
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxamate salts as novel agents for the restoration of marble and limestone substrates: case study of ammonium N-phenyloxamate†
Laura
Maiore
a,
M. Carla
Aragoni
a,
Gianfranco
Carcangiu
b,
Ombretta
Cocco
c,
Francesco
Isaia
a,
Vito
Lippolis
a,
Paola
Meloni
d,
Arianna
Murru
c,
Alexandra M. Z.
Slawin
e,
Enrica
Tuveri
a,
J. Derek
Woollins
e and
Massimiliano
Arca
*a
aDipartimento di Scienze Chimiche e Geologiche, S.S. 554 bivio per Sestu, 09042 Monserrato (Cagliari), Italy. E-mail: marca@unica.it
bConsiglio Nazionale delle Ricerche (CNR), Istituto di Scienze dell'Atmosfera e del Clima (ISAC), UOS di Cagliari c/o Dipartimento di Fisica, Università degli Studi di Cagliari, S.S. 554 bivio per Sestu, 09042 Monserrato (Cagliari), Italy
cDipartimento di Ingegneria Civile, Ambientale e Architettura, via Marengo 2, 09123 Cagliari, Italy
dDipartimento di Ingegneria Meccanica, Chimica e dei Materiali, via Marengo 2, 09123 Cagliari, Italy
eEaStCHEM School of Chemistry, University of St. Andrews, North Haugh, St. Andrews, Fife KY16 9ST, UK
First published on 19th January 2016
Abstract
The ammonium salt of N-phenyloxamic acid (AmPhOxam) was synthesised, characterised by FT-IR, FT-Raman, UV-Vis, 1H-NMR spectroscopic methods and single crystal X-ray diffraction, and evaluated as a protective and consolidating agent for calcareous stone substrates under mild conditions. Hydro-alcoholic solutions of AmPhOxam were tested for the treatment of naturally weathered white marble and biomicritic limestone. Mercury intrusion porosimetry, FT-NIR spectroscopy measurements and SEM microscopy showed the formation of a superficial protective layer of crystals of the corresponding monohydrated calcium salt, CaPhOxam, on both treated stones.
1. Introduction
The protection and consolidation of marbles and limestones of historical significance is a matter of current interest. In fact, polluted air, containing sulfur and nitrogen oxides, results in acidic rain capable of determining the dissolution of calcite in marble and limestone.1–4 This process causes visible roughness of the stone surface and the loss of carved details. Only a few inorganic compounds have been exploited to date for the protection and restoration of carbonate stones, mainly re-mineralizing agents based on second group metal hydroxides,5,6 CaCO3 itself,7 or inorganic salts such as diammonium hydrogen phosphate, (NH4)2(HPO4), a precursor of hydroxyapatite Ca10(PO4)6(OH)2,8,9 and calcium alkoxides.10,11 The formation of an artificial coating of pure calcium oxalate (CaC2O4, CaOx) is one of the most promising techniques for the protection of stone artefacts.12,13 This coating can be achieved by the application of ammonium oxalate, (NH4)2(C2O4) (AmOx), that reacts with calcium carbonate generating insoluble14 calcium oxalate (reaction 1), mainly in its monohydrated form Ca(C2O4)·H2O (whewellite; solubility product Ksp = 2.0 × 10−9).15,16| CaCO3(s) + (NH4)2(C2O4)(aq) → Ca(C2O4)(s) + 2NH3(g) + H2O(l) + CO2(g) | (1) |
| CaSO4(s) + (NH4)2(C2O4)(aq) → Ca(C2O4)(s) + (NH4)2SO4(aq) | (2) |
Recently, we reported an alternative procedure for the protection of marble and limestone substrates with calcium oxalate, consisting of the treatment with oxalic acid monomethyl ester derivatives (AmMeox), whose in situ hydrolysis generates the desired salt (reaction 3).19
| CaCO3(s) + (NH4)(CH3C2O4)(aq) → Ca(C2O4)(s) + NH3(g) + CH3OH(l) + CO2(g) | (3) |
Surprisingly, the strategy to improve AmOx performances by structural modifications on the oxalate anion is almost unexplored. Very recently, some of the authors reported on the treatment of calcareous stone samples with ammonium oxamate [(NH4)2(H2C2NO3), AmOxam].19 AmOxam indeed has a large solubility in water (∼130 g L−1, 1.22 M) and hydro-alcoholic mixtures as compared to AmOx, while its hydrated calcium salt, CaOxam, shows a low solubility (1.3 g L−1,19 corresponding to Ksp = 3.67 × 10−7). The treatment of naturally weathered Carrara marble and biomicritic limestone with AmOxam results in a coating of neo-formed calcium salt, CaOxam, displaying morphologies and thickness depending on salt concentration and solvent mixture. The formation of a crystalline coating accompanied by variations in stone porosity was observed, especially when hydro-alcoholic mixtures were used. The introduction of organic substituents on the oxamate anion could in principle represent a synthetic tool to lower the solubility of the corresponding protecting calcium salts deposited on the surface of historical stone substrates, and at the same time to increase the solubility of the corresponding salts in volatile and possibly non-toxic mixture of solvents for its application on the artefacts of interest.
Therefore, we turned to the functionalization of the oxamate anion, by introducing a phenyl substituent to the amidic group to give N-phenyloxamate (oxanilate) salts. We report here on the synthesis and characterization of ammonium N-phenyloxamate (AmPhOxam) and its investigation as a protection agent for historical calcareous stone substrates.
2. Experimental
2.1 Materials and methods
Reagents and solvents were purchased from Sigma-Aldrich and Carlo Erba Reagenti and used as received. Carrara marble samples were cut from a slab from the Monumental Cemetery of Bonaria (Cagliari, Italy). This slab, dating to the beginning of twentieth century, has always been exposed outdoors. Samples of biomicritic limestone were obtained from Cava Flore (Santa Caterina di Pittinuri, Oristano, Italy), by cutting a piece of the same stone into slices of about 1.0 × 0.4 × 0.4 cm. Petrographic examinations of the stone samples were carried out using a Zeiss Axioskop 40 optical microscope with Zeiss camera Axiocam HR, operating in PPTL (plain polarized transmitted light) and CPL (cross polarized light) mode. Colorimetric measurements were carried out on untreated and treated samples using a Konica Minolta CM-700d spectrophotometer (illuminant D65). CieLab75 color space has been adopted to evaluate changes in sample color. The instrument has been adjusted to repeat the measurement 6 times on each point: for each sample eight points have been identified and measured. Carbon, hydrogen, and nitrogen contents were determined with a PerkinElmer 240B elemental analyzer. FT-IR and FT-NIR spectra were recorded with a Thermo-Nicolet 5700 spectrometer at room temperature in KBr pellets with a KBr beam-splitter and KBr windows (4000–400 cm−1, resolution 4 cm−1) equipped with SABIR fiber optic diffuse reflectance probe (4000–7400 cm−1). FT-Raman measurements were carried out in the range 500–50 cm−1 (resolution: 2 cm−1) on a Bruker RFS100 FT-Raman spectrometer fitted with an InGaAs detector (RT) and a Nd-YAG laser (excitation wavelength: 1064 nm; scattering geometry 180°). UV-Vis absorption spectra were recorded on a Thermo Nicolet Evolution 300 spectrophotometer (190–1100 nm) at room temperature using quartz cuvettes with optical path length of 10.0 mm. Emission spectra were obtained at room temperature with a Varian Cary Eclipse Fluorescence spectrophotometer (Xe lamp) using quartz cuvettes with an optical path length of 10.0 mm. 1H-NMR spectra were recorded on a Varian INOVA-500 spectrometer. Solubility and Ksp values at 25 °C were evaluated spectrophotometrically on saturated solutions, after recording of a calibration curve on five samples. Single crystal X-ray diffraction data for AmPhOxam were collected with the St. Andrews Robotic diffractometer at 125(2) K.21 The structures were solved by direct methods with SHELXS-9722 and refined on F2 by using SHELXL2013.23 All relevant data and parameters of the X-ray measurements and refinements are given in Tables S1–S10 in ESI.† CCDC 1425298 (AmPhOxam). The stone samples were immersed in a freshly prepared AmPhOxam solution (50 mL) into a static batch for 24 h, washed with water, dried at room temperature for 4 days, and then kept into a thermostatic heater (40 °C) for 24 h. Porosimetric analyses were performed on treated and untreated stone samples with a Micrometrics Autopore IV 9500. SEM Microscopy investigation was performed with a Zeiss Evo LS15 equipped with a LaB6 filament as electron source.DFT24 theoretical calculations were performed with the commercial suite of software Gaussian 0925 on the anions oxamate (Oxam) and N-phenyloxamate (PhOxam) with the Becke3LYP three-parameter hybrid functional26–28 and 6-311G basis set.29,30 The nature of the energy minima of each optimised structure was verified by harmonic frequency calculations. A potential energy surface (PES) investigation was carried out for each oxamic acid derivative in order to evaluate the rotational barrier around the C–C bond and – in the case of PhOxam – the rotational barrier of phenyl rotation. Natural31 charge distributions were calculated at the optimised geometries at the same level of theory. All calculations were carried out on a E4 workstation equipped with four quad-core AMD Opteron processors and 16 GB of RAM and running the 64 bit version of the Ubuntu 12.04 Linux operating system.
Synthesis of AmPhOxam. A freshly prepared aqueous solution of NH4HCO3 (399.0 mg, 5.0 mmol, 50 mL) was added to an aqueous solution of N-phenyloxamic acid (826.0 mg, 5.0 mmol, 50 mL), and stirred at room temperature until the evolution of gaseous CO2 was complete (about 1 hour). Subsequently, the solution was evaporated at room temperature affording colourless crystals of AmPhOxam (824.3 mg, 90%). Melting point: 209–212 °C. Elem. anal. calcd for C8H10N2O3 (FW 182.18 g mol−1): C 52.74, H 5.53, N 15.38%; found C 52.55, H 5.38, N 15.07%. FT-MIR bands (ν): 3368 (s), 3244 (s), 3183 (s), 3055 (s), 3035 (s, broad), 2884 (s), 2843 (s), 1698 (s), 1617 (s), 1588 (s), 1520 (s), 1495 (m), 1468 (s), 1440 (s), 1418 (s), 1387 (s), 1365 (s), 1310 (m), 1293 (m), 1226 (w), 1181 (m), 1155 (w), 1078 (w), 1027 (w), 985 (w), 965 (w), 938 (w), 900 (w), 841 (w), 815 (w), 780 (m), 746 (s), 692 (s), 642 (m, broad), 596 (w), 532 (m), 485 (m) cm−1. FT-Raman (ν): 3055, 1599, 1524, 1449, 1225, 1029, 1000, 841, 782, 618, 278, 141, 101, 80 cm−1. 1H-NMR (D2O; δ): 7.56 (d, 2H, J = 8.3 Hz), 7.47 (dd, 2H, J = 7.7 and 7.4 Hz), 7.30 ppm (dd, 1H, J = 7.4 Hz). UV-vis [1.0 × 10−4 M aqueous solution, λ(ε)]: 194 (11
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 253), 196 (10934), 201 (shoulder, 9602), 255 nm (4940 M−1 cm−1). Emission (5.50 × 10−5 M aqueous solution; λexc = 253 nm; slit 20 × 20; λem): 367, 510 nm.
253), 196 (10934), 201 (shoulder, 9602), 255 nm (4940 M−1 cm−1). Emission (5.50 × 10−5 M aqueous solution; λexc = 253 nm; slit 20 × 20; λem): 367, 510 nm.
Synthesis of CaPhOxam. Solid Ca(OH)2 (9.1 mg, 0.125 mmol) was dissolved in an aqueous solution of N-phenyloxamic acid (40.8 mg, 0.250 mmol, 25 mL). In few minutes a white solid was formed, and the resulting suspension was stirred at room temperature for three hours. The white solid was separated by filtration affording 25.4 mg of CaPhOxam (55% yield). Melting point: 216–220 °C decomp. Elem. anal. calcd for C16H12CaN2O6H2O (386.37 g mol−1): C 49.74, H 3.65, N 7.25%; found C 50.78, H 2.54, N 7.72%. FT-NIR bands (ν): 6602 (w), 5992 (w), 5167 (s), 4967 (m), 4901 (ms), 4867 (m), 4761 (m), 4672 (m), 4644 (m), 4598 (ms), 4557 (s), 4309 (m), 4220 (m), 4062 (s), 4026 (s) cm−1. FT-Raman (ν): 3076, 3073, 1599, 1557, 1545, 1451, 1233, 1030, 105 cm−1. UV-vis absorption [1.2 × 10−4 M aqueous solution), λ(ε)]: 192 (14
326), 203 (shoulder, 7369), 254 nm (4681, M−1 cm−1). Emission (5.43 × 10−5 M aqueous solution; λexc = 253 nm; slit 2.5 × 2.5; λem): 361, 511 nm.
3. Results and discussion
3.1 Theoretical calculations
Density functional theory (DFT)25 has proved in recent years to be an invaluable tool in understanding the electronic properties of a plethora of compounds, providing reliable quantitative results in material science at an acceptable computational cost. In this context, theoretical DFT calculations demonstrated that the oxalate and oxamate anions could strongly interact with the crystal lattice of calcite through the carboxylate moiety.19 Therefore we turned to investigate the effect of introducing the phenyl N-substituent at the oxamate group on the electronic features of the free carboxylate in the phenyloxamate anion. Notably, the optimised metric parameters of the oxamate moiety in the N-phenyloxamate anion (PhOxam) are barely affected by the introduction of the phenyl substituent with respect to the unsubstituted oxamate (Oxam: C–C, 1.571; C–N, 1.368; average C–O, 1.270; PhOxam: C–C, 1.571; C–N, 1.386; average C–O, 1.268 Å). In analogy to the oxamate anion, a relaxed potential energy surface (PES) scan indicated the completely planar conformation as the most stable one (Fig. S1 in ESI†). In addition, the natural charge distribution32 showed that the terminal carboxyl group carries a negative charge (−0.804 e), whose absolute value is only slightly smaller than that calculated for Oxam at the same level of theory (−0.840 e), thus suggesting that the possibility to interact with the calcite surface through the carboxyl group19 is preserved.3.2 Synthesis and characterisation of AmPhOxam and CaPhOxam
The ammonium salt of N-phenyloxamic acid (NH4)(C8H10N2O3), AmPhOxam, was synthesised in almost quantitative yield by neutralization of the acid with a stoichiometric amount of (NH4)HCO3 in aqueous solution, in accordance with a previously reported method,19 and characterised by FT-IR, FT-Raman, 1H-NMR, and UV-Vis spectroscopy. Differently from AmOxam, UV-Vis spectroscopy measurements carried out at different concentrations (up to 0.1 mM) do not show any deviation from the Lambert–Beer law in diluted solutions. The single crystal X-ray diffraction structure of AmPhOxam (Fig. 1 and Tables S1–S10 in ESI†) shows the oxamate anion is almost perfectly planar [torsion 2.8(8)°], with the phenyl ring rotated by 39(1)°. The C–C bond distance [1.549(9) Å], close to that calculated at DFT level, indicates the absence of conjugation between the CO2− and PhNHCO moieties, analogously to what is observed in the case of ethyl N-phenyloxamate.32 A network of hydrogen bonds involving all the O-atoms of the oxamate and the ammonium cation is responsible for the crystal packing [N1⋯O10, 2.836; N1⋯O1a, 2.826; N1⋯O2a, 2.877; N1⋯O1b, 2.832; N1⋯O10i, 2.914; N1⋯O2i, 2.908 Å; a = x, 1 + y, z; b = 1 − x, 1/2 + y, 3/2 − z; i = x, −1/2 − y, −1/2 + z, Table S10 in ESI†]. AmPhOxam is very stable in aqueous solution and does not show any spontaneous hydrolysis reaction at room temperature. The hydrated calcium salt of N-phenyloxamic acid, CaPhOxam, was prepared by neutralization of the acid with a stoichiometric amount of Ca(OH)2 in aqueous suspension, and isolated as a white solid. Spectrophotometric measurements showed that in saturated aqueous solution the solubility of this salt at 25 °C amounts to 1.64 × 10−2 M (Ksp = 1.75 × 10−5). Single crystals obtained from the slow evaporation of a diluted aqueous solution of CaPhOxam were examined by X-ray diffraction, revealing that during the crystallization hydrolysis of the calcium salt occurred to give the trihydrate calcium oxalate (caoxite).33,34 No evidence of the formation of the mono- and di-hydrated salts (whewellite and weddellite, respectively) was observed.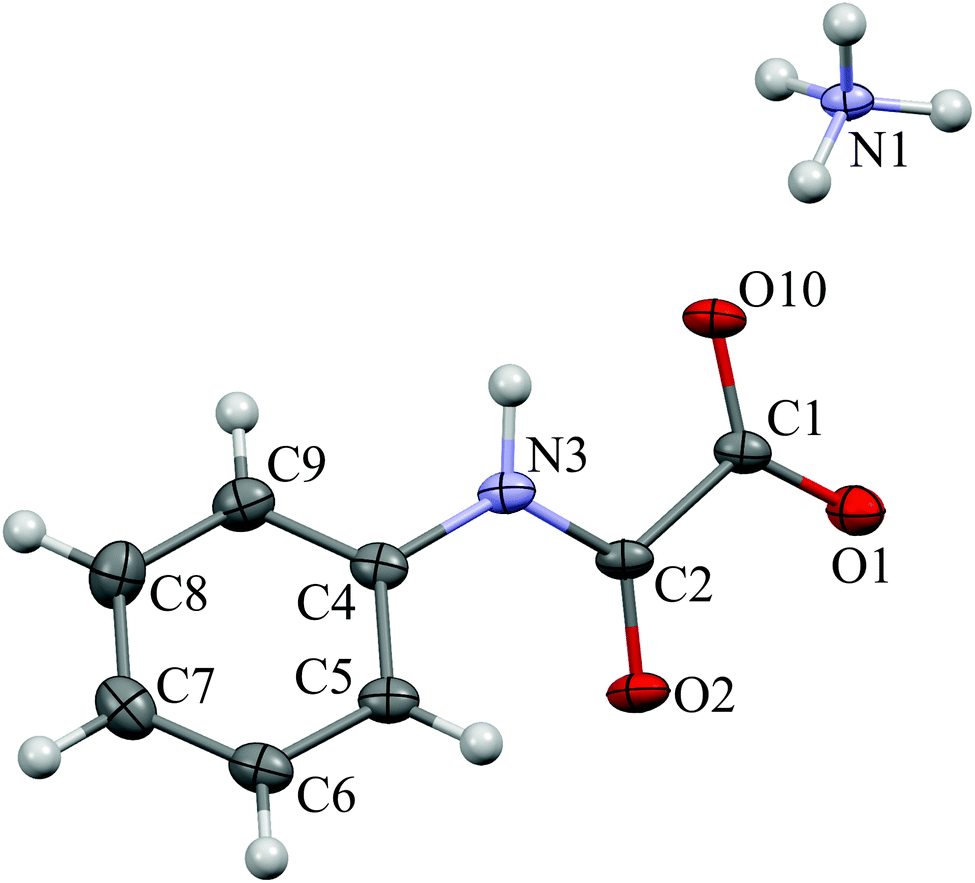 | ||
| Fig. 1 View of ammonium N-phenyloxamate (AmPhOxam) showing thermal displacement ellipsoid at 30% probability level. | ||
3.3 Treatment of calcareous stones with AmPhOxam
The filler/consolidating properties of AmPhOxam were assayed on two calcareous stones, a white Carrara marble coming from a slab of the Monumental Cemetery of Bonaria (Cagliari, Italy), naturally weathered for about one century, and a biomicritic limestone from a disused mining site in Santa Caterina di Pittinuri (Oristano, Italy). The stones were previously subject to petrographic examinations (Fig. S2 in ESI†). Optical microscopy observations in transmitted polarized light (TPL) on marble samples revealed a pseudo-polygonal microstructure with medium–large grains (100–150 μm), straight grain boundaries and 120° triple junctions. These microstructural features typically characterize many types of white Carrara Marble and we retain that the analysed samples could be ascribed to a type of white Carrara marble commercially known as “ordinario”.35–38 This type of marble, along with the so-called “statuario” and “statuarietto” varieties, were exploited in most of the artworks of the monumental cemetery of Cagliari. Biomicritic limestone from Santa Caterina di Pittinuri, quarried from a Miocenic formation, is a lithotype common not only in Western Sardinia but also in different countries overlooking the Mediterranean basin.39,40 A very similar stone is the so-called “Globigerina Limestone” from the island of Malta.41 The limestone, under petrographic microscope in PPTL mode, reveals an abundant microfossilifer fauna embedded in a micrite matrix; very scarce is the carbonatic cement. The biomicritic limestone exhibits low physical–mechanical properties. The most common decay forms are alveolar weathering, erosion, pulverization and crumbling.Samples of both stones were analysed and characterised before and after treatments with AmPhOxam 2.5% w/w in EtOH:H2O 1:3 v/v mixture by FT-IR spectroscopy, mercury porosimetry, X-ray powder diffraction, TG-DTA analyses and SEM microscopy. Mercury intrusion porosimetry (MIP) has been largely exploited to obtain qualitative and quantitative information on porous stones and the effect of their treatments with protection compounds,42–44 although its application to consolidate rocks has been criticised.45 Untreated marble samples featured an average total porosity value of about 7%, with pore sizes ranging between 0.06 and 60 μm, those in the range 1.0–10.0 μm being the most abundant. The high porosity is mainly due to thermal stress processes that caused irreversible deformations of the structure, resulting in a large variability of local porosity, as confirmed by SEM microscopy analysis (Fig. S3 in ESI,† top). Disunion between grains was evidenced in the cross sections of the samples, as well as fractures, the largest part having dimension in the range between 1.0 and 10.0 μm. However, the surface showed lack of chemical corrosion or sulfate deposition. Marble samples treated with a solution of AmPhOxam showed an increase in the total porosity and the average pore radius values (Table 1). The low porosity of untreated marble, resulting in not interconnected void, is possibly responsible for the very limited permeation of the AmPhOxam solution. As a consequence, the crystals nucleation and growth occur almost entirely on the surface developing a thick coating (up to 15 μm). Accordingly, SEM microscopy revealed the presence of a deposition of fine crystals on the surface of the stone (Fig. S3 in ESI,† middle and bottom). This coating is characterized by small “globular” crystal clusters covering all the surface of the sample. On this spongy coating many large tabular crystals, sometimes organized in clusters, were embedded. In our opinion the microstructural features of the coating might be responsible for the small increase in the porosity of the treated marble sample, that can be ascribed to a novel secondary porosity. FT-NIR spectra recorded on the surface of treated marble samples showed the diagnostic bands between 4000 and 5200 cm−1,19 in agreement with the deposition of the neo-formed calcium salt CaPhOxam (Fig. S4 in ESI†). Colorimetric measurements were performed on untreated and treated samples (Table S11 in ESI†). The obtained values show only slight changes (color difference ΔE = 3.76), not perceptible by naked eyes (ΔE < 5). Therefore the AmPhOxam treatment does not significantly modify the color of rock surfaces.
| Porosity (%) | Average pore radius (μm) | Total intrusion volume (mL g−1) | |
|---|---|---|---|
| Carrara white marble | |||
| Untreated | 6.71 | 2.53 | 0.03 |
| AmPhOxam 2.50% w/w | 7.41 | 2.36 | 0.03 |
| Variation % | 10 | −7 | 18 |
| Biomicritic limestone | |||
| Untreated | 30.60 | 0.44 | 0.18 |
| AmPhOxam 2.50% w/w | 25.16 | 0.37 | 0.14 |
| Variation % | −18 | −18 | −21 |
As compared to Carrara marble, untreated biomicritic limestone samples showed different morphologies and composition. X-ray powder diffraction analysis revealed the typical composition of biomicrites, made up of a microcrystalline calcite (micrite) and other mineralogical phases as impurities (ankerite, quartz, muscovite, dolomite, and montmorillonite). This stone has a very high porosity compared to marble but smaller pores (Table 1), most of which fall in the range 0.15–2.0 μm. SEM images (Fig. 3, top) revealed the typical structure made up of microcrystals of calcite (2–4 μm) and rare impurities of larger dimensions (about 100 μm). Notably, the large porosity of the biomicritic limestone characterized by interconnected micrometric and sub-micrometric pores, allows the AmPhOxam solution to effectively permeate the biomicritic sample forming new mineral phases both on the cortical portion (0.3 mm in depth) and on the surface of the sample, as highlighted by SEM investigations. In fact, the formation of a coating (about 4 μm thick) on the overall surface of limestone is observed, consisting of the agglomeration of thin needles (Fig. 3, middle) of CaPhOxam, as confirmed unambiguously by FT-NIR spectra recorded on the surface of the treated samples (Fig. S4 in ESI†). Accordingly, treatment of biomicritic limestone with AmPhOxam solution resulted in a marked decrease of both total porosity (about −18%) and average pore radius values (about −18%), suggesting the size reduction of medium–large pores (Fig. 2, bottom). It is worth noting that the treatments with AmPhOxam did not result in any appreciable chromatic variation (color difference ΔE = 2.79, Table S11 in ESI†).
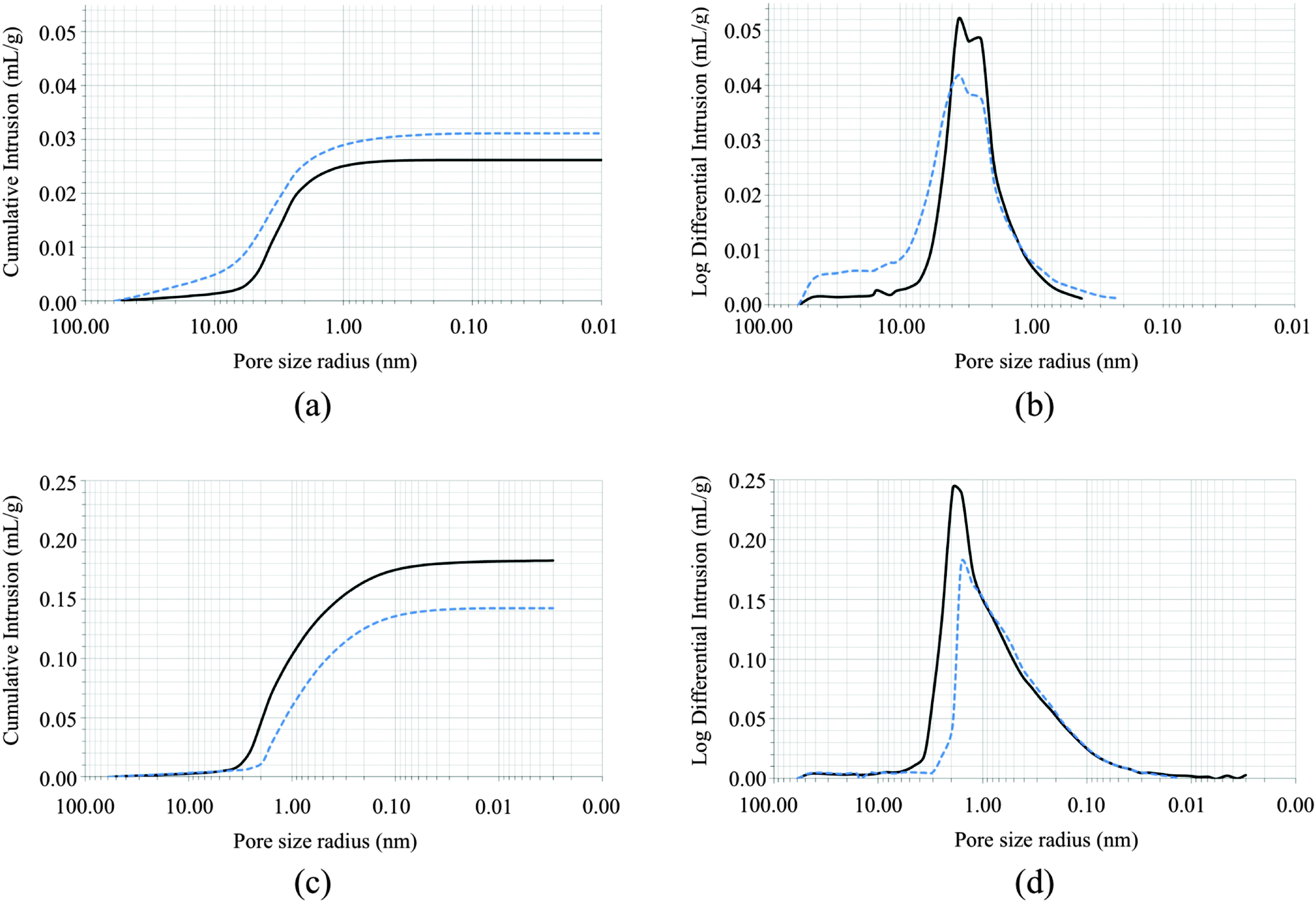 | ||
| Fig. 2 Cumulative intrusion (a and c) and differential intrusion as a function of the pore size radius (b and c) in Carrara marble (top; a and b) and biomicritic limestone (bottom; c and d) before (continuous line) and after (dotted line) treatment with AmPhOxam solutions (2.5% w/w EtOH:H2O 1:3 v/v mixture). | ||
 | ||
| Fig. 3 SEM images of biomicritic limestone before (top) and after treatment with AmPhOxam (2.5% w/w EtOH:H2O 1:3 v/v mixture, middle and bottom) in static batch at room temperature for 24 hours. | ||
Compared to AmOxam, the effects of the treatment with AmPhOxam solutions on the porosity network of both stones are remarkable. Actually, treatment with a 2.5% w/w solution of AmOxam in hydroalcoholic solution on Carrara marble resulted in a decrease both in the total porosity (−10%) and in the pore average radius value (−19%).19 On the contrary, the treatment of Carrara marble with AmPhOxam resulted in an increase in both parameters (Table 1 and Fig. 2).
As far as samples of biomicritic limestone are concerned, differently from AmOxam (porosity variation: 2%),19 the treatment with the novel compound AmPhOxam was effective on limestone. Furthermore, just one pattern of neo-crystallised CaPhOxam was observed on the surface of treated stones, while AmOxam produced two or three different patterns of superimposed crystals with dimensions falling in a very large range (<1–100 μm).19
4. Conclusion
In summary, the introduction of structural modifications on the oxamate anion proved a promising tool for the development of protective agents for calcareous stones. In fact, the deposition of CaPhOxam on the surface of both Carrara marble and biomicritic limestone can be considered as a possible protecting tool towards acidic agents, which represent one of the main causes of the degradation of artefacts exposed to atmospheric agents. A systematic variation in the substituents at the oxamate anion could in fact represent a strategy to reduce the water solubility of the calcium salts deposited on the surface and simultaneously tailor the solubility in solvent mixture different from pure water. Albeit preliminary, the results described here represent a first step towards the design and preparation of novel protection agents for limestone substrates. Further experiments aimed at fully characterising the physical properties of the treated surfaces are necessary to demonstrate the applicability of AmPhOxam to actual cases.Acknowledgements
M. A., L. M., and E. T. gratefully acknowledge RAS (Regione Autonoma della Sardegna) for financial support (P.O.R. Sardegna F.S.E. Operational Programme of Regione Autonoma della Sardegna, European Social Fund 2007–2013 – Axis IV Human Resources, Objective l.3, Line of Activity l.3.1 “Avviso di chiamata per il finanziamento di Assegni di Ricerca”. P. M. and G. C. acknowledge RAS for financial support (L.R. 7, 2010, Project CRP 27588 “I beni culturali e la prova del tempo: strategie innovative per la conservazione mediante il controllo cinetico di processi minerogenetici e inibitivi”). M. A. thanks the Fondazione Banco di Sardegna for financial support. The Comune di Cagliari and “Soprintendenza BAPSAE per le Province di Cagliari e Oristano” are kindly acknowledged for their support in the activities of the “Laboratorio Colle di Bonaria”.References
- R. J. Cheng, J. R. Hwu, J. T. Kim and S. M. Leu, Anal. Chem., 1987, 59, 104A–106A CAS.
- A. E. Charola, J. Chem. Educ., 1987, 64, 436–437 CrossRef CAS.
- P. A. Baedecker and M. M. Reddy, J. Chem. Educ., 1993, 70, 104–108 CrossRef CAS.
- P. Brimblecombe, Environment and Architectural Stone, in Stone in Architecture, ed. S. Siegesmund and R. Snethlage, Springer-Verlag, Berlin Heidelberg, 5th edn, 2014, ch. 5, pp. 317–347 Search PubMed.
- P. Baglioni, L. Dei, F. Piqué, G. Sarti and E. Ferroni, Stud. Conserv., 1997, 42, 43–54 CrossRef CAS.
- A. H. Church, Br. Pat. 220, 1862 Search PubMed.
- M. I. Daskalakis, A. Magoulas, G. Kotoulas, I. Catsikis, A. Bakolas, A. P. Karageorgis, A. Mavridou, D. Doulia and F. Rigas, J. Appl. Microbiol., 2013, 115, 409–423 CrossRef CAS PubMed.
- E. Sassoni, S. Naidu and G. W. Scherer, J. Cult. Herit., 2013, 12, 346–355 CrossRef.
- E. Sassoni and E. Franzoni, Appl. Phys. A: Mater. Sci. Process., 2014, A117, 1893–1906 CrossRef.
- M. Favaro, P. Tomasin, F. Ossola and P. A. Vigato, Appl. Organomet. Chem., 2008, 22, 698–704 CAS.
- F. Ossola, P. Tomasin, C. De Zorzi, N. ElHabra, M. Chiurato and M. Favaro, New J. Chem., 2012, 36, 2618–2624 RSC.
- B. Salvadori, D. Pinna and S. Porcinai, Environ. Sci. Pollut. Res. Int., 2014, 21, 1884–1896 CrossRef CAS PubMed.
- D. Pinna, B. Salvadori and S. Porcinai, Constr. Build. Mater., 2011, 25, 2723–2732 CrossRef.
- B. Doherty, M. Pamplona, R. Selvaggi, C. Miliani, M. Matteini, A. Sgamellotti and B. Brunetti, Appl. Surf. Sci., 2007, 253, 4477–4484 CrossRef CAS.
- E. V. Petrova, N. V. Gvozdev and L. N. Rashkovich, J. Optoelectron. Adv. Mater., 2004, 6, 261–268 CAS.
- C. Conti, C. Colombo, D. Dellasega, M. Matteini, M. Realini and G. Zerbi, J. Cult. Herit., 2011, 12, 372–379 CrossRef.
- E. Hansen, E. Doehne, J. Fidler, J. Larson, B. Martin, M. Matteini, C. Rodriguez-Navarro, E. S. Pardo, C. Price, A. de Tagle, J. M. Teutonico and N. Weiss, Rev. Conserv., 2003, 4, 13–25 CAS.
- D. Mudronja, F. Vanmeert, K. Hellemans, S. Fazinic, K. Janssens, D. Tibljas, M. Rogosic and S. Jakovljevic, Appl. Phys. A: Mater. Sci. Process., 2013, 111, 109–119 CrossRef CAS.
- L. Maiore, M. C. Aragoni, G. Carcangiu, O. Cocco, F. Isaia, V. Lippolis, P. Meloni, A. Murru, E. Tuveri and M. Arca, J. Colloid Interface Sci., 2015, 448, 320–330 CrossRef CAS PubMed.
- T. M. Cezar, J. Conserv. Museum Studies, 1998, 4, 6–10 CrossRef.
- A. L Fuller, L. A. S. Scott-Hayward, Y. Li, M. Bühl, A. M. Z. Slawin and J. D. Woollins, J. Am. Chem. Soc., 2010, 132, 5799–5802 CrossRef PubMed.
- G. M. Sheldricks, SHELXL-97, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- W. Koch and M. C. Holthausen, A Chemist's Guide to Density Functional Theory, Wiley-VCH, Weinheim, 2nd edn, 2002 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- E. V. García-Báez, C. Z. Gómez-Castro, H. Höpfl, F. J. Martínez-Martínez and I. I. Padilla-Martínez, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, o541–o543 Search PubMed.
- S. Deganello, A. K. Kampf and P. B. Moore, Am. Mineral., 1981, 66, 859–865 CAS.
- N. S. Blom, J. A. Kanters and W. M. M. Heijnen, Cryst. Struct. Commun., 1981, 10, 1283–1288 CAS.
- G. Molli, P. Conti, G. Giorgetti, M. Meccheri and N. Oesterling, J. Struct. Geol., 2000, 22, 1809–1825 CrossRef.
- E. Cantisani, R. Canova, F. Fratini, C. Manganelli Del Fa, R. Mazzuoli and G. Molli, Period. Mineral., 2000, 69(3), 257–268 Search PubMed.
- B. Leiss and T. Weiss, J. Struct. Geol., 2000, 22(11–12), 1731–1745 Search PubMed.
- S. Siegesmund, K. Ullemeyer, T. Weiss and E. K. Tschegg, Int. J. Earth Sci., 2000, 89, 170–182 CrossRef CAS.
- A. Assorgia, S. Barca, P. Mighela, A. Muntoni, G. Murgia, A. Porcu, R. Rizzo, G. Rombi and C. Spano, La successione vulcano-sedimentaria oligo-miocenica del settore compreso tra Bosa e Santa Caterina di Pittinuri (Sardegna centro-occidentale), in La “Fossa Sarda” nell'ambito dell'evoluzione geodinamica cenozoica del Mediterraneo occidentale, A. Assorgia, S. Barca and C. Spano (ed.), Villanovaforru, 19–22 June, 1997 Search PubMed.
- A. Cherchi, Appunti biostratigrafici sul Miocene della Sardegna (Italia), Inter. Néogéne Medit., Lyon-1971, Mem. B.R.G.M., 1974, vol. 78, pp. 433–445, Lyon.
- J. Cassar, Deterioration of the Globigerina Limestone of the Maltese Islands, in Natural Stone, Weathering Phenomena, Conservation Strategies and Case Studies, S. Siegesmund, T. Weiss, and A. Vollbrecht, Geological Society, London, Special Publication, No. 205, 2002, pp. 33–49 Search PubMed.
- N. Ashurst and J. Kelly, The analytic approach to stone: its cleaning, repair and treatment, in Conservation of Building and Decorative Stone, ed. F. J. Dimes and J. Ashurst, Routledge, 1998, vol. 2, pp. 240–243 Search PubMed.
- M. Laho, C. Franzen, R. Holzer and P. W. Mirwald, Pore and hygric properties of porous limestones: a case study from Bratislava, Slovakia, in Natural Stone Resources for Historical Monuments, ed. R. Přikryl and Á. Török, Geological Society of London, Special Publications 333, 2010, pp. 165–174 Search PubMed.
- K. Kovářová, R. Ševčík and Z. Weishauptová, Acta Geodyn. Geomater., 2012, 9, 541–549 Search PubMed.
- M. J. Mosquera, J. Pozo, L. Esquivias, T. Rivas and B. Silva, J. Non-Cryst. Solids, 2002, 311, 185–194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for AmPhOxam, potential energy surface calculated at DFT level, and FT-NIR spectra. CCDC 1425298. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj02505b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |