
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural studies of malonaldehyde–glyoxal and malonaldehyde–methylglyoxal etheno adducts of adenine nucleosides based on spectroscopic methods and DFT-GIAO calculations†
Kinga
Salus
,
Marcin
Hoffmann
*,
Bożena
Wyrzykiewicz
and
Donata
Pluskota-Karwatka
*
Adam Mickiewicz University in Poznań, Faculty of Chemistry, Umultowska 89b, 61-614 Poznań, Poland. E-mail: mmh@amu.edu.pl; donatap@amu.edu.pl
First published on 8th March 2016
Abstract
Etheno adducts are formed in the reactions of DNA bases with chloroacetaldehyde, with lipid peroxidation products, and also with metabolites of vinyl chloride and furan. The presence of such modifications in the genetic material may lead to errors in replication with consequences of mutations and even carcinogenesis. For an understanding of the biological significance of etheno adducts it is important to determine their structures. Structural identification is also essential for using these adducts as inflammatory or cancer biomarkers. This paper reports structural studies on two adducts formed in the reactions of malonaldehyde and glyoxal with adenosine (M1Gx-A), and malonaldehyde and methylglyoxal with 2′-deoxyadenosine (M1MGx-dA). NMR spectroscopy and theoretical methods have been used. DFT-GIAO calculations were performed at M06/6-311++G(2df,2pd), B3LYP/6-311++G(2df,2pd) and M06/6-31++G(d,p) levels both in the gas phase and taking into account the effect of solvents (water, methanol and DMSO) using PCM approximation. It has been shown that when M06 or B3LYP functionals with the 6-311++G(2df,2pd) basis set are used, 1H NMR chemical shifts very close to experimental values are obtained and that the results of GIAO calculations at the M06/6-31++G(d,p) level have a better correlation with measured 13C NMR chemical shift values. PCM improves the correlation of results in both cases.
Introduction
Etheno adducts, 1,N6-etheno-2′-deoxyadenosine, 3,N4-etheno-2′-deoxycytidine, 1,N2-etheno-2′-deoxyguanosine and N2,3-etheno-2′-deoxyguanosine, are structural modifications of DNA bases in which an ethylene unit bridges two nucleophilic sites. These derivatives may be formed as a result of interactions between the genetic material and lipid peroxidation carbonyl products.1–16Etheno adducts are also formed in the reactions of DNA bases with chloroacetic aldehyde.17 Furthermore, xenobiotics, such as furan,18 tetrahydrofuran19 and vinyl halides,20 generate etheno adducts after their biotransformation.
Etheno adducts exhibit promutagenic and procarcinogenic properties.21,22 The formation of etheno DNA adducts may affect the structure and stability of the double helix through disturbed base pairing23–26 or else cause changes in glycosidic bond conformation in the modified nucleoside.23 As a consequence, DNA metabolism (this affects particularly human polymerases27 and taq polymerases in PCR28) and metabolism of repair enzymes (particularly glycosylases29,30) are impaired. Etheno DNA adducts may lead to conservation of mutations and as a consequence to initiation of carcinogenesis or to cell apoptosis.27,31 When etheno RNA adducts are formed, impaired cellular processes related to mRNA and epigenetic control may occur.7
It is important to determine the structure of etheno adducts considering their potential use in biochemistry and medicine as biomarkers of oxidative stress, which enhances lipid peroxidation.32–34 Quantitative and qualitative etheno adduct biomonitoring may be an indicator of various pathologies, including a measure of cancer risk or an early stage of cancer.32–34
Glyoxal and methylglyoxal are endogenously formed DNA damaging agents.35,36 These extremely reactive electrophiles were identified as precursors of advanced glycation end products (AGEs) which comprise a structurally diverse class of DNA and protein modifications formed in living organisms.37,38 While protein AGEs are believed to be involved in the pathologies associated with aging, cancer, Alzheimer's disease and diabetes-related complications,39,40 little is known about the structural features of this type of DNA lesion. Aldehydes continuously generated during a number of cellular processes can coexist in biological tissues and can modify biopolymers in a synergistic manner.41,42
Recently, we synthesized etheno derivatives of adenine nucleosides formed in reactions with malonaldehyde and glyoxal (M1Gx-A), and with malonaldehyde and methylglyoxal, (M1MGx-dA).43 Data resulting from spectroscopic methods suggested that a dialdehyde methyl moiety was present at position C8. However, results of our most recent preliminary studies on M1Gx-A and M1MGx-dA reactivity toward nucleophilic biomolecules may indicate that the substituent can also be attached to C7. Therefore, structural studies of both adducts were undertaken in which computational together with experimental (NMR spectroscopy) methods were used. Quantum chemical methods provide useful information for determining structures of organic compounds, e.g. by predicting NMR chemical shifts. In this study the GIAO method was applied, which is currently the most widely used method in such investigations.44–58
Calculations of 1H and 13C NMR chemical shifts were performed for M1Gx-A and M1MGx-dA structures with a dialdehyde methyl substituent at C843 (Fig. 1). Absolute errors were determined by comparison between calculated and experimental values.35 The largest errors were seen for C7 and C8 atoms of the etheno ring. Therefore, we performed further thorough spectroscopic investigation and calculations for alternative structures with the dialdehyde methyl substituent at C7 (Fig. 2) and on the basis of the obtained results we found that in both adducts the dialdehyde methyl group was attached to this carbon atom.
 | ||
| Fig. 1 Structures of M1Gx-A and M1MGx-dA.43 | ||
 | ||
| Fig. 2 Alternative structures for M1Gx-A and M1MGx-dA. | ||
Experimental section
Materials
1,1,3,3-Tetramethoxypropane (TMP), 2′-deoxyadenosine monohydrate, adenosine, methylglyoxal solution (40 wt% in H2O), glyoxal solution (40 wt% in H2O), NH4HCO3, acetonitrile (gradient grade for chromatography), D2O (containing 0.05% TSP-d4), CD3OD (containing 0.03% TMS) and DMSO-d6 (containing 0.03% TMS) were purchased from Sigma-Aldrich.Chromatographic methods
Separation and purification of M1Gx-A and M1MGx-dA were carried out using an Agilent 1200 Series HPLC system consisting of a binary pump (G1312A), a vacuum degasser (G1379B), an autosampler (G1329A), a thermostatted column compartment (G1316A), a diode-array detector (UV; G1315B), a fraction collector (G1364C), and an Agilent ChemStation data handling program (Agilent Technologies). Isolation was performed under semi-preparative conditions: 5 μm and 10 × 250 mm (Hypersil BDS, Thermo Scientific) reversed-phase C18 column; flow was set to 3 mL min−1; the thermostat was set to 25 °C; 1.2 mM NH4HCO3 and ACN were used as eluents.General procedure for M1Gx-A and M1MGx-dA synthesis
The M1Gx-A and M1MGx-dA adducts were synthesized using a modified procedure developed earlier in our laboratory.43 Malonaldehyde was prepared in situ by the acidic hydrolysis of 1,1,3,3-tetramethoxypropane (TMP).59Nucleosides (adenosine (170 mg, 0.64 mmol) and 2′-deoxyadenosine monohydrate (170 mg, 0.63 mmol)) were dissolved in 15 mL of 0.5 M phosphate buffer at pH = 4.6. Two portions of TMP (550 mg, 3.3 mmol) were mixed with 2 mL of 0.1 M HCl and stirred at 50 °C for 15 min. Glyoxal (2 mL, 17 mmol) and methylglyoxal (4 mL, 26 mmol) were added separately to the hydrolyzed TMP. The pH of the resulting solutions was adjusted to 4.6 with 1 M NaOH. The malonaldehyde–glyoxal mixture was then added to the solution of adenosine, and the malonaldehyde–methylglyoxal mixture was combined with 2′-deoxyadenosine solution. The reactions were performed at 50 °C for 18 h. The mixtures were then concentrated and passed through a preparative C18 column. The column was eluted with H2O (300 mL) and with a gradient of 1% to 10% ACN in H2O. The fractions containing products were combined respectively, concentrated and subjected to further isolation using a semipreparative C18 column. M1Gx-A isolation was carried out in a gradient of 0% to 0.7% ACN for 19 min, and then of 0.7% to 15% ACN for 4 min. For the purification of M1MGx-dA the column was eluted with a gradient of 0% to 0.7% ACN for 21 min, and then of 0.7% to 20% ACN for 5 min. The solutions containing pure adducts were combined, evaporated to dryness and lyophilized. The resulting M1Gx-A (13 mg, yield: 3.6%) and M1MGx-dA (24 mg, yield: 6.7%) were characterized by NMR.
NMR spectroscopy
Samples were dissolved in D2O (internal standard: 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt, TSP-d4), CD3OD or DMSO-d6 (internal standard: tetramethylsilane, TMS). The experiments were performed at the temperature of 298 K. All spectra were acquired on a Bruker Avance III DRX system, operating at frequencies of 600.3 MHz (1H) and 150.9 MHz (13C). The spectrometer was equipped with a 5 mm triple-resonance inverse probe head [1H/31P/BB]. High-power 1H, 13C π/2 pulses of 9.00 and 15.00 μs, respectively, were used. 1D and 2D homo- and heteronuclear correlation experiments were carried out using pulse sequences from the Bruker pulse program library.Calculation methods
The initial structures of M1Gx-A, M1MGx-dA and NMR standards: tetramethylsilane (TMS) and 3-(trimethylsilyl)-2,2′,3,3′-tetradeuteropropionic acid (TSP-d4) were generated based on average bond lengths and valence angles; torsional angles for rotatable bonds were sampled randomly. All initial conformers of M1Gx-A (11 of enol and 11 of dialdehyde forms), M1MGx-dA (11 of enol and 13 of dialdehyde forms), TMS and TSP (1 of anion and 7 neutral forms) were optimized at the M06/6-31G(d) level.The lowest-energy conformers were selected for further research (one each for the enol and dialdehyde form for each adduct) and for the NMR standards. NMR shielding constants were calculated for the structures using GIAO at the following levels: M06/6-311++G(2df,2pd), B3LYP/6-311++G(2df,2pd) and M06/6-31++G(d,p). The calculations were performed for the compounds in the gas phase and also taking into account the effect of solvents (water, methanol and DMSO) using the polarizable continuum model (PCM). Geometry was optimized and GIAO calculations were performed using Gaussian 09.60 Calculations of 1H and 13C NMR chemical shifts (σcalc [ppm]) were based on the isotropic magnetic shield tensor (IMS) [ppm]. σcalc values [ppm] were derived as usual55 by subtracting IMS values for the test structures from the IMS value of the NMR standard (TSP for calculations in the gas phase and water or TMS for calculations in the gas phase, methanol and DMSO), that is: σcalc = IMSref − IMScalc.
The NMR chemical shifts calculated using quantum chemical methods were compared with the experimental values. Comparisons for the enol and dialdehyde forms of the studied compounds were made for each solvent. Absolute errors of NMR chemical shifts and mean absolute errors (MAEs) were calculated. The experimental and calculated results were also compared using linear regression.
Energy barriers for proton transfer in the β-dicarbonyl moieties in M1Gx-A and M1MGx-dA were calculated using the M06/6-31++G(d,p)//M06/6-31G(d) level.
Results and discussion
Synthesis of malonaldehyde–glyoxal and malonaldehyde–methylglyoxal etheno adducts of adenine nucleosides
M1Gx-A was formed in the reaction of malonaldehyde and glyoxal with adenosine, while M1MGx-dA was prepared in the reaction of malonaldehyde and methylglyoxal with 2′-deoxyadenosine43 (Fig. 3). The optimization of the synthesis conditions resulted in the product yields of 3.6% and 6.7%, respectively. | ||
| Fig. 3 Formation of M1Gx-A and M1MGx-dA. | ||
Mechanism for the formation of the studied adducts
M1Gx-A and M1MGx-dA belong to the group of conjugate adducts – the nucleoside modifications that comprise units derived from the condensation products of carbonyl compounds. The presence of malonaldehyde in the reaction mixtures results in the formation of dimers and trimers of this aldehyde, but also of conjugates with glyoxal and methylglyoxal, respectively. Multimeric adducts of malonaldehyde were proposed to be formed by reactions of this aldehyde oligomers with the DNA bases.61 An analogous mechanism was suggested for the formation of the nucleoside–malonaldehyde–acetaldehyde conjugate adducts62,63 and a similar mechanism is most likely valid also for the formation of M1Gx-A and M1MGx-dA. We proposed that these adducts are derived from the reactions of adenine nucleosides with the initially formed malonaldehyde–glyoxal and malonaldehyde–methylglyoxal conjugates, respectively. The reaction is thought to take place through a two-step addition, however the mechanism for this addition is yet to be clarified in further kinetics studies, which is beyond the scope of the current paper.Reactions between α,β-unsaturated aldehydes and 2′-deoxyguanosine are supposed to take place through Michael addition occurring by N2 or N1 of the nucleoside at the β-carbon atom followed by nucleophilic attack of N1 or N2 of dG at the carbon atom of the aldehyde group.64 The presence of a substituent at the β-carbon atom can preclude initial attack by N1.64 Reactions of α,β-unsaturated aldehydes with 2′-deoxyadenosine are suggested to occur by the addition of N6 of dA to the carbonyl carbon atom with subsequent ring closure by attack of N1 at C2 of the carbonyl compound.2,5
Due to the distinctive structure of malonaldehyde–glyoxal and malonaldehyde–methylglyoxal conjugates all plausible routes leading to the formation of M1Gx-A and M1MGx-dA should be considered. One mechanism that can be proposed, involves Michael-type addiction of N6 of the adenine nucleosides to the β-carbon atom followed by addition of the nucleoside N1 to the carbonyl group derived from glyoxal or methylglyoxal, respectively (ESI,† Fig. S1A). An alternative mechanistic explanation for M1Gx-A and M1MGx-dA formation is based on 1,4-addition of N1 to the conjugate and subsequent attack of N6 at the appropriate carbonyl carbon atom (ESI,† Fig. S1B). Preference of the first route would result in the formation of adducts with the dialdehyde methyl substituent at C8, while the second mechanism would give rise to compounds having this substituent at C7.
Discussion on the M1Gx-A and M1MGx-dA formation is additionally complicated by the fact that in both plausible mechanisms the opposite order of the addition reactions could not be ruled out (ESI,† Fig. S1C and D, respectively).
Determination of the dialdehyde methyl substituent position in M1Gx-A and M1MGx-dA on the basis of spectroscopic analysis
1H NMR, 13C NMR and correlation spectra (COSY, HSQC, HMBC and NOESY) were recorded for the M1Gx-A and M1MGx-dA adducts.The assignment of the appropriate signals from 13C NMR spectra to the etheno ring carbon atoms C7 and C8 was essential for the determination of the dialdehyde methyl group position. The assignment of the signal observed in the 13C NMR spectrum of M1Gx-A at δ 133.32 ppm (Table 1) to the carbon atom C8 was based on the one-bond correlation (HSQC) with the proton signal at δ 7.49 ppm (Table 1) and long-range correlations (HMBC) between this proton signal and carbon signals derived from C7, C1′′, CHO and C9a (Fig. 4A). The assignment of the signal at δ 121.55 ppm to the carbon atom C7 was achieved on the basis of correlation observed in the HMBC spectrum between this signal and the H-C5 proton signal (Table 1 and Fig. 4A), while no correlation were seen between the signal assigned to C8 and H-C5 signals (Fig. 4A). Furthermore, the NOESY spectrum of the adduct showed correlation between the aldehyde protons and the H-C5 proton (Fig. 4B). Therefore, the dialdehyde methyl substituent was assumed to be at position C7. This was additionally confirmed by the lack in the NOESY spectrum of M1Gx-A of correlation between the H-C5 proton and proton of the etheno ring. This correlation should be strong if the dialdehyde methyl group was bonded to the C8.
| δ(H) [ppm] | Multiplicity | J H,H [Hz] | δ(C) [ppm] | HMBC | |
|---|---|---|---|---|---|
| 2 CHO | 8.98 | s | 193.21 | C7 | |
| C1′′ | 110.58 | ||||
| C3a | 142.85 | ||||
| H-C2 | 8.46 | s | 143.77 | C3a, C9b, C1′ | |
| H-C5 | 8.60 | s | 139.89 | C3a, C9a, C9b, C7 | |
| H-C7 | 121.55 | ||||
| C8 | 7.49 | s | 133.32 | C7, C1′′, CHO, C9a | |
| C9a | 141.56 | ||||
| C9b | 125.13 | ||||
| H-C1′ | 6.21 | d | 5.43 | 91.90 | C2, C3a, C2′, C3′, C4′ |
| H-C2′ | 4.90 | t | 5.43 | 76.74 | C1′, C4′ |
| H-C3′ | 4.49 | dd | 4.18; 5.10 | 73.28 | C1′, C4′, C5′ |
| H-C4′ | 4.30 | dd | 4.18; 7.21 | 88.31 | C2′, C3′, C1′, C5′ |
| H-C5′a | 3.94 | dd | 3.02; 12.76 | 64.25 | C3′, C4′ |
| H-C5′b | 3.88 | dd | 4.25; 12.76 | C3′, C4′ |
 | ||
| Fig. 4 Major correlations in the HMBC spectrum (D2O) (A) and in the NOESY spectrum (DMSO-d6) (B) of M1Gx-A. | ||
In the etheno ring of M1MGx-dA neither C7 nor C8 is bonded to the proton and correlation observed in the HMBC spectrum between the H-C5 proton signal and one of these carbon atom signals is not sufficient to distinguish one of these atoms from the other (Table 2 and Fig. 5A). Therefore, correlations observed in the NOESY spectrum between the H-C5 proton and the aldehyde protons were crucial for the assignment of the signal at δ 117.65 ppm (Table 2 and Fig. 5B) to the C7 carbon atom bearing the dialdehyde methyl group.
| δ(H) [ppm] | Multiplicity | J H,H [Hz] | δ(C) [ppm] | HMBC | NOESY | |
|---|---|---|---|---|---|---|
| 2 CHO | 9.02 | s | 193.25 | C1′′, C7 | CH3, H-C5 | |
| C1′′ | 110.14 | |||||
| C3a | 142.01 | |||||
| H-C2 | 8.44 | s | 143.71 | C3a, C9b, C1′ | H-C1′, H-C2′a, H-C3′, H-C5′a, H-C5′b | |
| H-C5 | 8.54 | s | 139.08 | C3a, C9a, C9b, C7 | CHO | |
| C7 | 117.65 | |||||
| CH3 | 2.32 | s | 14.77 | C7, C8, C1′′, CHO | CHO | |
| C8 | 141.70 | |||||
| C9a | 142.12 | |||||
| C9b | 124.05 | |||||
| H-C1′ | 6.57 | t | 6.80 | 87.82 | C2, C3a, C2, C3′, C4′, C5′ | H-C2, H-C2′a, H-C2′b, H-C4′, H-C5′a, H-C5′b |
| H-C2′a | 2.92 | m | 6.80, 14.02 | 41.92 | C1′, C3′, C4′ | H-C1′, H-C2′b, H-C3′, H-C2 |
| H-C2′b | 2.63 | ddd | 3.92, 6.43, 14.02 | C3′, C4′ | H-C1′, H-C2′b, H-C3′, H-C4′ | |
| H-C3′ | 4.68 | dt | 3.73, 6.43 | 73.94 | C1′, C4′, C5′ | H-C2, H-C2′a, H-C2′b, H-C4′, H-C5′a, H-C5′b |
| H-C4′ | 4.18 | td | 3.59, 7.21 | 90.24 | C1′, C2′, C3′ | H-C1′, HC2′a, H-C3′, H-C5′a, H-C5′b |
| H-C5′a | 3.83 | dd | 3.59, 12.51 | 64.43 | C3′, C4′ | C-H1′, H-C2′b, H-C3′, H-C4′, H-C2 |
| H-C5′b | 3.79 | dd | 4.87; 12.51 | C3′, C4′ | C-H1′, H-C2′b, H-C3′, H-C4′, H-C2 |
 | ||
| Fig. 5 Major correlations in the HMBC spectrum (D2O) (A) and in the NOESY spectrum (DMSO-d6) (B) of M1MGx-dA. | ||
Based on the presence of respective correlations in the NOESY spectra, and also on the lack thereof, the structures of other substituted etheno adducts were determined.9,65 The position of the 2-oxobutyl group in the etheno ring of the 2′-deoxyadenosine derivative formed in a reaction between the nucleoside and 4-oxo-2-hexanal9 as well as the position of the methyl group in etheno adenosine derivatives was determined.65 The 4-oxobutyl substituent at position C7 was additionally confirmed by correlation between protons of the methylene group of the substituents bonded to the etheno ring and the adenosine N1 atom seen in the 1H–15N HMBC spectrum.9
Structural determination of M1Gx-A and M1MGx-dA based on comparison between calculated and experimental chemical shift values
Most of the M1Gx-A and M1MGx-dA conformers selected for GIAO calculations (Fig. 6) had syn conformation, even though it is generally assumed that the anti conformation prevails in DNA, in nucleosides and nucleotides. However, results of spectroscopic studies combined with calculations indicated that adenine nucleosides take both conformations at a similar level in solution.66 Furthermore, results based on quantum chemical calculations suggest that syn conformers of nucleoside derivatives are more stable in aqueous media.67 Etheno adducts reveal a tendency for syn conformation in double-strand DNA that facilitates the formation of Hoogsteen base pairs.23,68 Furthermore, etheno adducts in the syn conformation are more susceptible to DNA repair processes.69 | ||
| Fig. 6 Lowest-energy conformers of M1Gx-A ((A) – dialdehyde form; (B) – enol form) and of M1MGx-dA ((C) – dialdehyde form; (D) – enol form). | ||
GIAO calculations were carried out at the M06/6-311++G(2df,2pd), B3LYP/6-311++G(2df,2pd) and M06/6-31++G(d,p) levels. TMS and a neutral and anionic TSP form were standards in NMR chemical shift calculations. The calculations were performed for in vacuo molecules and using the PCM to take into account the effect of solvents (water, methanol and DMSO).
Computational results were compared with the experimental data. The comparison was based on the following parameters: absolute errors, mean absolute errors (MAEs) and coefficients of determination R2. Furthermore, the solvent effects on the correlation between experimental and theoretical results were determined.
The analysis of calculated 13C NMR chemical shifts for the dialdehyde forms of M1Gx-A and M1MGx-dA showed that the largest absolute errors occurred for the C1′′ atoms (up to 60 ppm) and C8 and CHO (up to 20 ppm) (see ESI,† Tables S9–S12 and S17–S20). The largest errors for the enol forms were seen for the CHO, C1′ and C2 atoms (up to 20 ppm) in M1Gx-A and CHO, C8 (up to 20 ppm) in M1MGx-dA (see ESI,† Tables S13–S16 and S21–S24). The largest absolute errors for 1H NMR chemical shifts for the dialdehyde and enol forms of M1Gx-A and M1MGx-dA occurred for the aldehyde protons (2 ppm) (see ESI,† Tables S9–S24). The R2 coefficients determined for calculations of 13C NMR chemical shifts for M1Gx-A and M1MGx-dA were 0.83–0.89 and 0.91–0.93, respectively, for the dialdehyde forms and 0.98–0.99 and >0.99, respectively, for the enol forms of both compounds. The R2 coefficients determined for calculations of 1H NMR chemical shifts for M1Gx-A and M1MGx-dA were 0.97–0.98 and 0.93–0.97, respectively, for the dialdehyde forms and 0.96–0.99 and 0.98–0.99, respectively, for the enol forms (see ESI,† Tables S9–S24). Comparison of R2 values for the calculations performed for the enol forms shows that they are closer to the experimental data. The calculated values with the best correlation with the spectroscopic data are shown in Tables 3 and 4, and also S26 and S27 in the ESI† (remaining data in ESI,† Tables S9–S28).
| δ exp(H) [ppm] | δ exp(C) [ppm] | δ calc(H) [ppm] | |Δ|δ(H) [ppm] | δ calc(C) [ppm] | |Δ|δ(C) [ppm] | |
|---|---|---|---|---|---|---|
| 2 CHO | 8.98 | 193.21 | 8.78 | 0.20 | 193.81 | 0.60 |
| C1′′ | 110.58 | 112.67 | 2.09 | |||
| C3a | 142.85 | 145.38 | 2.53 | |||
| H-C2 | 8.46 | 143.77 | 8.03 | 0.43 | 149.65 | 5.88 |
| H-C5 | 8.6 | 139.89 | 8.92 | 0.32 | 140.47 | 0.58 |
| C7 | 121.55 | 125.60 | 4.05 | |||
| C8 | 7.49 | 133.32 | 7.66 | 0.17 | 142.53 | 9.21 |
| C9a | 141.56 | 150.69 | 9.13 | |||
| C9b | 125.13 | 134.36 | 9.23 | |||
| H-C1′ | 6.21 | 91.9 | 5.98 | 0.23 | 100.73 | 8.83 |
| H-C2′a | 4.9 | 76.74 | 4.88 | 0.02 | 80.05 | 3.31 |
| H-C3′ | 4.49 | 73.28 | 4.63 | 0.14 | 80.80 | 7.52 |
| H-C4′ | 4.3 | 88.31 | 4.38 | 0.08 | 96.42 | 8.11 |
| H-C5′a | 3.94 | 64.25 | 3.81 | 0.13 | 69.75 | 5.50 |
| H-C5′b | 3.88 | 4.07 | 0.19 | |||
| MAE | 0.19 | MAE | 5.47 | |||
| R 2 | 0.9877 | R 2 | 0.9922 |
| δ exp(H) [ppm] | δ exp(C) [ppm] | δ calc(H) [ppm] | |Δ|δ(H) [ppm] | δ calc(C) [ppm] | |Δ|δ(C) [ppm] | |
|---|---|---|---|---|---|---|
| 2 CHO | 8.91 | 183.64 | 9.03 | 0.12 | 182.54 | 1.10 |
| C1′′ | 106.56 | 101.19 | 5.37 | |||
| C3a | 137.86 | 137.88 | 0.02 | |||
| H-C2 | 8.49 | 139.56 | 8.13 | 0.36 | 140.00 | 0.44 |
| H-C5 | 8.33 | 136.1 | 8.88 | 0.55 | 135.56 | 0.54 |
| C7 | 115.37 | 113.88 | 1.49 | |||
| CH3 | 2.18 | 14.16 | 2.61 | 0.43 | 9.92 | 4.24 |
| C8 | 138.42 | 145.70 | 7.28 | |||
| C9a | 138.68 | 141.07 | 2.39 | |||
| C9b | 122.33 | 125.20 | 2.87 | |||
| H-C1′ | 6.47 | 83.87 | 6.34 | 0.13 | 86.64 | 2.77 |
| H-C2′a | 2.74 | 39.71 | 3.49 | 0.75 | 38.12 | 1.59 |
| H-C2′b | 2.36 | 2.26 | 0.10 | |||
| H-C3′ | 4.44 | 70.68 | 4.20 | 0.24 | 75.13 | 4.45 |
| H-C4′ | 3.9 | 87.9 | 4.28 | 0.38 | 89.28 | 1.38 |
| H-C5′a | 3.63 | 61.65 | 3.99 | 0.36 | 64.31 | 2.66 |
| H-C5′b | 3.54 | 3.76 | 0.22 | |||
| MAE | 0.33 | MAE | 2.57 | |||
| R 2 | 0.9819 | R 2 | 0.9951 |
Similar to R2 values, the analysis of mean absolute errors (MAEs) for 1H NMR chemical shifts for M1Gx-A (Chart 1A) shows that error values are typically higher for the dialdehyde form. Computational results for the enol form of the adduct are much more similar to the experimental data other than results for the spectrum with DMSO-d6 as solvent. When a model for solvent was included in calculations, lower MAE values were obtained. The lowest MAE values (approx. 0.2 ppm, Chart 1A) were found with M06 and B3LYP functionals and the 6-311++G(2df,2pd) basis set for the enol form of the adduct when the results were compared to the NMR spectra measured using D2O and CD3OD as solvents.
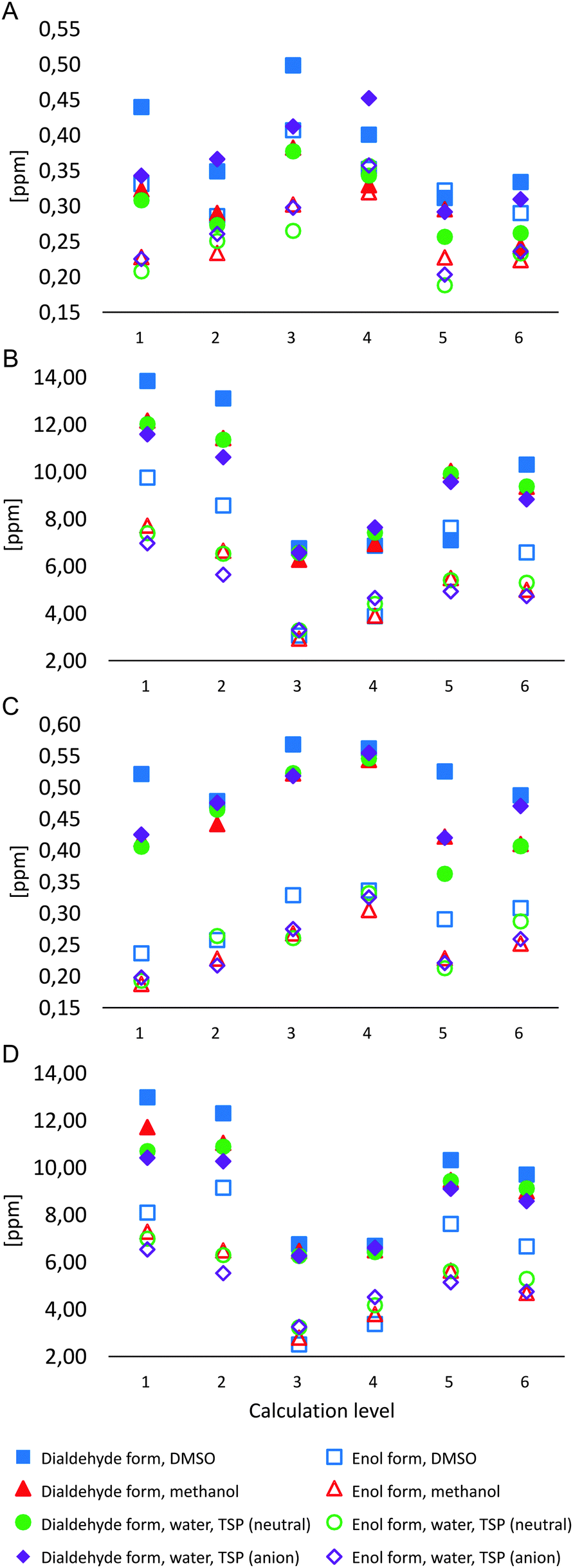 | ||
| Chart 1 Comparison of MAE values for chemical shift calculations: (A) 1H NMR, M1GxA; (B) 13C NMR, M1Gx-A; (C) 1H NMR, M1MGx-dA; (D) 13C NMR, M1MGx-dA; (1) M06/6-311++G(2df,2pd), PCM; (2) M06/6-311++G(2df,2pd), in vacuo; (3) M06/6-31++G(d,p) PCM; (4) M06/6-31++G(d,p), in vacuo; (5) B3LYP/6-311++G(2df,2pd) PCM; (6) B3LYP/6-311++G(2df,2pd), in vacuo. | ||
The comparison of experimental and calculated 13C NMR chemical shifts for M1Gx-A (Chart 1B) showed that mean absolute errors were higher for the dialdehyde form, similar to the 1H NMR chemical shifts. The best correlation with the experimental data derived from NMR spectra recorded using D2O, CD3OD and DMSO-d6 as solvents was seen for the calculations based on the M06/6-31++G(d,p) level for the enol form of M1Gx-A with solvent effects included (MAE = 3 ppm, Chart 1B).
The comparison of MAE values for 1H NMR chemical shift calculations for M1MGx-dA shows significant separation of results for the dialdehyde and the enol form (Chart 1C). Calculated results for the enol form are more similar to the spectroscopic data. Calculations that include solvent effects show even better correlation with the experimental data than those based on the same methods for the compound in the gas phase. Similar to M1Gx-A, the lowest MAE values (approx. 0.2 ppm, Chart 1C) were obtained using M06 and B3LYP functionals and the 6-311++G(2df,2dp) basis set including PCM for the solvent when the results were compared to the NMR spectra recorded using D2O and CD3OD. Slightly higher MAEs were obtained when the calculated results based on the above parameters were compared with the experimental results with DMSO-d6 as solvent (Chart 1C).
The comparison of MAE values for the 13C NMR chemical shifts calculated and measured for the M1MGx-dA adduct shows clear separation of results for the dialdehyde and the enol form (Chart 1D). The chart shows that calculations for the enol form at the M06/6-31++G(d,p) level including PCM for the solvent provided results closest to the experimental data (MAE of 2.5–3.5 ppm). The comparison shows that DMSO is the solvent that yields the best match between the experimental and the computational results (MAE = 2.57 ppm) (Chart 1D).
The largest differences between the experimental and the computational results concerned the β-dicarbonyl moiety in both adducts. The computational results for the dialdehyde forms significantly differed from experimental data especially for the shielding constant of the C1′′ atom. The computational results for the enol forms were much closer to the experimental data. Furthermore, one signal with integration 2 from the aldehyde protons was seen in the 1H NMR spectra and one signal from the aldehyde carbon atoms in the 13C NMR spectra. This proves that the experimental NMR chemical shifts within those parts of the adducts were averaged. This most likely results from the enol form being dominant for the compounds and more rapid conversions between the two possible enol forms than the relaxation of excited hydrogen and carbon nuclei.70–72 The calculated energy barriers for proton transfer in the β-dicarbonyl moieties are 1.8 kcal mol−1 for M1Gx-A and 1.5 kcal mol−1 for M1MGx-dA. Literature reports are available that confirm that the energy barriers for proton transfer in similar systems are low.73,74 The energy barrier for proton transfer in malonaldehyde calculated using ab initio methods is similar to the values obtained for M1Gx-A and M1MGx-dA,73 while the results of semiempirical methods seem to be very high.75 The results of the crystallographic investigation of compounds with a β-dicarbonyl system also confirm the averaged position of the hydrogen atom between the two oxygen atoms.76
It is noted that the experimental results (D2O) were obtained with TSP-d4 as the NMR standard. The standard was also used in chemical shift calculations. TSP may have a neutral or an anionic form. Both structures were optimized and subjected to GIAO calculations using the same methods as the adducts studied. Subsequently, IMS values for both the neutral and anionic TSP form were used to determine NMR chemical shifts of the adducts. Better correlation with the experimental data was seen with the anionic TSP form as the NMR standard in calculations for 13C NMR chemical shifts. The neutral form of the standard, in turn, provided better correlation with spectroscopic data for 1H NMR chemical shifts. More thorough investigation would be needed to determine the actual TSP form present in the studied solutions, and it goes beyond the scope of this project. It is noted, however, that TSP was used in literature reports for NMR experiments whose results were compared to computational results with TMS as the reference.77,78 This may be due to the fact that NMR chemical shifts for both TMS and TSP are by definition δH = 0 ppm and δC = 0 ppm. Experiments prove, however, that minor differences occur between chemical shifts of the standards, being approx. 0.1 ppm for 1H NMR and approx. 0.2 ppm for 13C NMR depending on the solvent.79–82 The conformers of TSP in the neutral and anionic form used in GIAO calculations with the calculated IMS values are listed in ESI,† Fig. S2, S3 and Table S8.
Conclusions
It is now commonly agreed that exocyclic etheno adducts together with other oxidative DNA damage could play an important role in the multistage process of carcinogenesis. The use of such adducts as biomarkers offers a promising tool in studies on cancer etiology and prevention, particularly for human neoplasms in which the causative factors and mechanisms are still poorly understood.83 Therefore detailed knowledge of DNA adduct structure is extremely important.Experimental methods (NMR spectroscopy) together with quantum chemical calculations were applied for the structural studies of substituted malonaldehyde–glyoxal and malonaldehyde–methylglyoxal etheno adducts of adenine nucleosides (M1Gx-A and M1MGx-dA, respectively). It was found on the basis of the results that in both adducts the dialdehyde methyl substituent was attached to C7 of the etheno rings, in contrast to the earlier report.43 Our results as well as those derived from the literature9,65 clearly indicate that correlation spectra, especially NOESY, are necessary for the determination of the substitution position in the etheno rings of the adducts among spectroscopic techniques. However NOE data are not always pivotal.2 Our studies demonstrate that quantum chemical calculations provide valuable information that helps in solving the structural problems associated with the presence of the substituent at C7 or C8 in substituted etheno adducts.
Based on the comparison of data derived from NMR spectra recorded in three solvents (D2O, CD3OD and DMSO-d6) with computational results in the gas phase and using PCM, the optimum computational method for the compounds was selected. The experimental 1H NMR chemical shift values had the highest correlation with computational results for M06 or B3LYP functionals with the 6-311++G(2df,2pd) basis set. However, 13C NMR chemical shift calculations had the highest correlation with GIAO results at the M06/6-31++G(d,p) level. PCM improved the correlation of results in both cases.
Furthermore, computational results for the enol forms of adduct structures were closer to the experimental data. Therefore, the enol form was suggested to prevail in solution for the compounds studied. Furthermore, the averaging of 1H and 13C NMR chemical shifts indicated rapid proton transfer between both enol forms of the compounds. This resulted from a low energy barrier for proton transfer in the β-dicarbonyl system. The calculated energy barrier for proton transfer is 1.8 kcal mol−1 for M1Gx-A and 1.5 kcal mol−1 for M1MGx-dA.
List of abbreviations
| MA | Malonaldehyde |
| MAE | Mean absolute error |
| M1Gx | Conjugate of one molecule of malonaldeyde and one molecule of glyoxal |
| M1MGx | Conjugate of one molecule of malonaldeyde and one molecule of methylglyoxal |
| M1Gx-A | 7-(Diformylmethyl)-3-(β-D-ribofuranosyl)imidazo[2,1-i]purine |
| M1MGx-dA | 7-(Diformylmethyl)-8-methyl-3-(2′-deoxy-β-D-ribofuranosyl)imidazo[2,1-i]purine |
| TMP | 1,1,3,3-Tetramethoxypropane |
| TMS | Tetramethylsilane |
| TSP-d4 | 3-(Trimethylsilyl)propionic-2,2,3,3-d4 acid |
Acknowledgements
This research was supported in part by PL-Grid Infrastructure (http://www.plgrid.pl/en).References
- D. Rindgen, M. Nakajima, S. Wehrli, K. Xu and I. A. Blair, Chem. Res. Toxicol., 1999, 12, 1195–1204 CrossRef CAS PubMed.
- S. H. Lee, D. Rindgen, R. H. Bible, E. Hajdu and I. A. Blair, Chem. Res. Toxicol., 2000, 13, 565–574 CrossRef CAS PubMed.
- D. Rindgen, S. H. Lee, M. Nakajima and I. A. Blair, Chem. Res. Toxicol., 2000, 13, 846–852 CrossRef CAS PubMed.
- Y. Fu, R. G. Nath, M. Dyba, I. M. Cruz, S. R. Pondicherry, A. Fernandez, C. L. Schultz, P. Yang, J. Pan, D. Desai, J. Krzeminski, S. Amin, P. P. Christov, Y. Hara and F.-L. Chung, Free Radical Biol. Med., 2014, 73, 12–20 CrossRef CAS PubMed.
- M. Pollack, T. Oe, S. H. Lee, M. V. Silva Elipe, B. H. Arison and I. A. Blair, Chem. Res. Toxicol., 2003, 16, 893–900 CrossRef CAS PubMed.
- Y. Kawai, K. Uchida and T. Osawa, Free Radical Biol. Med., 2004, 36, 529–541 CrossRef CAS PubMed.
- P. Zhu, S. H. Lee, S. Wehrli and I. A. Blair, Chem. Res. Toxicol., 2006, 19, 809–817 CrossRef CAS PubMed.
- S. H. Lee and I. A. Blair, Chem. Res. Toxicol., 2000, 13, 698–702 CrossRef CAS PubMed.
- K. Kawai, P.-H. Chou, T. Matsuda, M. Inoue, K. Aaltonen, K. Savela, Y. Takahashi, H. Nakamura, T. Kimura, T. Watanabe, R. Sawa, K. Dobashi, Y.-S. Li and H. Kasai, Chem. Res. Toxicol., 2010, 23, 630–636 CrossRef CAS PubMed.
- V. M. Carvalho, P. Di Mascio, I. P. de Arruda Campos, T. Douki, J. Cadet and M. H. G. Medeiros, Chem. Res. Toxicol., 1998, 11, 1042–1047 CrossRef CAS PubMed.
- V. M. Carvalho, F. Asahara, P. Di Mascio, I. P. de Arruda Campos, J. Cadet and M. H. G. Medeiros, Chem. Res. Toxicol., 2000, 13, 397–405 CrossRef CAS PubMed.
- A. P. M. Loureiro, P. Di Mascio, O. F. Gomes and M. H. G. Medeiros, Chem. Res. Toxicol., 2000, 13, 601–609 CrossRef CAS PubMed.
- S. H. Lee, J. A. Arora, T. Oe and I. A. Blair, Chem. Res. Toxicol., 2005, 18, 780–786 CrossRef CAS PubMed.
- I. G. Minko, I. D. Kozekov, T. M. Harris, C. J. Rizzo, R. S. Lloyd and M. P. Stone, Chem. Res. Toxicol., 2009, 22, 759–778 CrossRef CAS PubMed.
- S. H. Lee, T. Oe and I. A. Blair, Chem. Res. Toxicol., 2002, 15, 300–304 CrossRef CAS PubMed.
- S. H. Lee, M. V. Silva Elipe, J. S. Arora and I. A. Blair, Chem. Res. Toxicol., 2005, 18, 566–578 CrossRef CAS PubMed.
- N. K. Kochetkov, V. N. Shibaev and A. A. Kost, Tetrahedron Lett., 1971, 12, 1993–1996 CrossRef.
- M. C. Byrns, C. C. Vu and L. A. Peterson, Chem. Res. Toxicol., 2004, 17, 1607–1613 CrossRef CAS PubMed.
- A. P. M. Loureiro, I. P. de Arruda Campos, O. F. Gomes, E. P. M. Possari, P. Di Mascio and M. H. G. Medeiros, Chem. Res. Toxicol., 2005, 18, 290–299 CrossRef CAS PubMed.
- F. P. Guengerich and V. D. Raney, J. Am. Chem. Soc., 1992, 114, 1074–1080 CrossRef CAS.
- K. Linhart, H. Bartsch and H. K. Seitz, Redox Biol., 2014, 3, 56–62 CrossRef CAS PubMed.
- J. Nair, A. Barbin, I. Velic and H. Bartsch, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1999, 424, 59–69 CrossRef CAS PubMed.
- G. Shanmugam, I. D. Kozekov, F. P. Guengerich, C. J. Rizzo and M. P. Stone, Chem. Res. Toxicol., 2011, 24, 1071–1079 CrossRef CAS PubMed.
- A. B. Guliaev, J. Sági and B. Singer, Carcinogenesis, 2000, 21, 1727–1736 CrossRef CAS PubMed.
- P. K. Sahu, C.-W. Kuo and S.-L. Lee, J. Phys. Chem. B, 2007, 111, 2991–2998 CrossRef CAS PubMed.
- G. Shanmugam, I. D. Kozekov, F. P. Guengerich, C. J. Rizzo and M. P. Stone, Biochemistry, 2010, 49, 2615–2626 CrossRef CAS PubMed.
- L. Zhao, M. G. Pence, P. P. Christov, Z. Wawrzak, J.-Y. Choi, C. J. Rizzo, M. Egli and F. P. Guengerich, J. Biol. Chem., 2012, 287, 35516–35526 CrossRef CAS PubMed.
- S. C. Srivastava, S. K. Raza and R. Misra, Nucleic Acids Res., 1994, 22, 1296–1304 CrossRef CAS PubMed.
- M. Goto, K. Shinmura, Y. Matsushima, K. Ishino, H. Yamada, Y. Totsuka, T. Matsuda, H. Nakagama and H. Sugimura, Free Radical Biol. Med., 2014, 76, 136–146 CrossRef CAS PubMed.
- A. Winczura, A. Czubaty, K. Winczura, K. Masłowska, M. Nałcz, D. A. Dudzińska, M. Saparbaev, K. Staroń and B. Tudek, DNA Repair, 2014, 22, 1–11 CrossRef CAS PubMed.
- E. Speina, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2003, 531, 205–217 CrossRef CAS PubMed.
- H. Bartsch and J. Nair, Eur. J. Cancer, 2000, 36, 1229–1234 CrossRef CAS PubMed.
- S. J. Sturla, Curr. Opin. Chem. Biol., 2007, 11, 293–299 CrossRef CAS PubMed.
- S. M. Rappaport, H. Li, H. Grigoryan, W. E. Funk and E. R. Williams, Toxicol. Lett., 2012, 213, 83–90 CrossRef CAS PubMed.
- G. Aldini, I. Dalle-Donne, R. M. Facino, A. Milzani and M. Carini, Med. Res. Rev., 2007, 27, 817–868 CrossRef CAS PubMed.
- T. Shibamoto, J. Pharm. Biomed. Anal., 2006, 41, 12–25 CrossRef CAS PubMed.
- G. E. Wuenschell, D. Tamae, A. Cercillieux, R. Yamanaka, C. Yu and J. Termini, Biochemistry, 2010, 49, 1814–1821 CrossRef CAS PubMed.
- T. Wang, R. Kartika and D. A. Spiegel, J. Am. Chem. Soc., 2012, 134, 8958–8967 CrossRef CAS PubMed.
- M. Kalousová, T. Zima, V. Tesar, S. Dusilová-Sulková and J. Skrha, Mutat. Res., 2005, 579, 37–46 CrossRef PubMed.
- C. L. Price and S. C. Knight, Trends Endocrinol. Metab., 2009, 20, 312–317 CrossRef CAS PubMed.
- R. Rolla, D. Vay, E. Mottaran, M. Parodi, N. Traverso, S. Aricó, M. Sartori, G. Bellomo, L. W. Klassen, G. M. Thiele, D. J. Tuma and E. Albano, Hepatology, 2000, 31, 878–884 CrossRef CAS PubMed.
- D. J. Tuma, M. L. Kearley, G. M. Thiele, S. Worrall, A. Haver, L. W. Klassen and M. F. Sorrell, Chem. Res. Toxicol., 2001, 14, 822–832 CrossRef CAS PubMed.
- D. Pluskota-Karwatka, A. J. Pawłowicz, M. Bruszyńska, A. Greszkiewicz, R. Latajka and L. Kronberg, Chem. Biodiversity, 2010, 7, 959–974 CAS.
- J. C. Facelli, J. Phys. Chem. B, 1998, 102, 2111–2116 CrossRef CAS.
- H. M. E. Dong Hui Kim, Bull. Korean Chem. Soc., 2000, 21, 148–150 Search PubMed.
- S. Olejniczak and M. J. Potrzebowski, Org. Biomol. Chem., 2004, 2, 2315 CAS.
- A. Bagno, F. Rastrelli and G. Saielli, Chem. – Eur. J., 2006, 12, 5514–5525 CrossRef CAS PubMed.
- S. N. Azizi and C. Esmaili, World Appl. Sci. J., 2009, 7, 559–566 CAS.
- M. Bugaj, P. A. Baran, P. Bernatowicz, P. Brożek, K. Kamieńska-Trela, A. Krówczyński and B. Kamieński, Magn. Reson. Chem., 2009, 47, 830–842 CrossRef CAS PubMed.
- M. Karabacak, M. Cinar and M. Kurt, Spectrochim. Acta, Part A, 2009, 74, 1197–1203 CrossRef PubMed.
- B. Wang, A. T. Dossey, S. S. Walse, A. S. Edison and K. M. Merz, J. Nat. Prod., 2009, 72, 709–713 CrossRef CAS PubMed.
- F. A. A. Mulder and M. Filatov, Chem. Soc. Rev., 2010, 39, 578–590 RSC.
- H.-D. Xie, Y.-P. Li, K.-X. Qiu, B. Liu and Y.-P. Chen, Chem. Res. Chin. Univ., 2010, 26, 1016–1019 CAS.
- E. J. Borkowski, Open Nat. Prod. J., 2012, 5, 1–6 CrossRef CAS.
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS PubMed.
- M. Corredor, J. Bujons, À. Messeguer and I. Alfonso, Org. Biomol. Chem., 2013, 11, 7318 CAS.
- F. J. Melendez, A. Degollado, M. E. Castro, N. A. Caballero, J. A. Guevara-García and T. Scior, Inorg. Chim. Acta, 2014, 420, 149–158 CrossRef CAS.
- T. J. Aímola, D. J. P. Lima, L. C. Dias, C. F. Tormena and M. A. B. Ferreira, Org. Biomol. Chem., 2015, 13, 2140–2145 Search PubMed.
- K. Stone, A. Uzieblo and L. J. Marnett, Chem. Res. Toxicol., 1990, 3, 467–472 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- K. Stone, M. B. Ksebati and L. J. Marnett, Chem. Res. Toxicol., 1990, 3, 33–38 CrossRef CAS PubMed.
- D. Pluskota-Karwatka, F. Le Curieux, T. Munter, R. Sjöholm and L. Kronberg, Chem. Res. Toxicol., 2005, 18, 300–307 CrossRef CAS PubMed.
- D. Pluskota-Karwatka, A. J. Pawłowicz and L. Kronberg, Chem. Res. Toxicol., 2006, 19, 921–926 CrossRef CAS PubMed.
- D. W. Boerth, E. Eder, S. Hussain and C. Hoffman, Chem. Res. Toxicol., 1998, 11, 284–294 CrossRef CAS PubMed.
- P. Virta, T. Holmström, T. Munter, T. Nyholm, L. Kronberg and R. Sjöholm, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 85–98 CAS.
- H. Rosemeyer, G. Toth, B. Golankiewicz, Z. Kazimierczuk, W. Bourgeois, U. Kretschmer, H. P. Muth and F. Seela, J. Org. Chem., 1990, 55, 5784–5790 CrossRef CAS.
- T. Grabarkiewicz and M. Hoffmann, J. Mol. Model., 2006, 12, 205–212 CrossRef CAS PubMed.
- G. Shanmugam, I. D. Kozekov, F. P. Guengerich, C. J. Rizzo and M. P. Stone, Chem. Res. Toxicol., 2008, 21, 1795–1805 CrossRef CAS PubMed.
- G. Shanmugam, A. K. Goodeneough, I. D. Kozekov, F. P. Guengerich, C. J. Rizzo and M. P. Stone, Chem. Res. Toxicol., 2007, 20, 1601–1611 CrossRef CAS PubMed.
- S. L. Baughcum, R. W. Duerst, W. F. Rowe, Z. Smith and E. B. Wilson, J. Am. Chem. Soc., 1981, 103, 6296–6303 CrossRef CAS.
- M. Tkadlecová, J. Havlíček, P. Matějka, J. Fähnrich, V. Král and K. Volka, J. Mol. Struct., 2001, 563–564, 497–501 CrossRef.
- E. Iglesias, Curr. Org. Chem., 2004, 8, 1–24 CrossRef CAS.
- G. Karlstrom, B. Jonsson, B. Roos and H. Wennerstrom, J. Am. Chem. Soc., 1976, 98, 6851–6854 CrossRef CAS.
- S. J. Grabowski, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2006, 102, 131 RSC.
- G. Buemi and C. Gandolfo, J. Chem. Soc., Faraday Trans. 2, 1989, 85, 215–227 RSC.
- G. K. H. Madsen, B. B. Iversen, F. K. Larsen, M. Kapon, G. M. Reisner and F. H. Herbstein, J. Am. Chem. Soc., 1998, 120, 10040–10045 CrossRef CAS.
- S. A. Samsonov, S. Theisgen, T. Riemer, D. Huster and M. T. Pisabarro, BioMed Res. Int., 2014, 2014, 1–11 CrossRef PubMed.
- M. G. Siskos, V. G. Kontogianni, C. G. Tsiafoulis, A. G. Tzakos and I. P. Gerothanassis, Org. Biomol. Chem., 2013, 11, 7400 CAS.
- R. E. Hoffman, Magn. Reson. Chem., 2006, 44, 606–616 CrossRef CAS PubMed.
- F. van de Velde, L. Pereira and H. S. Rollema, Carbohydr. Res., 2004, 339, 2309–2313 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Pure Appl. Chem., 2008, 80, 59–84 CrossRef CAS.
- B. Singer and H. Bartsch, Exocyclic DNA Adducts in Mutagenesis and Carcinogenesis, IARC Sci. Publ. No 150, IARC, Lyon, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nj02835c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |