
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile solvolysis of a surprisingly twisted tertiary amide†
Aaron J.
Bloomfield
*ab,
Subhajyoti
Chaudhuri
bc,
Brandon Q.
Mercado
a,
Victor S.
Batista
ab and
Robert H.
Crabtree
ab
aDepartment of Chemistry, Yale University, 225 Prospect St., New Haven, CT 06510, USA. E-mail: aaron.bloomfield@yale.edu
bEnergy Sciences Institute, Yale University, 520 West Campus Drive, West Haven, CT 06516, USA
cYale School of Engineering and Applied Science, Yale University, 10 Hillhouse Ave, New Haven, Connecticut 06520-8107, USA
First published on 5th January 2016
Abstract
A bicyclo[2.2.2]octane derivative containing both a tertiary amide and a methyl ester (1) was shown crystallographically to adopt a conformation in which the amide is in the cis configuration, which is sterically disfavored, but electronically favored. The steric strain induces a significant torsion (15.9°) of the amide, thereby greatly increasing the solvolytic lability of the amide to the extent that we see competitive amide solvolysis in the presence of the normally more labile methyl ester also present in the molecule.
1. Introduction
Amide bonds are typically robust1 and easily synthesized,2,3 making this linkage popular among synthetic chemists, and common in biomolecules. Amides that are distorted, especially by pyramidalization of the nitrogen, or by torsion of the carbonyl C–N bond, are a subject of interest because of their increased lability.4–6 There is also evidence that enzymes twist amide substrates to enhance the rate of C–N bond cleavage,7,8 so studies on such amides are relevant to this enzymatic activation.9–11 Bicyclic lactams, especially those which place the nitrogen at the bridgehead position, appear to be a favorite structural motif for preparing and studying twisted amides,9–14 due to the geometry enforced by Bredt's rule. However some acyclic amides are distorted only by steric interactions of large substituents.11,15,16There are several reports indicating that secondary aryl amides (RCONHAr) exist primarily in the trans conformation, while the corresponding methylated amides (RCONMeAr) are more stable in the cis conformation.17–22 However, there is no clear explanation of this phenomenon. Additionally, the influence of the steric strain induced by the change in conformational preference has not been studied in detail before.
In the course of our own studies, we have found that the 3° N-methyl amide, 1, adopts the cis conformation, despite the significant steric interaction that would be alleviated by adoption of the trans conformation, which is preferred in the case of the unmethylated 2° amide analog (2). Instead, to accommodate this conformation, the molecule distorts, twisting the amide about the carbonyl–N bond, and skewing the bicyclic system. These distortions result in high lability of the amide under both acidic and alkaline conditions (Scheme 1). In this paper, we report the preparation and solvolysis of three bicyclooctyl carboxamides (1–3), compare their structures to those of other reported amides, and investigate the observed trends using computational methods. Additionally, we discuss a possible explanation for the cis-preference of N-alkyl-N-aryl amides.
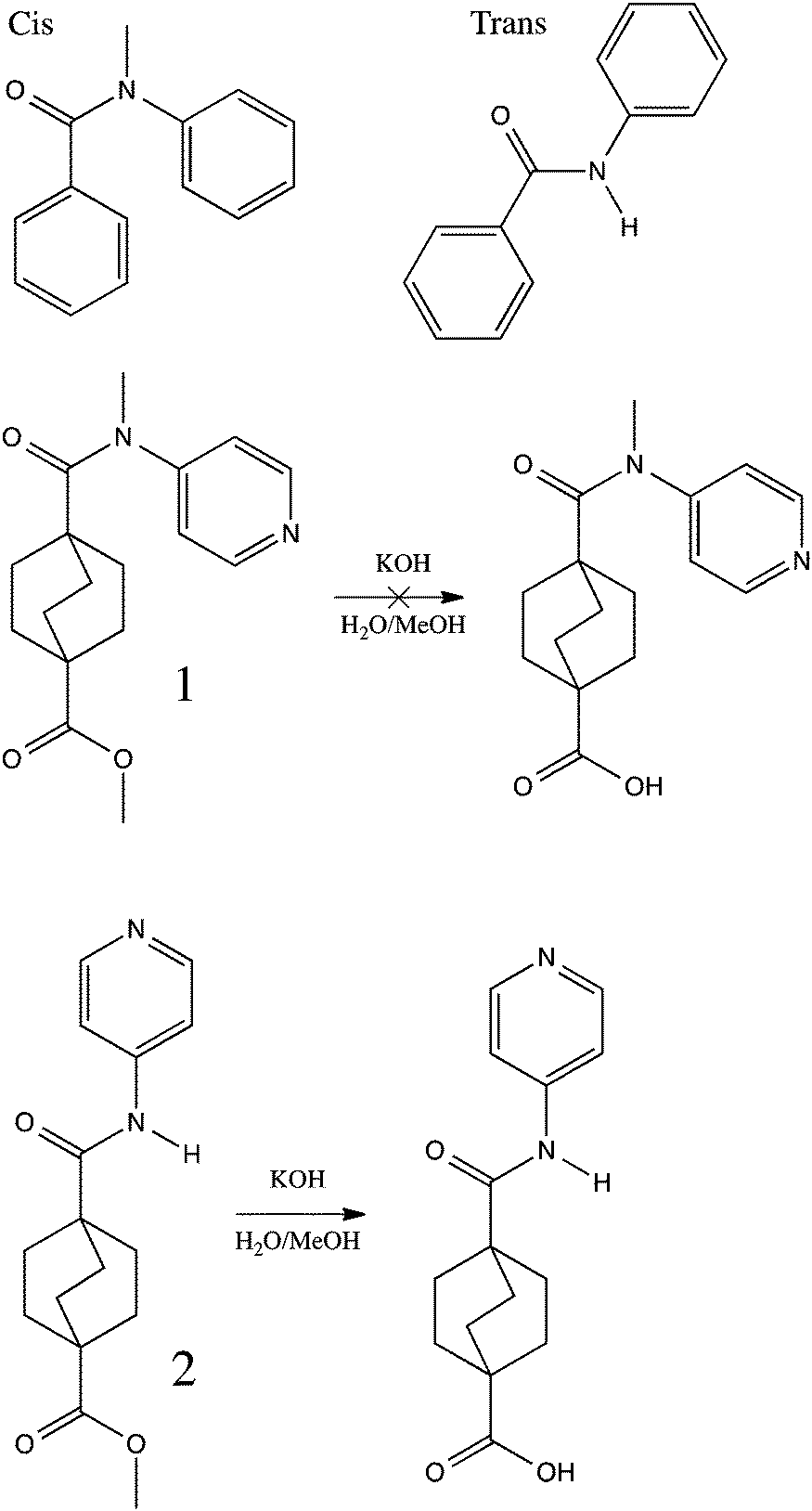 | ||
| Scheme 1 Pictorial definition of cis and trans amide geometries, and illustration of the failed solvolysis of ester 1 and successful solvolysis of ester 2. | ||
2. Experimental
2.1 Synthesis of amides
Amides 1, 2, and 3 were all synthesized using the same protocol, involving the reaction of the appropriate amine with the activated acid chloride prepared from the combination of 4-(methoxycarbonyl)bicyclo[2.2.2]octane-1-carboxylic acid and oxalyl chloride (see ESI† for synthetic details.)2.2 Solvolysis of amides
Solutions (100 mM, 600 μL) of the each of the amides in deuteromethanol were prepared in 1-dram vials, and then transferred to 5 mm NMR tubes. For each, sample, a 1H NMR spectrum (8 scans) was taken at 22 °C. Then the tube was ejected from the spectrometer. A solution of sodium t-butoxide in deuteromethanol (300 mM, 200 μL) was added in one portion. The NMR tube was turned up-side-down and right-side-up 3 times to mix the contents before being inserted into the spectrometer, for 4 successive 1H NMR spectra. The first spectrum was finished approximately 5 minutes after butoxide addition, the next three occurred at 20, 40 and 60 minutes after butoxide addition. This process was repeated for samples of each compound. (See spectra on pages S7–S9, ESI†).3. Results and discussion
3.1 Synthesis and structure of bicyclooctyl amides
Small dyad molecules containing an N-methylated amide linker (1) or a secondary amide linker (2) were synthesized with the goal of unmasking the pendant ester to a carboxylic acid. However, we found that the N-methylated analog was significantly less stable, and the ester could not be cleaved selectively under alkaline or acidic conditions.X-ray diffraction studies (Fig. 1) demonstrated that 2° amide, 2, adopts the expected trans conformation, while the N-methylated analogue, 1, adopts a cis conformation, at least in the solid state, despite the significant steric strain brought on by the interaction of the pyridyl ring with the bicyclic system. This interaction is ameliorated by distortion both of the amide, which adopts a non-planar geometry, and of the bicyclic system, for which the C–C bond connecting the bridgehead and carbonyl is tilted away from the pyridyl ring by almost 10°. The crystal structure of 2 shows no significant distortion in either the amide or bicyclic system.
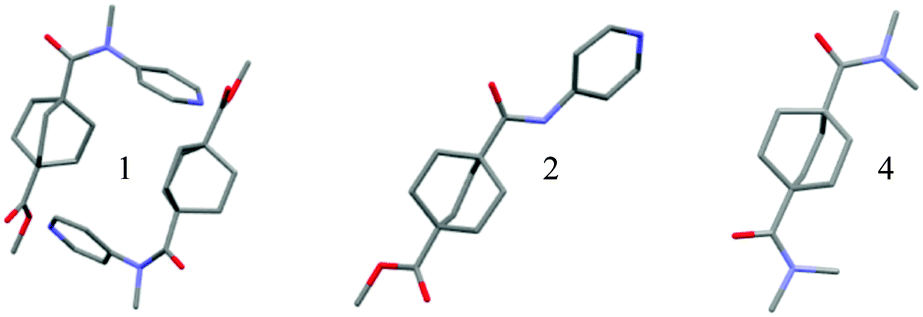 | ||
| Fig. 1 Crystal structures of bicyclooctylcarboxamides. Crystal Structure of 1, 2, and 4, with hydrogen atoms omitted for clarity. The structure of 4 was obtained from the Cambridge Crystallographic Data Center (IDUMUP)23 co-crystallized with hydroquinone (omitted for clarity). | ||
Similarly, the reported crystal structure of N,N,N′,N′-tetramethyl-1,4-bicyclooctyldicarboxamide (4)23 shows no significant distortions, indicating minimal steric repulsion between the bicyclic system and promixal N-methyl group. (Table 1 shows a comparison of bridgehead and amide geometry for compounds 1, 2 and 4.) Additionally, the ester terminus of the bicyclic systems in both 1 and 2 maintain high symmetry.
| Bridgehead bond anglesa | α | |τ|c | ||
|---|---|---|---|---|
| a Bridgehead angles defined as ∠C1C2C3, ∠C1C2C4 and ∠C1C2C5 for amides and ∠C7C10R3, ∠C8C10R3 and ∠C9C10R3 for esters; listed in decreasing order; not same numbering scheme as in ESI. b Torsion angle α = ∠R2R1NC1. c Twist parameter |τ| = |∠C2C1NR2 + ∠OC1NR1|/2. d Compound is co-crystallized with hydroquinone, one amide is engaged in hydrogen bonding (indicated here with –H), and one amide is not. | ||||
| 4 | 113.4(2)°, 112.2(2)°, 110.0(3)° | 178.7(5)° | 4.0° |
|
| 4–Hd | 112.7(3)°, 112.5(2)°, 109.3(3)° | 178.2(4)° | 0.4° | |
| 1–1 Amide | 119.3(3)°, 108.0(3)°, 105.9(3)° | 164.4(5)° | 15.9° | |
| 1–2 Amide | 118.3(3)°, 108.1(3)°, 107.1(3)° | 160.8(4)° | 14.7° | |
| 1–1 Ester | 113.3(3)°, 110.6(3)°, 108.2(3)° | NA | NA | |
| 1–2 Ester | 112.6(3)°, 110.1(3)°, 110.0(3)° | NA | NA | |
| 2 Amide | 111.1(1)°, 111.1(1)°, 109.8(1)° | 173(1)° | 3.1° | |
| 2 Ester | 111.7(1)°, 110.3(1)°, 109.4(1)° | NA | NA | |
The crystal structures of other amides (5–7)24,25 containing the same nitrogen substitution pattern as 1 show the same cis geometry as well as similar distortions of the amide, where the extent of distortion appears to be determined by the steric demands of the carbonyl substituent (Table 2). The amides containing ferrocenyl (5) and bicyclooctyl (1) groups are the most twisted of the series. The amide geometries of 1 and 5–7 are only slightly pyramidalized, with the sum of bond angles about the nitrogen is within 3° of 360°, in the case of 1, and within 1° for the rest, indicating near planarity. While slightly more distorted than amide 8,26 which has a bond angle sum of 359.2°, 1 is significantly less pyramidal than amides 926 and 10a–c,16,27 with bond angle sums as low as 347°, indicating a significantly greater degree of sp3 character (328.4° would be the ideal sum for a perfect tetrahedral center).26 Comparison of the χN values also indicates only a modest deviation from planarity in amide 1, as compared to amides 9 and 10a–c, but still significantly pyramidalized compared to amides 2–8.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Amide | CSD code | ω 1 (°) | ω 2 (°) | ω 3 (°) | ω 4 (°) | |τ| (°) | |χC| (°) | |χN| (°) | a + b + c (°) |
| a Two chemically identical, but crystallographically distinct structures appear in the unit cell. b Compound is co-crystallized with hydroquinone, one amide is engaged in hydrogen bonding (indicated here with –H), and one amide is not. c Both cis and trans geometries appear in the same molecule. d Both cis and trans conformers appear in the same unit cell. | |||||||||
| 1-1 | 1423262 | −27.7 | −4.0 | 170.8 | 157.6 | 15.9 | 5.3 | 18.5 | 357.8 |
| 1–2 | 1423262 | −27.3 | −2.0 | 174.5 | 156.2 | 14.7 | 3.5 | 21.8 | 356.7 |
| 2 | 1423261 | 7.5 | −1.3 | 179.8 | −173.6 | 3.1 | 1.1 | 7.3 | 359.6 |
| 4 | IDUMUP23 | 4.8 | 3.4 | −176.8 | −175.1 | 4.1 | 0.1 | 1.6 | 360.0 |
| 4-Hb | IDUMUP23 | 1.2 | −2.0 | 179.1 | 179.9 | 0.4 | 1.1 | 0.3 | 360.0 |
| 5 | PEZXAV24 | 20.5 | 9.3 | −169.0 | −161.2 | 14.9 | 1.7 | 9.5 | 359.4 |
| 6 | REWHUX25 | 17.7 | 7.7 | −168.4 | −166.2 | 12.7 | 3.9 | 6.1 | 359.7 |
| cis-7c | REWJIN25 | 19.2 | 10.2 | 170.5 | 164.5 | 14.7 | 3.7 | 9.7 | 359.6 |
| trans-7c | REWJIN25 | −19.1 | −5.9 | −168.5 | −162.1 | 12.5 | 1.2 | 7.6 | 359.3 |
| 8 | PETYIW26 | −0.7 | −13.3 | NA | NA | 7.0 | NA | NA | 359.2 |
| 9-1a | PEVDUP26 | 38.7 | −12.7 | NA | NA | 13.0 | NA | NA | 347.1 |
| 9-2a | PEVDUP26 | 29.1 | −15.4 | NA | NA | 6.9 | NA | NA | 350.1 |
| 10a-1a | RIRLUZ16 | 32.1 | −2.4 | −179.8 | −150.4 | 14.9 | 2.5 | 31.9 | 351.9 |
| 10a-2a | RIRLUZ16 | 33.5 | −0.6 | −178.7 | −148.4 | 16.8 | 1.9 | 32.2 | 352.3 |
| trans-10bd | SUQBEL16 | −7.1 | −17.6 | 160.7 | −171.2 | 5.3 | 1.7 | 12.2 | 354.4 |
| cis-10bd | SUQBEL16 | 13.5 | −6.0 | 174.2 | −166.8 | 3.7 | 0.3 | 19.3 | 357.0 |
| 10c | YITQEX27 | −11.1 | 11.3 | −167.9 | 168.0 | 0.1 | 0.9 | 23.2 | 355.7 |
| 11 | WUPBEO28 | 45.7 | 22.5 | −151.3 | −140.5 | 34.1 | 6.2 | 17.0 | 357.8 |

Instead of pyramidalization, distortion of the amide linkages in 1 and 5–7 manifests as a twist of the carbonyl–nitrogen bond. The amide in 1 is twisted from planarity by 15.9°, placing it among the most twisted 5% of the 41575 reported acyclic (non-lactam) amide structures in the CCDC (Fig. 2).28–30 (Mean 4.94° Std. Dev. 4.70°) There are examples of much more twisted outliers, such as 11, at 34.1°!31 This population excludes bicyclic lactams, which can achieve much more significant twist angles, as demonstrated by Kirby et al., who reported an example of the extreme case in which a completely orthogonal geometry is enforced by the bicycle.32
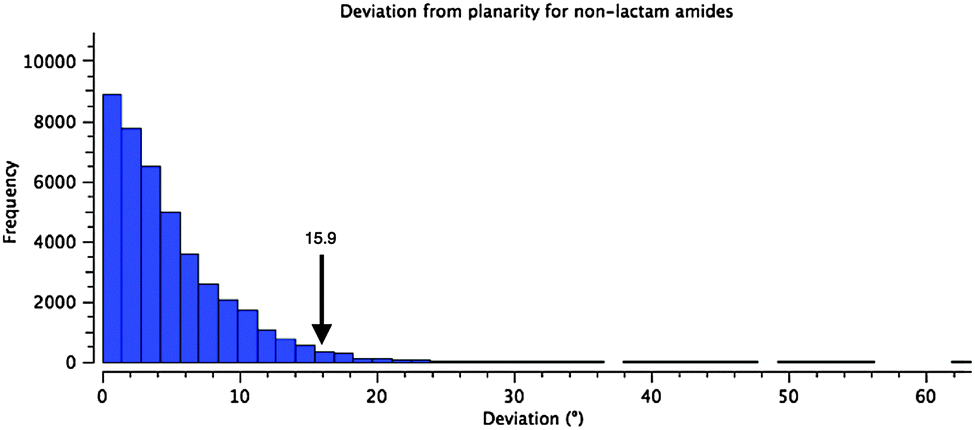 | ||
| Fig. 2 Histogram showing the distribution of torsional angles in 41575 reported acyclic amide structures in the CCDC (R < 0.1). The arrow indicates the value found for 1. | ||
There is no clear correlation relating the twist angles, and pyramidalizations among the examples in Table 2, nor is there a striking trend in the 13C chemical shifts or IR stretching frequencies of the carbonyl groups in amides 1–3. However, the extent of the difference in solvolytic lability between amide 1 and amides 2 and 3 indicates a substantial difference in the character of the amide group.
3.2 Solvolysis studies
Since solvolytic rate enhancement is observed for many distorted amides,4–6,31–34 this distortion is most likely responsible for the amide linkage in 1 being cleaved competitively with the methyl ester. To test this, 100 mM solutions of 1, 2, and the N,N-dimethylamide analogue (3) in deuteromethanol, were subjected to sodium t-butoxide at 22 °C in an NMR spectrometer, and several successive 1H NMR spectra were taken.In all three cases, the methyl ester was >80% trans-esterified by the one-hour mark, providing the deuteromethyl ester and free methanol. In the case of 1, solvolysis of the amide also occurred at nearly the same rate. No amide solvolysis was observed for either 2 or 3, indicating that the lability of 1 is not due to either the 4-pyridyl substituent or the 3° amide alone. (Data in ESI†.) In addition to base-mediated solvolysis, the amide group in 1 was found to be labile under mildly acidic conditions. Exposure to a saturated aqueous solution of ammonium chloride was sufficient to decompose 1, while 2 and 3 were robust under these conditions.
These results are consistent with a destabilization of the ground state of the strained amide, as proposed by Mujika et al.33 With no protons alpha to the carbonyl, it is unlikely that the solvolysis proceeds by formation of a ketene, as observed by Hutchby et al., who reported pH-neutral methanolysis of enolizable, strained tertiary amides possessing electron-withdrawing alpha-substituents.34
The precise mechanism for the base-mediated methanolysis is not easy to specify since it involves several explicit solvent molecules, but it is likely that nucleophilic attack to form the tetrahedral intermediate is rate limiting.35 The twisted geometry of 1 increases electrophilic character of the carbonyl carbon, reducing the activation energy for nucleophilic attack, while relief of steric strain is expected to provide additional driving force for both the conversion from the amide to the tetrahedral intermediate and the cleavage of the C–N bond.
3.3 Quantum chemistry calculations
Gas-phase ab initio calculations, using both B3LYP and ωB97XD functionals with a 631+G(2d,p) basis set, were used to optimize the geometry and calculate the heats of formation of both trans and cis conformations of amides 1 and 2, as well as of two truncated model structures (12 and 13). High performance at a low computational cost has made Kohn–Sham density functional theory36,37 (KS-DFT) the most widely used computational method. However, the exchange correlation energy functional Exc, which is a key component of KS-DFT still needs to be approximated. Both B3LYP & ωB97XD are hybrid functionals that incorporate Hartree–Fock (HF) exchange term in the functional form to reduce the self-interaction error, leading to more accurate energy calculations.The calculations indicate a clear thermodynamic preference for the trans conformer in both 2° amides, and for the cis conformer in both methylated amides. In all cases the relative free energy values calculated using both B3LYP and ωB97XD functionals agree on which conformer is preferred. However, the magnitude of the calculated relative free energies differ by a significant margin in the case of the full molecules. In these cases, values from ωB97XD calculations are expected to be more accurate, because ωB97XD includes empirical dispersion terms and features range separation (22% HF exchange in short range & 100% HF exchange in long range compared to B3LYP which has a 20% HF exchange throughout). Interestingly, ΔG values calculated using ωB97XD for the conformers of the methylated amides, 1 and 12, are very similar (3.87 and 2.53 kcal mol−1), whereas the two 2° amides (2 and 13) have ΔG values that differ by almost 5 kcal mol−1 depending on the method chosen.
Calculation of the dipole moments of each of these molecules provides some understanding of one of the contributing factors of the electronic preference. For each of the four amides analysed (1, 2, 12, and 13), the cis conformers have significantly smaller dipole moments than the trans conformer. The 3° amides adopt the less polar geometry, while the 2° amides adopt the more polar geometry. The difference in overall dipole moment calculated for cis and trans conformers of 1 (1.39 D) is significantly less than the change in dipole moment between the two conformers of 2 (−4.06 D). In contrast, the truncated amides, 12 and 13 show the opposite trend, the difference in dipole moments between the two conformers of 12 (4.33 D) being significantly larger than the difference between the conformers of 13 (1.39 D).
Analysis of the molecular orbital energies and topologies of the conformers of the amides shown in Scheme 2 indicates that the HOMO−1 may play an important role in the difference between the conformers of amides 1 and 2, while the analogous orbitals in amides 12 and 13 is the HOMO (Scheme 3). It appears that the topologies of the HOMOs in the cis and trans isomers of 13 are essentially identical, while the HOMOs of the cis and trans isomers of 12 differ substantially in the interaction between the methyl group and the arene. We attribute this difference to the fact that a hydrogen substituent does not have any occupied p orbitals, while the methyl group has several occupied orbitals with p character. The HOMO of trans-12 shows a nodal plane bisecting the N-methyl bond, while the HOMO of cis-12 shows continuity of the wavefunction across the N-methyl bond and the N-aryl bond.
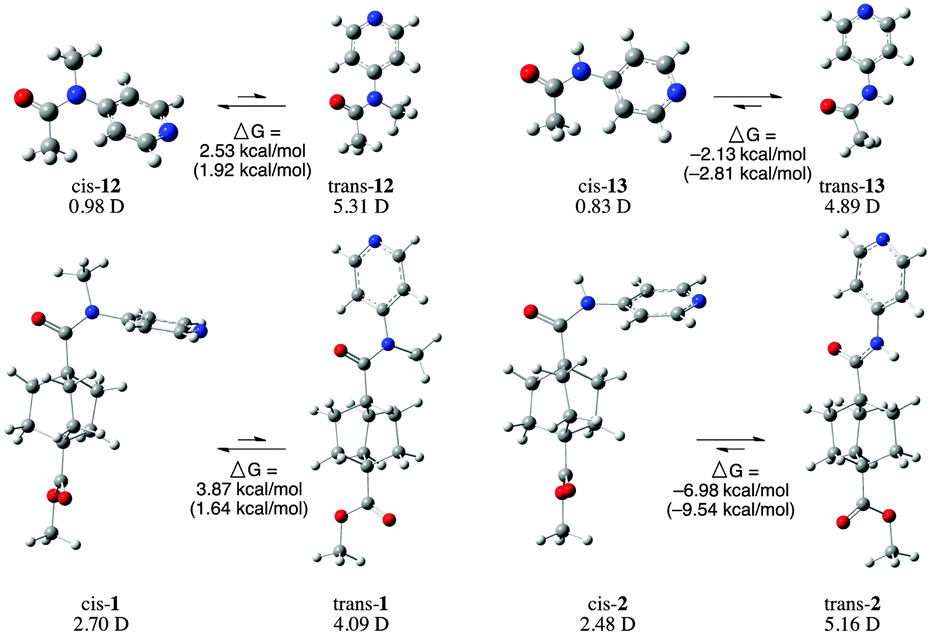 | ||
| Scheme 2 Optimized structures of cis and trans conformers of 1, 2, and truncated forms 12 and 13. (ΔG values calculated with ωB97XD functional shown without parentheses, and those calculated with B3LYP shown in parentheses.) Dipole moments calculated with B3LYP shown under each structure. | ||
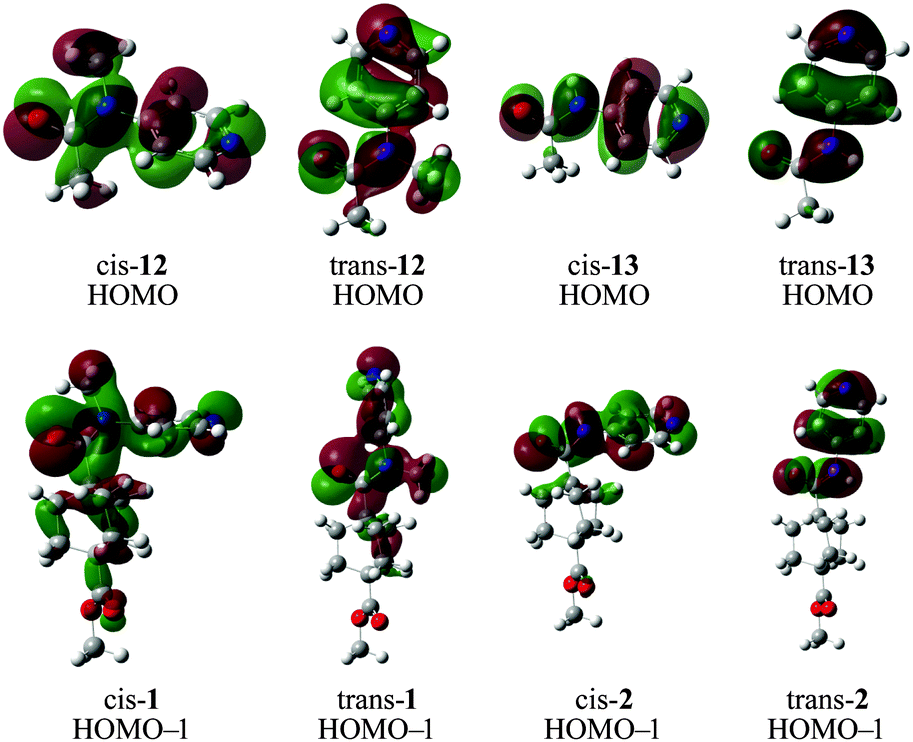 | ||
| Scheme 3 Optimized structures of cis and trans conformers of 1, 2, and truncated forms 12 and 13, with superimposed visualizations of the HOMO−1 molecular orbital, calculated using B3LYP. | ||
This trend is also evident in the HOMO−1 orbitals in the conformers of 1 and 2. The HOMO−1 orbitals of both cis- and trans-2 are quite similar (although there is a slight twist in cis-2, which reduces the similarity somewhat), while the analogous orbitals in cis- and trans-1 possess substantially different topologies. The HOMO−1 orbital in cis-1 has a continuous lobe across the carbonyl, nitrogen, methyl- and aryl-substituents, while the HOMO−1 in trans-1 appears more localized.
Unfortunately, the energies of these orbitals do not appear to make a dramatic contribution to the energies of the conformers. The HOMOs of cis-12 and cis-13 are 2.636 kcal mol−1 and 2.108 kcal mol−1 (respectively) lower in energy than the HOMOs of their corresponding trans conformers, indicating a very modest contribution to the conformational preference. However, the HOMO−1 orbitals of cis-1 and cis-2 are 1.939 kcal mol−1 and 7.072 kcal mol−1 lower in energy than their corresponding trans isomers, indicating an opposing contribution to the observed effect. However, the sum of the energies of all occupied MOs in cis-1 is lower than the same sum in trans-1, while the opposite trend holds for 2.
3.4 A proposed explanation of the conformational preference
A1,3 strain between the aryl and methyl substituents is arguably the factor that tips the aryl system out of conjugation with the amide. The resulting molecular orbitals are substantially higher in energy when the amide is in the trans conformation than in the cis conformation.A survey of over 1000 3° N-alkyl-N-aryl amide crystal structures in the CCDC supports this notion (Fig. 3).38 These structures can be divided into four general classes based on their geometry: Class I (orthogonal-cis), Class II (coplanar-trans), Class III (orthogonal-trans), and Class IV (coplanar-cis). Most of these 3° amide structures fall into Class I. All examples in which the aryl substituent is within 20° of coplanarity with the amide system also adopt the trans conformation, falling into Class II rather than Class IV, which is conspicuously empty, not even populated by formamides, for which no significant steric effects would disfavour the cis-planar geometry. In most examples of Class II structures, a small ring connects the alkyl and aryl substituents, enforcing the planar geometry.
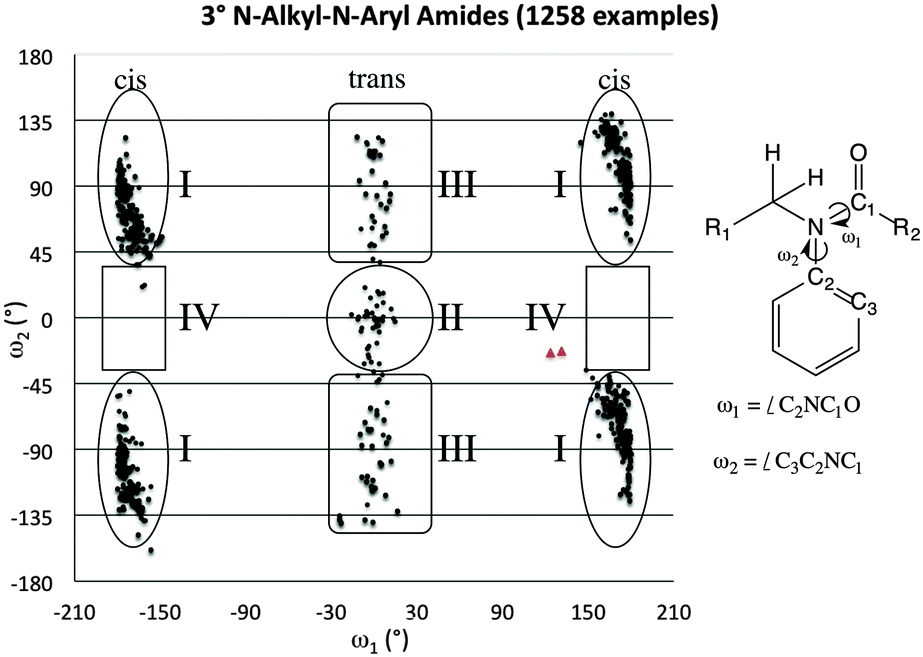 | ||
| Fig. 3 Scotterplot showing the distribution of two torsional angles in 1258 reported acyclic amide structures in the CCDC. The red triangles indicate the values found for 1. | ||
For almost all examples of Class III 3° amides, the geometry can be attributed to extreme steric repulsion between the arene and the fragment bonded to the carbonyl, as in the case of trans-N-methyl-2,2′,6,6′-tetramethylbenzanilide (YEGJEY).39 Other 3° amides of Class III appear to be heavily influenced by π-stacking interactions, as is the case for trans-N-(2,6-dimethylphenyl)-N-methylpentafluorobenzamide (DIBGAX).40
A survey of N-aryl 2° amides (Fig. 4) indicates that over 80% fall in to Class II, while just under 20% are in Class III.41 Limiting the scope to N-(2,6-disubstituted-aryl) 2° aryl amides42 focuses in on 2° amides with substantial A1,3 interactions that force an orthogonal orientation of the aromatic substituent (Fig. 5). Unlike the distribution of 3° amides analysed in Fig. 3, there is a strong preference for Class III over Class I geometries. The only Class I 2° amide appearing in this survey (14, POXWIJ, Fig. 6)43 represents an extreme case. Also, although there are no significant hydrogen-bonding interactions to be seen in the crystal structure, the large number of polar bonds present might affect the geometry electrostatically. The few examples of co-planar Class II amides in this survey appear to be enforced by a hydrogen-bonding interaction between the oxygen of the amide and a hydrogen bond donor ortho substituent such as a phenol (e.g., YISDEK).44
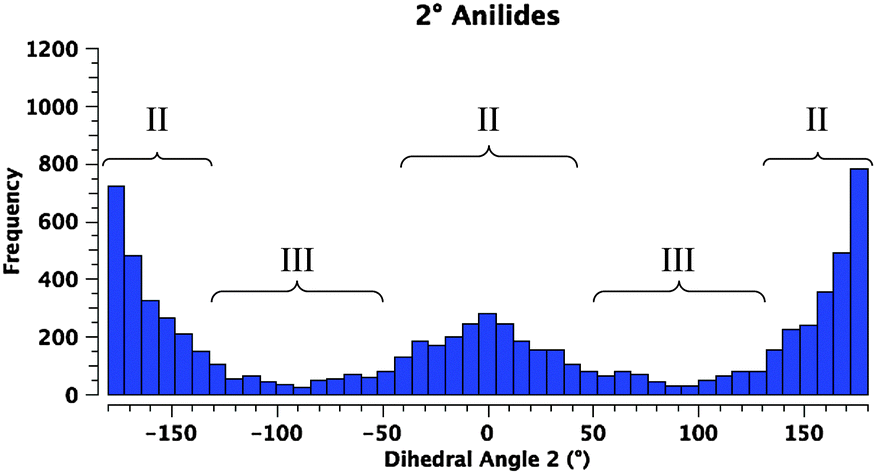 | ||
| Fig. 4 Histogram showing the distribution of torsional angles in 41575 reported acyclic 2° N-aryl amide structures in the CCDC. | ||
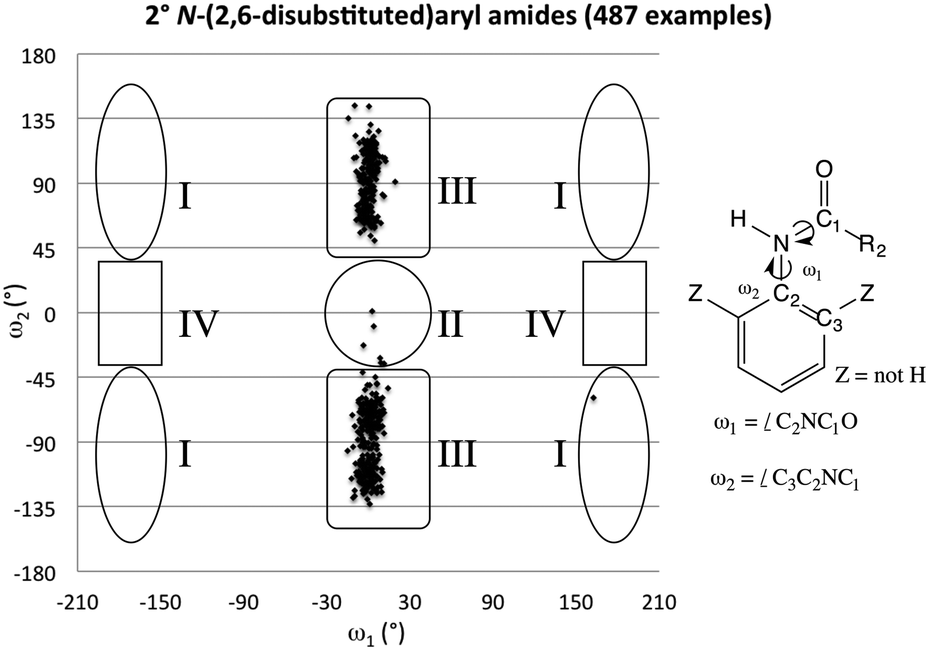 | ||
| Fig. 5 Scotterplot showing the distribution of two torsional angles in 487 reported acyclic 2° N-(2,6-disubstituted)aryl amide structures in the CCDC. | ||
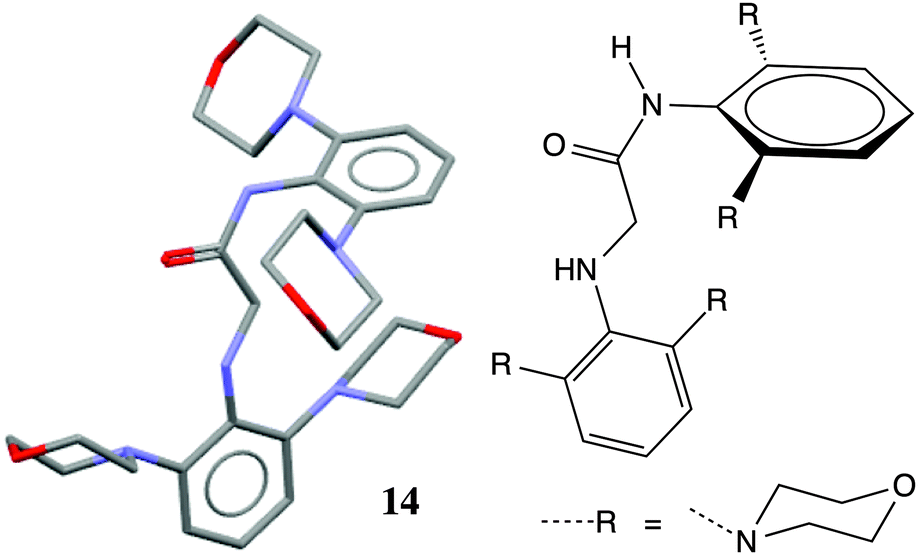 | ||
| Fig. 6 Crystal structure of amide 14 with hydrogen atoms omitted for clarity. | ||
Enforcing a coplanar arrangement of the amide and aryl substituent in 3° N-alkyl-N-aryl amides thus seems to be sufficient to favor a Class II geometry, unless overcome by extreme steric repulsion or stacking interactions. If the steric repulsion is not quite enough to favor Class III However, even when forced out of plane, 2° amides are not predisposed to adopt a Class I geometry.
Altogether, it appears that the N-alkyl substituent in N-alkyl-N-aryl amides leads to a cis geometry because of two factors. A1,3 interactions push the N-aryl substituent out of the plane of the amide, and then the sp3 orbitals facilitate mixing of orbitals on both sides of the nitrogen in the amide. This mixing only occurs when the arene is orthogonal to the amide, and is not possible for 2° amides, whose H atoms do not provide the necessary orbitals.
4. Conclusions
The increased lability of 3° amide 1 with respect to its demethylated 2° amide analog (2) is a result of the difference in conformation of two amides. The cis geometry adopted by 1 induces a substantial 15° twist of the amide bond, a very slight pyramidalization of the nitrogen atom, and a distortion of the bicyclooctyl system, which together cause the amide to be as susceptible to base-mediated methanolysis as the remote methyl ester group in the molecule.Surveys of thousands of amide structures registered in the CCDC indicated that 1 is among the top 5% most twisted amides (excluding lactams), and constitutes an outlier among Class I amide geometries (as defined above).
Analysis of these amide structures indicates that for N-alkyl-N-aryl amides in which the aryl substituent is orthogonal to the amide system, the cis geometry is strongly favored (I ≫ IV). However, in cases for which the aryl substituent is coplanar with the amide, the trans geometry is weakly favored, but easily overcome by steric effects or hydrogen bonding (II ≥ III). The trans geometry is overwhelmingly favored in all cases of 2° N-aryl amides. Even when the aryl substituent is forced out of plane, 2° amides retain this trans geometry, adopting a Class III geometry rather than Class I, in all but one reported case.
Our comparison of the molecular orbitals of 1, 2, 12, and 13 indicates that the contribution of the sp3 orbitals on the N-methyl substituent dramatically alters the topology of high-lying occupied molecular orbitals, as compared to the analogous orbitals of the analogous 2° amides. The orbitals most affected by the substitution also appear to be highly sensitive to the cis/trans conformation of the amide, suggesting a possible relationship. However, analysis of the orbital energies indicates that this is not a complete explanation of the phenomenon.
Despite prior reports indicating a preference for a cis geometry in N-alkyl-N-aryl amides, the magnitude of the influence of the methyl substituent on amides 1 and 2 was not anticipated. Future synthetic endeavors will have to take this effect into account. Perhaps future researchers may take advantage of this lability, and use a bulky 3° amide as a masked carboxylic acid, or a temporary linkage between two detachable species.
Acknowledgements
The authors would like to thank Professors Gary Brudvig and Charles Schmuttenmaer for productive conversations. This work was funded by a generous donation from the TomKat Charitable Trust, and also supported as part of the Argonne-Northwestern Solar Energy Research (ANSER) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Numbers DE-PS02-08ER15944 and DE-SC0001059 (V. S. B. and R. H. C.). V. S. B. acknowledges high performance computing time from NERC and from the computational work was supported by the facilities and staff of the Yale University Faculty of Arts and Sciences High Performance Computing Center and by the National Science Foundation under grant number CNS08-21132 that partially funded acquisition of these facilities.Notes and references
- T. Yamana, Y. Mizukami, A. Tsuji, Y. Yasuda and K. Masuda, Chem. Pharm. Bull., 1972, 20, 881 CrossRef CAS.
- A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- A. J. Bennet, Q.-P. Wang, H. Slebocka-Tilk, V. Somayaji and R. S. Brown, J. Am. Chem. Soc., 1990, 112, 6383 CrossRef CAS.
- K. E. Laidig and R. F. W. Bader, J. Am. Chem. Soc., 1991, 113, 6312 CrossRef CAS.
- X. Lopez, J. I. Mujika, G. M. Blackburn and M. Karplus, J. Phys. Chem. A, 2003, 107, 2304 CrossRef CAS.
- A. Romanelli, A. Shekhtman, D. Cowburn and T. W. Muir, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6397 CrossRef CAS PubMed.
- M. Schuman, X. Lopez, M. Karplus and V. Gouverneur, Tetrahedron, 2001, 57, 10299 CrossRef CAS.
- V. Somayaji and R. S. Brown, J. Org. Chem., 1986, 51, 2676 CrossRef CAS.
- Q. Wang, A. J. Bennet and R. S. Brown, Can. J. Chem., 1990, 68, 1732 CrossRef CAS.
- Q.-P. Wang, A. J. Bennet, R. S. Brown and B. D. Santarsiero, J. Am. Chem. Soc., 1991, 113, 5757 CrossRef CAS.
- K. Tani and B. M. Stoltz, Nature, 2006, 441, 731 CrossRef CAS PubMed.
- M. Szostack and J. Aubé, Org. Biomol. Chem., 2011, 9, 27 Search PubMed.
- R. Szostak, J. Aubé and M. Szostak, J. Org. Chem., 2015, 80, 7095 CrossRef PubMed.
- G. Yamamoto, H. Murakami, N. Tsubai and Y. Mazaki, Chem. Lett., 1997, 605 CrossRef CAS.
- G. Yamamoto, N. Tsubai, H. Murakami and Y. Mazaki, Chem. Lett., 1997, 1295 CrossRef CAS.
- H. B. Thompson and K. M. Hallberg, J. Phys. Chem., 1963, 67, 2486 CrossRef.
- B. F. Pedersen, Acta Chem. Scand., 1967, 21, 1415 CrossRef CAS.
- A. Itai, Y. Toriumi, N. Tomioka, H. Kagechika, I. Azumaya and K. Shudo, Tetrahedron Lett., 1989, 30, 6177 CrossRef CAS.
- I. Azumaya, H. Kagechika, Y. Fujiwara, M. Itoh, K. Yamaguchi and K. Shudo, J. Am. Chem. Soc., 1991, 113, 2833 CrossRef CAS.
- S. Saito, Y. Toriumi, N. Tomioka and A. Itai, J. Org. Chem., 1995, 60, 4715 CrossRef CAS.
- R. Yamasaki, A. Tanatani, I. Azumaya, S. Saito, K. Yamaguchi and H. Kagechika, Org. Lett., 2003, 5, 1265 CrossRef CAS PubMed.
- B. M. Foxman, D. J. Guarrera, R. Pai, C. Tassa and J. C. Warner, Cryst. Eng., 1999, 2, 55 CrossRef CAS.
- B. Kashyap and P. Phukan, RSC Adv., 2013, 3, 15327 RSC.
- I. Okamoto, M. Terashima, H. Masu, M. Nabeta, K. Ono, N. Morita, K. Katagiri, I. Azumaya and O. Tamura, Tetrahedron, 2011, 67, 8536 CrossRef CAS.
- Y. Otani, O. Nagae, Y. Naruse, S. Inagaki, M. Ohno, K. Yamaguchi, G. Yamamoto, M. Uchiyama and T. Ohwada, J. Am. Chem. Soc., 2003, 125, 15191 CrossRef CAS PubMed.
- G. Yamamoto, F. Nakajo and Y. Mazaki, Bull. Chem. Soc. Jpn., 2001, 74, 1973 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- A search of the CCDC for 3° amides in which the carbonyl C–N bond is not included in a ring (non-lactam), and excluding structures containing more than 3 atoms bonded to the nitrogen of the amide and structures with other heteroatoms on the carbons bonded to the amide nitrogen. Structure UBUMAH was thrown out of the results due to poor refinement (R > 2) and unreasonably high deviation (88°) for dimethylformamide solvent included in unit cell of a coordination compound.
- J. Clayden, Y. J. Y. Foricher and H. K. Lam, Eur. J. Org. Chem., 2002, 3558 CrossRef CAS.
- S. A. Glover and A. A. Rosser, J. Org. Chem., 2012, 77, 5492 CrossRef CAS PubMed.
- A. J. Kirby, I. V. Komarov and N. Feeder, J. Chem. Soc., Perkin Trans. 2, 2001, 522 RSC.
- J. I. Mujika, J. M. Mercero and X. Lopez, J. Am. Chem. Soc., 2005, 127, 4445 CrossRef CAS PubMed.
- M. Hutchby, C. E. Houlden, M. F. Haddow, S. N. G. Tyler, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2012, 51, 548 CrossRef CAS PubMed.
- Y. Xiong and C.-G. Zhan, J. Phys. Chem. A, 2006, 110, 12644 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- A search of the CCDC for 3° amides in which the carbonyl C–N bond is not included in a cycle (non-lactam), the amide nitrogen is bonded to a phenyl ring and an aliphatic carbon that has at least two hydrogen atoms on it.
- I. Azumaya, K. Yamaguchi, H. Kagechika, S. Saito, A. Itai and K. Shudo, Yakugaku Zasshi, 1994, 114, 414 CAS.
- S. L. Cockroft, J. Perkins, C. Zonta, H. Adams, S. E. Spey, C. M. R. Low, J. G. Vinter, K. R. Lawson, C. J. Urch and C. A. Hunter, Org. Biomol. Chem., 2007, 5, 1062 CAS.
- A search of the CCDC for 2° amides in which the carbonyl C–N bond is not included in a cycle (non-lactam), the amide nitrogen is bonded to a phenyl ring. Structure XUHWON was thrown out because the amide is coordinated to a Lewis acid.
- A search of the CCDC for 2° amides in which the carbonyl C–N bond is not included in a cycle (non-lactam), the amide nitrogen is bonded to a phenyl ring which contains non-hydrogen substituents at the 2 and 6 positions. Structure XUHWON was thrown out because the amide is coordinated to a Lewis acid.
- Y. Irie, Y. Koga, T. Matsumoto and K. Matsubara, Eur. J. Org. Chem., 2009, 2243 CrossRef CAS.
- K. C. Nicolaou, Y. Tang, J. Wang, A. F. Stepan, A. Li and A. Montero, J. Am. Chem. Soc., 2007, 129, 14850 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis, solvolysis and X-ray crystallographic studies as well as spectroscopic data and computational details. CCDC 1423261 and 1423262. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj02449h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |