
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the formation of gold nanoparticles from [AuIIICl4]− and a non-classical reduced polyoxomolybdate as an electron source: a quantum mechanical modelling and experimental study†
Zhongling
Lang
a,
Isabel Maicas
Gabas
b,
Xavier
López
*a,
Anna
Clotet
a,
Jesús M.
de la Fuente
c,
Scott G.
Mitchell
*c and
Josep M.
Poblet
*a
aDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, c/Marcel·lí Domingo 1, 43007 Tarragona, Spain. E-mail: josepmaria.poblet@urv.cat
bInstituto de Nanociencia de Aragón (INA), Universidad de Zaragoza, 50018 Zaragoza, Spain
cInstituto de Ciencia de Materiales de Aragón-CSIC/Universidad de Zaragoza, Spain. E-mail: scott@unizar.es
First published on 5th January 2016
Abstract
Polyoxometalate (POM)-mediated reduction and nucleation mechanisms in nanoparticle (NP) syntheses are still largely unknown. We carried out comprehensive theoretical analysis using density functional theory (DFT) to gain insight into the molecular and electronic changes that occur during the reduction of HAuIIICl4 with the Kabanos-type polyoxomolybdate, [Na{(MoV2O4)3(μ2-O)3(μ2-SO3)3(μ6-SO3)}2]15−. In the system presented herein the electrons are supplied by the POM, making the computational thermodynamic analysis more feasible. Our results reveal that this particular POM is a multi-electron source and the proton-coupled electron transfer (PCET) greatly promotes the reduction process. Based on the energy and molecular orbital studies of the intermediate species the reduction of AuIII to AuI is shown to be thermodynamically favourable, and a low HOMO–LUMO gap of the POM–Au superstructure is advantageous for electron transfer. By modelling the reduction of three couples of AuIII → AuI by the same POM unit, it is proposed that the reduced polyoxomolybdate is finally fully oxidised. The subjacent idea of using the Kabanos POM was confirmed by comprehensive experimental characterisation of POM-stabilised gold nanoparticles (AuNPs@POM). Present theoretical analysis suggests that protons have a significant influence on the final AuI to Au0 reduction step that ultimately leads to colloidal AuNPs@POM.
Introduction
Metal nanoparticles (NPs) (or nanocrystals), such as Ag, Au, Pd, and Pt possess an array of fascinating physicochemical properties (e.g. superparamagnetism, surface plasmons and radiation-to-heat transduction) that are not present in the corresponding bulk materials.1 These size-dependent properties lend them a wide variety of applications in different scientific, technological and biomedical fields such as chemical catalysis2 and materials science,3 but also biomedical and biotechnological applications such as photothermal therapy,4 optoacoustic imaging5 as well as colorimetric and thermal biosensors.6 AuNP syntheses are particularly well known due to their applications in diagnosis, drug delivery, imaging, treatment and theranostics.7–13 The size, shape and dispersion of NPs govern their properties,14–16 thus optimising and understanding preparation methods and elucidating the nucleation mechanisms have become core research topics.17–19 Their final shapes and sizes are determined by a combination of thermodynamic (temperature and reduction potential) and kinetic factors (reactant concentration, diffusion and solubility), as well as the presence and nature of their capping (or stabilising) agents.The recent application of polyoxometalates (abbreviated POMs for any form belonging to this family of compounds) to the synthesis and stabilisation of AuNPs has opened up the door to environmentally friendly and energy saving methods since the redox properties of POMs allow them to be recycled and reused.20–22 POMs are anionic metal-oxide molecular clusters typically formed by early-row transition metals (Mo, W, V, Nb, and Ta) that exhibit fascinating and applicable physical and chemical properties.23–30 Importantly, numerous POMs can act as effective electron-transfer catalysts due to their unique ability to reversibly accept and release electrons. Several recent reviews by Nadjo,31 Mitchell32 and Weinstock33 have pointed out that POMs or POM-based materials can act both as reducing agents and as stabilisers for the preparation of metallic (such as Au, Ag, Pd, and Pt) nanostructures. This motivated us to investigate the use of POMs with ‘in-built’ reduction capabilities for the preparation of AuNPs under mild reaction conditions.34 In spite of these advances, these processes face significant challenges due to a poor understanding of their reducing and stabilising mechanisms coupled with the limited number of POMs that have been explored for this purpose. The role played by theoretical chemistry in the field of POMs in the last two decades35,36 has opened up a possibility to significantly understand the present research.
Small two-electron reduced (MoIV) thiometalates have been used to produce AuNPs.37 We recently identified a highly-reduced POM, the Kabanos structure, [Na{(MoV2O4)3(μ2-O)3(μ2-SO3)3(μ6-SO3)}2]15−, that could potentially be used as a reducing agent and as an efficient multidentate capping ligand in the preparation of AuNPs (Fig. 1).38 This is because this non-classical POM contains two layers of hexanuclear polyoxomolybdenum(V) sulfite anions with all of its Mo atoms in a one-electron reduced oxidation state (MoV). Recently, some of us have found that this POM can be used to synthesize pseudo-spherical AuNPs@POMs that demonstrate significant cell internalisation capacity in vitro and antiproliferative effects in tumoural cells.39
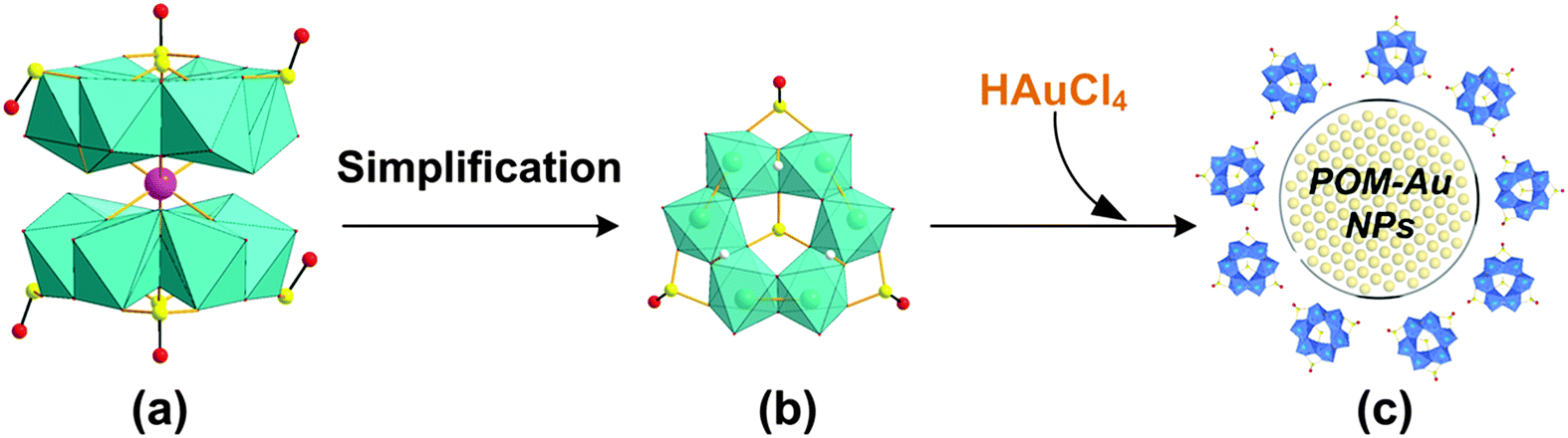 | ||
| Fig. 1 Relevant species acting in the HAuCl4 reduction: (a) dimeric form [Na{(MoV2O4)3(μ2-O)3(μ2-SO3)3(μ6-SO3)}2]15−, (b) simplified protonated monomeric form [(MoV2O4)3(μ2-OH)3(μ2-SO3)3(μ6-SO3)]5− used for modelling and (c) a schematic view of the final AuNP with the resulting POM stabilising the AuNP core (AuNPs@POM). Colour code: green octahedra – reduced MoO6, yellow – S, magenta – Na, red – O, blue octahedra – oxidised MoO6. | ||
We herein report computational evidence of the role of the Kabanos POM in the formation of AuNPs as well as experimental data for AuNPs@POM. The computational part of the research clarifies the role of the POM in the reduction of AuCl4− ions, and allows us to propose a route for the complete reduction of AuIII. We have focused essentially on understanding the thermodynamics of the processes until the full oxidation of the anions and on the capacity of the two anions to approach each other in order to be ready for electron transfer from the reduced polyoxometalate to gold species. It is worth mentioning that the reaction does not require electrochemical reduction of gold (all of the electrons are supplied by the Kabanos POM itself), which greatly simplifies the computational analysis of the thermodynamic aspects of the reaction.
Methods
Computational details
All geometric and electronic properties were determined by density functional theory (DFT) calculations implemented in the Gaussian 09 software package.40 In the first step, we optimized the geometries of all the compounds using the B3LYP functional41,42 and double-ζ type basis sets: LANL2DZ for Mo and Au, and 6-31G(d,p) for non metal atoms (O, S, Cl, and H).43–46 On top of these structures we re-optimized the geometries using large triple-ζ type basis sets: 6-311++G(d,p) for O, S, Cl and H, and def2-TZVPP for Au and Mo,47,48 together with dispersion effects using the B3LYP-D3(BJ) approach.49 These accurate structures furnished total electronic energies (Eelec) and molecular orbitals. Using the double-ζ type basis sets we performed molecular frequency calculations to get estimates of the thermal correction to the Gibbs free energy (ΔGcorr). This parameter, combined with Eelec from the large basis set calculations, gives the thermal free energy in solution (Gsol). In all the calculations, the solvation effects were introduced to mimic an aqueous solution by using the ‘self-consistent reaction field’ based on the polarizable continuum model (PCM).50Synthesis and characterisation of POM-stabilised gold nanoparticles (AuNPs@POM)
Single crystals of (NH4)15{Na[(Mo2O4)6(μ2-SO3)3(μ6-SO3)]2}·5H2O (0.05 g, 0.019 mmol) were added to 100 mL of 0.667 mM HAuCl4 (aq) at pH 5 under magnetic stirring. Following the addition of POM, the aqueous HAuCl4 solution showed a significant variation in its colour, changing first from pale yellow to colourless and finally to dark purple after 30 min stirring at room temperature. The colloidal solution was centrifuged at 5000 rpm for 15 min in order to separate the colloidal material from the solvent. The resulting supernatant was discarded and the dark precipitate that was isolated by the initial centrifugation was resuspended in MilliQ water and washed two more times using the same centrifugation protocol before being filtered through a 0.2 μm Millipore filter. At the time of writing the resulting colloidal material has been stable in solution for more than 18 months. For analyses involving dried particles, the suspended particles were precipitated by centrifugation and lyophilized.UV-vis spectroscopy analysis of the resuspended colloidal material showed one distinctive LSPR band at 535 nm. In agreement with UV-vis spectroscopy, SEM and TEM analyses confirmed the presence of pseudo-spherical (polyhedral) AuNPs with a diameter of 29 ± 4 nm and a fairly homogeneous size distribution. The FTIR spectrum of AuNPs@POM at room temperature was dominated by bands at 1075 (SO32−); 983 and 962 (Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) from terminal oxygen atoms; 923, 890, 862, 855 and 840 (SO32−); 791, 738 and 635 cm−1 (Mo–O–Mo or Mo–O–S) from vibrations involving edge and corner sharing oxygen atoms. ICP (atomic%): Au 97.69, Mo 2.08 and S 0.19. XPS (eV): 84.4 (4f7/2, Au(0)), 88.4 (4f5/2, Au(0)), 169.4 (2p3/2, S(IV)), 170.5 (2p1/2, S(IV)), 233.0 (3d5/2, Mo(VI)), 236.2 (3d3/2, Mo(VI), 531.0 (1s, O). XPS (atomic%): Au 40.6, Mo 5.4 and S 0.3. PXRD: (1 1 1), (2 0 0), (2 2 0) and (3 1 1) corresponding to FCC for Au (JCPDS-International Centre for Diffraction Data-2000 file no. 04-0487).
O) from terminal oxygen atoms; 923, 890, 862, 855 and 840 (SO32−); 791, 738 and 635 cm−1 (Mo–O–Mo or Mo–O–S) from vibrations involving edge and corner sharing oxygen atoms. ICP (atomic%): Au 97.69, Mo 2.08 and S 0.19. XPS (eV): 84.4 (4f7/2, Au(0)), 88.4 (4f5/2, Au(0)), 169.4 (2p3/2, S(IV)), 170.5 (2p1/2, S(IV)), 233.0 (3d5/2, Mo(VI)), 236.2 (3d3/2, Mo(VI), 531.0 (1s, O). XPS (atomic%): Au 40.6, Mo 5.4 and S 0.3. PXRD: (1 1 1), (2 0 0), (2 2 0) and (3 1 1) corresponding to FCC for Au (JCPDS-International Centre for Diffraction Data-2000 file no. 04-0487).
Many mild reducing agents are available for the synthesis and stabilisation of AuNPs; however, POMs are known to stabilise noble metal NPs by reduction of aqueous solutions of metal salts under mild reaction conditions31–33 and, as mentioned previously, serve as an ideal starting point (an appropriate model) for our purposes. Several POMs containing MoV centres (POMs with ‘in-built’ reducing capabilities) were screened for the synthesis. Kabanos-type POM [Na{(MoV2O4)3(μ2-O)3(μ2-SO3)3(μ6-SO3)}2]15− was selected for several reasons: the resulting AuNPs@POM were stable in solution (for almost two years at the time of writing), the reaction was simple, rapid and high yielding. These particular AuNPs were also suitable for in vitro studies (results presented elsewhere).39
Firstly, AuNPs@POM were formed by adding 50 mg of Kabanos POM single crystals to 100 mL of 0.667 mM HAuCl4(aq) at pH 5 under magnetic stirring. The pale yellow colour of the AuIII chloroauric acid solution quickly became transparent upon addition of the POM before turning dark red several minutes later, indicating the slow kinetics involved in the formation of colloidal Au0 suspensions. UV-vis spectroscopy analysis of the colloidal material showed a dominant localised surface plasmon resonance (LSPR) band at 535 nm corresponding to polyhedral gold nanoparticles (Fig. 2a and see ESI,† Fig. S1). The colloidal material was washed with Milli-Q water to remove unwanted by-products (a minor amount triangular gold nanoprisms) and reagents via several centrifugation/resuspension steps. Powder X-ray diffraction (PXRD) patterns of the polyhedral gold nanoparticles isolated from the reduction of HAuCl4 with Kabanos showed a 2θ Bragg match with the face centred cubic structure of gold, (1 1 1) (2 0 0) (2 2 0) (3 1 1) (Fig. 2b). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) characterisation of AuNPs@POM was used to obtain additional information on the size and shape of the nanoparticles (Fig. S2 and S3 (ESI†), respectively). In agreement with UV-vis spectroscopy, pseudo-spherical (polyhedral) nanoparticles with a mean diameter of 29 nm were observed. Atomic resolution Cs corrected STEM-HAADF was used to obtain more detailed information on the structure of the particles, clearly highlighting their polyhedral structure (Fig. 2c and Fig. S4, ESI†).
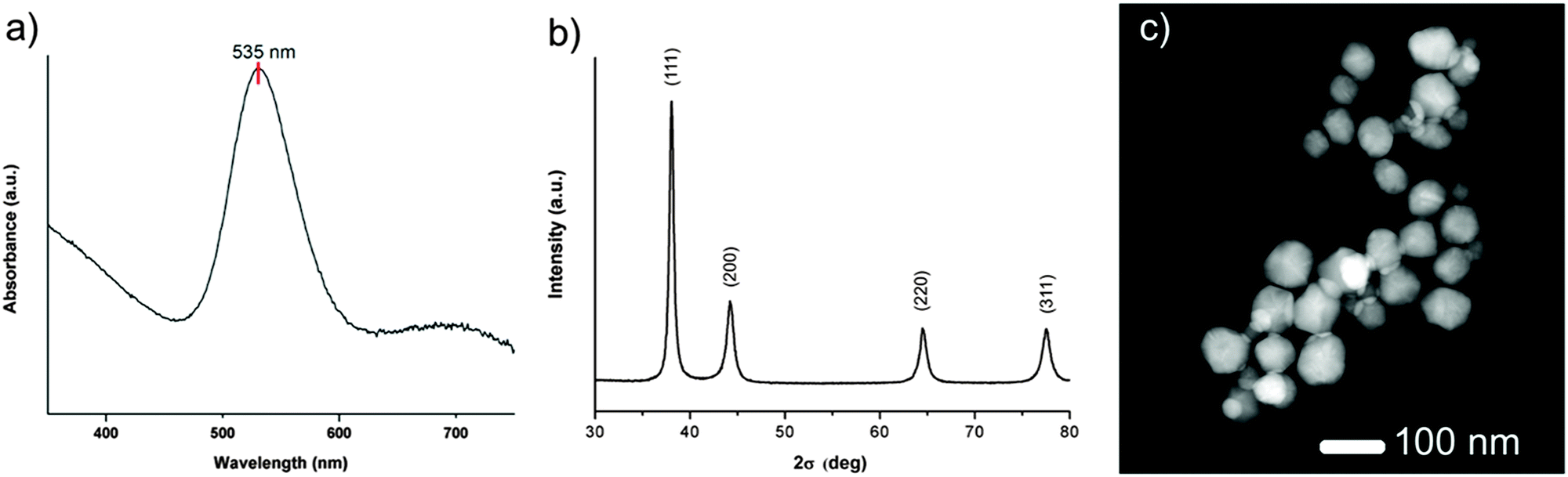 | ||
| Fig. 2 (a) UV-vis absorption spectrum showing the dominant LSPR band of the polyhedral gold nanoparticles at 535 nm; (b) powder X-ray diffraction of AuNPs@POM with a 2σ Bragg match with the face centred cubic structure of gold, (1 1 1) (2 0 0) (2 2 0) (3 1 1); (c) high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) images of polyhedral AuNPs@POM. | ||
The role of the POM in stabilising the AuNPs was determined via several spectroscopic methods: X-ray photoelectron spectroscopy (XPS), energy dispersive X-ray (EDX) spectroscopy, Fourier-transform infrared (FTIR) spectroscopy and inductively coupled plasma-atomic emission spectroscopy (ICP-AES). ICP elemental analysis of the bulk material determined the relative atomic concentration to be 97.69% Au, 2.08% Mo, 0.19% S and 0.05% Na. The FTIR spectrum of AuNPs@POM at room temperature provided evidence of SO32− at 1075 cm−1 and terminal MoO vibrational stretching bands at 983 and 962 cm−1, while Mo–O–Mo vibrations involving edge and corner sharing oxygen atoms are evident at 791, 738, 635 and 609 cm−1, where there are also some overlap with the ν(S–O) stretching vibration bands (Fig. S5, ESI†). EDX analysis of a solid sample of AuNPs@POM also provided additional confirmation of the presence of the POM coating on the AuNP surface (Fig. S6, ESI†). XPS affords information on the atomic concentration and oxidation state of the elements present on the surface of crystalline samples, where the typical depth of analysis is ca. 5 nm (3–10 nm). Fig. S7 (ESI†) shows the general spectrum of AuNPs@POM along with high resolution Mo(3d) and Au(4f) spectra. The Mo 3d peak is composed of a well-resolved spin–orbit doublet at 236.2 and 233.0 eV (splitting of 3.2 eV) for Mo 3d3/2 and 3d5/2, respectively. Such values and splitting are typical of MoVI atoms and in agreement with other literature examples.51 The latter of these reported examples showed the presence of mostly MoVI but some residual MoV. Therefore, the presence of MoVI—and absence of MoV—in our AuNPs@POM provide further evidence of the complete oxidation of MoV centres during the reduction of AuIII to Au0. The clean spectrum shows that the Au 4f peak also comprises of a spin–orbit doublet at 88.4 and 84.7 eV (splitting of 3.7 eV) for 4f5/2 and 4f7/2, respectively. These values are commensurate with reported literature examples,34 but at slightly elevated energies, possibly indicating the presence of Au2O3, which could be expected at the nanoparticle surface. We should therefore emphasize that the interface between the stabilising POM and AuNPs is still largely unknown, due to the Au peaks in the XPS data, which are suggestive of Au2O3. Note that for all the aforementioned analyses, bulk samples of AuNPs@POM had been washed, filtered and dried as described in the Methods section (above).
Results and discussion
Selection of the POM model
For a detailed understanding of the role of POM in the reduction of AuCl4− ions, DFT calculations were performed for the process until complete reduction of AuIII. In the first step of our computational study, we focused on the selection of the POM model. The Kabanos structure (Fig. 1a) [Na{(MoV2O4)3(μ2-O)3(μ2-SO3)3(μ6-SO3)}2]15−, features a dimeric structure (abbreviated as {dimeric-H3S4Mo6} because the Kabanos anion would be protonated under the experimental reaction conditions used in this research) that was simplified to the monomeric form, {H3S4Mo6} without interstitial Na+, shown in Fig. 1b, which carries a total charge of −5 in the three-fold protonated form. This simplification is aimed at minimising the computational resources necessary to correctly describe the effect of the POM on the mechanism of formation of AuNPs. After geometry optimisation of both forms, the unprotonated {dimeric-S4Mo6} structure features a distance of 9.082 Å (to be compared with 7.905 Å obtained from X-ray data) between the two S atoms placed at the centre of each monomer, whereas the average Mo–Oμ2 distance (1.976 Å) is 0.17 Å shorter than the experimental one (2.146 Å). Only the inclusion of six protons on μ2-O generates optimised structures in excellent agreement with the X-ray data (7.965 vs. 7.905 Å). It is worth mentioning that previous studies on isostructural compounds—based on C, P and As—suggest that the three bridging μ2-O atoms are typically protonated.52–55 Therefore, we conclude that it is more reasonable to consider the current complex to be six-fold protonated in acidic solution. As expected, when only monomeric {H3S4Mo6} was optimised, the geometrical parameters reproduced the X-ray data with accuracy.The energies and compositions of the frontier molecular orbitals are examined to evaluate the appropriateness of the {H3S4Mo6} model for exploring the role of the Kabanos structure in the formation of metallic gold. It is important to note that negligible differences were observed at the B3LYP/6-31G**/LANL2DZ level on comparing the frontier orbitals—the highest occupied and the lowest unoccupied ones (HOMOs and LUMOs, respectively, Fig. S8, ESI†). The calculated orbital energies show that the energy gap between HOMO and HOMO-2 (for the monomer) is only 0.12 eV and between HOMO and HOMO-5 (for the dimer) is 0.28 eV. This phenomenon clearly demonstrates that it is possible to sequentially remove six electrons from the monomer (twelve from the dimer) during the redox cycle. In addition, a sine qua non condition for the reaction POMn− + Aum+ → POM(n−1)− + Au(m−1)+ to occur is that the POM features an adequate oxidation potential so that electron transfer towards the gold salt is a favourable process. We calculated the oxidation energy for {H3S4Mo6} and {dimeric-H3S4Mo6} to be 0.22 and 0.24 eV (vs. NHE), respectively. Further calculations (triple-ζ level basis set) on the monomer show that orbital energies are overall down shifted less than 0.3 eV and that the HOMO–LUMO energy gap remains almost unchanged. In summary, the above analysis shows that the monomer model behaves similar to {dimeric-H3S4Mo6} and, hence, it can be used to explore the reduction mechanism of gold. The following theoretical discussion is therefore based entirely on the role of the monomeric {H3S4Mo6} species.
Possible mechanisms of {H3S4Mo6}-mediated AuIII to AuI reduction
Since the [AuCl4]− species is not stable in aqueous solution and always undergoes spontaneous hydrolysis, the existence of intermediates of the type [AuClxOH4−x]− is largely pH dependent. In the experiments reported herein the pH was adjusted close to 5, thus in our calculations we have only considered the most representative [AuIIICl3OH]− species as the reactant under these conditions.56,57 In the pathway towards the AuNPs, the fundamental questions we wished to solve were: how do the [AuIIICl3OH]− ions and the POM interact? And by which mechanism are the electrons transferred from {H3S4Mo6} to AuIII? Despite a considerable number of theoretical reports on the growth of AuNPs, the role of POMs has never been addressed until now.The following discussion goes through the initial reaction stages, when the first gold unit approaches the fully reduced POM5− and gains two electrons, from AuIII to AuI. Considering explicitly the molecular charges for a better understanding of the electron transfer processes, the associated general mechanism for such initial steps can be proposed as:
| {H3S4Mo6}5− + [AuIIICl3OH]− → {H3S4Mo6}5−–[AuIIICl3OH]− (precursor complex) |
| {H3S4Mo6}5−–[AuIIICl3OH]− → {H3S4Mo6}5−–[AuIIICl2OH] + Cl− (Cl substitution) |
| {H3S4Mo6}5−–[AuIIICl2OH] + Cl− → {H3S4Mo6}4−–[AuIICl2OH]− + Cl− (1st electron transfer) |
| {H3S4Mo6}4−–[AuIICl2OH]− + Cl− → {H2S4Mo6}4−–[AuICl2]− + Cl− + H2O (2nd electron transfer) |
It must be mentioned that further oxidation of {H2S4Mo6}4− can occur if more [AuIIICl3OH]− molecules react with it given that four Mo(d1) electrons are still present in {H2S4Mo6}4−. The associated thermodynamic data are discussed later on.
Because both the gold complex (like the substrate) and {H3S4Mo6}5− are anionic species, the interaction between them should be weak. Most probably, hydrogen bonding patterns in aqueous solution prevail. The corresponding binding mode between POM and [AuIIICl3OH]− is depicted in Fig. 3. The optimised structures show two strong hydrogen bonds formed between the –OH ligand of [AuCl3OH]− and H (and O) from {H3S4Mo6}5−, and Au approaching the O between two MoV. Also weak Cl⋯H bonds are formed that must be helpful for the interaction. Fig. 4 shows the computed free energy profile (G, relative to the reactants) following the reduction process. Unlike classical reducing agents, such as citrate,58,59 the highly reduced {H3S4Mo6}5− POM structure offers multiple reduction sites (in the present case six MoV centres). Hence, once the substrate is attached to the POM surface (precursor complex, A), the electron transfer may lead to different configurations, a fact that is herein assumed. We found that the formation of A—[AuIIICl3OH]− adsorption on the bottom of the POM mainly by hydrogen bonding—is slightly exothermic even if the separate parts are both anions, with a reaction free energy of −6.5 kcal mol−1. Because the release of Cl− from the substrate is easier than that of OH− before electron transfer, the first reaction step is subject to Cl− elimination from [AuIIICl3OH]− associated with attachment to the POM, giving species B. This step was also found to be thermodynamically favourable with an associated energy of −1.3 kcal mol−1 referred to A. On the other hand, releasing the OH− group costs about 60 kcal mol−1, making this hypothetical process unlikely.
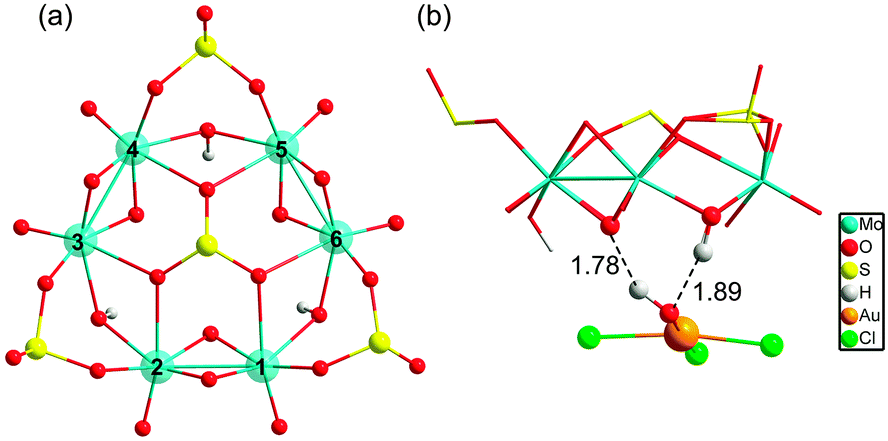 | ||
| Fig. 3 (a) Top view of [(MoV2O4)3(μ2-OH)3(μ2-SO3)3(μ6-SO3)]5−, Mo(d1) centres of the POM are numbered 1–6. (b) The most plausible binding mode for [AuIIICl3OH]− attaching on the POM surface, via strong O⋯H and weak Cl⋯H interactions. | ||
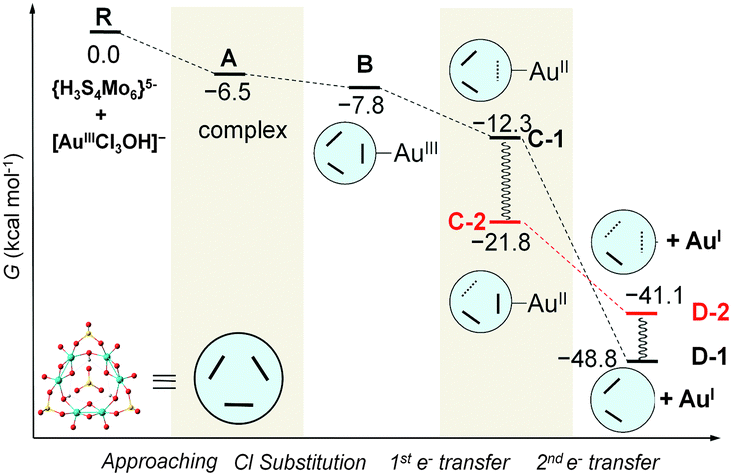 | ||
| Fig. 4 Computed free energy profile (in kcal mol−1, values relative to reactants) associated with the gold reduction process. We use the schematic notation defined for the POM monomer, {H3S4Mo6}5−, with thick lines denoting Mo–Mo bonds and dotted lines denoting one-electron Mo–Mo bonds. Species C-2 is assumed to rearrange from C-1. Species D-1 has lost two electrons from the same Mo–Mo bond, whereas D-2 has two one-electron Mo–Mo bonds. Geometrical details of the computed intermediates are shown in Fig. 5. | ||
Reduction of AuIII should occur through intramolecular MoV → AuIII electron transfer. Obviously, there are six electron-providing centres, as shown in Fig. 3a, the attaching MoV(1)–MoV(2) group being the logical site when the distance effect is considered. The first electron transfer from the POM to AuIII leads to species C-1, with ΔG = −12.3 kcal mol−1. C-1 exhibit a diradical nature, with spin densities of 0.96 e on one Mo2 group and 0.90 e on the [AuIICl2OH]− moiety, namely with unpaired electrons, respectively, on one MoIV–MoV group and on AuII. Following C-1, a more exergonic process is obtained for the reduction of AuII to AuI. The proton-coupled electron transfer (PCET) is a reaction mechanism that is thought to be common in redox reactions.60–62 In our case, we observe that when 2e-oxidation occurs in the POM, an intramolecular hydrogen bond cleavage takes place and this hydrogen is transferred to the –OH of the gold complex with associated H2O release and formation of the linear [AuICl2]− species. The proton transfer forming H2O significantly promotes the AuII to AuI reduction. On the basis of C-1, the continuous reduction by Mo from the Mo1–Mo2 group will lead to the 2e-reduced product D-1, with ΔG ≈ −48 kcal mol−1 from reactants at the present level of calculation. On the other hand, we also considered one more isomeric structure of D-1, giving [AuICl(H2O)] instead of [AuICl2]−, which is 3.6 kcal mol−1 less stable than D-1. In other words, [AuICl(H2O)] can covert to [AuICl2]− by exchanging H2O with Cl− in solution. Furthermore, calculations show that the AuII → AuI step is thermodynamically more advantageous than AuIII → AuII, suggesting that AuII could be a short-lived intermediate. This fact can also be ascribed to the favourable evolution from d9 to d10 in this step.
C-1 may undergo electron rearrangement in the POM moiety due to the available electrons from the other Mo–Mo groups. If one electron is transferred from Mo3–Mo4 to the Mo1–Mo2 group to form C-2, the energy changes by −9.5 kcal mol−1, which we attribute to the stronger binding between the [AuIICl2OH]− group and the almost intact Mo2O2 group in C-1 (see Fig. 3). In the last step of the energy profile, a 2e-unpaired configuration, D-2, was computed based on the possibility that the electrons are transferred from Mo1–Mo2 and Mo3–Mo4 groups, one each, at variance with D-1, in which the two electrons have migrated to the gold atom from the same Mo–Mo group. We found that D-1 is more stable than D-2 by about 7 kcal mol−1 (−48.8 vs. −41.1 kcal mol−1), indicating that two electrons coming from the same Mo–Mo group and breaking this Mo–Mo bond, is preferred over two electrons coming from different Mo–Mo groups. Alternative to the direct B → C-1 → D-1 pathway, in which electrons flow between the closest sites of the POM to Au, the results also suggest that the electronic rearrangement C-1 → C-2 is thermodynamically favoured and could supply the second electron to AuII to form D-2. From the present data we cannot say with total confidence which pathway (1 or 2) is the most effective.
To analyse the structural evolution in the POM5−–AuIIICl2OH → POM3−–AuICl2 process, Fig. 5 focuses on the binding region of intermediates B → C-1 → D-1. Addition of the first electron to AuIIICl2OH does not cause significant variations in Au–Cl and Au–OH bond lengths, with variations being less than 0.2 Å. However, a two-electron transfer from the POM to AuIIICl2OH can pull one chlorine ligand or water group (H release from the POM to the –OH group to form H2O) out of the AuI centre. From intermediate B to D-1, the Au–Ob distance increases to 2.919 Å, evidencing a very weak interaction between the POM and [AuICl2]−. The short distance between O (–OH in the Au complex) and H (in the POM) in B and C, of around 1.4–1.5 Å, indicates strong hydrogen interaction. From B to C, the electron transfer induces the hydrogen bonding to become much stronger. After the second POM → Au electron transfer, the proton is completely relocated to release a H2O molecule. Concerning the geometry of B, the Mo–Mo bond is stretched to 2.652 Å, notably longer than the initial 2.560 Å in the free closed-shell POM. However, allowing for an open-shell configuration, we obtain C-1 with metal–metal bond breaking and spontaneous electron transfer. We found that further electron loss from the same Mo2 group elongates dramatically this Mo–Mo distance to 3.503 Å in species D-1. In the latter system, oxidation of the two MoV from the same group leads to hydrogen bond cleavage and transfer to the OH of the gold complex, which possibly produces a SO2 unit detachment. One of the S–O bonds is clearly stretched to 2.118 Å, implying that SO2 units have a natural tendency to leave as the POM looses electrons.
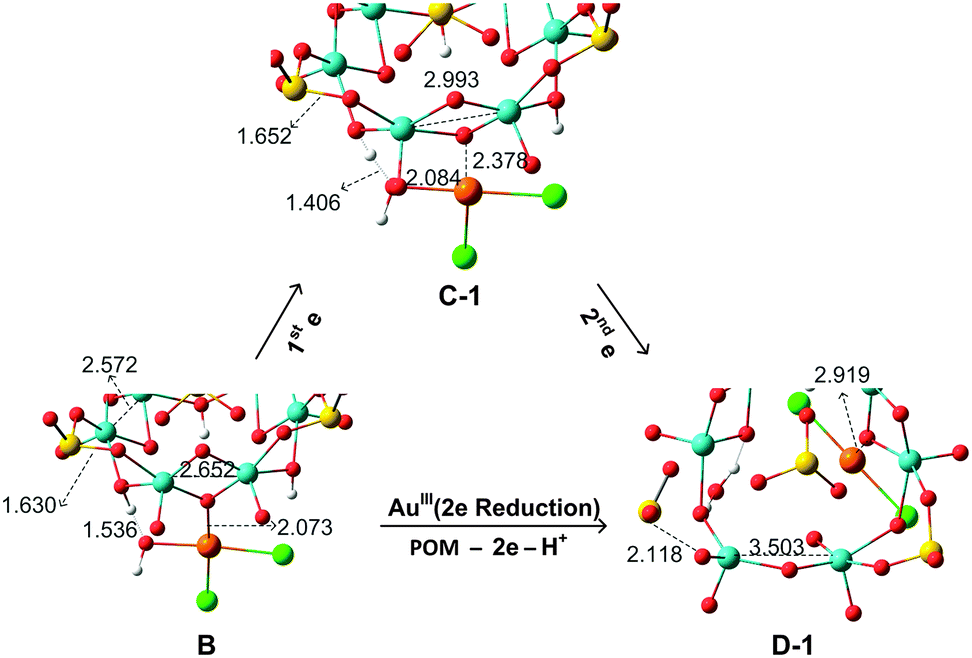 | ||
| Fig. 5 Main structural changes (distances in Å) for the intermediate structures B → C-1 → D-1. Hydrogen bonds and Au–O interactions are fundamental in the formation of the intermediates. | ||
A relevant point for the following discussion is the identification of the strong driving force for gold reduction. Besides the energies of the intermediate species, their molecular orbitals contain very useful information concerning the reaction mechanism, especially for redox processes. To go into the details of the reaction, we analysed the most important orbitals of intermediates B, C-1 and D-1, shown in Fig. 5.
When the orbital energies of B are compared with those of the free {H3S4Mo6} moiety one notices that the addition of AuIIICl2OH increases the energy of the HOMO by 0.02 eV only, and that of HOMOs in B are more localized on each Mo–Mo group than in {H3S4Mo6} (Fig. S9, ESI†), which acts as the electron source during the reduction process. At variance with the possible electron donation from Mo–Mo groups with participation in HOMO and HOMO−1, the HOMO−2 also shows contributions from AuIIICl2OH and some bonding interaction between Au and O. It is also remarkable that the LUMO of B resembles much that of AuIIICl2OH and the connected Mo1–Mo2 unit, showing an antibonding character between the Au and O atoms which would promote Au–O bond cleavage by electron occupation upon reduction. Considering the overlap between the HOMO−2 and LUMO, the electron transfer is reliable via these two orbitals separated by 2.0 eV. We cannot neglect the contribution from the HOMO to electron donation even if the electron density on Mo1–Mo2 is very small. B → C-1 involves a one-electron transfer from the HOMO−2 into the AuIII moiety. In C-1, 0.44 e can be attributed to Au (mainly dx2−y2 character) and 0.36 e are shared by two Cl and H2O ligands. The spin density representation of C-1 (Fig. 6) clarifies this electron distribution. The unrestricted formalism applied to the electronic open shell gives HOMO–LUMO gaps for α and β orbitals of 2.26 and 2.01 eV, respectively. α-LUMO has no contribution from the [AuIICl2OH]− group and leads to 2.26 eV, making the α-HOMO → α-LUMO electron transfer unlikely. However, the composition and energy of β-LUMO resembles much that in B, suggesting that the second electron will go into this orbital to form D-1via α-HOMO → β-LUMO electron transfer, with a smaller energy gap of 1.90 eV. Despite the fact that this energy gap is very similar to the one in B, the second electron transfer is thermodynamically much more favourable in comparison to the first one.
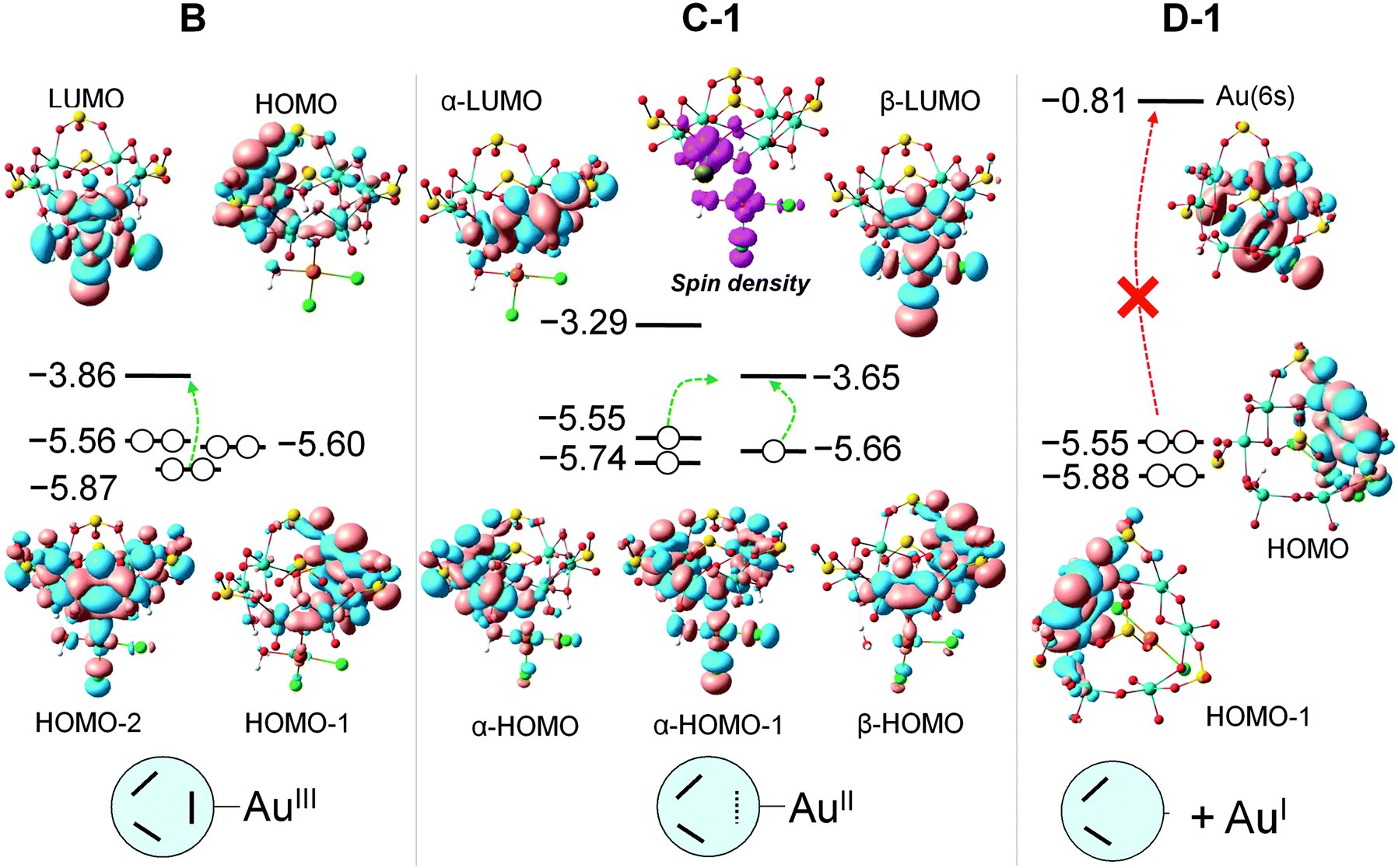 | ||
| Fig. 6 Computed orbital energies (values in eV) involved in the main electron transitions (dashed green arrows) during the reduction process of AuIII to AuI. The spin density representation for species C-1 (magenta) corresponds to the triplet state. | ||
The formation of D-1 can take place either by direct electron transfer to the β-LUMO, as mentioned. However, a different thermodynamically stable conformation was found in step C (species C-2, Fig. 4). This can be produced after internal electron reorganization from Mo3–Mo4 (α-HOMO or β-HOMO) to Mo1–Mo2 due to the low energy of the β-LUMO, with concomitant geometric rearrangement. The C-1 → C-2 process has ΔG = −9.5 kcal mol−1. Indeed, the α-HOMO is the result of the first reduction, and the next occupied orbital (α-HOMO−1) lies 0.19 eV below, indicating the strong preference of the [AuIICl2OH]− unit to accept the electrons to form a stable doubly occupied orbital, that is, the HOMO−1 in D-1. Therefore, a two-electron transfer can take place from the POM to the Au unit, causing complete cleavage of the Au–O bond to minimize the electron–electron repulsions between the two anions (AuCl2− and POM4−).
AuI to Au0 step
As mentioned above, [AuCl4]− can be easily reduced to form the AuCl2− species. To achieve the first seed in the formation of AuNPs, namely one Au0 atom, AuI should be further reduced. It features a stable d10 electronic configuration; hence, the additional electron must be accommodated in the 6s orbital. As shown in Fig. 6, the unoccupied orbital labelled ‘Au(6s)’ is the lowest AuI orbital but it is not the lowest empty orbital available. The gap between the POM-localised HOMO and the lowest Au-like empty orbital is very large (4.7 eV). At variance with the first two reduction steps (AuIII → AuII → AuI), the AuI + e → Au0 process is expected to be very difficult. So, the question of how AuI intermediates can be reduced to Au0 arises. The calculated results proved that two or three [AuICl2]− units can, from thermodynamic considerations, form dimeric or trimeric species when dispersion corrections are applied to the calculations. We found exothermic nucleation energies of −4.7 and −3.9 kcal mol−1 to form gold dimer and trimer species, respectively. If dispersion corrections are not considered, repulsive coulombic interactions are dominating giving an endothermic process of 2.8 kcal mol−1. Therefore, inclusion of dispersion corrections is required in order to correctly describe the incipient processes.Moreover, the computed reduction energies (REs, or electron affinities in solution) for the [AuIIICl4]−/[AuICl2]− and [AuICl2]−/Au0 processes are −5.21 and −2.60 eV, respectively. RE = −5.21 eV is in good agreement with the experimental value (−5.17 eV). However, the computational estimate for [AuICl2]−/Au0 is far from the experimental −5.39 eV. We found this discrepancy to be caused mainly by the cohesion energy missing of solid gold when only the single Au atom is calculated. As presented in a recently published study, the disproportionation reaction 3[AuICl2]− → AuCl4− + 2Au(s) + 2Cl− was one possible route for the reduction of AuI.63 It is true that the formation of AuNPs will be promoted by this way with the increase of the AuI and Au0 species. However, for the first or initial reduction of AuI, the effects of the oxidant and environment should be taken into account. We argue that the endergonic reaction energy (estimated to be ∼70 kcal mol−1) was obtained when only the POM and [AuICl2]− species are considered to react. This finding led us to consider and include additional effects that may affect the thermochemistry, such as the presence of other species in solution. Notably, in our reaction conditions Na+, NH4+ and H3O+ coexist with the POM and Aun+ species. Thus, either one NH4+, Na+ or H3O+ molecule was explicitly added to the [AuICl2]− model calculations. The originally computed AuI electron affinity in solution (−2.60 eV with only [AuCl2]− in the model) remain unchanged when NH4+ (−2.88 eV) or Na+ (−2.63 eV) is considered. However, including one H3O+ molecule in the calculations has a large effect in the electron affinity (−3.40 eV).
To analyse the energy demands associated with this reduction process, we choose the initial {S4Mo6}5− species to interact with the [AuCl2]− dimer surrounded by one H3O+ (Fig. 7), which makes the reaction exergonic, ΔG = −4.5 kcal mol−1. In addition, the formation of a Au–Au bond promotes the reduction of AuI to Au0.64 Instead, including Na+ in the reaction still makes the process unfavourable, with ΔG = 30.1 kcal mol−1. We have to remark that even the concentration of H3O+ is quite small in solution; we believe that H3O+ promotes the AuI species to approach each other, helps the charge balance on the new gold cluster and stabilizes the released Cl− in the initial steps. As soon as the first Au0 species is formed, it is expected that the gold reduction process will be less energy demanding as the gold nanoparticle increases in size. We remark here that Au/AuI(solid) has a positive redox potential of 1.68 V vs. NHE from Lange's Handbook of Chemistry, whereas the value for atomic Au/AuI is negative (−1.5 V vs. NHE), explaining that only the first steps must be aided by H3O+.65,66
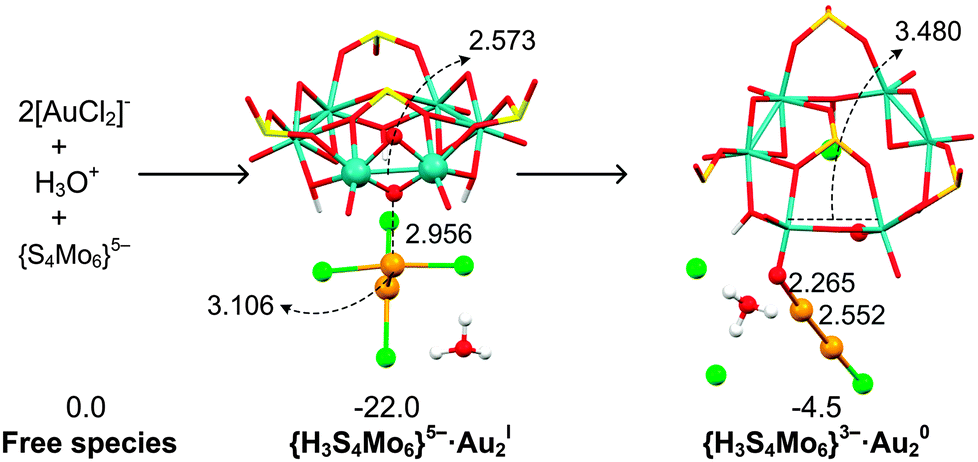 | ||
| Fig. 7 Structural (in Å) and energetic (in kcal mol−1) variations during the reduction of the [AuICl2]− dimer by the initial {H3S4Mo6} in the presence of one H3O+. | ||
Because the AuIII species is pH-dependent, [AuCl3OH]− itself has been considered instead of [AuCl4]−, since the experimental pH is 5. Following the crucial role of protons, we also explored if the protonated AuI dimer can be reduced by a two-electron-oxidised Kabanos anion. In fact, the computed reaction free energy for this step is roughly the same as that of the initial reduced structure. From this viewpoint, it is evident that the two-electron-oxidised Kabanos can be further oxidised, and we deduce that the reduction of AuI to Au0 can occur by the co-action of POM and H3O+ at the first stage.
Persistence of POM activity until full oxidation
To explore further the POM-related redox processes, one can successively introduce the second and third [AuIIICl3OH]− units for reaction with the partially oxidised {H2S4MoVI2MoV4}4− compound. Our goal is to analyse on-going electron transfer from it to other gold units until complete oxidation to {S4MoVI6}2−, as shown in Chart 1.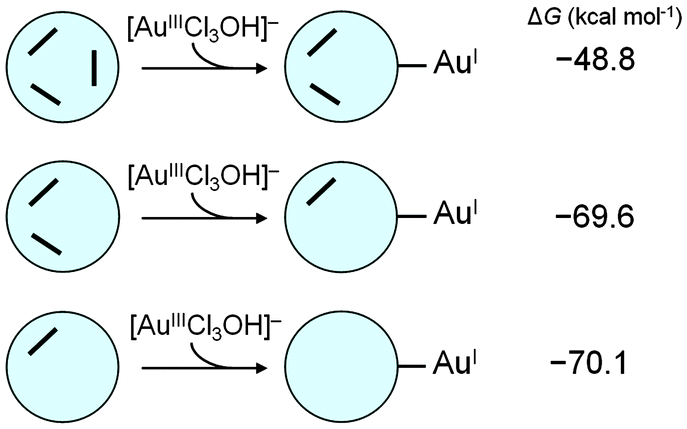 | ||
| Chart 1 Computed free energies (in kcal mol−1) associated with the second and third [AuCl3OH]− reductions based on the 2e-oxidised {H2S4MoVI2MoV4}4−. | ||
It has been stated above that the 2e-reduction product D-1 (with two electrons coming from one Mo–Mo group) is more stable than the open shell product. Indeed, it is a reasonable assumption that some [AuIIICl3OH]− groups may attach to the POM at the beginning of the reaction. We chose D-1 as the starting point for the successive processes to simplify the electronic state, and the reduction of the second [AuIIICl3OH]− was studied considering the routes with oxidation of MoV from the same group only. These calculations indicate that addition and reduction of the second AuIII to AuI by the partially oxidised {H2S4MoVI2MoV4}4− is exergonic, with ΔG = −69.6 kcal mol−1. Also, the final state in Chart 1 (formation of the third AuI unit) is obtained after a very favourable step, with ΔG = −70.1 kcal mol−1 if the four-electron-oxidised POM, {HS4MoVI4MoV2}3−, is considered as the reducing agent for this step. It is worth mentioning that successive POM–Au electron transfers are increasingly exergonic compared with the fully reduced POM, which may be attributed to the concomitant favourable SO2 release observed as {S4Mo6} looses electrons. The calculations show that it is highly reasonable that the reduced molybdosulphate POM can provide six electrons in the presence of AuIII. This fact supports the experimental XPS data (Fig. S7, ESI†) obtained for AuNPs@POM, which reveal the presence of only MoVI on the surface of the AuNP, corresponding to the total oxidation of the POM.
As deduced from the structural change analysis associated with the first 2e-reduction of [AuCl3OH]−, one SO2 unit is very likely to be released from the POM. Interestingly, three SO2 units are completely detached after reaction with two more [AuCl3OH]− complexes to convert all six MoV to MoVI. The present calculations strongly suggest that the fully oxidised oxomolybdate that might be attached to AuNPs may be viewed as a re-organisation of the initial structure with partial loss of SO2, namely the [SMoVI6O21]2− structure. We propose that it is ring-shaped with a central SO3 unit weakly bound to the external framework, reminiscent of the previously reported [SeMo6O21]2− (Fig. S10, ESI†).67 The total energy associated with the loss of three SO2 units has been estimated to be −26 kcal mol−1 (referenced to the fully oxidised POM intermediate), confirming that SO2 release takes place spontaneously as electrons are removed from the POM. On the other hand, the hypothetical process of removing one SO2 unit from the fully reduced {H3S4Mo6} is computed to be 55.5 kcal mol−1. Such a largely endothermic process evidences that SO2 loss occurs as electrons are removed from the POM unit. The proposed oxidised structure is presumably stable owing to the large HOMO–LUMO gap of 4.68 eV. These results are experimentally supported. From the ICP data obtained for AuNPs@POM (Au 97.69%, Mo 2.08% and S 0.19%, Methods section), we determined a molar ratio Mo/S = 3.65, a value much larger than the one deduced from the initial reduced form of the POM (Mo/S = 1.5), and between 6/1 and 6/2 (loose of three and two SO2 units, respectively). Thus, the experimental and theoretical parts allow concluding that at least two SO2 have been detached from the POM.
Conclusions
Quantum chemical calculations were applied to study the formation mechanism of highly stable AuNPs obtained from an aqueous solution containing [AuIIICl4]− and an electron-rich POM structure. The principal aim of this project was to understand the first steps of AuNP formation using a POM ({H3S4Mo6}) model, not to improve or develop new synthetic methodologies. This work discloses a mechanistic route for the complete reduction of a AuIII salt to Au0 using an ‘in built’ electron source, the Kabanos POM ([Na{(MoV2O4)3(μ2-O)3(μ2-SO3)3(μ6-SO3)}2]15−), which provides several electrons. The experimental observation and characterisation of the resulting polyoxometalate-stabilised nanoparticles confirm that this POM is a suitable electron source. The reduction process is greatly driven by the coupled electron and proton transfer process. Both theoretical and experimental data presented show that all MoV centres of the Kabanos POM were completely oxidised to MoVI with a concomitant loss of SO2 units from the original Kabanos form, as ICP data and DFT calculations suggest. Considering the AuI to Au0 step a key process, the calculations indicate that the AuI complex is not tightly attached to the POM surface and, in combination with a high HOMO(POM)-6s (AuI) energy gap, we suggest that the POM is not directly responsible for the AuI → Au0 step. Instead, a AuI dimer is formed before the first Au0 atom is generated; the presence of H3O+ in solution boosts this step without further oxidation of the POM. This is commensurate with the most favourable experimental conditions encountered when the pH is varied from neutral to acidic. Gaining a deeper understanding of the mechanisms of AuNP formation is crucial for optimising their size and shape-dependent physicochemical properties and therefore essential for harnessing their full potential in subsequent applications.Acknowledgements
The authors are grateful to COST Action CM1203 “Polyoxometalate Chemistry for Molecular Nanoscience (PoCheMoN)” for supporting this work, and also to the Spanish Government (Grant no. CTQ2011-29054), Generalitat de Catalunya (Grant no. 2014SGR199) and Xarxa de Referència en Química Teòrica i Computacional, XRQTC). This work has been funded by the Fundación General CSIC (Programa ComFuturo) and ERC-Starting Grant 239931-NANOPUZZLE. Dr Alfonso Ibarra (LMA-UNIZAR) provided invaluable advice and assistance with the electron microscopy data.Notes and references
- V. V. Mody, R. Siwale, A. Singh and H. R. Mody, J. Pharm. BioAllied Sci., 2010, 2, 282 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC.
- L. He, M. D. Musick, S. R. Nicewarner, F. G. Salinas, S. J. Benkovic, M. J. Natan and C. D. Keating, J. Am. Chem. Soc., 2000, 122, 9071 CrossRef CAS.
- M. Pérez-Hernández, P. del Pino, S. G. Mitchell, M. Moros, G. Stepien, B. Pelaz, W. Parak, E. Gálvez, J. Pardo and J. M. de la Fuente, ACS Nano, 2015, 9, 52–61 CrossRef PubMed.
- C. Bao, N. Beziere, P. del Pino, B. Pelaz, G. Estrada, F. Tian, V. Ntziachristos, J. M. de la Fuente and D. Cui, Small, 2012, 9, 68 CrossRef PubMed.
- E. Polo, P. del Pino, B. Pelaz, V. Grazu and J. M. de la Fuente, Chem. Commun., 2013, 49, 3676 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. N. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739 CrossRef CAS PubMed.
- M. V. Yezhelyev, X. Gao, Y. Xing, A. Al-Hajj, S. Nie and R. M. O'Regan, Lancet Oncol., 2006, 7, 657 CrossRef CAS PubMed.
- S. Lal, S. E. Clare and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842 CrossRef CAS PubMed.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885 RSC.
- M. Colombo, S. Carregal-Romero, M. F. Casula, L. Gutiérrez, M. P. Morales, I. B. Böhm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Chem. Soc. Rev., 2012, 41, 4306 RSC.
- G. Y. Chen, H. L. Qiu, P. N. Prasad and X. Y. Chen, Chem. Rev., 2014, 114, 5161 CrossRef CAS PubMed.
- R. Bardhan, S. Lal, A. Joshi and N. J. Halas, Acc. Chem. Res., 2011, 44, 936 CrossRef CAS PubMed.
- M. Grzelczak, J. P. Perez-Juste, P. Mulvaney and L. M. Liz-Marz-an, Chem. Soc. Rev., 2008, 37, 1783 RSC.
- S. J. Soenen, P. Rivera-Gil, J.-M. Montenegro, W. J. Parak, S. C. De Smedt and K. Braeckmans, Nano Today, 2011, 6, 446 CrossRef CAS.
- B. Pelaz, V. Grazú, A. Ibarra, C. Magen, P. del Pino and J. M. de la Fuente, Langmuir, 2012, 28, 8965 CrossRef CAS PubMed.
- A. Roldán, S. González, J. M. Ricart and F. Illas, ChemPhysChem, 2009, 10, 348 CrossRef PubMed.
- T. Yao, Z. H. Sun, Y. Y. Li, Z. Y. Pan, H. Wei, Y. Xie, M. Nomura, Y. Niwa, W. S. Yan, Z. Y. Wu, Y. Jiang, Q. H. Liu and S. Q. Wei, J. Am. Chem. Soc., 2010, 132, 7696 CrossRef CAS PubMed.
- D. Toroz and S. Corni, Nano Lett., 2011, 11, 1313 CrossRef CAS PubMed.
- B. Keita, I. M. Mbomekalle, L. Nadjo and C. Haut, Electrochem. Commun., 2004, 6, 978 CrossRef CAS.
- A. Dolbecq, J.-D. Compain, P. Mialane, J. Marrot, F. S. Secheresse, B. Keita, L. R. B. Holzle, F. Miserque and L. Nadjo, Chem. – Eur. J., 2009, 15, 733 CrossRef CAS PubMed.
- S. G. Mitchell and J. M. de la Fuente, Eur. J. Inorg. Chem., 2013, 5517 CrossRef CAS.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, New York, 1983 Search PubMed.
- A. Müller and M. T. Pope, Polyoxometalate Chemistry: From Topology via Self-Assembly to Applications, Kluwer, 2001 Search PubMed.
- D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105 RSC.
- C. Busche, L. Vilà-Nadal, J. Yan, H. N. Miras, D. L. Long, V. P. Georgiev, A. Asenov, R. H. Pedersen, N. Gadegaard, M. M. Mirza, D. J. Paul, J. M. Poblet and L. Cronin, Nature, 2014, 515, 545 CrossRef CAS PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572 RSC.
- S.-X. Guo, Y. Liu, C.-Y. Lee, A. M. Bond, J. Zhang, Y. V. Geletii and C. L. Hill, Energy Environ. Sci., 2013, 6, 2654 CAS.
- J. J. Baldoví, S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño and H. Prima-García, Chem. Commun., 2013, 49, 8922 RSC.
- Y. Nishimoto, D. Yokogawa, H. Yoshikawa, K. Awaga and S. Irle, J. Am. Chem. Soc., 2014, 136, 9042 CrossRef CAS PubMed.
- B. Keita, T. B. Liu and L. Nadjo, J. Mater. Chem., 2009, 19, 19 RSC.
- S. G. Mitchell and J. M. de la Fuente, J. Mater. Chem., 2012, 22, 18091 RSC.
- Y. F. Wang and I. A. Weinstock, Chem. Soc. Rev., 2012, 41, 7479 RSC.
- G. J. Zhang, B. Keita, R. N. Biboum, F. Miserque, P. Berthet, A. Dolbecq, P. Mialane, L. Catala and L. Nadjo, J. Mater. Chem., 2009, 19, 8639 RSC.
- X. López, P. Miró, J. J. Carbó, A. Rodríguez-Fortea, C. Bo and J. M. Poblet, Theor. Chem. Acc., 2011, 128, 393 CrossRef.
- X. López, J. J. Carbó, C. Bo and J. M. Poblet, Chem. Soc. Rev., 2012, 41, 7537 RSC.
- B. Keita, R. N. Biboum, I. M. Mbomekallé, S. Floquet, C. Simonnet-Jégat, E. Cadot, F. Miserque, P. Berthet and L. Nadjo, J. Mater. Chem., 2008, 18, 3196 RSC.
- M. J. Manos, J. D. Woollins, A. M. Z. Slawin and T. A. Kabanos, Angew. Chem., Int. Ed., 2002, 15, 2801 CrossRef.
- I. M. Gabas, G. Stepien, M. Moros, S. G. Mitchell and J. M. de la Fuente, New J. Chem., 2016 Search PubMed , accepted.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09W, Revision A02; Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 CrossRef CAS.
- D. Feller, J. Comput. Chem., 1996, 17, 1571 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase and J. Liand T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- H. K. Daima, P. R. Selvakannan, A. E. Kandjani, R. Shukl, S. K. Bhargava and V. Bansal, Nanoscale, 2014, 6, 758 RSC.
- M. J. Manos, A. D. Keramidas, J. D. Woollins, A. M. Z. Slawin and T. A. Kabanos, J. Chem. Soc., Dalton Trans., 2001, 3419 RSC.
- E. Cadot, A. Dolbecq, B. Salignac and F. Sécheresse, Chem. – Eur. J., 1999, 5, 2396 CrossRef CAS.
- A. Leclaire, A. Guesdon, F. Berrah, M. M. Borel and B. Raveau, J. Solid State Chem., 1999, 145, 291 CrossRef CAS.
- A. Leclaire, C. Biot, H. Rebbah, M. M. Borel and B. Raveau, J. Mater. Chem., 1998, 8, 439 RSC.
- D. V. Goia and E. Matijevic, Colloids Surf., A, 1999, 146, 139 CrossRef CAS.
- S. Wang, K. Qian, X. Z. Bi and W. X. Huang, J. Phys. Chem. C, 2009, 113, 6505 CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, J. Phys. Chem., 1953, 57, 670 CrossRef CAS.
- M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004 CrossRef CAS PubMed.
- See special issue devoted to reviews on proton-coupled electron transfer, Chem. Rev. 2010, 110, 6937.
- A. Sartorel, P. Miró, E. Salvadori, S. Romain, M. Carraro, G. Scorrano, M. D. Valentin, A. Llobet, C. Bo and M. Bonchio, J. Am. Chem. Soc., 2009, 131, 16051 CrossRef CAS PubMed.
- K. Theilacker, H. B. Schlegel, M. Kaupp and P. Schwerdtfeger, Inorg. Chem., 2015, 54, 9869 CrossRef CAS PubMed.
- For aurophilic interactions see H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- S. Mosseri, A. Henglein and E. Janata, J. Phys. Chem., 1989, 93, 6791 CrossRef CAS.
- E. Gachard, H. Remita, J. Khatouri, B. Keita, L. Nadjo and J. Belloni, New J. Chem., 1998, 1257 RSC.
- D. Yang, S. Li, P. Ma and J. Niu, Inorg. Chem., 2013, 52, 14034 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of Experimental methods and full characterisation of AuNPs@POM (SEM, HRTEM, STEM, FTIR, EDX and XPS). DFT calculations: computed frontier molecular orbitals for dimeric and monomeric forms, 3D representations of the frontier molecular orbitals of {H3S4Mo6}, AuCl4− and the {H3S4Mo6}–AuCl3 adduct, and structures and frontier orbitals of the monomeric reactant and the final POM product. See DOI: 10.1039/c5nj02773j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |