
Facile synthesis of arylthiophenyl-functionalized diketopyrrolopyrrole derivatives via direct C–H arylation: characterization and utilization in organic electronic devices†
Akshaya Kumar
Palai‡
a,
Amit
Kumar§
a,
Kyoseung
Sim
a,
Jaehyuk
Kwon
a,
Tae Joo
Shin¶
b,
Soonmin
Jang
c,
Sungwoo
Cho
d,
Seung-Un
Park
a and
Seungmoon
Pyo
*a
aDepartment of Chemistry, Konkuk University, 120 Neungdong-ro, Gwangjin-gu, Seoul 143-701, Republic of Korea. E-mail: pyosm@konkuk.ac.kr; Fax: +82-2-34365382; Tel: +82-2-450-3397
bPohang Accelerator Laboratory, Pohang, 790-784, Republic of Korea
cDepartment of Chemistry, Sejong University, 209 Neungdong-ro, Gwangjin-gu, Seoul 143-747, Republic of Korea
dCenter for Core Research Facility, Daegu Gyeongbuk Institute of Science & Technology, Daegu 711-873, Republic of Korea
First published on 23rd October 2015
Abstract
A series of symmetrically arylthiophenyl-functionalized diketopyrrolopyrrole (DPP) derivatives were synthesized via palladium-catalyzed direct C–H arylation to develop an active material for organic electronic devices. The properties of DPPs could be tuned by simple variation of the end groups such as t-butylphenyl, cyanophenyl and methoxynaphthyl. The effect of the substituent on the optical, electrochemical, and thermal properties of DPPs was evaluated using UV-visible spectroscopy, cyclic voltammetry, and thermogravimetric analysis. The morphology and molecular packing of thin-films of DPPs were analyzed by atomic force microscopy (AFM), density functional theory (DFT) calculations, and two-dimensional grazing incidence X-ray diffraction (2D-GIXD) experiments. Utilization of the synthesized DPPs as channel materials in organic field-effect transistors was demonstrated.
1. Introduction
Organic electronic devices such as organic field-effect transistors (OFETs),1–5 organic photovoltaics (OPVs),6–8 dye sensitized solar cells,9–11 and organic light-emitting diodes (OLEDs)12–14 have undergone remarkable innovation in recent years; nevertheless, the development of new organic π-conjugated small molecular and polymeric materials with highly desirable properties remains a major challenge. While regioregularity15 and polydispersity16 issues encountered by polymeric materials may deleteriously impact the performance of polymer-based devices, small molecular materials offer several promising advantages such as well-defined structure, minimized batch to batch synthetic variation, and relatively easy purification.Small molecular materials may strategically be obtained by starting with an easily accessible chromophore that can be modified through a simple synthetic approach to achieve tunable opto-electronic properties. In this respect, versatile chemical modification of the diketopyrrolopyrrole (DPP) core can easily be achieved by the introduction of various aromatic blocks at the 2,5 position of the DPP core, leading to tunable optical properties via π–π intermolecular interactions. In addition, various end-capping groups can be introduced onto the aromatic blocks to further tune the molecular properties, such as the optical bandgap, and the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels. Alkyl chains can also be introduced onto the N-atoms of the lactam unit to control the solubility in common organic solvents, the degree of crystallinity, and the molecular packing.17–19 Due to the amenability of DPP to structural engineering and the inherent stability of this molecule, the development of novel analogues remains an active pursuit.20,21
Among the reports of small molecular semiconductors based on the DPP core for organic electronics applications,22–29 Wurthner et al. reported the optical and redox properties of a series of bisthiophene-functionalized DPPs,30 Nguyen et al. evaluated the influence of structural variation on the solid-state properties of a series of DPP-based oligophenylenethiophenes,31 and Sonar et al. reported the synthesis and properties of DPP-based derivatives functionalized with electron withdrawing end capping groups.32 Our group has also reported carbazole as well as alkylthiophene-functionalized DPP chromophores for OFETs.33–35 However, these DPP-based molecules were invariably synthesized via Stille, Suzuki, or Negishi cross-coupling reactions that involve the preparation of organometallic precursor reagents. Direct C–H arylation has recently emerged as a new synthetic approach for the synthesis of DPP dyes.36–38 C–H activation is of great importance in conjugated material synthesis as it requires fewer synthetic steps and is a clean and efficient protocol.39
In this report, a series of symmetrically functionalized molecular DPP derivatives are designed and synthesized via direct C–H arylation by altering the end-group in the terminal aromatic units of DPP (Scheme 1). A comprehensive study of these DPP derivatives is carried out based on the analysis of their structural, optical, electrochemical, and thermal properties. Atomic force microscopy (AFM), density functional theory (DFT) calculations, and two-dimensional grazing incidence X-ray diffraction (2D-GIXD) are utilized for the analysis of the morphology and molecular packing of the DPP thin-films. The effect of the introduced end-capping groups on the electrical properties of the DPPs is assessed using organic field-effect transistors employing the DPP thin-films as a channel component.
 | ||
| Scheme 1 Synthesis of DPPa–cvia direct C–H arylation. | ||
2. Experimental
2.1. Materials and characterization
All starting materials were purchased from Sigma Aldrich or from local chemical companies. 2,5-Dihexadecyl-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione and 3,6-bis(5-bromothiophen-2-yl)-2,5-dihexadecylpyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione were synthesized according to a previously reported procedure.33,40Thin layer chromatography (TLC) was performed on glass plates, precoated with silica gel 60 F254 (Merck). Column chromatography was performed on silica gel 60 (0.063–0.200 mm, 70–230 mesh, Merck). 1H NMR measurements were conducted on a Bruker Avance 400 MHz NMR spectrometer using deuterated chloroform (CDCl3) and tetramethylsilane (TMS) as a solvent and an internal standard, respectively. High-resolution electrospray ionization-Fourier transform (ESI-FT) mass spectrometry was carried out using a Thermo LTQ-Orbitrap XL mass spectrometer. Elemental analyses were conducted using a Thermo Scientific elemental analyzer (FLASH 2000). TGA analysis was conducted using a TA instrument (TGA 7, Perkin Elmer) at a heating rate of 10 °C min−1 under nitrogen gas flow. UV-visible measurements were performed in CHCl3 (10−5 M) using a conventional quartz cell (path length 10 mm) on a Varian Cary 500 spectrometer. Thin films (30 nm) for UV-visible absorption measurements were prepared on quartz substrates via thermal vacuum deposition. Cyclic voltammetry experiments were performed using an Electrochemical Interface & Impedance Analyzer (IVIUM Technologies) in an electrolyte solution of 0.05 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) in dry dichloromethane. A three electrode cell was used in all experiments. A glassy carbon electrode (coated with the thin film of DPPs) was used as a working electrode, a platinum mesh as a counter electrode, and Ag/AgNO3 (0.01 M) as a reference electrode (scan rate: 50 mV s−1). The CV curves were calibrated using the ferrocene/ferrocenium (Fc/Fc+) redox couple. The energy level of Fc/Fc+ was assumed to be −4.8 eV with respect to a vacuum. Density functional theory (DFT) calculation was performed at the B3LPY/6-31G(d,p) level, using SPARTAN (ver. 10).41 Tapping mode atomic force microscopy (AFM) (nanoscope IIIa, Digital Instruments) was used to evaluate the morphology of the semiconductor film. Two-dimensional grazing incidence X-ray diffraction (2D-GIXD) experiments were conducted using an incident X-ray energy of 11.24 keV (λ = 1.103 Å) at the PLS-II 9A U-SAXS beam line of Pohang Accelerator Laboratory in Korea.
2.2. Synthesis of DPP derivatives (DPPa–c) via direct C–H arylation
The typical procedure for direct C–H arylation of the three new DPPs is as follows: 2,5-dihexadecyl-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione (100 mg, 0.133 mmol), K2CO3 (4.5 equiv., 0.60 mmol), Pd(OAc)2 (10 mol%), and pivalic acid (01 equiv., 0.133 mmol) were placed in a round-bottomed Schlenk flask. Under an argon atmosphere, anhydrous dimethyl acetamide (DMA, 4 mL) was added, followed by addition of mono-bromoaryl reagents (0.4 mmol) into the aforementioned reaction flask. The mixture was stirred at 100–110 °C for 3–6 h under an inert atmosphere, then cooled to room temperature, and poured into brine solution (100 mL), and the precipitate obtained was extracted with chloroform (3 × 25 mL). The organic layer was collected, dried over MgSO4, and concentrated under reduced pressure. The residue was treated with 100 mL of methanol, and the obtained precipitate was filtered and purified by column chromatography (silica gel, 50% petroleum ether in chloroform as an eluent) to obtain the desired product.2.3. Device fabrication and characterization
Organic field-effect transistors (OFETs) were fabricated in bottom-gate top-contact geometry on an indium-tin oxide (ITO, gate electrode) coated glass substrate with cross-linked poly(4-vinylphenol) (Ci = 61.04 pF mm−2) as a gate dielectric. Our previous publications provide a detailed description of the preparation of the dielectric layer and the fabrication of the OFET devices.40,42 A simple description of the device fabrication is as follows: for p-type OFETs, the gate dielectric layer was prepared by spin-coating the precursor solution at a rate of 3000 rpm for 30 s; the assembly was baked on a hot plate in air. A 30 nm thick DPPa coating was deposited at a rate of ca. 0.2 Å s−1 as a channel layer under a high vacuum (5 × 10−6 Torr) over the gate dielectric. The substrate temperatures for thin-film deposition of DPPa and DPPb were 25 and 120 °C, respectively. Finally, for the formation of source and drain electrodes, 50 nm gold was deposited on the top of the DPPa film using a shadow mask; the defining channel length (L) and width (W) were 50 and 1000 μm, respectively; deposition was performed at a rate of ca. 0.3 Å s−1 under high vacuum (5 × 10−6 Torr). The same procedure has been applied to the formation of n-type OFETs with the DPPb (50 nm) film deposited at substrate temperature of 120 °C as a channel layer. An in situ quartz crystal corrected with density parameters was used to monitor the thickness of the deposited films. Electrical characterization of the devices was performed at room temperature under ambient conditions using an HP semiconductor parameter analyzer (HP 4155B).3. Results and discussion
3.1. Synthesis and thermal properties
The synthetic strategy used herein is based on the introduction of an electron withdrawing or donating end capping moiety onto the aromatic terminal group based on a recently reported efficient synthetic route for small organic materials.37,43,44 Using this strategy, three new small molecular DPPs (DPPa–c) were developed as active materials for organic electronics. Scheme 1 shows the palladium-catalyzed direct C–H arylation route used in the synthesis of the DPP derivatives starting from alkylated DPP (1) and appropriate bromoarenes.First, compound (1) containing two solubilizing hexadecyl groups at lactam nitrogen atoms was synthesized according to the literature procedure.33 Subsequently, DPPa was readily obtained with the yield of 88% by using the reported synthetic procedure37 where Pd(OAc)2, K2CO3, and pivalic acid (PivOH) in anhydrous dimethylacetamide (DMA) were used as a catalytic system. The reaction was carried out at 110 °C for 3 h. DPPb having an electron-withdrawing end-capping moiety (cyanophenyl) was synthesized with a yield of 79% through the same reaction route as DPPa at a relatively lower reaction temperature (100 °C), because aryl-bromide reagents with an electron-withdrawing moiety are usually more active towards direct C–H arylation reactions than corresponding electron-donor containing derivatives. Similar reaction at the lower temperature (i.e. 100 °C) has also been reported for a series of DPP derivatives with electron-withdrawing benzene derivatives.37
It should be noted that, we also tried to synthesize DPPbvia Suzuki coupling reaction of 4-cyanophenylboronic acid with 3,6-bis(5-bromothiophen-2-yl)-2,5-dihexadecylpyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione. However, a desired product was obtained only with 8% yield. A similar observation has also been reported by the Wurthner group while preparing another cyano end-capped DPP derivative (7f).30 These results suggest that a direct C–H arylation route could be employed effectively for the synthesis of molecular DPP derivatives, especially with a strong electron-withdrawing moiety. For the synthesis of DPPc, we first carried out the reaction under the same conditions as DPPa. However, a sticky product was obtained with a lower yield (27%). As a next approach, we have carried out the same reaction at a lower temperature (100 °C) for a longer time (6 h), and a dirk solid product was obtained with a yield of 42%. All the reactions were monitored by thin layer chromatography (TLC). The crude products were purified via column chromatography. The chemical structures of the DPPs were characterized by 1H NMR spectroscopy, high resolution mass spectroscopy (HRMS) (see ESI,† Fig. S1–S3) and elemental analysis. All the diketopyrrolopyrrole derivatives (DPPa–c) were soluble in common organic solvents such as chloroform, o-dichlorobenzene, and toluene.
The thermal stability of the DPPs was evaluated using thermogravimetric analysis (TGA) at a heating rate of 10 °C min−1 under a nitrogen atmosphere. The respective thermograms are shown in Fig. 1. The thermal decomposition temperatures (Td) (5% weight loss) of DPPa, DPPb, and DPPc were 423, 394, and 411 °C, respectively (Table 1), clearly reflecting sufficient thermal stability of DPPs for thermal vacuum deposition for the formation of a thin-film.
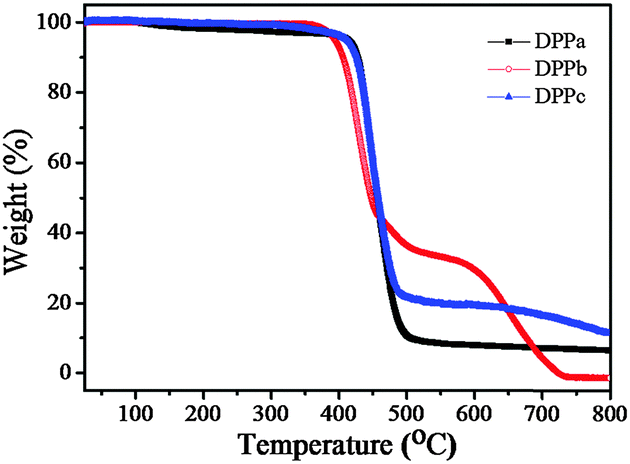 | ||
| Fig. 1 TGA thermograms of DPPa–c with a heating rate of 10 °C min−1 under a N2 atmosphere. | ||
| DPP dyes | λ abs (nm) | λ onset (nm) | E g,opt (eV) | T d (°C) | HOMOe (eV) | LUMOf (eV) | |
|---|---|---|---|---|---|---|---|
| Solutiona | Filmb | ||||||
| a UV-vis data of DPP dyes recorded in chloroform solution. b UV-vis data of DPP derivative films. c Calculated from absorption onsets of the DPP derivative films (Eg,opt = 1240/λonset eV). d Obtained from TGA measurements (temperature at 5% weight loss). e Calculated based on EHOMO = −e(Eonset,ox − Eonset,Fc/Fc+) − 4.8 eV, where Eonset,ox and Eonset,Fc/Fc+ (−0.03 V) are the onset oxidation potentials of DPP samples and ferrocene, respectively. f Estimated from ELUMO = EHOMO + Eg,opt. g The values are obtained from ref. 33. h Measured from the reduction potential (Eonset,red). ELUMO = −e(Eonset,red − Eonset,Fc/Fc+) − 4.8 eV. i Estimated from EHOMO = ELUMO − Eg,opt. | |||||||
| 1(DPP-R16) | — | 345, 485, 557, 610 | 656 | 1.89 | — | −5.32 | −3.43 |
| DPPa | 335, 396, 562 (s), 602 | 558, 585 (s), 636 (s) | 708 | 1.75 | 423 | −5.12 | −3.37 |
| DPPb | 350, 402 (s), 570 (s), 608 | 540 (s), 616, 658 (s) | 733 | 1.69 | 394 | −5.34i | −3.65h |
| DPPc | 352, 410, 576 (s), 616 | 570 (s), 652 (s), 719 | 795 | 1.56 | 411 | −4.91 | −3.35 |
3.2. Optical and electrochemical properties
The optical properties of DPPa–c were investigated via UV-vis absorption spectroscopy, and the spectra were recorded in the solution and the thin-film state.Fig. 2a shows the absorption spectra of DPPa, DPPb, and DPPc in CHCl3 (10−5 M). Absorption bands were observed in the UV as well as the visible/near IR region for all samples. Compared to the λmax (547 nm)33 of compound 1 (Scheme 1) with no end-capping group, there was a bathochromic shift of λmax for all the DPPs. The largest shift was observed for DPPc (λabs: 616 nm) with a methoxynaphthyl end-capping group, and the smallest shift was observed for DPPa (λabs: 602 nm) bearing t-butylphenyl end-capping groups. A similar effect was reported by Wurthner et al.30Fig. 2b shows the normalized absorption spectra of the DPPa–c thin-films (30 nm), prepared over quartz substrates via thermal vacuum deposition. A strong red shift in the absorption was observed for all DPPs compared to the corresponding solution spectra. In addition, the absorption peaks in the solid-state spectra were broader with a shoulder in the longer wavelength region. This red shift is due to the strong intermolecular interaction in the thin-film solid state. As observed in the solution spectra, the largest red shift in absorption was observed for DPPc (λabs: 719 nm), which may be derived from the extended conjugation within the molecule due to the presence of methoxynaphthyl as the end-capping group. The absorption band-edges (λonset) of the DPPa–c thin-films were 708, 733, and 795 nm, respectively, corresponding to optical bandgaps (Eg,opt) of 1.75, 1.69, and 1.56 eV. A similar effect of the naphthalene moiety on Eg,opt was previously observed in DPP-based copolymers (1.59–1.45 eV).45,46 The absorption maxima (λabs) and Eg,opt of the DPPs are summarized in Table 1.
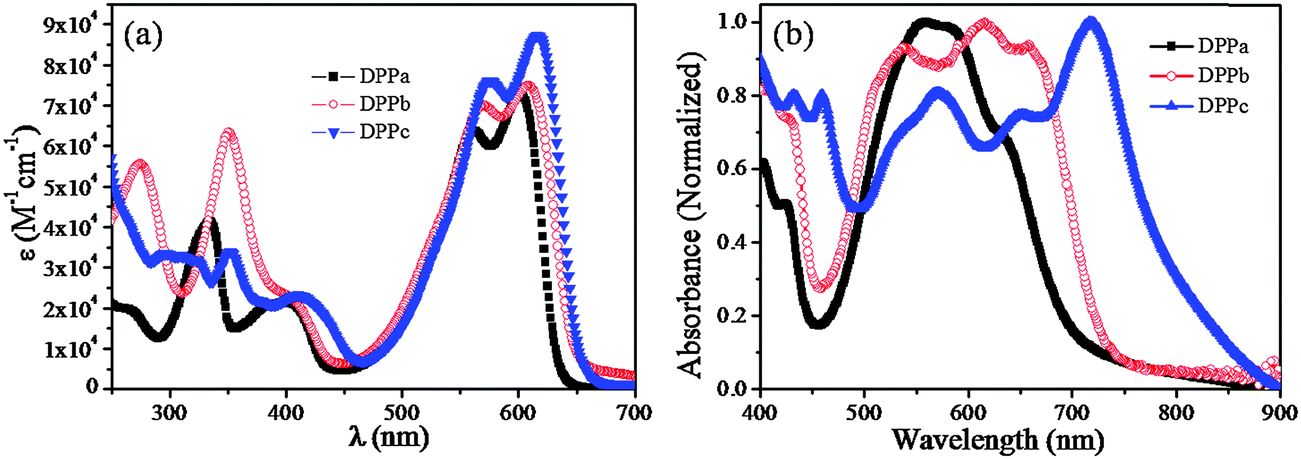 | ||
| Fig. 2 UV-visible absorption spectra of DPPa–c in (a) solution (in CHCl3) and (b) thin-film (30 nm) prepared over quartz crystals by thermal vacuum deposition. | ||
The electrochemical properties of DPPa–c were evaluated based on cyclic voltammetry (CV) using a film of materials on glassy carbon as a working electrode in dry dichloromethane solution containing 0.05 M Bu4NPF6. The CVs were recorded versus the potential of Ag/AgNO3, which was calibrated using the ferrocene–ferrocenium (Fc/Fc+) redox couple.
The cyclic voltammograms of DPPa–c are shown in Fig. 3. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of DPPa, DPPb, and DPPc were estimated from the corresponding oxidation, reduction onset and optical band gap (see Table 1, footnotes), and are summarized in Table 1. DPPb shows the highest HOMO energy level (−4.91 eV) among the studied materials. It is also interesting to find that the HOMO energy level decreases with the increase of the electron-withdrawing ability of the end-capping group. The HOMO level follows the order DPPc > DPPa > DPPb. A similar trend has also been reported for a set of DPP derivatives.37 The LUMO (−3.65 eV) and the HOMO (−5.34 eV) energy level for DPPb are found to be relatively lower compared to those of DPPa and DPPc. The deep-lying energy levels of DPPb may be responsible for electron dominant charge transport behavior in devices.
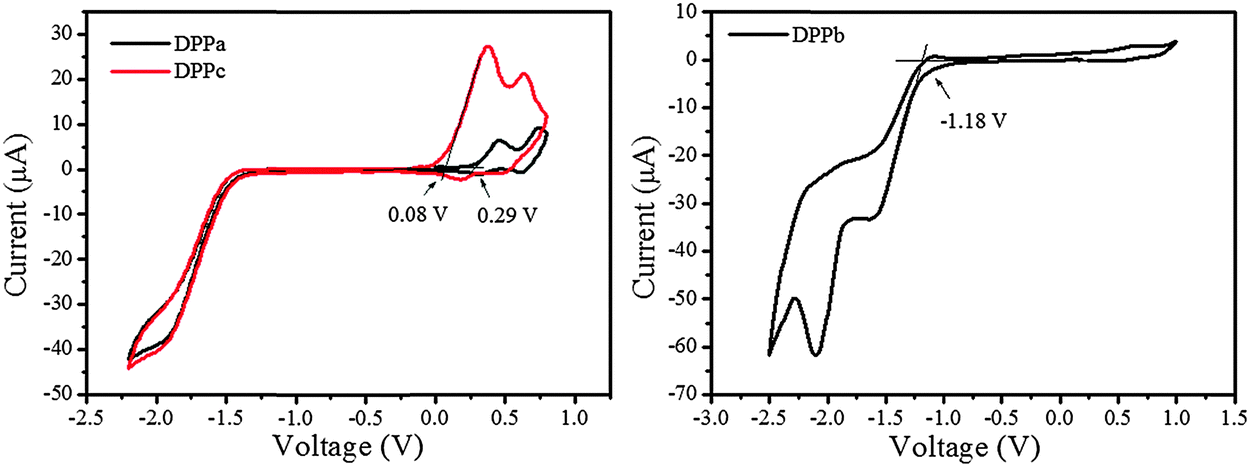 | ||
| Fig. 3 Cyclic voltammograms of DPPa–c in dry dichloromethane solution using Fc/Fc+ as reference; a scan rate of 50 mV s−1 with 0.05 M Bu4NPF6 as the supporting electrolyte. | ||
3.3. AFM and XRD analysis
The morphology of the DPP thin-films deposited on cross-linked poly(4-vinylphenol) (CL-PVP), used as a gate dielectric for OFET devices, was characterized by atomic force microscopy (AFM). The respective images are shown in Fig. 4a–c. The AFM image of DPPa indicates that the film comprised numerous closely packed, small, granular structures with a root mean square (RMS) roughness value of 2.26 nm. The DPPb film comprised larger grains with a RMS roughness value of 3.59 nm. Both films had quite good crystalline structures with contiguous well packed grains on the surface. However, DPPc comprised discontiguous grains with an island-like structure and a RMS roughness value of 4.94 nm. | ||
| Fig. 4 Tapping-mode AFM height images of the (a) DPPa, (b) DPPb, and (c) DPPc thin-films. | ||
Two-dimensional grazing incidence X-ray diffraction (2D-GIXD) was used to gain further insight into the molecular packing of the DPPs on CL-PVP in the thin-films; the corresponding GIXD images and out-of-plane (qz) profiles are shown in Fig. 5a–c.
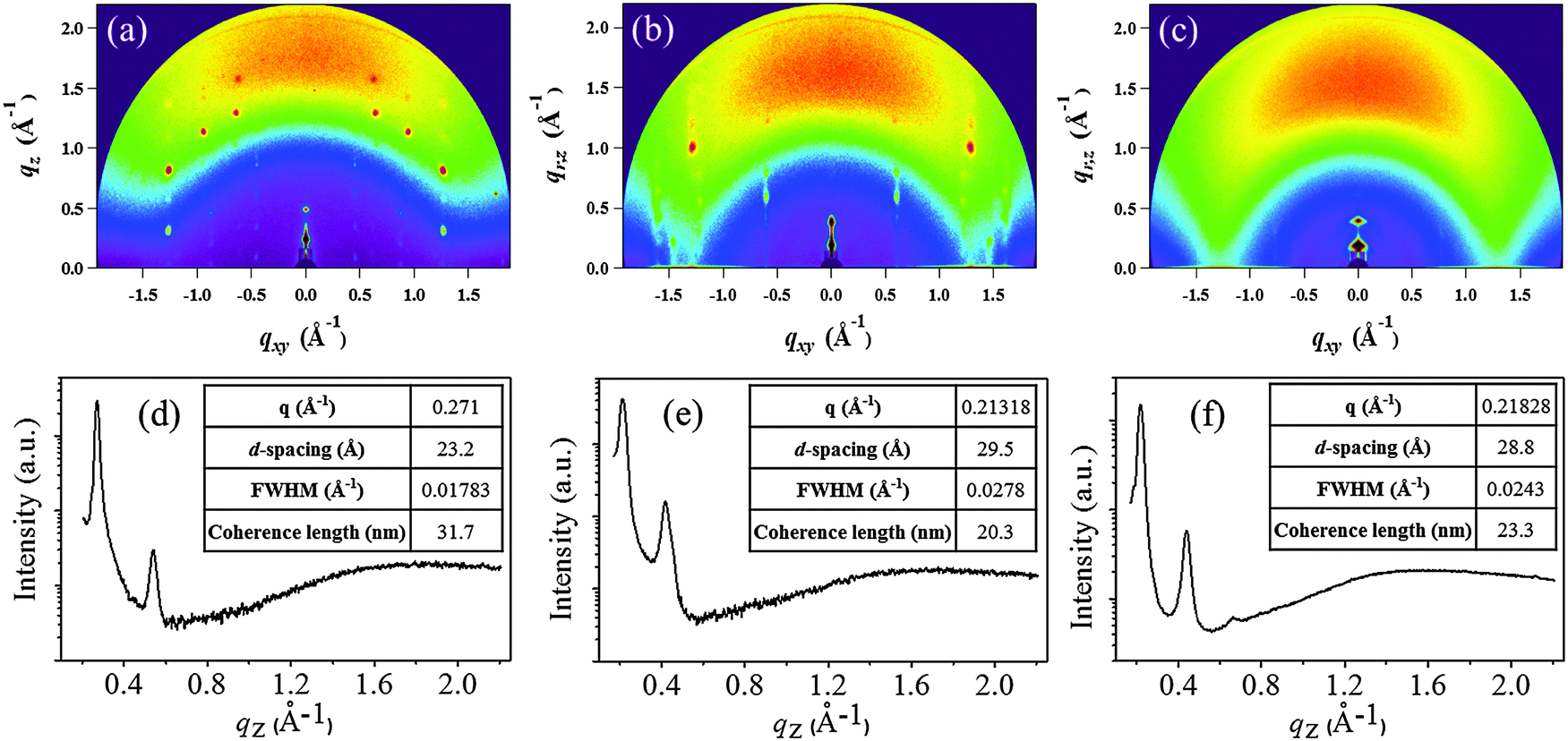 | ||
| Fig. 5 2D-GIXD images (a–c) of DPPa, DPPb, and DPPc thin-films (30 nm) deposited on the CL-PVP/ITO substrate. (d–f) Respective out-of-plane (qz) profiles extracted from 2D-GIXD images. | ||
Fig. 5a and b show a number of strong diffraction spots extended to a higher q-range, suggesting that the DPPa and DPPb thin-films have a crystalline nature. On the other hand, the GIXD pattern of the DPPc thin-film (Fig. 5c) exhibits broad and diffuse peaks, indicative of relatively lower crystallinity than DPPa and DPPb. The corresponding out-of-plane 1D-GIXD profiles of the DPPa, DPPb, and DPPc thin-films at qxy = 0 (qz-profiles) are plotted in Fig. 5d–f. The qz-profiles of all the DPPs show two distinct out-of-plane peaks, assigned to the 1st and 2nd order diffraction peaks of the (h00) plane, respectively. The first diffraction peak at qz = 0.271 Å−1 for the DPPa film corresponds to a d-spacing of 23.20 Å. This d-spacing is consistent with the calculated (from DFT) conjugation length (23.96 Å, ESI,† Fig. S4a), which may implies that the DPPa molecules are stacked on the gate dielectric surfaces via t-butyl groups; i.e., possesses end-on orientation (ESI,† Fig. S5a). A diffraction peak was observed at qz = 0.213 Å−1 for the DPPb film, corresponding to a d-spacing of 29.50 Å. This spacing is longer than the conjugation length (21.06 Å, ESI,† Fig. S4b) and is shorter than the length along the fully extended alkyl chain direction (ca. 43.86 Å, ESI,† Fig. S4b). This suggests that the side alkyl chains (–C16H33) are closely interdigitated in adjacent layers and are packed perpendicular to the CL-PVP surface, i.e., possess edge-on orientation (ESI,† Fig. S5b). Similarly, a (100) peak was observed at 0.218 Å−1 for the DPPc film having a larger conjugated unit (naphthalene), corresponding to a d-spacing of 28.8 Å. In addition, broad and dispersed peaks were observed in the qz (out-of-plane) direction, suggesting that the molecules are arranged in a random fashion (or have a wide distribution). Thus, the intermolecular packing of DPPc may be very poor, which is unfavorable to charge transport, leading to poor performance of the organic electronic devices.
3.4. OFET properties
Evaluation of the electrochemical and thermal properties of the DPPs demonstrated that these materials were suitable for use as an active material component for organic electronic devices. In order to explore the charge transport properties of the DPPs, organic field-effect transistors (OFETs) were fabricated using a polymeric gate dielectric layer; this layer is required for advanced flexible devices. The device performance was analyzed, where the profiles of the output and transfer characteristics were recorded under the ambient laboratory conditions. The drain–source current (IDS) in the saturation region can be described by eqn (1):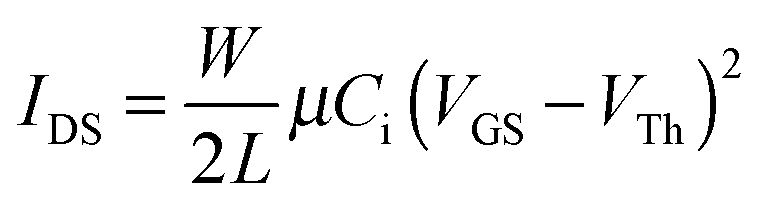 | (1) |
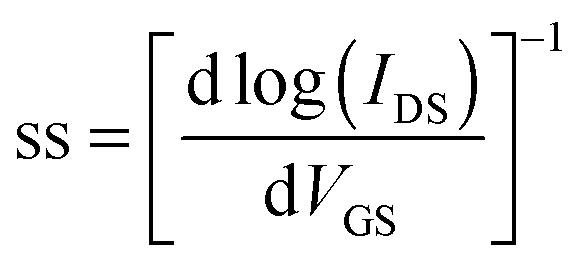 | (2) |
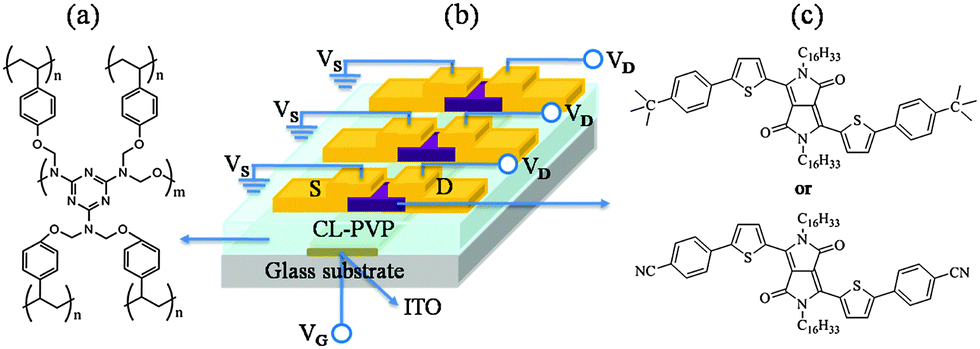 | ||
| Fig. 6 Chemical structure of (a) cross-linked poly(4-vinylphenol) (CL-PVP), (b) schematic of the OFET device structure, and (c) the chemical structure of DPPa (top) and DPPb (bottom). | ||
The output (IDSvs. VDS) and transfer (IDSvs. VGS) characteristics of the devices are plotted in Fig. 7. The output curves were recorded at gate voltages (VGS) of 0 to ±60 V at ±10 V increments while sweeping VDS from 0 to −60 V for p-channel operation and from 0 to +40 V for n-channel operation. The transfer characteristics were measured by sweeping VGS from 20 V to −60 V at a fixed VDS of −60 V (p-channel) and by sweeping VGS from 0 V to 60 V with a fixed VDS of 60 V (n-channel). The OFET based on DPPa (Fig. 7a and b) as a channel material showed typical p-channel charge transport behavior with a moderate transition from linear to saturation regimes. On the other hand, although there was some leakage current that should be studied further either by an optimization of device fabrication conditions or device structure engineering, the initial result of the device based on DPPb with a cyanophenyl end-capping group as a channel material showed n-channel charge transport behavior (Fig. 7c and d).
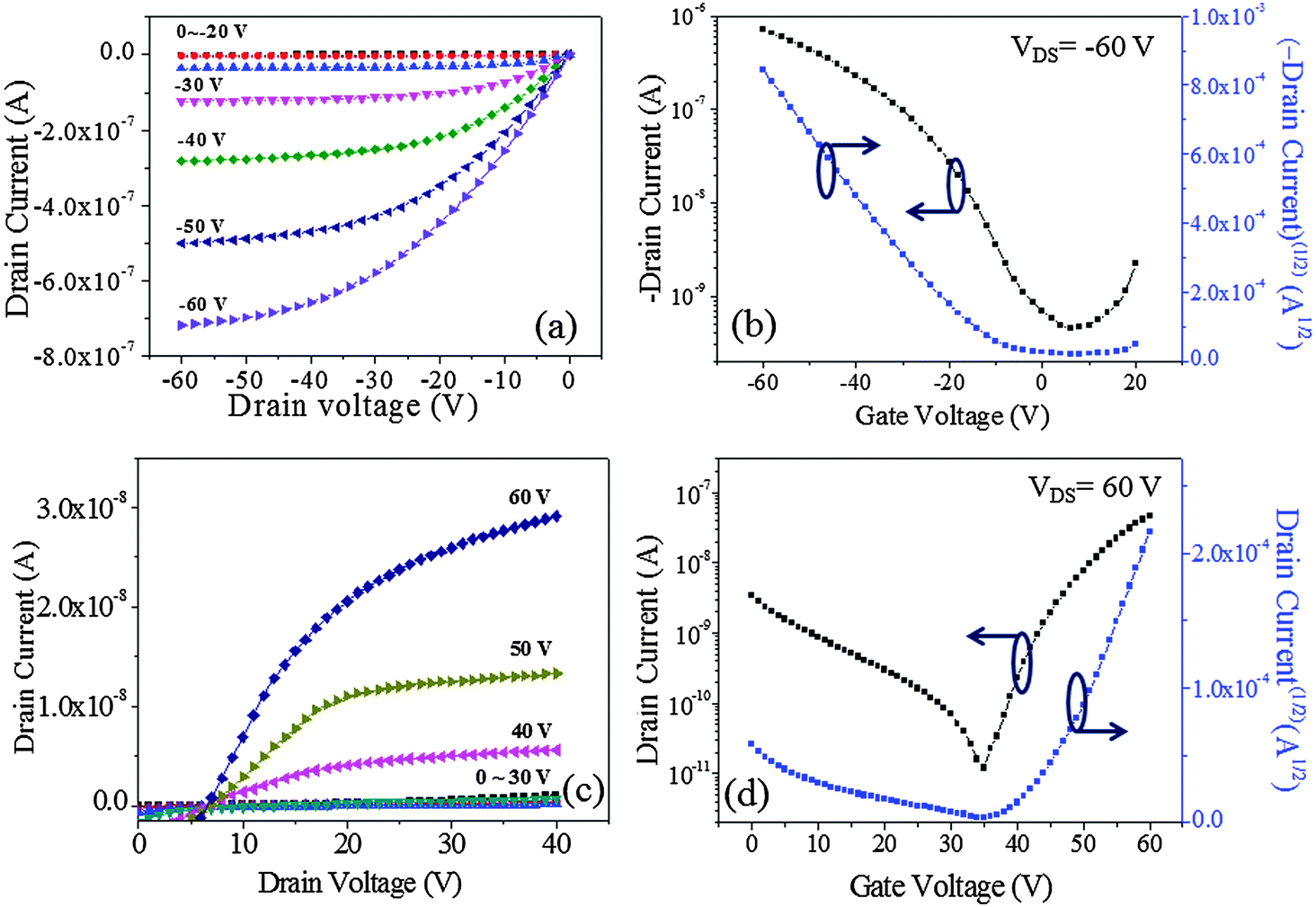 | ||
| Fig. 7 Output (a and c) and transfer (b and d) curves of the devices with DPPa and DPPb, respectively. | ||
The maximum hole mobility (μh), threshold voltage (VTh), sub threshold swing (ss), and current on-to-off ratio (ION/IOFF) of the devices with DPPa were estimated to be 5.91 × 10−3 cm2 V−1 s−1, −13.88 V, 13.89 V dec−1, and 1.6 × 103, respectively. The electron mobility (μe), VTh, ss, and ION/IOFF ratio of the device with DPPb were 3.4 × 10−3 cm2 V−1 s−1, 44.1 V, 3.8 V dec−1, and 6.9 × 102, respectively. Long-term stability of the device based on p-channel with DPPa was investigated by measuring the device after 100 days of storage under ambient conditions. The device still showed typical hole-dominant transport behaviors as shown in Fig. S6 (ESI†).
4. Conclusions
2,5-Dihexadecyl-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione was successfully arylated to generate three new diketopyrrolopyrrole derivatives (DPPa–c) using Pd-catalyzed C–H functionalization. All of the diketopyrrolopyrrole derivatives exhibit broad photo-absorption, narrow band-gaps (1.56–1.75 eV), highest occupied molecular orbital levels (−5.34 to −4.91 eV), and good thermal stability (Td = 394–423 °C). The electrochemical properties and intermolecular packing (edge-on or face-on) in the thin-films of the DPPs were influenced by the end-capping group, thereby affecting the charge transport behavior. Introduction of the cyanophenyl end-capping unit gave rise to a lowest unoccupied molecular orbital energy level of −3.65 eV for DPPb, which showed typical n-type characteristics with an electron mobility (μe) of 3.4 × 10−3 cm2 V−1 s−1 in the bottom-gate top-contact organic field effect transistor geometry with a polymer gate dielectric. The highest hole mobility of 5.9 × 10−3 cm2 V−1 s−1 was achieved with the use of the t-butylphenyl end-capping group (DPPa) in the same device configuration under ambient conditions.Acknowledgements
This work was supported by Konkuk University's research support program for its faculty on sabbatical leave in 2015.References
- X. Qi, S. Zou, X. Liu, W. Hao, H. Zhang, Z. Zang, H. Zhang, J. Gao and W. Hu, New J. Chem., 2015, 39, 1045–1050 RSC.
- X. Zhan, J. Zhang, S. Tang, Y. Lin, M. Zhao, J. Yang, H. Zhang, Q. Peng, G. Yu and Z. Li, Chem. Commun., 2015, 51, 7156–7159 RSC.
- A. Lorenzoni, M. Muccini and F. Mercuri, RSC Adv., 2015, 5, 11797–11805 RSC.
- L. Torsi, M. Magliulo, K. Manoli and G. Palazzo, Chem. Soc. Rev., 2013, 42, 8612–8628 RSC.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS PubMed.
- A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
- A. Mishra and P. Bauerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- A. K. Palai, S. P. Mishra, A. Kumar, R. Srivastava, M. N. Kamalasanan and M. Patri, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 832–841 CrossRef CAS.
- A. Kumar, R. Srivastava, S. S. Bawa, D. Singh, K. Singh, G. Chauhan, I. Singh and M. N. Kamalasanan, J. Lumin., 2010, 130, 1516–1520 CrossRef CAS.
- R. Steyrleuthner, R. D. Pietro, B. A. Collins, F. Polzer, S. Himmelberger, M. Schubert, Z. Chen, S. Zhang, A. Salleo, H. Ade, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2014, 136, 4245–4256 CrossRef CAS PubMed.
- G. L. Gibson, D. Gao, A. A. Jahnke, J. Sun, A. J. Tilley and D. S. Seferos, J. Mater. Chem. A, 2014, 2, 14468–14480 CAS.
- Y. Zhang, X.-D. Dang, C. Kim and T.-Q. Nguyen, Adv. Energy Mater., 2011, 1, 610–617 CrossRef CAS.
- X. Zhang, L. J. Richter, D. M. DeLongchamp, R. J. Kline, M. R. Hammond, I. McCulloch, M. Heeney, R. S. Ashraf, J. N. Smith, T. D. Anthopoulos, B. Schroeder, Y. H. Geerts, D. A. Fischer and M. F. Toney, J. Am. Chem. Soc., 2011, 133, 15073–15084 CrossRef CAS PubMed.
- S. Loser, C. J. Bruns, H. Miyauchi, R. P. Ortiz, A. Facchetti, S. I. Stupp and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 8142–8145 CrossRef CAS PubMed.
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684–1710 CAS.
- S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039–3051 RSC.
- Y. Wang, Q. Huang, Z. Liu and H. Li, RSC Adv., 2014, 4, 29509–29513 RSC.
- Y. Qiao, Y. Guo, C. Yu, F. Zhang, W. Xu, Y. Liu and D. Zhu, J. Am. Chem. Soc., 2012, 134, 4084–4087 CrossRef CAS PubMed.
- L. Wang, X. Zhang, H. Tian, Y. Lu, Y. Geng and F. Wang, Chem. Commun., 2013, 49, 11272–11274 RSC.
- P. Sonar, E. L. Williams, S. P. Singh, S. Manzhos and A. Dodabalapur, Phys. Chem. Chem. Phys., 2013, 15, 17064–17069 RSC.
- X. Lin, Y. Tani, R. Kanda, K.-i. Nakayama and S. Yagai, J. Mater. Chem. A, 2013, 1, 14686–14691 CAS.
- J. Dhar, N. Venkatramaiah, A. Anitha and S. Patil, J. Mater. Chem. C, 2014, 2, 3457–3466 RSC.
- X. Y. Shen, Y. J. Wang, H. Zhang, A. Qin, J. Z. Sun and B. Z. Tang, Chem. Commun., 2014, 50, 8747–8750 RSC.
- J.-H. Huang, K.-J. Jiang, F. Zhang, W. Wu, S.-G. Li, L.-M. Yang and Y.-L. Song, RSC Adv., 2014, 4, 16906–16912 RSC.
- H. Burckstummer, A. Weissenstein, D. Bialas and F. Wurthner, J. Org. Chem., 2011, 76, 2426–2432 CrossRef PubMed.
- C. Kim, J. Liu, J. Lin, A. B. Tamayo, B. Walker, G. Wu and T.-Q. Nguyen, Chem. Mater., 2012, 24, 1699–1709 CrossRef CAS.
- P. Sonar, G.-M. Ng, T. T. Lin, A. Dodabalapur and Z.-K. Chen, J. Mater. Chem., 2010, 20, 3626–3636 RSC.
- A. K. Palai, J. Lee, S. Das, J. Lee, H. Cho, S.-U. Park and S. Pyo, Org. Electron., 2012, 13, 2553–2560 CrossRef CAS.
- A. K. Palai, J. Lee, M. Jea, H. Na, T. J. Shin, S. Jang, S.-U. Park and S. Pyo, J. Mater. Sci., 2014, 49, 4215–4224 CrossRef CAS.
- A. K. Palai, J. Lee, T. J. Shin, A. Kumar, S.-U. Park and S. Pyo, Chem. Commun., 2014, 50, 8845–8848 RSC.
- A. D. Hendsbee, J.-P. Sun, L. R. Rutledge, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 4198–4207 CAS.
- S.-Y. Liu, M.-M. Shi, J.-C. Huang, Z.-N. Jin, X.-L. Hu, J.-Y. Pan, H.-Y. Li, A. K.-Y. Jen and H.-Z. Chen, J. Mater. Chem. A, 2013, 1, 2795–2805 CAS.
- S.-Y. Liu, W.-Q. Liu, J.-Q. Xu, C.-C. Fan, W.-F. Fu, J. Ling, J.-Y. Wu, M.-M. Shi, A. K.-Y. Jen and H.-Z. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 6765–6775 CAS.
- (a) J. Areephong, A. D. Hendsbee and G. C. Welch, New J. Chem., 2015, 39, 6714–6717 RSC; (b) J. Zhang, D.-Y. Kang, S. Barlow and S. R. Marder, J. Mater. Chem., 2012, 22, 21392–21394 RSC.
- A. K. Palai, H. Cho, S. Cho, T. J. Shin, S. Jang, S.-U. Park and S. Pyo, Org. Electron., 2013, 14, 1396–1406 CrossRef CAS.
- Spartan'10, Wavefunction Inc., Irvine, CA, USA, 2011 Search PubMed.
- A. K. Palai, S. Kim, H. Shim, S. Cho, A. Kumar, J. Kwon, S.-U. Park and S. Pyo, RSC Adv., 2014, 4, 41476–41482 RSC.
- S. E. Kazzouli, J. Koubachi, N. E. Brahmi and G. Guillaumet, RSC Adv., 2015, 5, 15292–15327 RSC.
- F. Shibahara and T. Murai, Asian J. Org. Chem., 2013, 2, 624–636 CrossRef CAS.
- B. J. Kim, H.-S. Lee, J. S. Lee, S. Cho, H. Kim, H. J. Son, H. Kim, M. J. Ko, S. Park, M. S. Kang, S. Y. Oh, B. S. Kim and J. H. Cho, J. Phys. Chem. C, 2013, 117, 11479–11486 CAS.
- P. Sonar, T. R. B. Foong and A. Dodabalapur, Phys. Chem. Chem. Phys., 2014, 16, 4275–4283 RSC.
- W. S. Yoon, S. K. Park, I. Cho, J. Oh, J. H. Kim and S. Y. Park, Adv. Funct. Mater., 2013, 23, 3519–3524 CrossRef CAS.
- M. Stolar and T. Baumgartner, Phys. Chem. Chem. Phys., 2013, 15, 9007–9024 RSC.
- Y. Kim, C. E. Song, E.-J. Ko, D. Kim, S.-J. Moon and E. Lim, RSC Adv., 2015, 5, 4811–4821 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nj02631h |
| ‡ Present address: Laboratory for Advanced Research in Polymeric Materials (LARPM), Central Institute of Plastics Engineering and Technology, Bhubaneswar 751 024, Odisha, India. |
| § Present address: Amity Institute of Advanced Research and Studies (Materials and Devices), Amity University, Noida, UP-201303, India. |
| ¶ Present address: UNIST Central Research Facilities & School of Natural Science, UNIST, Ulsan 689-798, Republic of Korea. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |