
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and structural chemistry of bicyclic hexaaza-dithia macrocycles containing pendant donor groups†
Mathias
Gressenbuch
,
Ulrike
Lehmann
and
Berthold
Kersting
*
Institut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: b.kersting@uni-leipzig.de; Fax: +49 (0)341-97-36199
First published on 25th January 2016
Abstract
A short and efficient synthesis of a series of macrobicyclic aza-thioethers with pendant allyl (8, 13, 14), cyanethyl (15), 3-aminopropyl (16), 2-methoxyacetyl (17, 19), 2-methoxyethyl (18, 20), and tert-butyloxycarbonyl substituents (22, 23) has been achieved. The parent macrobicycles 1 and 2 are readily alkylated without overalkylation and without affecting the masked thiolate functions. The protocol is also feasible for the synthesis of macrobicycles with different alkyl groups on the benzylic and central nitrogen atoms of the linking diethylene triamine units. The identity of the compounds was substantiated using ESI MS, FT-IR, 1H-NMR, and 13C-NMR spectroscopy. The crystal and molecular structures of six compounds (8, 15, 17·3DMSO, 19·2DMSO·2H2O, 20 and 23) were additionally solved. The macrocycles are rather flexible and can adopt folded or stepped conformations. The ability of the compounds to form inclusion complexes with DMSO is also demonstrated. The crystal structures are governed by extensive inter- and intramolecular CH⋯π interactions.
Introduction
The synthesis of macrocyclic ligands with pendant donor arms is highly desirable in view of a range of potential applications such as catalysis, selective cation binding, biomimetic chemistry, and radionuclide therapy.1–5 Thus, the chemistry of aliphatic polyaza-macrocycles has been well investigated as has the chemistry of their corresponding thia analogs. Many monocyclic macrocycles with side arms terminated with N, O, S, or P donor groups have been prepared and their coordination chemistry investigated.6,7 Surprisingly, the chemistry of multidentate macrocycles with mixed N and S donor functions has received much less attention.8–10 This is true in particular for the families of amine-thioether ligands containing aromatic thioether groups.11–13 An early example is the 14-membered aromatic N2S2 macrocycle, which was described by Lindoy and co-workers.14,15 Until now only few more ligands of this sort have been reported in the literature.16–23Our group has reported the synthesis of the macrobicyclic azathioethers 124 and 225 and of some alkylated derivatives 3–5 (Fig. 1).26,27 More recently, we have reported the first examples of bicyclic aminothioethers bearing pendant hydroxyethyl groups (6, 7).28 The structures of the free macrocycles 3,294,255,27 and 628 have been determined. The macrocycles adopt a folded conformation in which the two aryl rings and the alkyl residues forming a cavity. However, none of these structures are clathrate, nor inclusion, complexes. As part of this program, we sought to extend our exploration to other variants of this versatile ligand system. To our knowledge, there are no systematic investigations on such hexaaza-thioether macrocycles. We have obtained a series of new macrobicyclic compounds bearing olefinic, nitrile, amine, and methoxy groups in place of the alkyl functions. Herein, we describe their synthesis and solid state structures. The effect of the pendant groups on the structural features is discussed, and compared with those of the parent ligand systems 1–3.
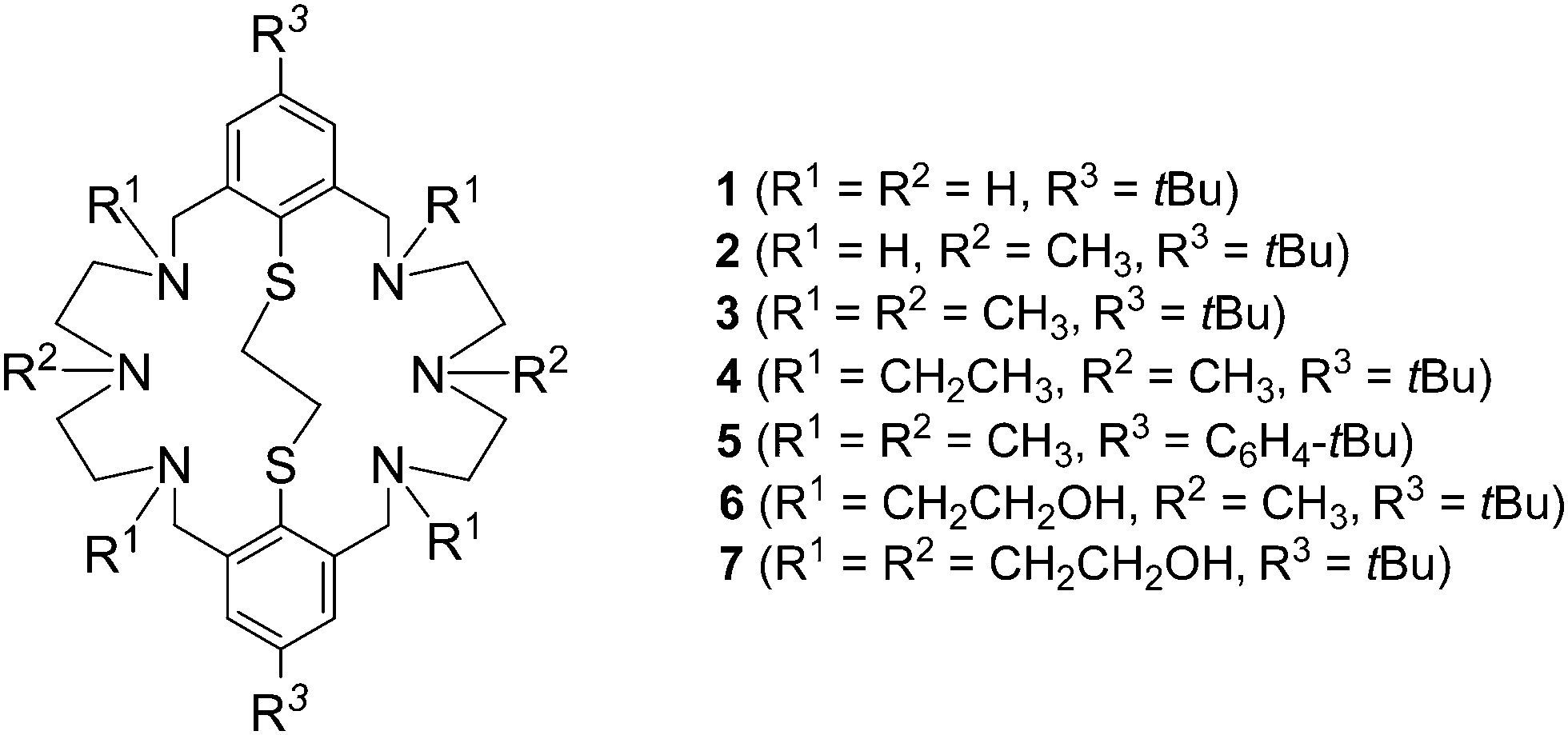 | ||
| Fig. 1 Formula of macrobicyclic aza-thioethers 1–7. | ||
Experimental section
Materials and physical measurements
The bicyclic aza-thioethers 124 and 2,25 1,2-bis(4-tert-butyl-2,6-diformylthioethane) (10),24 bis(2-phthalimidoethyl)amine30 (11), and tert-butyl-bis(2-aminoethyl)carbamate (21)31 were prepared according to literature. Melting points were determined with an Electrothermal IA9000 series instrument using open glass capillaries and are uncorrected. Mass spectra were obtained using the positive ion electrospray ionization modus (ESI) on a FT-ICR-MS Bruker Daltronics APEX II instrument. NMR spectra were recorded on a Bruker DRX-600, Bruker DRX-400 or Varian Mercury plus 400 spectrometer. Chemical shifts refer to solvent signals. The atom labels used to assign the NMR signals are not identical with those used in the X-ray structures. Elemental analyses were carried out with a VARIO EL – elemental analyzer.Synthesis and analysis of compounds
The corresponding spectra for the IR, 1H-NMR, 13C-NMR are included in the ESI,† for each of the synthetized compounds. A summary of the obtained results are shown here.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 7.50 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 31.60 (C(CH3)3), 34.98 (C(CH3)3), 35.82 (SCH2)2, 50.89 ((NCH2CH2)2N), 51.71 ((NCH2CH2)2N), 56.88 (ArCH2NCH2CHCH2), 57.51 (ArCH2), 59.23 (NCH2CHCH2), 116.99 (ArCH2NCH2CHCH2), 117.46 (NCH2CHCH2), 124.86 (ArC-3,3′), 128.18 (ArC-2,2′), 136.35 (ArCH2NCH2CHCH2), 136.34 (CHCH2), 143.71 (ArC-1), 151.40 (ArC-4). This compound was additionally characterized by X-ray crystallography.
CH2), 7.37 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 31.39 (C(CH3)3), 34.76 (C(CH3)3), 36.69 (SCH2)2, 42.94 (CH3), 51.16 ((NCH2CH2)2N), 55.17 ((NCH2CH2)2N), 56.58 (NCH2CHCH2), 57.91 (ArCH2), 117.50 (CHCH2), 125.73 (ArC-3,3′), 129.31 (ArC-2,2′), 135.42 (CHCH2), 143.78 (ArC-1), 151.24 (ArC-4). This compound was additionally characterized by X-ray crystallography.
CH2), 3.67 (t, 3J = 6.4 Hz, 4 H, (NCH2CH2)2N, 4.98–5.08 (dd, 1 H, 3J = 16.4 Hz, 3J = 10.4 Hz, CHCH2), 5.56–5.63 (m, 2 H, CHCH2), 7.60–7.67 (m, 8 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 36.30 (NCH2CH2)2N), 51.90 (NCH2CH2)2N, 57.03 (CH2CHCH2), 118.47 (CHCH2), 123.59 (ArC-2,2′), 132.73 (ArC-1,1′), 133.96 (ArC-3,3′), 135.56 (CHCH2), 168.74 (CO).
CH2), 5.67 (m, 1 H, CHCH2), 5.93 (m, 2 H, CHCH2). 13C{1H}-NMR (100 MHz, D2O): δ = 34.0 (H2NCH2), 49.6 ((CH2)2N), 56.3 (NCH2), 124.8 (CHCH2), 128.2 (CHCH2).
CH2), 5.17 (m, 1 H, CHCH2), 5.89 (m, 2 H, CHCH2). 13C{1H}-NMR (100 MHz, CD3OD): δ = 38.57 (H2NCH2), 55.96 ((CH2)2N), 57.12 (NCH2), 116.70 (CHCH2), 135.27 (CHCH2).
CH2), 7.50 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 31.60 (C(CH3)3), 34.98 (C(CH3)3), 35.82 (SCH2)2, 50.89 ((NCH2CH2)2N), 51.71 ((NCH2CH2)2N), 56.88 (ArCH2NCH2CHCH2), 57.51 (ArCH2), 59.23 (NCH2CHCH2), 116.99 (ArCH2NCH2CHCH2), 117.46 (NCH2CHCH2), 124.86 (ArC-3,3′), 128.18 (ArC-2,2′), 136.35 (ArCH2NCH2CHCH2), 136.34 (CHCH2), 143.71 (ArC-1), 151.40 (ArC-4). This compound was additionally characterized by X-ray crystallography.
CH2), 7.37 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 31.39 (C(CH3)3), 34.76 (C(CH3)3), 36.69 (SCH2)2, 42.94 (CH3), 51.16 ((NCH2CH2)2N), 55.17 ((NCH2CH2)2N), 56.58 (NCH2CHCH2), 57.91 (ArCH2), 117.50 (CHCH2), 125.73 (ArC-3,3′), 129.31 (ArC-2,2′), 135.42 (CHCH2), 143.78 (ArC-1), 151.24 (ArC-4). This compound was additionally characterized by X-ray crystallography.
CH2), 3.67 (t, 3J = 6.4 Hz, 4 H, (NCH2CH2)2N, 4.98–5.08 (dd, 1 H, 3J = 16.4 Hz, 3J = 10.4 Hz, CHCH2), 5.56–5.63 (m, 2 H, CHCH2), 7.60–7.67 (m, 8 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 36.30 (NCH2CH2)2N), 51.90 (NCH2CH2)2N, 57.03 (CH2CHCH2), 118.47 (CHCH2), 123.59 (ArC-2,2′), 132.73 (ArC-1,1′), 133.96 (ArC-3,3′), 135.56 (CHCH2), 168.74 (CO).
CH2), 5.67 (m, 1 H, CHCH2), 5.93 (m, 2 H, CHCH2). 13C{1H}-NMR (100 MHz, D2O): δ = 34.0 (H2NCH2), 49.6 ((CH2)2N), 56.3 (NCH2), 124.8 (CHCH2), 128.2 (CHCH2).
CH2), 5.17 (m, 1 H, CHCH2), 5.89 (m, 2 H, CHCH2). 13C{1H}-NMR (100 MHz, CD3OD): δ = 38.57 (H2NCH2), 55.96 ((CH2)2N), 57.12 (NCH2), 116.70 (CHCH2), 135.27 (CHCH2).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v) and a solution of 1,2-bis(4-tert-butyl-2,6-diformylphenylthio)ethane 10 (1.08 g, 2.30 mmol) in CH2Cl2 (500 mL) were added simultaneously over the course of 3 h into a EtOH/CH2Cl2 (600 mL, 1:3 v:v) solvent mixture. After the resulting mixture was stirred for further 2 d, the CH2Cl2 solvent was removed under reduced pressure. Sodium borohydride (690 mg, 18.24 mmol) was added and the mixture was stirred at r.t. for another 2 h. The excess reducing agent was destroyed by adding HCl conc (final pH = 1). The mixture was subsequently evaporated to dryness, re-dissolved in CH2Cl2/H2O (100 mL, 1:1 v:v), and the pH was adjusted to ∼13 with aqueous KOH (5 M). After stirring for 2 h, the layers were separated and the aqueous phase was extracted with dichloromethane (4 × 150 mL). The organic fractions were combined and dried with anhydrous K2CO3. Evaporation of the solvent gave a foam, which was recrystallized from EtOH to give 1.41 g (88%) of the title compound. Found: C 68.90, H 9.29, N 12.29, S 9.11; C40H64N6S2 (693.11) requires: C 69.32, H 9.31, N 12.13, S 9.25. IR (KBr): ν/cm−1 = 3424 m, 3261 m, 2955 vs, 2902 s, 2867 s, 2803 vs, 1599 m, 1577 m, 1451 vs, 1417 s, 1366 s, 1330 w, 1293 w, 1262 m, 1230 w, 1205 m, 1149 s, 1094 s, 1054 s, 1029 s, 921 m, 897 w, 804 s. 1H-NMR (300 MHz, CDCl3): δ = 1.30 (s, 18 H, C(CH3)3), 2.81 (s, 4 H, SCH2), 2.96 (m, 8 H, (NCH2CH2)2N), 3.14 (m, 8 H, (NCH2CH2)2N), 3.30 (d, J = 6.6 Hz, 4 H, CH2CHCH2), 4.12 (s, 8 H, ArCH2), 5.21 (m, 2 H, CHCHH), 5.23 (m, 2 H, CHCHH), 5.90 (m, 2 H, CHCH2), 6.32 (s (br), 4 H, NH), 7.72 (s, 4 H, ArH). 13C{1H}-NMR (75 MHz, CDCl3): δ = 31.36 (C(CH3)3), 35.20 (C(CH3)3), 37.58 (SCH2)2, 49.02 ((NCH2CH2)2N), 50.31 ((NCH2CH2)2N), 51.41 (ArCH2), 60.20 (NCH2CHCH2), 119.32 (CHCH2), 128.34 (ArC-3,3′), 130.07 (ArC-2,2′), 134.56 (CHCH2), 140.16 (ArC-1), 154.18 (ArC-4).
:1 CH2Cl2/CH3CN solvent mixture. Evaporation of the CH2Cl2 provided a colorless solid, which was filtered and dried in air. Yield: 7.08 g (7.6 mmol, 99%), colorless solid. M.p. 170–172 °C. Found: C 66.83, H 7.92, N 17.82; C52H74N12S2 (931.37) requires: C 67.06, H 8.01, N 18.05. m/z (ESI+, MeOH): C52H74N12S2 (930.56) [M + H+]+ calcd: 931.57; found 931.56. IR (KBr): ν/cm−1 = 3426 vw, 3054 m, 2951 vs, 2818 vs, 2246 vs ν(CN), 1593 m, 1556 vw, 1477 m, 1464 s, 1434 s, 1424 s, 1405 m, 1381 m, 1356 s, 1328 m,1292 m, 1276 s, 1248 m, 1215 m, 1171 w, 1139 s, 1110 vs, 1047 s, 1005 m, 987 m, 961 m, 947 m, 887 s, 832 w, 801 vw, 774 m, 733 m, 704 vw, 681 vw, 650 w, 596 w. 1H-NMR (300 MHz, CDCl3): δ = 1.33 (s, 18 H, ArC(CH3)3), 2.38 (m, 8 H, CH2CN),2.40 (m, 4 H, CH2CN), 2.66 (m, 8 H, (NCH2CH2)2N), 2.67 (s, 4 H, SCH2), 2.68 (m, 8 H, (NCH2CH2)2N), 2.73 (m, 8 H, CH2CH2CN), 2.78 (m, 4 H, CH2CH2CN), 3.76 (s, 8 H, ArCH2), 7.53 (s, 4 H, ArH). 13C{1H}-NMR (75 MHz, CDCl3): δ = 17.03 (CH2CN), 17.38 (CH2CN), 31.46 (C(CH3)3), 35.00 (C(CH3)3), 36.16 (SCH2), 47.3 (ArCH2NCH2CH2CN), 51.82 (CH2CH2CN), 52.39 ((NCH2CH2)2N), 52.96 ((NCH2CH2)2N), 63.1 (ArCH2N), 118.90 (ArCH2NCH2CH2CN), 118.96 (CN), 125.20 (ArC-3,3′), 128.17 (ArC-2,2′), 142.71 (ArC-1), 152.26 (ArC-4). This compound was additionally characterized by X-ray crystallography.
:1) solution afforded colorless crystals. Found: C 64.76, H 9.89, N 8.65, S 6.50; C52H92N6O6S2 (961.46) requires: C 64.96, H 9.65, N 8.74, S 6.67. m/z (ESI+, MeOH): C52H92N6O6S2 (960.55) [M + H+]+ calcd: 961.66; found 961.7. IR (KBr): ν/cm−1 = 3425 s, 2953 vs, 2871 vs, 2816 vs, 2336 vw, 1954 vw, 1667 m, 1594 m,1559 w, 1451 vs, 1404 s, 1362 vs, 1297 s, 1261 m, 1199 s, 1119 vs, 1080 vs, 1018 s, 958 m, 927 m, 894 m, 781 w, 733 w, 704 w, 647 w, 558 w. 1H-NMR (600 MHz, 400 K, DMSO-d6): δ = 1.32 (s, 18 H, C(CH3)), 2.64 (t, 3J = 5.3, 8 H, NCH2CH2OCH3), 2.73 (m, 4 H, NCH2CH2OCH3)), 2.75 (s, 4 H, SCH2), 2.85 (m, 16 H, NCH2CH2N), 3.28 (s, 12 H, OCH3), 3.31 (s, 6 H, OCH3), 3.41 (t, 3J = 5.3, 8 H, CH2OCH3), 3.60 (m, 4 H, CH2OCH3), 3.78 (s, 8 H, ArCH2), 7.53 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 31.32 (C(CH3)3), 34.68 (C(CH3)3), 35.57 (SCH2), 50.82 (NCH2CH2OCH3), 50.92 ((NCH2CH2)2N), 52.74 (ArCH2NCH2CH2OCH3), 54.68 ((NCH2CH2)2N), 58.17 (ArCH2N), 58.92 (OCH3), 58.97 (OCH3), 70.50 (CH2OCH3), 70.82 (CH2OCH3), 125.72 (ArC-3,3′), 128.55 (ArC-2,2′), 142.30 (ArC-1), 151.87 (ArC-4).
:1) solution afforded colorless crystals. Found: C 66.13, H 9.78, N 9.41, S 7.22; C48H84N6O4S2 (873.36) requires: C 66.01, H 9.69, N 9.62, S 7.34. m/z (ESI+, MeOH): C48H84N6O4S2 (872.60) [M + H+]+ calcd: 873.61; found 873.60. IR (KBr): ν/cm−1 = 3425 s, 2953 vs, 2871 vs, 2816 vs, 2336 vw, 1954 vw, 1667 m, 1594 m, 1559 w, 1451 vs, 1404 s, 1362 vs, 1297 s, 1261 m, 1199 s, 1119 vs, 1080 vs, 1018 s, 958 m, 927 m, 894 m, 781 w, 733 w, 704 w, 647 w, 558 w. 1H-NMR (600 MHz, 400 K, DMSO-d6): δ = 1.30 (s, 18 H, C(CH3)3), 2.25 (s, 6 H, NCH3), 2.50 (s(br), 8 H, (NCH2CH2)2NCH3), 2.60 (t, 3J = 9.8, 8 H, NCH2CH2OCH3), 2.68 (s (br), 8 H, (NCH2CH2)2NCH3), 2.79 (s, 4 H, SCH2), 3.28 (s, 12 H, OCH3), 3.39 (t, 3J = 9.8, 8 H, NCH2CH2OCH3), 3.80 (s, 8 H, ArCH2), 7.47 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 31.43 (C(CH3)3), 34.87 (C(CH3)3), 36.16 (SCH2), 43.42 (NCH3), 52.35 ((NCH2CH2)2N), 53.37 (ArCH2NCH2CH2OCH3), 58.57 (ArCH2N), 58.93 (OCH3), 71.50 (CH2OCH3), (one CH2 signal not observed), 125.70 (ArC-3,3′), 128.79 (ArC-2,2′), 143.79 (ArC-1), 151.44 (ArC-4).
:1 v/v) at 0 °C was added a solution of 1,2-bis(4-tert-butyl-2,6-diformylphenylthio)ethane (3.34 g, 7.11 mmol) in CHCl3 (500 mL) over the course of 8 h. After the resulting mixture was stirred for further 2 d, the CHCl3 solvent was removed under reduced pressure. Sodium borohydride (2.15 g, 56.88 mmol) was added and the mixture was stirred at r.t. for 18 h. The mixture was evaporated to dryness, then redissolved in CH2Cl2/H2O (100 mL, 1:1, v/v), and the pH was adjusted to ∼13 with aqueous KOH (5 M). After stirring for 2 h, the layers were separated and the aqueous phase was extracted with CH2Cl2 (4 × 50 mL). The organic fractions were combined and dried with anhydrous MgSO4. Evaporation of the solvent gave an oil, which crystallized from EtOH (10 ml) after standing for 4 weeks. Yield: 1.20 g (1.47 mmol, 21%). M.p. 136–138 °C. Found: C 64.75, H 8.58, N 9.75; C44H72N6O4S2 0.5 EtOH (813.21 + 23.04) requires: C 64.63, H 9.04, N 10.05. m/z (ESI+): 813.4 (M + H+). IR (KBr): ν/cm−1 = 3443 s, 3290 m ν(NH), 2965 s, 2927 s, 2865 m, 1693 s ν(CO), 1597 w, 1478 s, 1458 s, 1411 s, 1365 s, 1249 m, 1228 w, 1173 s, 1154 s, 1091 w, 1043 w, 966 w, 886 w, 858 w, 823 w, 774 w. 1H-NMR (400 MHz, CDCl3): δ = 1.29 (s, 18 H, ArC(CH3)3), 1.45 (s, 18 H, C(CH3)3), 2.96 (t, 3J = 6 Hz, 8 H, (NCH2CH2)2N), 3.20 (s, 4 H, SCH2), 3.49 (t, 3J = 6 Hz, 8 H, (NCH2CH2)2N), 3.97 (s, 8 H, ArCH2N), 7.28 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 28.6 (C(CH3)3), 31.4 (ArC(CH3)3), 34.7 (ArC(CH3)3), 37.1 ((ArSCH2)2), 47.8 ((NCH2CH2)2N), 49.3 (ArCH2N), 53.8 ((NCH2CH2)2N), 79.7 (C(CH3)3), 126.4 (ArC-3,3), 130.0 (ArC-2,2), 144.8 (ArC-1), 152.2 (ArC-4), 156.0 (CO).
:1 v/v) was added. After stirring for 2 h, the layers were separated and the aqueous phase was extracted with CH2Cl2 (4 × 25 mL). The organic fractions were combined and dried with anhydrous MgSO4. Evaporation gave the crude product, which was purified by recrystallization from CH2Cl2/EtOH (1:1 v/v). Yield: 910 mg (1.05 mmol, 75%), colorless crystals. Mp 217–219 °C. Found: C 65.93, H 9.23, N 9.59; C48H80N6O4S2 (869.32) requires: C 66.32, H 9.28, N 9.67. m/z (ESI+, MeOH): C48H80N6O4S2 (868.57) [M + H+]+ calcd: 869.58; found 869.50. IR (KBr): ν/cm−1 = 3444 m, 2965 s, 2857 m, 2802 s, 2716 w, 1688 s (ν(CO)), 1569 w, 1557 w, 1479 s, 1456 s, 1416 s, 1393 m, 1365 s, 1293 m, 1273 m, 1249 m, 1227 m, 1209 m, 1161 s, 1125 m, 1109 m, 1046 m, 1035 m, 1019 m, 984 w, 970 w, 953 w, 912 w, 905 w, 891 m, 864 w, 838 w, 814 w, 804 w, 772 w, 701 w, 685 w, 635 w, 546 w. 1H-NMR (400 MHz, CDCl3): δ = 1.30 (s, 18 H, ArC(CH3)3), 1.48 (s, 18 H, C(CH3)3), 2.08 (m, 12 H, NCH3), 2.69 (m, 8 H, (NCH2CH2)2N), 3.20 (s, 4 H, SCH2), 3.40 (m, 8 H, (NCH2CH2)2N), 3.71 (s, 8 H, ArCH2N), 7.28 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 28.6 (C(CH3)3), 31.4 (ArC(CH3)3), 34.7 (ArC(CH3)3), 37.1 (SCH2), 41.3 (NCH3), 45.6 ((NCH2CH2)2N), 56.7 ((NCH2CH2)2N), 61.9 (ArCH2N), 79.5 (C(CH3)3), 126.8 (ArC-3,3), 131.4 (ArC-2,2)), 143.8 (ArC-1), 150.9 (ArC-4), 155.7 (CO). This compound was additionally characterized by X-ray crystallography.
:1, v:v) and a solution of 1,2-bis(4-tert-butyl-2,6-diformylphenylthio)ethane 10 (1.08 g, 2.30 mmol) in CH2Cl2 (500 mL) were added simultaneously over the course of 3 h into a EtOH/CH2Cl2 (600 mL, 1:3 v:v) solvent mixture. After the resulting mixture was stirred for further 2 d, the CH2Cl2 solvent was removed under reduced pressure. Sodium borohydride (690 mg, 18.24 mmol) was added and the mixture was stirred at r.t. for another 2 h. The excess reducing agent was destroyed by adding HCl conc (final pH = 1). The mixture was subsequently evaporated to dryness, re-dissolved in CH2Cl2/H2O (100 mL, 1:1 v:v), and the pH was adjusted to ∼13 with aqueous KOH (5 M). After stirring for 2 h, the layers were separated and the aqueous phase was extracted with dichloromethane (4 × 150 mL). The organic fractions were combined and dried with anhydrous K2CO3. Evaporation of the solvent gave a foam, which was recrystallized from EtOH to give 1.41 g (88%) of the title compound. Found: C 68.90, H 9.29, N 12.29, S 9.11; C40H64N6S2 (693.11) requires: C 69.32, H 9.31, N 12.13, S 9.25. IR (KBr): ν/cm−1 = 3424 m, 3261 m, 2955 vs, 2902 s, 2867 s, 2803 vs, 1599 m, 1577 m, 1451 vs, 1417 s, 1366 s, 1330 w, 1293 w, 1262 m, 1230 w, 1205 m, 1149 s, 1094 s, 1054 s, 1029 s, 921 m, 897 w, 804 s. 1H-NMR (300 MHz, CDCl3): δ = 1.30 (s, 18 H, C(CH3)3), 2.81 (s, 4 H, SCH2), 2.96 (m, 8 H, (NCH2CH2)2N), 3.14 (m, 8 H, (NCH2CH2)2N), 3.30 (d, J = 6.6 Hz, 4 H, CH2CHCH2), 4.12 (s, 8 H, ArCH2), 5.21 (m, 2 H, CHCHH), 5.23 (m, 2 H, CHCHH), 5.90 (m, 2 H, CHCH2), 6.32 (s (br), 4 H, NH), 7.72 (s, 4 H, ArH). 13C{1H}-NMR (75 MHz, CDCl3): δ = 31.36 (C(CH3)3), 35.20 (C(CH3)3), 37.58 (SCH2)2, 49.02 ((NCH2CH2)2N), 50.31 ((NCH2CH2)2N), 51.41 (ArCH2), 60.20 (NCH2CHCH2), 119.32 (CHCH2), 128.34 (ArC-3,3′), 130.07 (ArC-2,2′), 134.56 (CHCH2), 140.16 (ArC-1), 154.18 (ArC-4).
:1 CH2Cl2/CH3CN solvent mixture. Evaporation of the CH2Cl2 provided a colorless solid, which was filtered and dried in air. Yield: 7.08 g (7.6 mmol, 99%), colorless solid. M.p. 170–172 °C. Found: C 66.83, H 7.92, N 17.82; C52H74N12S2 (931.37) requires: C 67.06, H 8.01, N 18.05. m/z (ESI+, MeOH): C52H74N12S2 (930.56) [M + H+]+ calcd: 931.57; found 931.56. IR (KBr): ν/cm−1 = 3426 vw, 3054 m, 2951 vs, 2818 vs, 2246 vs ν(CN), 1593 m, 1556 vw, 1477 m, 1464 s, 1434 s, 1424 s, 1405 m, 1381 m, 1356 s, 1328 m,1292 m, 1276 s, 1248 m, 1215 m, 1171 w, 1139 s, 1110 vs, 1047 s, 1005 m, 987 m, 961 m, 947 m, 887 s, 832 w, 801 vw, 774 m, 733 m, 704 vw, 681 vw, 650 w, 596 w. 1H-NMR (300 MHz, CDCl3): δ = 1.33 (s, 18 H, ArC(CH3)3), 2.38 (m, 8 H, CH2CN),2.40 (m, 4 H, CH2CN), 2.66 (m, 8 H, (NCH2CH2)2N), 2.67 (s, 4 H, SCH2), 2.68 (m, 8 H, (NCH2CH2)2N), 2.73 (m, 8 H, CH2CH2CN), 2.78 (m, 4 H, CH2CH2CN), 3.76 (s, 8 H, ArCH2), 7.53 (s, 4 H, ArH). 13C{1H}-NMR (75 MHz, CDCl3): δ = 17.03 (CH2CN), 17.38 (CH2CN), 31.46 (C(CH3)3), 35.00 (C(CH3)3), 36.16 (SCH2), 47.3 (ArCH2NCH2CH2CN), 51.82 (CH2CH2CN), 52.39 ((NCH2CH2)2N), 52.96 ((NCH2CH2)2N), 63.1 (ArCH2N), 118.90 (ArCH2NCH2CH2CN), 118.96 (CN), 125.20 (ArC-3,3′), 128.17 (ArC-2,2′), 142.71 (ArC-1), 152.26 (ArC-4). This compound was additionally characterized by X-ray crystallography.
:1) solution afforded colorless crystals. Found: C 64.76, H 9.89, N 8.65, S 6.50; C52H92N6O6S2 (961.46) requires: C 64.96, H 9.65, N 8.74, S 6.67. m/z (ESI+, MeOH): C52H92N6O6S2 (960.55) [M + H+]+ calcd: 961.66; found 961.7. IR (KBr): ν/cm−1 = 3425 s, 2953 vs, 2871 vs, 2816 vs, 2336 vw, 1954 vw, 1667 m, 1594 m,1559 w, 1451 vs, 1404 s, 1362 vs, 1297 s, 1261 m, 1199 s, 1119 vs, 1080 vs, 1018 s, 958 m, 927 m, 894 m, 781 w, 733 w, 704 w, 647 w, 558 w. 1H-NMR (600 MHz, 400 K, DMSO-d6): δ = 1.32 (s, 18 H, C(CH3)), 2.64 (t, 3J = 5.3, 8 H, NCH2CH2OCH3), 2.73 (m, 4 H, NCH2CH2OCH3)), 2.75 (s, 4 H, SCH2), 2.85 (m, 16 H, NCH2CH2N), 3.28 (s, 12 H, OCH3), 3.31 (s, 6 H, OCH3), 3.41 (t, 3J = 5.3, 8 H, CH2OCH3), 3.60 (m, 4 H, CH2OCH3), 3.78 (s, 8 H, ArCH2), 7.53 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 31.32 (C(CH3)3), 34.68 (C(CH3)3), 35.57 (SCH2), 50.82 (NCH2CH2OCH3), 50.92 ((NCH2CH2)2N), 52.74 (ArCH2NCH2CH2OCH3), 54.68 ((NCH2CH2)2N), 58.17 (ArCH2N), 58.92 (OCH3), 58.97 (OCH3), 70.50 (CH2OCH3), 70.82 (CH2OCH3), 125.72 (ArC-3,3′), 128.55 (ArC-2,2′), 142.30 (ArC-1), 151.87 (ArC-4).
:1) solution afforded colorless crystals. Found: C 66.13, H 9.78, N 9.41, S 7.22; C48H84N6O4S2 (873.36) requires: C 66.01, H 9.69, N 9.62, S 7.34. m/z (ESI+, MeOH): C48H84N6O4S2 (872.60) [M + H+]+ calcd: 873.61; found 873.60. IR (KBr): ν/cm−1 = 3425 s, 2953 vs, 2871 vs, 2816 vs, 2336 vw, 1954 vw, 1667 m, 1594 m, 1559 w, 1451 vs, 1404 s, 1362 vs, 1297 s, 1261 m, 1199 s, 1119 vs, 1080 vs, 1018 s, 958 m, 927 m, 894 m, 781 w, 733 w, 704 w, 647 w, 558 w. 1H-NMR (600 MHz, 400 K, DMSO-d6): δ = 1.30 (s, 18 H, C(CH3)3), 2.25 (s, 6 H, NCH3), 2.50 (s(br), 8 H, (NCH2CH2)2NCH3), 2.60 (t, 3J = 9.8, 8 H, NCH2CH2OCH3), 2.68 (s (br), 8 H, (NCH2CH2)2NCH3), 2.79 (s, 4 H, SCH2), 3.28 (s, 12 H, OCH3), 3.39 (t, 3J = 9.8, 8 H, NCH2CH2OCH3), 3.80 (s, 8 H, ArCH2), 7.47 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 31.43 (C(CH3)3), 34.87 (C(CH3)3), 36.16 (SCH2), 43.42 (NCH3), 52.35 ((NCH2CH2)2N), 53.37 (ArCH2NCH2CH2OCH3), 58.57 (ArCH2N), 58.93 (OCH3), 71.50 (CH2OCH3), (one CH2 signal not observed), 125.70 (ArC-3,3′), 128.79 (ArC-2,2′), 143.79 (ArC-1), 151.44 (ArC-4).
:1 v/v) at 0 °C was added a solution of 1,2-bis(4-tert-butyl-2,6-diformylphenylthio)ethane (3.34 g, 7.11 mmol) in CHCl3 (500 mL) over the course of 8 h. After the resulting mixture was stirred for further 2 d, the CHCl3 solvent was removed under reduced pressure. Sodium borohydride (2.15 g, 56.88 mmol) was added and the mixture was stirred at r.t. for 18 h. The mixture was evaporated to dryness, then redissolved in CH2Cl2/H2O (100 mL, 1:1, v/v), and the pH was adjusted to ∼13 with aqueous KOH (5 M). After stirring for 2 h, the layers were separated and the aqueous phase was extracted with CH2Cl2 (4 × 50 mL). The organic fractions were combined and dried with anhydrous MgSO4. Evaporation of the solvent gave an oil, which crystallized from EtOH (10 ml) after standing for 4 weeks. Yield: 1.20 g (1.47 mmol, 21%). M.p. 136–138 °C. Found: C 64.75, H 8.58, N 9.75; C44H72N6O4S2 0.5 EtOH (813.21 + 23.04) requires: C 64.63, H 9.04, N 10.05. m/z (ESI+): 813.4 (M + H+). IR (KBr): ν/cm−1 = 3443 s, 3290 m ν(NH), 2965 s, 2927 s, 2865 m, 1693 s ν(CO), 1597 w, 1478 s, 1458 s, 1411 s, 1365 s, 1249 m, 1228 w, 1173 s, 1154 s, 1091 w, 1043 w, 966 w, 886 w, 858 w, 823 w, 774 w. 1H-NMR (400 MHz, CDCl3): δ = 1.29 (s, 18 H, ArC(CH3)3), 1.45 (s, 18 H, C(CH3)3), 2.96 (t, 3J = 6 Hz, 8 H, (NCH2CH2)2N), 3.20 (s, 4 H, SCH2), 3.49 (t, 3J = 6 Hz, 8 H, (NCH2CH2)2N), 3.97 (s, 8 H, ArCH2N), 7.28 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 28.6 (C(CH3)3), 31.4 (ArC(CH3)3), 34.7 (ArC(CH3)3), 37.1 ((ArSCH2)2), 47.8 ((NCH2CH2)2N), 49.3 (ArCH2N), 53.8 ((NCH2CH2)2N), 79.7 (C(CH3)3), 126.4 (ArC-3,3), 130.0 (ArC-2,2), 144.8 (ArC-1), 152.2 (ArC-4), 156.0 (CO).
:1 v/v) was added. After stirring for 2 h, the layers were separated and the aqueous phase was extracted with CH2Cl2 (4 × 25 mL). The organic fractions were combined and dried with anhydrous MgSO4. Evaporation gave the crude product, which was purified by recrystallization from CH2Cl2/EtOH (1:1 v/v). Yield: 910 mg (1.05 mmol, 75%), colorless crystals. Mp 217–219 °C. Found: C 65.93, H 9.23, N 9.59; C48H80N6O4S2 (869.32) requires: C 66.32, H 9.28, N 9.67. m/z (ESI+, MeOH): C48H80N6O4S2 (868.57) [M + H+]+ calcd: 869.58; found 869.50. IR (KBr): ν/cm−1 = 3444 m, 2965 s, 2857 m, 2802 s, 2716 w, 1688 s (ν(CO)), 1569 w, 1557 w, 1479 s, 1456 s, 1416 s, 1393 m, 1365 s, 1293 m, 1273 m, 1249 m, 1227 m, 1209 m, 1161 s, 1125 m, 1109 m, 1046 m, 1035 m, 1019 m, 984 w, 970 w, 953 w, 912 w, 905 w, 891 m, 864 w, 838 w, 814 w, 804 w, 772 w, 701 w, 685 w, 635 w, 546 w. 1H-NMR (400 MHz, CDCl3): δ = 1.30 (s, 18 H, ArC(CH3)3), 1.48 (s, 18 H, C(CH3)3), 2.08 (m, 12 H, NCH3), 2.69 (m, 8 H, (NCH2CH2)2N), 3.20 (s, 4 H, SCH2), 3.40 (m, 8 H, (NCH2CH2)2N), 3.71 (s, 8 H, ArCH2N), 7.28 (s, 4 H, ArH). 13C{1H}-NMR (100 MHz, CDCl3): δ = 28.6 (C(CH3)3), 31.4 (ArC(CH3)3), 34.7 (ArC(CH3)3), 37.1 (SCH2), 41.3 (NCH3), 45.6 ((NCH2CH2)2N), 56.7 ((NCH2CH2)2N), 61.9 (ArCH2N), 79.5 (C(CH3)3), 126.8 (ArC-3,3), 131.4 (ArC-2,2)), 143.8 (ArC-1), 150.9 (ArC-4), 155.7 (CO). This compound was additionally characterized by X-ray crystallography.
Crystallography
Suitable single crystals of compounds 8, 15, 17·3DMSO, 19·2DMSO·2H2O, 20, and 23 were selected and mounted on the tip of a glass fibre using perfluoropolyether oil. The data sets for 8, 17·3DMSO, 19·2DMSO·2H2O, and 20 were collected at 183(2) K using a STOE IPDS-2 diffractometer, while those for 15 and 23 were collected on a STOE IPDS-1 diffractometer at 213(2) K. Graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) was used throughout. The data were processed with the programs XAREA.32 Selected details of the data collection and refinement are given in Table 1. The structures were solved by direct methods33 and refined by full-matrix least-squares techniques on the basis of all data against F2 using SHELXL-97.34 PLATON was used to search for higher symmetry.35 All non-hydrogen atoms were refined anisotropically, except for those of some disordered solvate molecules. Disorder was modelled using split atom models with restrained Cl–O, O⋯O, C–C, and C⋯C distances using appropriate SADI instructions implemented in the SHELXL software package. Graphics were produced with Ortep3 for Windows and PovRAY.| Compound | 8 | 15 | 17·3DMSOa | 19·2DMSO·2H2Ob | 20 | 23 |
|---|---|---|---|---|---|---|
| a Three DMSO solvate molecules were located from the Fourier map but one was heavily disordered such that it was removed from the structure. b The DMSO and H2O solvate molecules were located from the Fourier map but were heavily disordered such that they were all removed from the structure, see Experimental section. c R 1 = Σ∥Fo| − |Fc∥/Σ|Fo|. d wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2. | ||||||
| Formula | C52H80N6S2 | C52H74N12S2 | C56H92N6O14S4 | C48H76N6O8S2 | C48H84N6O4S2 | C48H80N6O4S2 |
| Diffractometer | IPDS-2 | IPDS-1 | IPDS-2 | IPDS-2 | IPDS-2 | IPDS-1 |
| T/K | 180(2) | 213(2) | 180(2) | 180(2) | 180(2) | 213(2) |
| Formula weight [g mol−1] | 853.34 | 931.35 | 1201.60 | 929.27 | 873.33 | 869.30 |
| Space group | C2/c | I2/a |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
I2/a | C2/c | P21/c |
| a, Å | 27.300(2) | 19.481(4) | 12.894(2) | 18.934(3) | 27.580(4) | 13.007(3) |
| b, Å | 10.0280(6) | 9.905(2) | 13.432(2) | 10.294(3) | 9.948(2) | 10.269(2) |
| c, Å | 20.646(1) | 26.740(5) | 20.463(3) | 31.857(5) | 19.845(3) | 19.285(4) |
| α, deg | 90.00 | 90.00 | 91.86(2) | 90 | 90 | 90 |
| β, deg | 111.911(4) | 99.08(3) | 100.24(2) | 105.36(2) | 109.50(2) | 97.50(3) |
| γ, deg | 90.00 | 90.00 | 106.80(2) | 90 | 90 | 90 |
| V, Å3 | 5243.9(5) | 5095(2) | 6476(2) | 5987(2) | 5132(2) | 2553.7(9) |
| Z | 4 | 4 | 2 | 4 | 4 | 2 |
| D calcd, g cm−3 | 1.081 | 1.214 | 1.200 | 1.031 | 1.130 | 1.131 |
| Cryst. size, mm3 | 0.25 × 0.20 × 0.10 | 0.15 × 0.15 × 0.15 | 0.23 × 0.23 × 0.10 | 0.39 × 0.23 × 0.18 | 0.15 × 0.27 × 0.18 | 0.20 × 0.15 × 0.05 |
| μ(Mo Kα), mm−1 | 0.140 | 0.153 | 0.205 | 0.136 | 0.149 | 0.150 |
| R 1 (R1 all data) | 0.0547 (0.0754) | 0.0427 (0.0618) | 0.0506 (0.0951) | 0.0458 (0.0566) | 0.0677 (0.0875) | 0.0360 (0.1038) |
| wR2d (wR2 all data) | 0.1458 (0.1552) | 0.1029 (0.1078) | 0.1019 (0.1127) | 0.1321 (0.1387) | 0.2012 (0.2136) | 0.0446 (0.0519) |
| Max, min peaks, e Å−3 | 0.654/−0.394 | 0.505/−0.264 | 0.260/−0.426 | 0.431/−0.228 | 1.598/−0.519 | 0.147/−0.132 |
| CCDC | 1436363 | 1436364 | 1436365 | 1436366 | 1436367 | 1436368 |
In the crystal structure of 8 two allyl (N1, C18, C19, C20; C21, C22, C23) and one ethylene group (C12, C13) were found to be disordered over two sites. The site occupancies of one allyl and one ethyl group were fixed (0.74/0.26). The site occupancies of the other allyl group were refined (0.55/0.45). In the crystal structure of 17·3DMSO one DMSO solvate molecule (S5, O15, C57, C58) was found to be heavily disordered and was therefore removed from the structure (and the corresponding Fo) with the SQUEEZE procedure implemented in the PLATON program suite. Removing the DMSO molecule led to a solvent accessible void of 257 Å3, in good agreement with the space needed by one DMSO molecules. The solvate molecules in 19·2DMSO·2H2O were also found to be heavily disordered and were therefore removed utilizing the SQUEEZE procedure. This led to solvent accessible voids of 628 Å3, attributed to the space needed for two DMSO and two H2O molecules.
Results and discussion
Synthesis
Scheme 1 depicts the synthetic procedures for compounds 8, 9, and 15–20. The reaction of 1 with allylbromide in the presence of NEt3 in ethanol furnished the bicycle 8 in good yields (>81%). To prevent overalkylation the reaction was carried out at 0 °C. Under similar conditions, the dimethylated precursor 2 reacted preferentially in the benzylic position providing the corresponding tetraallylated system 9 (75%) as colorless needles after recrystallization from CHCl3/EtOH. The reductive amination of tetraaldehyde 10 with N1-(3-aminopropyl)-N1-methylpropane-1,3-diamine 13 under medium-dilution conditions provided the bis-allylated macrocycle 14 in excellent yield (Scheme 2).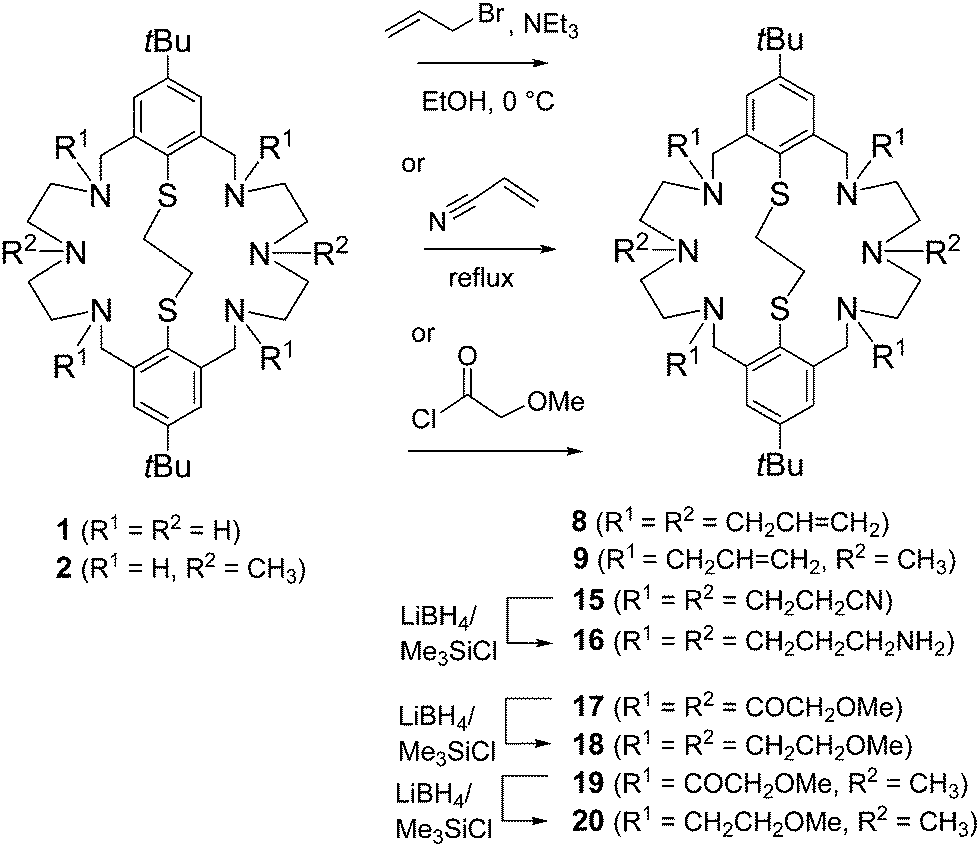 | ||
| Scheme 1 Synthesis of compounds 8, 9, and 15–20. | ||
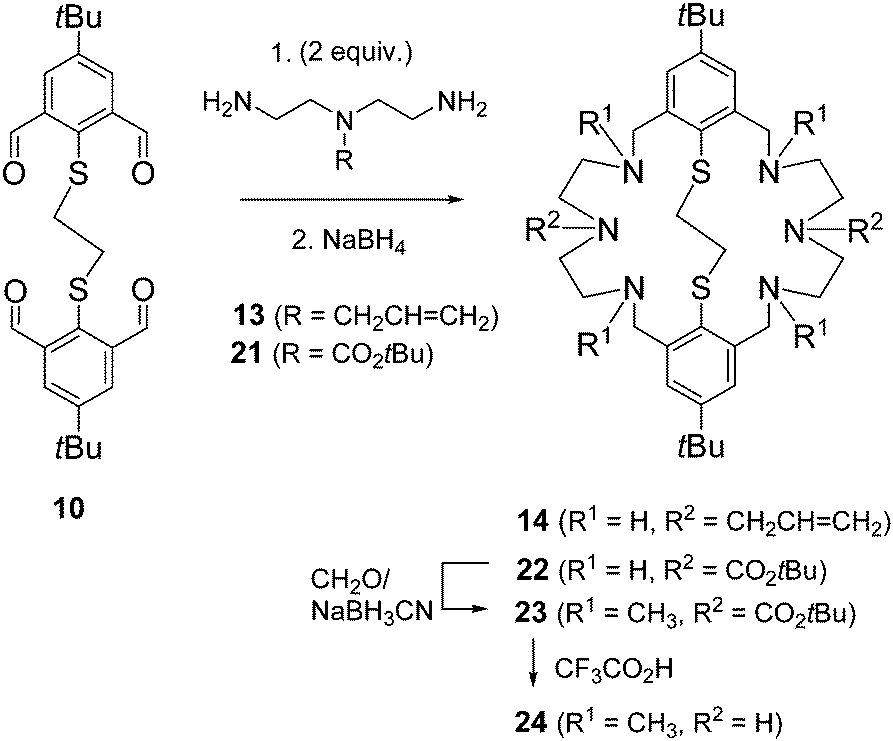 | ||
| Scheme 2 Synthesis of compounds 14 and 22–24. | ||
The second ligand system was prepared according to a protocol used for the cyanethylation of tetraazacycloalkanes.36 Thus, Michael addition of 1 to acrylonitrile led quantitatively to the hexacyanethylated product 15, which can be easily purified by recrystallization. It was reported that the nitrile functions of tetra(2-cyanoethyl)tetraazacycloalkanes can be reduced to the corresponding primary amines by reduction with LiAlH4, diborane or H2-RANEY®-Nickel.37–39 In our hands, the nitrile 15 failed to react in this fashion. Therefore, an alternative protocol involving reduction with LiBH4/Me3SiCl was employed.40 This sequence provided the hexa(3-aminopropylated) macrocycle 16 in moderate to good yields.
The route used for the synthesis of the amino-thioethers with methoxymethyl substituent is depicted in Scheme 1. A reaction sequence similar to that developed for similar N6S2-type macrocycles bearing “innocent” alkyl groups was employed. Key-step of this procedure is the acylation of 1 with 2-methoxyacetyl chloride. Thus, in reaction with 2-methoxyacetyl chloride the amide 17 was generated quantitatively and then reduced to 18 with LiBH4/Me3SiCl. As an illustration of the utility of this sequence, the N,N′-dimethyl derivative 2 was also quantitatively derivatized giving the bicyclic macrocycles 19 and 20, respectively.
So far only the precursors 1 and 2 had been utilized for functionalization. In reactions with 1 all six NH donors are derivatized, while modifications of 2 involved only the benzylic NH donors. We decided to develop a method that allows the selective functionalization of the two central NH donors. In an orienting experiment, the reductive amination of the tetraaldehyde 1 with tert-butyl-bis(2-aminoethyl)carbamate 21 was undertaken. This provided the desired macrocycle 22, albeit in low yield. Having succeeded with the preparation of 22, alkylation of the N-benzyl functions and deprotection of the carbamoyl groups could be examined. Indeed, 22 readily underwent reductive methylation with formaldehyde and NaBH3CN to give the tetramethylated derivative 23 in 75% yields, which was fully characterized including X-ray crystallography. Finally, deprotection of 23 with trifluoroacetic acid gave the desired amine 24.
All new compounds were characterized by elemental analysis, IR, 1H and 13C NMR spectroscopy. Some compounds were further characterized by X-ray crystallography, in order to study their host–guest chemistry.
Crystal structures
Fig. 2 displays the molecular structure of the hexaallylated macrocycle 8. The molecule has crystallographically imposed C2 symmetry, and adopts a folded conformation. Unlike in the permethylated derivative 3,29 the two aromatic rings are essentially coplanar, but are twisted about the S1⋯S1′ vector (torsional angle C1–S1⋯S1′–C1a = 37.8°), attributed to steric interactions between the tert-butyl groups. The allyl residues are all oriented away from the cavity. There are no specific intermolecular interactions in 8. The C–S bonds are of length 1.783(1) Å (S1–C1, S1′–C1′). Virtually the same distances are seen in 3.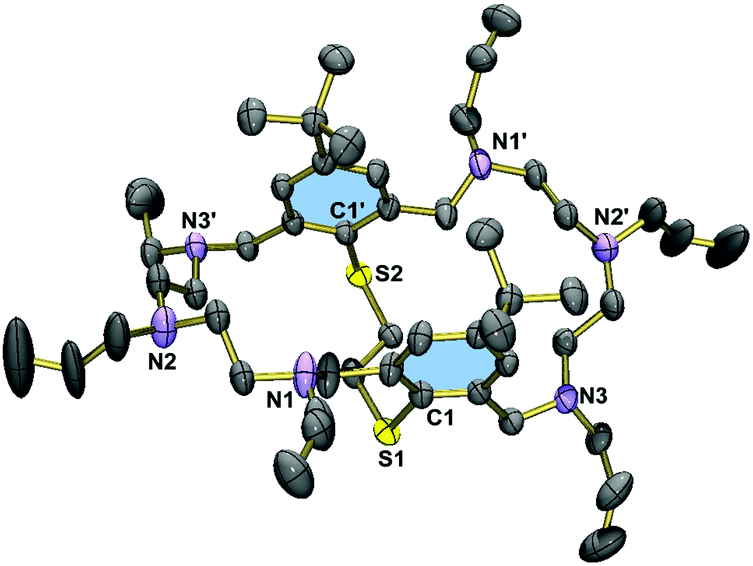 | ||
| Fig. 2 Molecular structure of 8 in the crystal with atomic numbering for key atoms. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. Symmetry code used to generate equivalent atoms: 1 − x, y, 0.5 − z. | ||
Fig. 3 displays the molecular structure of the hexanitrile 15. The macrobicycle adopts a folded conformation, which is similar but not identical to that seen in 8. Here, the two phenyl rings plane are bent into the cleft formed by the macrocycle, at an interplanar angle of 19°. The structure is stabilised by two intermolecular CH⋯π interactions as indicated by relatively short distances between the methylene groups and the aromatic rings (C11⋯centroid(aromatic ring) = 3.823 Å).41 In contrast to the hexaallylated macrocycle, molecules of 15 are connected via intermolecular CH2⋯NC interactions (N4⋯H17b′′ = 3.013, N5⋯H25b′′ 2.517, N6⋯H19b′′ 2.735 Å). These interactions lead to a three-dimensional network. The structure of the tetramethoxyethylated aza-thioether 20 is very similar to that of 15 (when neglecting the different N-substituents). However, the tilting of the two aryl rings is not so pronounced (5°) and the C11⋯centroid distances are longer at 3.875 Å.
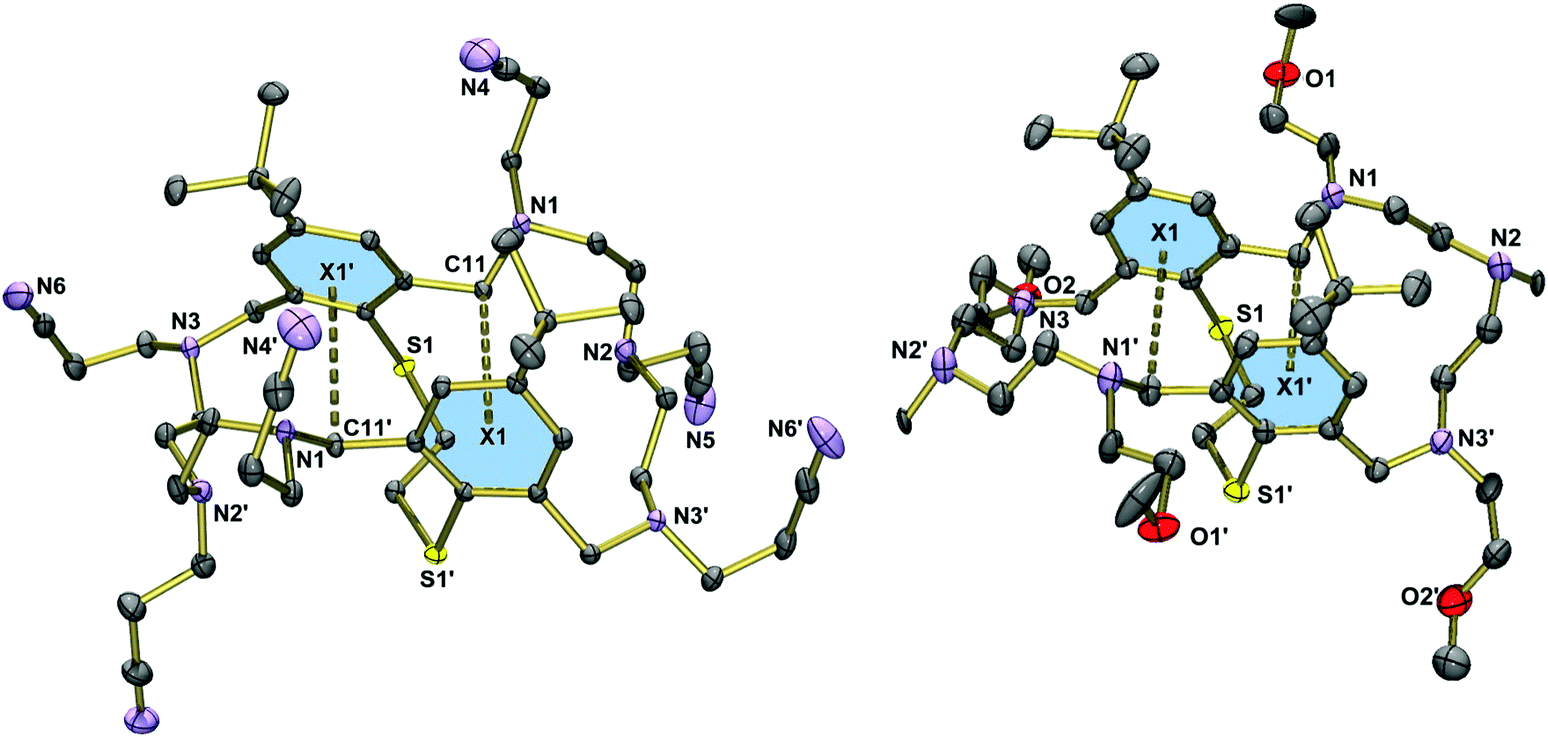 | ||
| Fig. 3 Left: Molecular structure of 15 in the crystal with atomic numbering for key atoms and centroids of aromatic rings. Thermal ellipsoids are set at 30% probability. Symmetry code used to generate equivalent atoms: 1/2 − x, y, 1 − z. Right: Molecular structure of 20 in the crystal. Hydrogen atoms have been omitted for clarity. Symmetry code: −x, y, 1/2 − z. CH⋯π interactions indicated by dashed lines. | ||
Hexa(2-methoxyacetylated) macrobicycle 17 crystallizes from DMSO with three solvate molecules. Fig. 4 shows the structure of the macrobicycle, which forms an inclusion complex with a DMSO molecule. The guest molecule is held in place by a CH⋯π interaction of length 3.823 Å (C11⋯centroid(aromatic ring)). The other two DMSO molecules are enclathrated in the voids of the structure. The structure of 17·3DMSO should be compared with that of the tetra(2-methoxyacetylated) derivative 19·2DMSO·2H2O (Fig. 4, right). This compound crystallizes also with solvate molecules, but does not form an inclusion complex. The two phenyl rings in 19 are coplanar as in the hexanitrile 15. However, the distance between the two best planes through the benzene rings is much larger at 5.087 Å. As a consequence, the phenyl rings are not involved in intermolecular CH⋯π interactions with the adjacent benzyl group (C11⋯centroid(aromatic ring) = 4.802 Å). Clearly, removal of two methoxyacetyl residues exerts more conformational flexibility on the macrocycle.
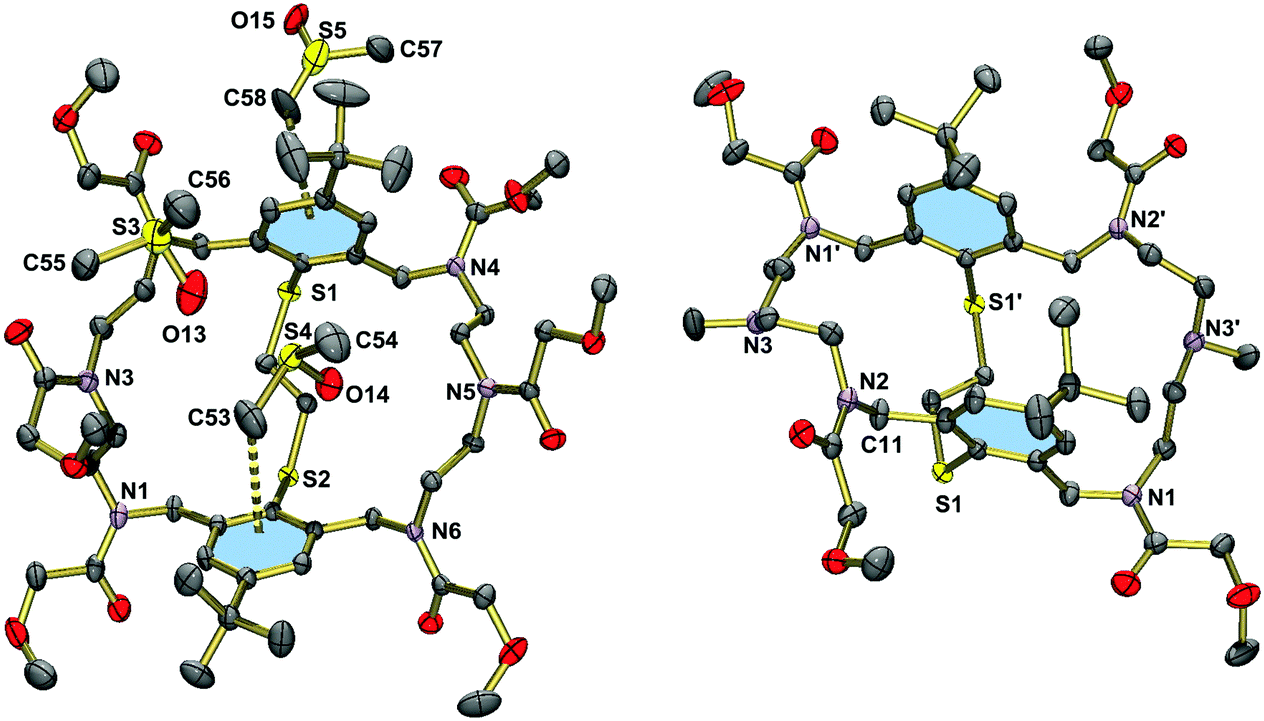 | ||
| Fig. 4 Left: Molecular structure of 17·3DMSO in the crystal. The atom N3 is obscured by a DMSO molecule (O(13)S(3)C(56)C(57)). The DMSO molecule O(15)S(5)C(58)C(59) is disordered over two positions. Only one orientation is displayed. Right: Molecular structure of 19·2DMSO·2H2O in the crystal. Thermal ellipsoids are set at 50% probability. Hydrogen atoms have been omitted for clarity. | ||
Fig. 5 displays the structure of the protected macrobicycle 23, which has crystallographically imposed inversion symmetry. Unlike in the above structures, the thioether adopts a stepped conformation, presumably a consequence of the steric requirements of the N-carbamate groups. As a consequence, the macrocycles are engaged in intermolecular CH⋯π interactions. The corresponding CH⋯π distances at 3.428 Å (C11⋯centroid(aromatic ring)) are significantly shorter than in 15 or 20. This compound crystallizes without guest molecules.
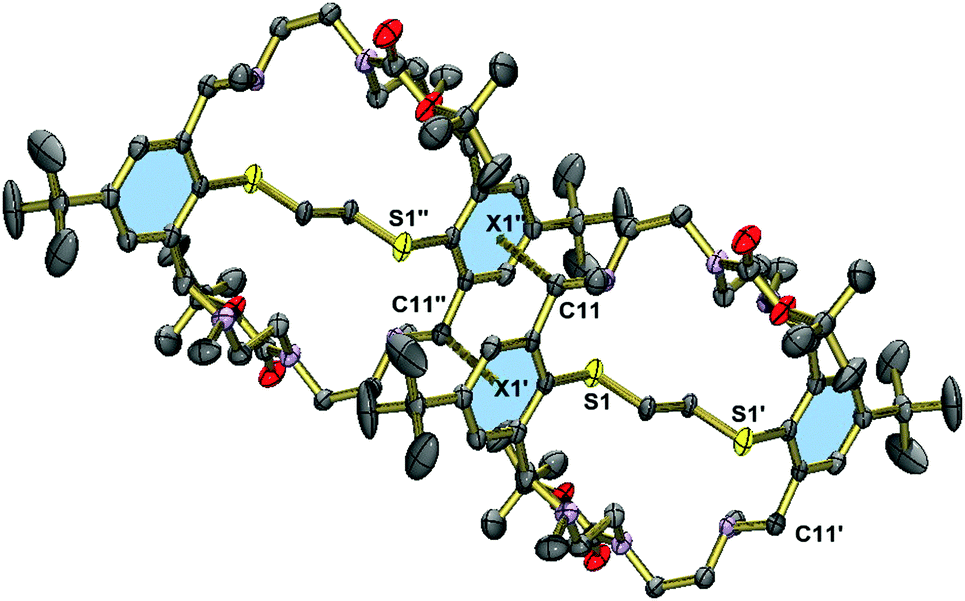 | ||
| Fig. 5 Molecular structure of compound 23 in the crystal showing the intermolecular CH⋯π interactions. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å]: C1–S1 1.789(3) Å. Symmetry codes: −x, 1 − y, −z (′); −x, 2 − y, −z (′′). | ||
Conclusion
Overall, a short and efficient protocol for the functionalization of bicyclic aza-thioethers has been described. All six secondary amine functions of the parent macrobicycles 1 and 2 are readily alkylated without overalkylation and without affecting the masked thiolate functions. The protocol is also feasible for the synthesis of macrobicycles with different alkyl groups on the benzylic and central nitrogen atoms of the linking diethylene triamine units, such that these derivatives are also now available. Six of the twelve new compounds were obtained in crystalline form, such that their molecular structures could be determined. In the solid state the macrobicycles can adopt a stepped or a folded conformation. The structures appear to be primarily governed by inter- and intramolecular CH2⋯π interactions (involving the benzylic methylene groups and the aromatic rings) rather due to steric effects played by the N alkyl functions. The observation that DMSO, which is a good CH donor, can form an inclusion complex held in place by a CH3⋯π interaction would be consistent with this in view.Acknowledgements
We are thankful to Prof. Dr H. Krautscheid for providing facilities for X-ray crystallographic measurements. This work was supported by the Deutsche Forschungsgemeinschaft (DFG Research unit 1154, “Towards Molecular Spintronics”).References
- E. C. Constable, Coordination Chemistry of Macrocyclic Compounds, Oxford University Press, Oxford, 1999, ch. 5 Search PubMed.
- L. Lindoy, The chemistry of macrocyclic ligand complexes, Cambridge University Press, Cambridge, 1990, ch. 6 Search PubMed.
- D. H. Busch, Acc. Chem. Res., 1978, 11, 392–400 CrossRef CAS.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, John Wiley & Sons, Weinheim, 6th edn, 1999 Search PubMed.
- (a) M. Schröder and V. Lippolis, in Macrocyclic Chemistry, Current Trends and Future Perspectives, ed. K. Gloe, Springer, Dordrecht, The Netherlands, 2005, ch. 5, p. 67 Search PubMed; (b) L. Tei, A. Bencini, A. J. Blake, V. Lippolis, A. Perra, B. Valtancoli, C. Wilson and M. Schröder, Dalton Trans., 2004, 1934–1944 RSC.
- (a) K. P. Wainwright, Coord. Chem. Rev., 1997, 166, 35–90 CrossRef CAS; (b) K. P. Wainwright, Adv. Inorg. Chem., 2001, 52, 293–334 CrossRef CAS.
- P. V. Bernhardt and G. A. Lawrence, Coord. Chem. Rev., 1990, 104, 297–343 CrossRef CAS.
- D. S. C. Black and I. A. McLean, Chem. Commun., 1968, 1004–1006 RSC.
- B. Dietrich, J. M. Lehn and J. P. Sauvage, Chem. Commun., 1970, 1055–1056 RSC.
- (a) U. Kallert and R. Mattes, Inorg. Chim. Acta, 1991, 180, 263–269 CrossRef CAS; (b) K. Wasielewski and R. Mattes, Z. Anorg. Allg. Chem., 1993, 619, 158–162 CrossRef CAS.
- I. Bertini, L. Sacconi and G. P. Speroni, Inorg. Chem., 1972, 11, 1323–1326 CrossRef CAS.
- R. C. Coombes, J.-P. Costes and D. E. Fenton, Inorg. Chim. Acta, 1983, 77, 173–174 CrossRef.
- D. A. Nation, M. R. Taylor and K. P. Wainwright, J. Chem. Soc., Dalton Trans., 1992, 1557–1562 RSC.
- L. F. Lindoy and R. J. Smith, Inorg. Chem., 1981, 20, 1314–1316 CrossRef CAS.
- H. Koyama and I. Murase, Bull. Chem. Soc. Jpn., 1977, 50, 895–897 CrossRef CAS.
- (a) T. Yamamura, M. Tadokora and R. Kuroda, Chem. Lett., 1989, 1245–1246 CrossRef CAS; (b) T. Yamamura, M. Tadokoro, K. Tanaka and R. Kuroda, Bull. Chem. Soc. Jpn., 1984, 66, 1984–1990 CrossRef.
- J. W. L. Martin, G. J. Organ, K. P. Wainwright, K. D. V. Weerasuria, A. C. Willis and S. B. Wild, Inorg. Chem., 1987, 26, 2963–2968 CrossRef CAS.
- C. J. Hinshaw, G. Peng, R. Singh, J. T. Spence, J. H. Enemark, M. Bruck, J. Kristowski, S. L. Merbs, R. B. Ortega and P. A. Wexler, Inorg. Chem., 1989, 28, 4483–4491 CrossRef CAS.
- Y. Sun, C. S. Cutler, A. E. Martell and M. J. Welch, Tetrahedron, 1999, 55, 5733–5740 CrossRef CAS.
- N. Ehlers, D. Funkemeier and R. Mattes, Z. Anorg. Allg. Chem., 1994, 620, 796–800 CrossRef CAS.
- M. W. A. Steenland, P. Westbroek, I. Dierck, G. G. Herman, W. Lippens, E. Temmerman and A. M. Goeminne, Polyhedron, 1999, 18, 3417–3424 CrossRef CAS.
- I. M. Vasilescu, D. J. Bray, J. K. Clegg, L. F. Lindoy, G. V. Meehan and G. Wei, Dalton Trans., 2006, 5115–5117 RSC.
- (a) M. C. Aragoni, M. Arca, A. Bencini, C. Caltagirone, A. Garau, F. Isaia, M. E. Light, V. Lippolis, C. Lodeiro, M. Mameli, R. Montis, M. C. Mostallino, A. Pintus and S. Puccioni, Dalton Trans., 2013, 42, 14516–14530 RSC; (b) K. Zargoosh, M. Shamsipur, M. Hosseini, C. Caltagirone and V. Lippolis, J. Lumin., 2012, 132, 2126–2129 CrossRef CAS; (c) M. Mameli, V. Lippolis, C. Caltagirone, J. L. Capelo, O. N. Faza and C. Lodeiro, Inorg. Chem., 2010, 49, 8276–8286 CrossRef CAS PubMed; (d) M. Mameli, M. C. Aragoni, M. Arca, M. Atzori, A. Bencini, C. Bazzicalupi, A. J. Blake, C. Caltagirone, F. A. Devillanova, A. Garau, M. B. Hursthouse, F. Isaia, V. Lippolis and B. Valtancoli, Inorg. Chem., 2009, 48, 9236–9249 CrossRef CAS PubMed; (e) M. C. Aragoni, M. Arca, A. Bencini, A. J. Blake, C. Caltagirone, A. Decortes, F. Demartin, F. A. Devillanova, E. Faggi, L. S. Dolci, A. Garau, F. Isaia, V. Lippolis, L. Prodi, C. Wilson, B. Valtancoli and N. Zaccheroni, Dalton Trans., 2005, 2994–3004 RSC; (f) C. Caltagirone, A. Bencini, F. Demartin, F. A. Devillanova, A. Garau, F. Isaia, V. Lippolis, P. Mariani, U. Papke, L. Tei and G. Verani, Dalton Trans., 2003, 901–909 RSC.
- (a) B. Kersting and G. Steinfeld, Chem. Commun., 2001, 1376–1377 RSC; (b) M. H. Klingele, G. Steinfeld and B. Kersting, Z. Naturforsch., 2001, 56b, 901–907 Search PubMed; (c) M. H. Klingele and B. Kersting, Z. Naturforsch., 2001, 56b, 437–439 Search PubMed.
- M. Gressenbuch and B. Kersting, Eur. J. Inorg. Chem., 2007, 90–102 CrossRef CAS.
- M. Gressenbuch, V. Lozan, G. Steinfeld and B. Kersting, Eur. J. Inorg. Chem., 2005, 2223–2234 CrossRef CAS.
- T. Gregor, C. F. Weise, V. Lozan and B. Kersting, Synthesis, 2007, 3706–3712 CAS.
- M. Gressenbuch and B. Kersting, Dalton Trans., 2009, 5281–5283 RSC.
- G. Siedle and B. Kersting, Z. Anorg. Allg. Chem., 2003, 629, 2083–2090 CrossRef CAS.
- C. Y. Ng, R. J. Motekaitis and A. E. Martell, Inorg. Chem., 1979, 18, 2982–2986 CrossRef CAS.
- (a) M. P. Hay, F. B. Pruijn, S. A. Gamage, H. D. S. Liyanage, M. S. Kovacs, A. V. Patterson, W. R; Wilson, J. M. Brown and W. A. Denny, J. Med. Chem., 2004, 47, 475–488 CrossRef CAS PubMed; (b) M. A. Ilies, W. A. Seitz, B. H. Johnson, E. L. Ezell, A. L. Miller, E. B. Thompson and A. T. Balaban, J. Med. Chem., 2006, 49, 3872–3887 CrossRef CAS PubMed.
- Stoe & Cie, X-AREA and X-RED 32; V1.35, Stoe & Cie, Darmstadt, Germany, 2006 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXL-97, Computer program for crystal structure refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, PLATON – A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2000 Search PubMed.
- K. P. Wrainwright, J. Chem. Soc., Dalton Trans., 1980, 2117–2120 RSC.
- K. P. Wrainwright, J. Chem. Soc., Dalton Trans., 1983, 1149–1152 RSC.
- R. J. Bergeron and J. R. Garlich, Synthesis, 1984, 782–784 CrossRef CAS.
- C. Gros, H. Chollet, A. K. Mishra and R. Guilard, Synth. Commun., 1996, 26, 35–47 CrossRef CAS.
- A. Giannis and K. Sandhoff, Angew. Chem., 1989, 101, 220–222 ( Angew. Chem., Int. Ed. Engl. , 1989 , 28 , 218–220 ) CrossRef CAS.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., 2003, 115, 1244–1287 ( Angew. Chem., Int. Ed. , 2003 , 42 , 1210–1250 ) CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, IR and MS spectra for new compounds. Crystallographic data in CIF format for 8, 15, 17·3DMSO, 19·2DMSO·2H2O, 20 and 23. CCDC 1436363–1436368. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj03197d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |