Wallis, J. Phys. Chem. A, 2012, 116, 1435–1444 CrossRef CAS PubMed.
- R. A. Cormanich, R. Rittner, D. O'Hagan and M. Bühl, J. Phys. Chem. A, 2014, 118, 7901–7910 CrossRef CAS PubMed.
- C. F. Matta, N. Castillo and R. J. Boyd, J. Phys. Chem. A, 2005, 109, 3669–3681 CrossRef CAS PubMed.
- D. Chopra, T. S. Cameron, J. D. Ferrara and T. N. G. Row, J. Phys. Chem. A, 2006, 110, 10465–10477 CrossRef CAS PubMed.
- V. R. Hathwar and T. N. G. Row, Cryst. Growth Des., 2011, 11, 1338–1346 CAS.
- M. S. Pavan, K. D. Prasad and T. N. G. Row, Chem. Commun., 2013, 49, 7558–7560 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 981582–981594. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj03211c |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence