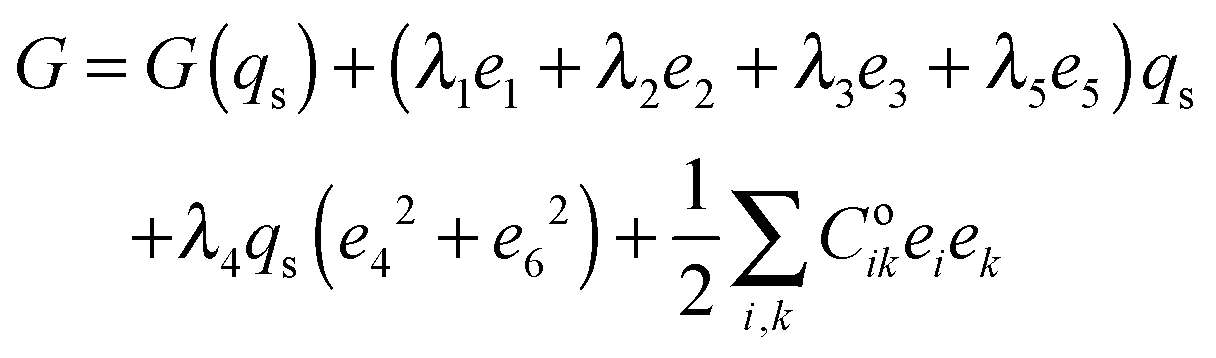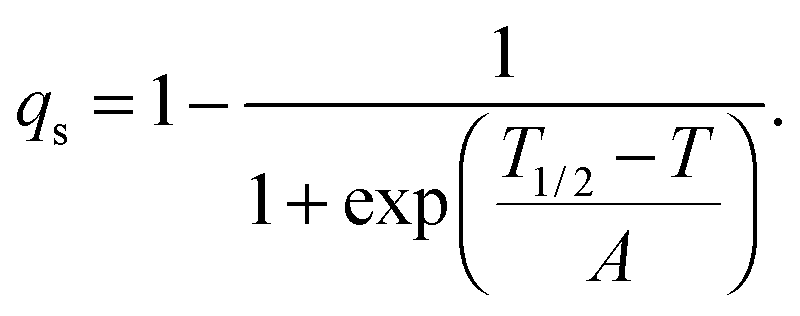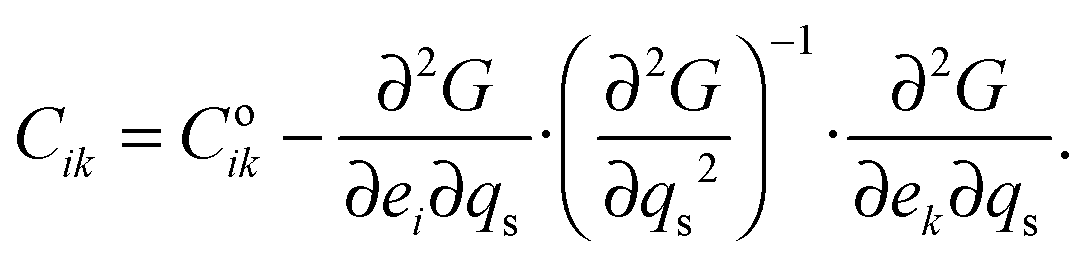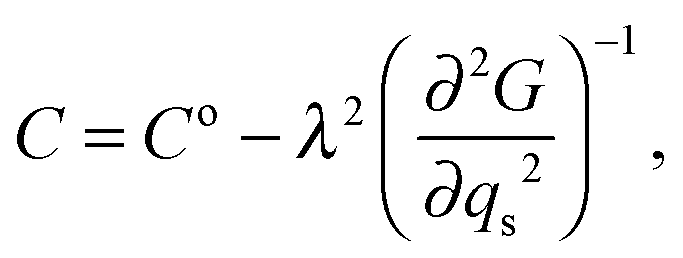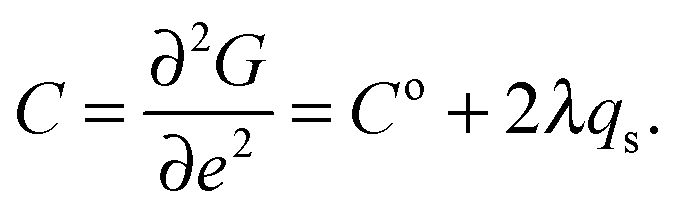DOI:
10.1039/C5NJ02359A
(Paper)
New J. Chem., 2016,
40, 2466-2478
Structural and spectroscopic characterisation of the spin crossover in [Fe(abpt)2(NCS)2] polymorph A†
Received
(in Victoria, Australia)
21st September 2015
, Accepted 8th January 2016
First published on 8th January 2016
Abstract
A crystallographic and solid state spectroscopic study of the spin crossover behaviour of [Fe(abpt)2(NCS)2] (abpt = 4-amino-3,5-bis(pyridin-2-yl)-1,2,4-triazole) polymorph A is reported. Structural features including crystallographic cell parameters, bond lengths and distortion parameters are monitored between 375 K and 30 K and crystal structures are reported at seven temperatures across the spin transition. In addition, the light induced excited spin state trapping (LIESST) metastable high spin structure, HS*, is reported at 30 K by continuous irradiation with a 670 nm, 5 mW CW laser during the data collection. Relaxation of the HS* state at 30 K with the laser switched off is found to occur within ∼4000 s in accordance with the literature. High pressure single crystal datasets are also reported to examine the effect of pressure on the spin transition. Single crystal variable temperature UV-Vis spectroscopy and resonant ultrasound spectroscopy support the crystallographic evidence relating to the spin crossover transition presented herein. Strain analysis of the lattice parameters yields the temperature dependence of the spin order parameter, indicating strong spin–lattice coupling to give a volume strain of up to ∼4% and a shear strain of up to ∼1.5%. These, in turn, are responsible for changes in elastic constants by up to ∼35%.
Introduction
Spin crossover is a reversible spin state transition of a metal centre which can occur upon change in temperature, pressure,1 magnetic field or through light irradiation.2 Spin crossover is a relatively common phenomenon for octahedral 3d4–3d7 metal centres and has been extensively studied in a large range of systems.3 A significant proportion of the spin crossover compounds that have been studied are octahedral Fe(II) complexes with nitrogen donor ligands,3 although spin crossover is also commonly observed in six-coordinate Fe(III)4 or Co(II) complexes.5,6 A number of potential applications have been suggested for spin crossover complexes, e.g. molecular switches, data storage7,8 and liquid crystals. Spin crossover is a very complex process which can occur in both solution and the solid state and involves the switching of magnetic, optical and structural properties. In solution there is no cooperativity as molecules are far apart and transitions tend to be gradual. However, in the solid state the potential for cooperativity results in a wide range of different behaviours, from abrupt transitions with hysteresis (strong cooperativity) to gradual transitions with no hysteresis (weak or no cooperativity), and can include single or multistep spin transition9 processes. Factors such as inclusion of solvent in the lattice, intermolecular interactions and counterion type are all known to influence the spin transition behaviour. Given the complex nature of the process it is particularly interesting to study compounds with more than one polymorph and which show different spin crossover behaviour.10 Comparing the structures and spin crossover processes in such species can provide significant insight into the phenomenon.
Four polymorphs of [Fe(abpt)2(NCS)2] (abpt = 4-amino-3,5-bis(pyridin-2-yl)-1,2,4-triazole) (A–D) have been reported previously. Three of these, A,11C12,13 and D,12,14 have been shown to undergo temperature induced spin crossover of at least one of the Fe(II) centres in the asymmetric unit from high spin (HS) to low spin (LS) upon cooling. On the other hand, polymorph B does not undergo a thermal spin transition at ambient pressure, but has been shown to undergo thermal spin crossover at pressures >4.4 kbar.15 The room temperature crystal structures of the three polymorphs which undergo temperature induced spin crossover have been established and while A has one independent Fe(II) centre in the asymmetric unit, C and D both contain two independent Fe(II) centres. The first magnetic susceptibility measurements on a single crystal sample of polymorph A indicated an incomplete spin transition from HS to LS with ∼23% of the molecules remaining HS in the ‘LS’ structure.11 A more recent publication using a different sample preparation method has shown that this polymorph can undergo a virtually complete spin transition upon cooling.12 In polymorphs C and D only one Fe centre undergoes a spin transition and the other remains HS upon cooling.12 All three polymorphs displaying thermal spin crossover behaviour also show light induced excited spin state trapping (LIESST) at low temperature. For C, T1/2 = 86 K and upon low temperature photoexcitation it forms a commensurate modulated LIESST state with four crystallographically independent Fe centres.13 For DT1/2 = 162 K and, in addition to displaying LIESST, the compound also displays photoinduced linkage isomerism with the NCS ligand changing from N to S bound.14 For A, which has a thermal spin crossover that occurs without hysteresis, T1/2 was initially reported as 180 K,11 but has been remeasured as T1/2 = 188 K12 for single crystals prepared using a different method. TLIESST = 40 K and up to approximately 30 K a significant fraction of the HS* state can be maintained for >1000 s; as the temperature is increased above 30 K this fraction decreases rapidly. Although the thermally induced spin crossover shows no hysteresis, a light induced thermal hysteresis (LITH)16 associated with the HS* state has been identified from the photomagnetic data.11 LS and HS* state structures for A have not been characterised previously and these are reported herein alongside a detailed crystallographic study of structural changes occurring during the spin transition and examination of the HS* relaxation behaviour of this polymorph. The results are supported by single crystal variable temperature UV-Vis spectroscopy and resonant ultrasound spectroscopy (RUS). An analysis of changes in the lattice parameters reveals strong coupling of both volume and shear strains with the spin state order parameter which, in turn, gives rise to large anomalies in elastic properties.
Experimental
Synthesis
Synthesis of [Fe(abpt)2(NCS)2] was carried out using a slow diffusion method which has been previously reported to produce crystals of polymorph A which undergo a virtually complete HS to LS transition upon cooling.12 Precursor materials were used without further purification, solvents degassed and all manipulations carried out under a nitrogen atmosphere. FeSO4·7H2O (1 mmol, 0.278 g) and KNCS (2 mmol, 0.194 g) were stirred in MeOH (10 ml) for 15 min. Pale yellow insoluble K2SO4 precipitate was removed by filtration and deionised H2O (10 ml) added to the remaining clear solution. abpt ligand (2 mmol, 0.477 g) was dissolved in MeOH (20 ml) and transferred to a narrow (<5 cm) Schlenk tube. The Fe2+/2(NCS)− solution was carefully injected underneath the abpt solution to form a lower layer. A coloured band containing the target complex immediately formed at the interface between the two layers. Within 1–4 weeks single crystals of A, B and D suitable for X-ray diffraction studies were formed and separated under the microscope.12 Crystals of A were also formed using a methanol–chloroform solution which favours the formation of this polymorph,15 however the quality of the crystals formed was lower than that obtained from methanol–water.
X-ray crystallography
Variable temperature and LIESST measurements.
X-ray diffraction data were collected on a Bruker Smart 1K CCD diffractometer using graphite monochromised Mo-Kα (λ = 0.71073 Å) radiation. Datasets below 105 K were collected using an Oxford Cryosystems HeliX,17 while those above 105 K were collected using an Oxford Instruments open flow N2 Cryostream for cooling. Data collection was carried out using the SMART software,18 integration was performed using SAINT19 and multi-scan absorption corrections were applied to all datasets using SADABS.20 The structures were solved by direct methods in SHELXS21 and refined by full matrix least squares on F2 in SHELXL21 using Olex2.22 All non-hydrogen atoms were refined anisotropically and all hydrogen atoms were located geometrically and refined using a riding model with the exception of the hydrogen atoms on N6 which were located in the difference map. The LIESST structure at 30 K was obtained by irradiating the crystal in situ using a 670 nm, 5 mW CW laser. The crystal was irradiated for 60 minutes prior to and also continued throughout the data collection to try to establish and maintain a photostationary state, i.e. the metastable LIESST HS* structure. The same crystal was used for all data collections below 300 K, including the full datasets at 270, 210, 180, 165, 150, 120, 30 K and 30 K under laser irradiation. A second crystal was used to collect the data between 300 K and 375 K illustrated in the variable temperature plots. Temperature calibration of the Cryostream and HeliX was carried out using phase transition materials benzil (83.5 K)23 and NH4H2PO4 (148 K).24 Offsets of −3 K and +3 K were observed for the Cryostream and HeliX respectively; i.e. a temperature set at 105 K corresponds to a temperature of 102 K or 108 K at the crystal surface for Cryostream or HeliX measurements respectively.25 This offset is taken into account in the variable temperature plots where the transition profile is discussed. Full structure determinations were carried out at twenty-four temperatures; seven of these structures (30, 120, 150, 165, 180, 210 and 270 K) are published herein.
LIESST HS* relaxation.
LIESST HS* structure relaxation measurements were carried out by continuous collection of omega scans (scan width 0.4°) consisting of 50 frames at alternating phi positions of 0° and 90° immediately after switching off the laser. The program SMARTreduce,26 a script to harvest reflections, perform cell indexation and least squares refinement automatically in SMART,18 was used to iteratively determine a unit cell from each consecutive set of two runs. This allowed the evolution of the unit cell dimensions as a function of time to be monitored.
High pressure.
A single crystal was mounted in a modified Merrill-Bassett type diamond anvil cell (DAC) custom built at Durham University using tungsten carbide backing plates with an opening angle of 85° and type Ia Boehler-Almax diamond anvils with 0.8 mm culets. Stainless steel gaskets were pre-indented and drilled to give a gasket chamber of ∼0.3 mm diameter and 0.15 mm depth and paraffin oil was used as the pressure transmitting medium. A small ruby chip was used to measure the pressure in the DAC using the Ruby R1 fluorescence method.27 Fluorescence measurements were carried out before and after data collection to ensure there was no change in pressure during the experiment. The pressure can alter if insufficient time has been given for the pressure in the DAC to equilibrate prior to data collection. The laser used for fluorescence measurements can induce a spin transition, therefore exposure was minimised as much as possible, although because the pressure measurement is almost always taken above TLIESST any structural change is short lived. Room temperature (296 K) single crystal X-ray diffraction data were collected at ambient pressure, 1.4(2) kbar and 5.1(2) kbar. Data collection was carried out using XIPHOS II,28 part of the XIPHOS diffraction facility.29 This diffractometer, custom built for high pressure studies, is equipped with an Incoatec Ag-Kα (λ = 0.56086 Å) IμS source.30 Data collection was carried out using the Bruker APEX2 software suite,31 integration was performed using SAINT32 and multi-scan absorption corrections were applied to all datasets using SADABS.20 ECLIPSE33 was used to generate dynamic mask files to exclude areas of the diffraction pattern which contain no data due to the body of the DAC from the integration. The structures were solved by direct methods in SHELXS21 and refined by full matrix least squares on F2 in SHELXL21 using Olex2.22 All of the non-hydrogen atoms were refined anisotropically and all hydrogen atoms were located geometrically and refined using a riding model with the exception of the hydrogen atoms on N6 which were located in the difference map.
Variable temperature UV-Vis transmission spectroscopy
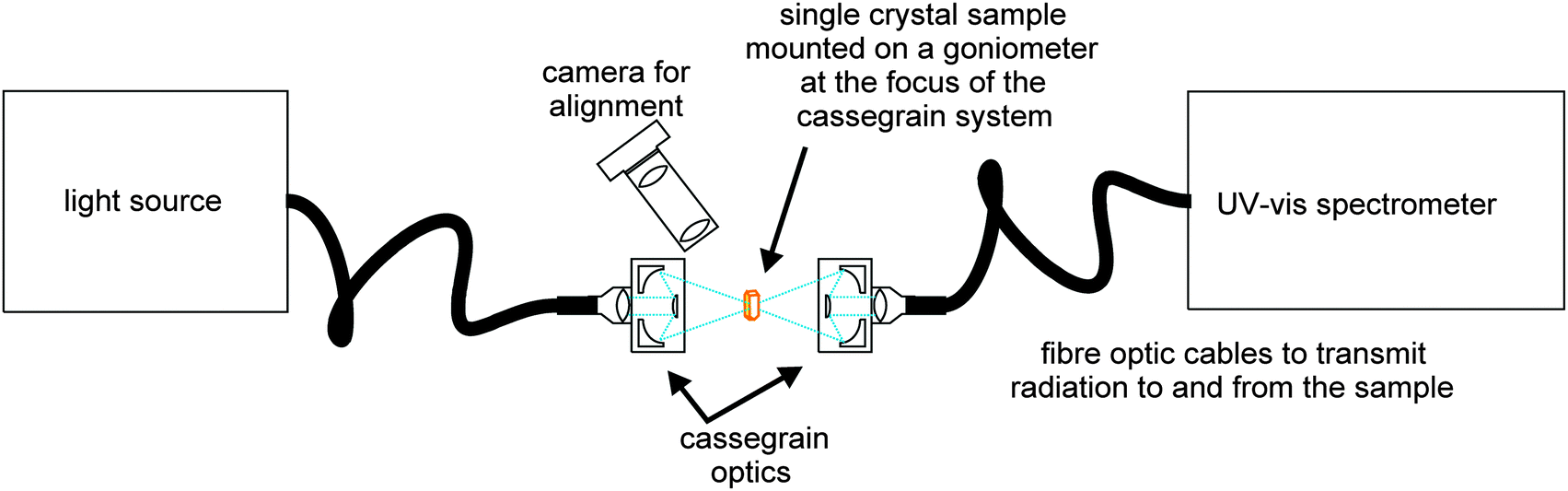
The single crystal UV-Vis setup used (Fig. 1) is based on a cassegrain system (Bruker) which utilises fibre optic cables to transmit radiation from a broadband light source (Hamamatsu UV-VIS fibre light source, L10290) to the sampling optics and from the optics into a UV-Vis spectrometer [Andor Shamrock SR-303i imaging spectrograph using 150 lines per mm grating groove density (specifications quote a resolution of 0.88 nm at a 500 nm centre wavelength) coupled with a Newton EMCCD camera]. The sample temperature was controlled using a non-liquid N2 Cryostream (Oxford Cryosystems, Cobra).
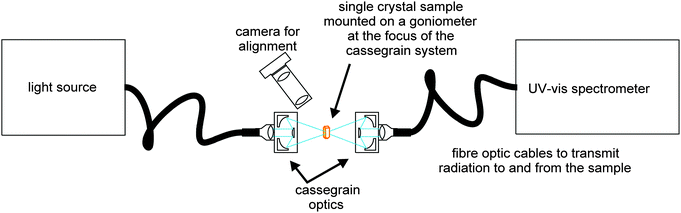 |
| | Fig. 1 Schematic of the cassegrain system used to collect UV-Vis spectra from single crystals. | |
A single crystal fragment of <100 × 100 × 25 μm was mounted using perfluoropolyether oil on a UV-Vis 100 μm aperture MicroLoop (MiTeGen, LLC) which was fixed on an XRD goniometer head. The crystals were visualised using a high-magnification zoom lens system mounted on a CMOS camera (Thorlabs) and positioned at the focus of the cassegrain system in the orientation giving the cleanest spectroscopic signal.
The area sampled by the UV-Vis radiation is controlled by the focus size of the cassegrain optics which is determined by the core size of the input fibre optic cable; for the crystals presented here the spot size was ∼50 μm in diameter. Spectra were collected using Andor's Solis software with a 20 ms exposure. Background (no light source) and reference (light source on a MicroLoop without sample) spectra were collected and data were presented by the Solis software in absorbance mode. OriginPro software34 was used for further data processing. Raw data were smoothed using a Savitzky–Golay filter (window size 13 points, polynomial order 2). The % transmittance, but not the spectral shape, is dependent on the orientation of the crystal within the light beam, and therefore raw spectra were normalised.
Resonant ultrasound spectroscopy
The principle of the resonant ultrasound spectroscopy (RUS) technique is quite straightforward.35 A sample with dimensions of ∼1 mm is held lightly between two piezoelectric transducers. The first transducer is driven by a frequency synthesizer at constant amplitude across a range of ultrasonic frequencies (0.1–2 MHz) which in turn causes the sample to resonate at particular frequencies. The second transducer acts as a signal detector which records the response of the sample in terms of its displacement when it is vibrated across the frequency range. Vibrational frequencies detected represent the frequency of normal modes of the sample. The square of a given peak frequency is directly proportional to the elastic constants associated with that normal mode.35 Low temperature measurements presented here were obtained using a single crystal of [Fe(abpt)2(NCS)2] polymorph A with irregular shape (∼0.4 mm in diameter, 0.0003 g) in a helium flow cryostat which has been described by McKnight et al.36 Data were collected with the sample chamber filled with a few mbar of helium to allow heat exchange between sample and cryostat. All spectra were transferred to the software package Igor Pro (WaveMetrics) for analysis. Peak positions and widths at half height were determined for a selection of peaks by fitting with an asymmetric Lorentzian function. The mechanical quality factor, Q, was calculated using the relationship Q = f/Δf, where f is the peak frequency and Δf is the width of the peak at half its maximum height. The inverse of the quality factor, Q−1, is a measure of acoustic dissipation (energy loss) in the sample.
Results
Thermal spin crossover
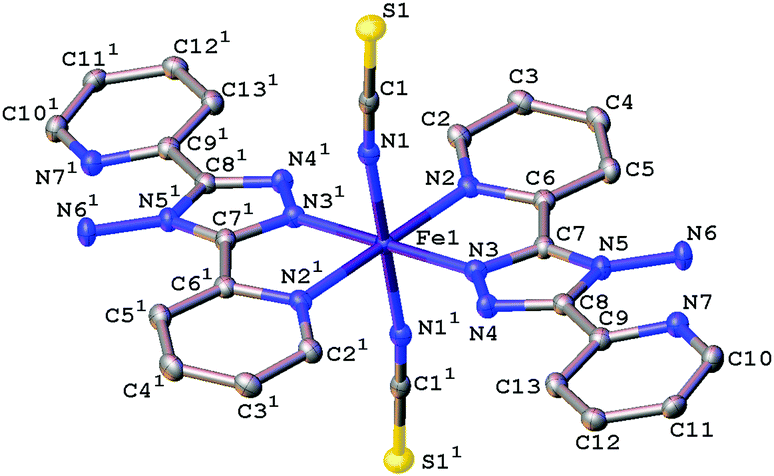
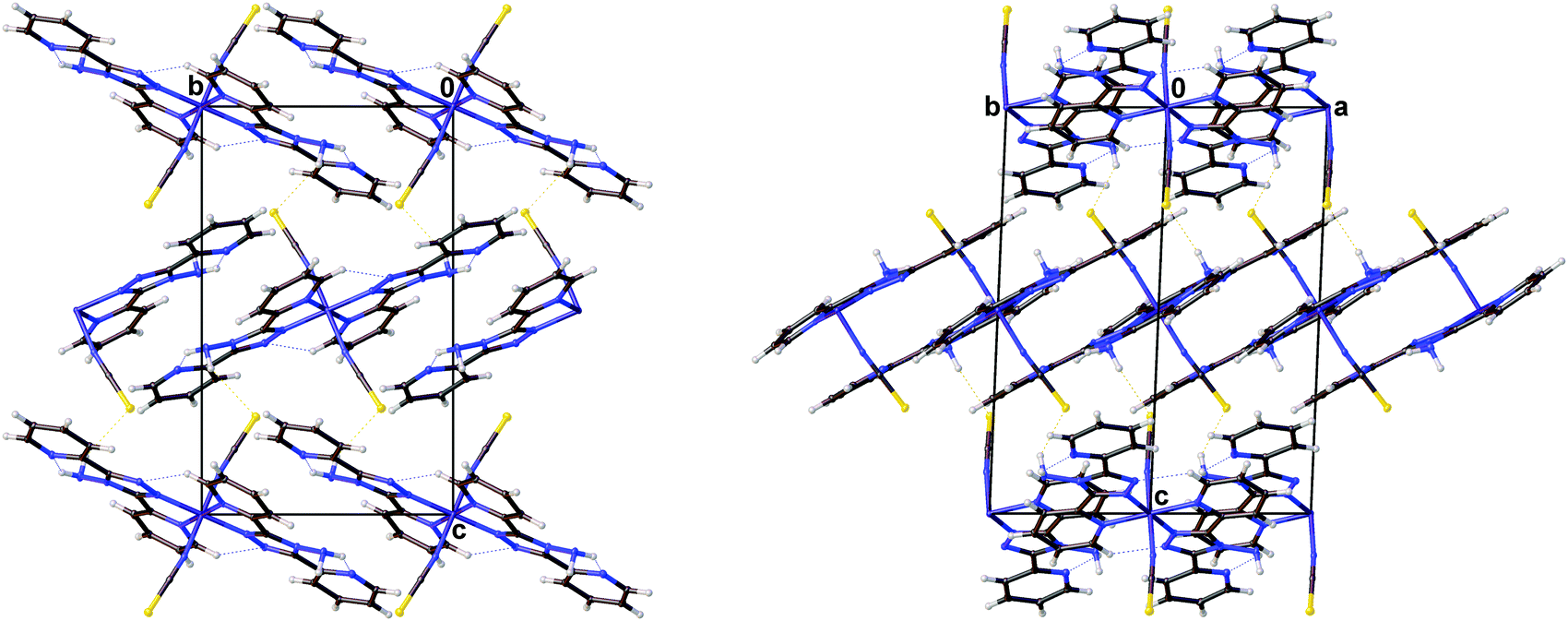
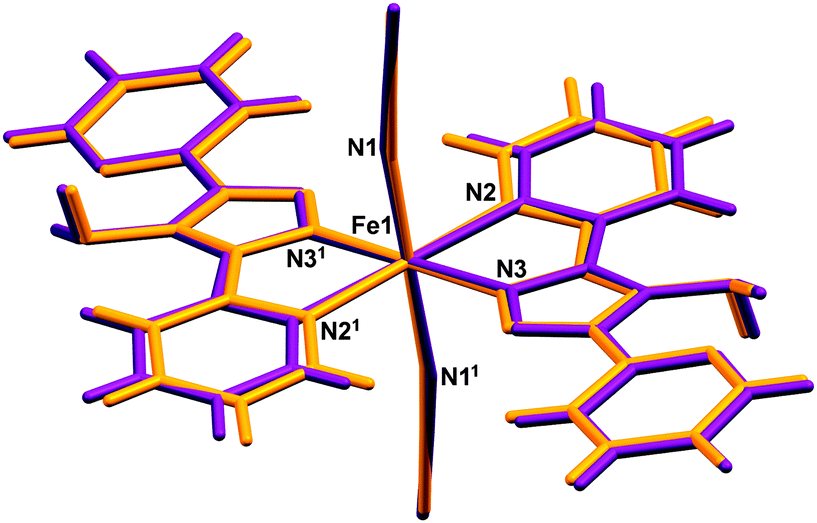
The magnetic data for polymorph A indicates that it undergoes thermal spin crossover without hysteresis between ∼202 K and 100 K11 (remeasured ∼250 K and 160 K),12 with T1/2 = 180 K11 (remeasured T1/2 = 188 K)12 and a residual HS fraction, γHS, of 23%11 (remeasured γHS ∼ 0%)12 at low temperature. The sample preparation method differs in the two reports11,12 which could explain the slight difference in the observed transition profile and completeness. The transition has been reported as gradual with moderate cooperativity between molecules11 playing a role in the propagation of the spin transition.37 Full crystallographic structure determinations upon cooling through the transition are reported from the same crystal at seven temperatures: 270, 210, 180, 165, 150, 120 and 30 K (Table 1 and Table S1, ESI†). The structure of A at 293 K11 has been previously reported and is consistent with the HS structure discussed here. All of the structures were solved in the monoclinic space group P21/n with half a molecule in the asymmetric unit (Z′ = 0.5) (Fig. 2). Each Fe(II) centre has 6 coordinated nitrogen atoms: one from each NCS group and two from each abpt ligand (one pyridyl nitrogen and one triazole nitrogen). An intramolecular hydrogen bond exists between N6–H6⋯N7, for which D–H = 0.88(2) Å, H⋯A = 2.16(2) Å, D⋯A = 2.853(2) Å, ∠DHA = 135(2)° at 30 K and D–H = 0.93(2) Å, H⋯A = 2.12(2) Å, D⋯A = 2.847(3) Å, ∠DHA = 135(2)° at 270 K. In addition, a weak C2–H2⋯N4#1 (#1 = −x + 1, −y + 1, −z + 1) interaction was also identified (Table S2, ESI†). The structures all contain π–π interactions between pairs of counterpart abpt pyridyl groups on adjacent molecules [centroid (N2, C2–C6) to centroid (N7#2, C9#2–C13#2, #2 = 1 − x, 2 − y, 1 − z) distance of ∼3.6 Å with an offset of ∼1.3–1.4 Å at all temperatures] creating a 1-dimensional chain through the structure which can be viewed along the (110) direction. Indeed the spin transition has been linked to the presence of these π–π interactions.12 Molecules along the c-axis direction are rotated by ∼40° from each other, molecules along the a-axis direction are in the same orientation as each other (Fig. 3). The differences between the HS, LS and HS* structures are discussed in more detail below.
Table 1 Crystal data and refinement results for [Fe(abpt)2(NCS)2] polymorph A at 270 K, 30 K and 30 K after irradiation
| Spin state (temperature) |
HS (270 K) |
LS (30 K) |
HS*, LIESST (30 K) |
| Empirical formula |
C26H20FeN14S2 |
C26H20FeN14S2 |
C26H20FeN14S2 |
| Formula weight |
648.53 |
648.53 |
648.53 |
|
λ (Å) |
0.71073 |
0.71073 |
0.71073 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/n |
P21/n |
|
a (Å) |
8.5367(4) |
8.3411(4) |
8.4505(4) |
|
b (Å) |
10.2206(5) |
9.9442(5) |
10.1273(5) |
|
c (Å) |
16.4268(8) |
16.1668(8) |
16.3033(8) |
|
β (°) |
93.899(1) |
93.112(1) |
93.491(1) |
|
V (Å3) |
1429.92(12) |
1338.99(11) |
1392.66(12) |
|
Z
|
2 |
2 |
2 |
| Temperature (K) |
270(2) |
30(2) |
30(2) |
|
D
c (Mg m−3) |
1.506 |
1.609 |
1.547 |
|
μ (mm−1) |
0.719 |
0.768 |
0.738 |
|
F(000) |
664 |
664 |
664 |
| Crystal size (mm3) |
0.36 × 0.30 × 0.26 |
0.36 × 0.30 × 0.26 |
0.36 × 0.30 × 0.26 |
|
θ range for data collection (°) |
2.35–28.28 |
2.41–28.28 |
2.37–28.29 |
| Ranges of h, k, l |
−11 ≤ h ≤ 11, −13 ≤ k ≤ 10, −21 ≤ l ≤ 16 |
−10 ≤ h ≤ 11, −13 ≤ k ≤ 9, −21 ≤ l ≤ 15 |
−10 ≤ h ≤ 11, −13 ≤ k ≤ 10, −21 ≤ l ≤ 16 |
| Refl. collected |
9716 |
9018 |
9399 |
|
R
int
|
0.0301 |
0.0370 |
0.0293 |
| Data/parameters |
3554/202 |
3318/202 |
3453/202 |
| Absorption coef. min/max |
0.697/1.000 |
0.787/1.000 |
0.471/0.743 |
| GooF (F2) |
1.034 |
1.040 |
1.049 |
| Final R1 [I > 2σ(I)] |
0.0362 |
0.0341 |
0.0311 |
| wR2 [I > 2σ(I)] |
0.0845 |
0.0797 |
0.0727 |
| Largest diff. peak/hole (e Å−3) |
0.41/−0.37 |
0.73/−0.52 |
0.37/−0.33 |
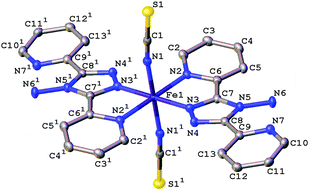 |
| | Fig. 2 Ellipsoid plot of [Fe(abpt)2(NCS)2] polymorph A at 120 K with the ellipsoids depicted at 50% probability level. The asymmetric unit (asu) consists of half a molecule and 1 represents the other half of the molecule which is symmetry generated by the operator 1 − x, 1 − y, 1 − z. Hydrogen atoms have been omitted for clarity. | |
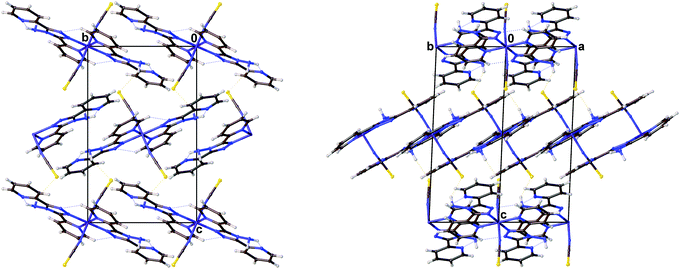 |
| | Fig. 3 Diagrams showing the packing of [Fe(abpt)2(NCS)2] polymorph A at 30 K viewed along the (left) (100) and (right) (110) directions. | |
Variable temperature data collections at 30, 120, 150, 165, 180, 210, 270 K and unit cell parameter changes
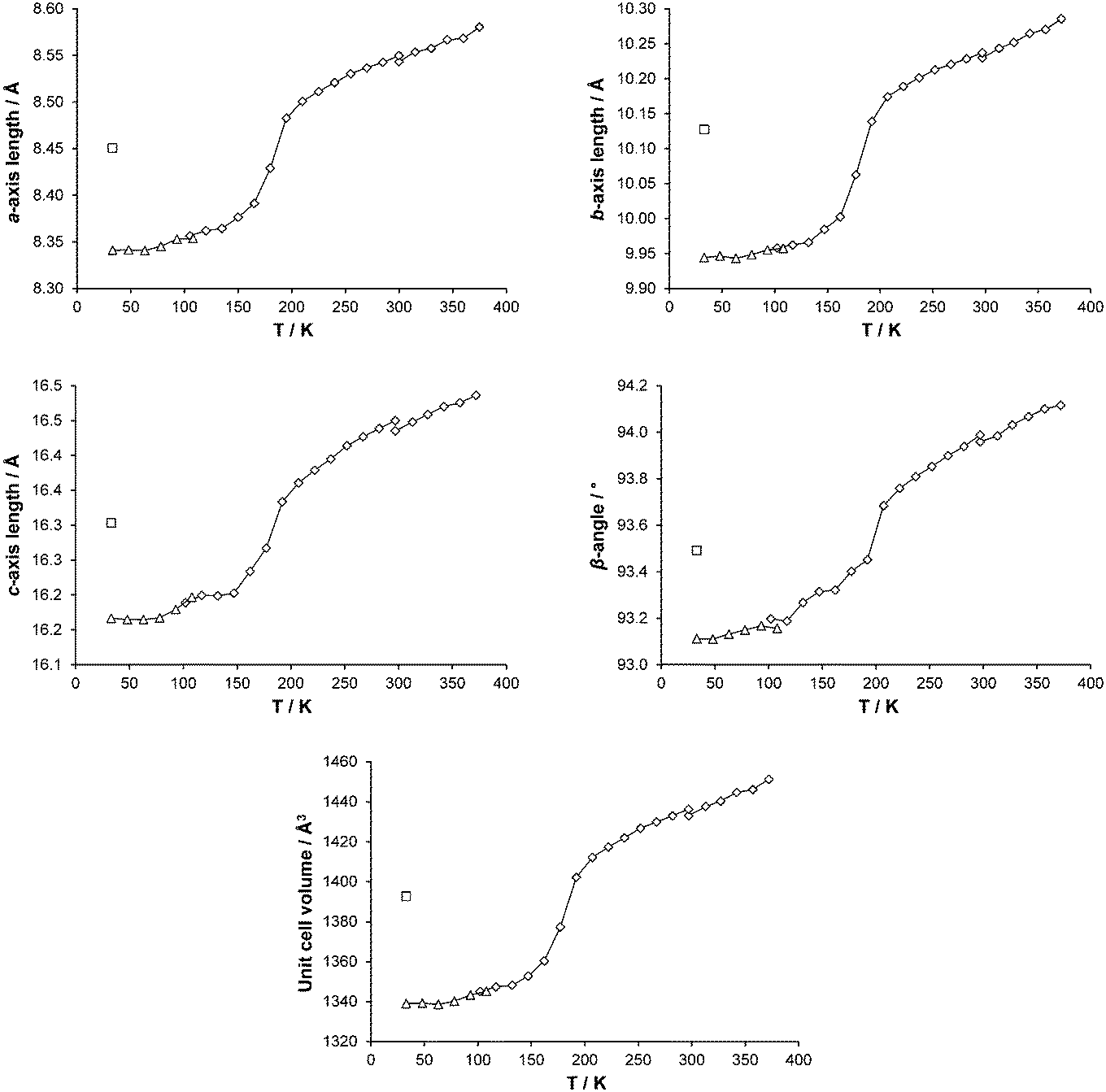
Cell parameters were monitored over the temperature range 375–30 K on two different crystals (crystal 1, 300–30 K; crystal 2, 375–300 K). As can be seen from Fig. 4, all the cell axes lengths and the β-angle decrease as the temperature decreases. Although only small changes are seen in the axes lengths and β-angle, overall they follow the same pattern as that observed in the magnetic data11 and the occurrence of the spin transition can clearly be seen.
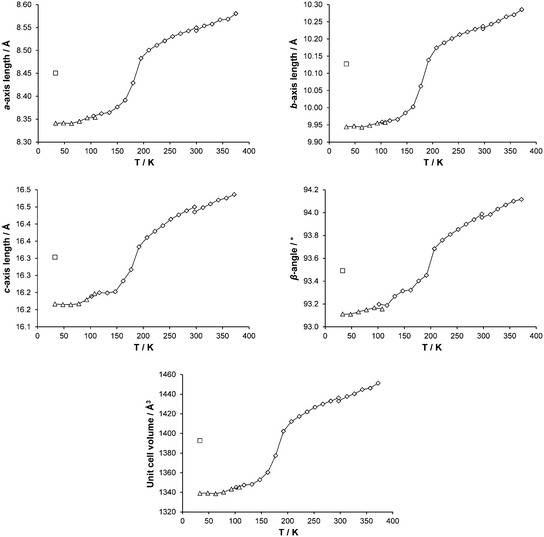 |
| | Fig. 4 Changes in cell parameters for [Fe(abpt)2(NCS)2] polymorph A with temperature are shown as points connected by a line: ◇ and △ denote data collected using the Cryostream or HeliX respectively and □ show corresponding LIESST, HS* cell parameters. The temperature scale has been adjusted to show temperature at the crystal surface for both Cryostream and HeliX measurements: see Experimental section, Variable temperature and LIESST measurements, for details. Error bars are included but obscured by the data markers. | |
The unirradiated structures at the two extremes of the measured temperature range, i.e. 30 K and 270 K, were examined and although very similar there are some clear differences between the structures (Fig. 5). The Fe–N bond lengths range from 2.131(2) to 2.215(2) Å at 270 K and 1.952(2) to 2.010(2) Å at 30 K. This ∼0.2 Å decrease in the Fe–N distances with decreasing temperature is consistent with the structure having undergone the expected spin crossover from HS (270 K) to LS (30 K) Table 2. In addition to a decrease in the Fe–N bond lengths upon cooling, the N2–Fe1–N3 bond angle increases from 74.8(3)° to 79.98(6)° with a concomitant decrease in the N2–Fe1–N3#1 (#1 = −x + 1, −y + 1, −z + 1) bond angle from 105.24(6)° to 100.02(6)°. This results in a shortening of the D⋯A distance associated with the weak C2–H2⋯N4#1 (#1 = −x + 1, −y + 1, −z + 1) interaction giving a trend consistent with that associated with the magnetic data (Table S2 and Fig. S2, ESI†).
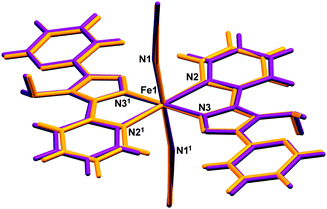 |
| | Fig. 5 Overlay of (purple) HS, 270 K and (orange) LS, 30 K structures of [Fe(abpt)2(NCS)2] polymorph A. The asu consists of half a molecule and 1 represents the other half of the molecule which is symmetry generated by the operator 1 − x, 1 − y, 1 − z. | |
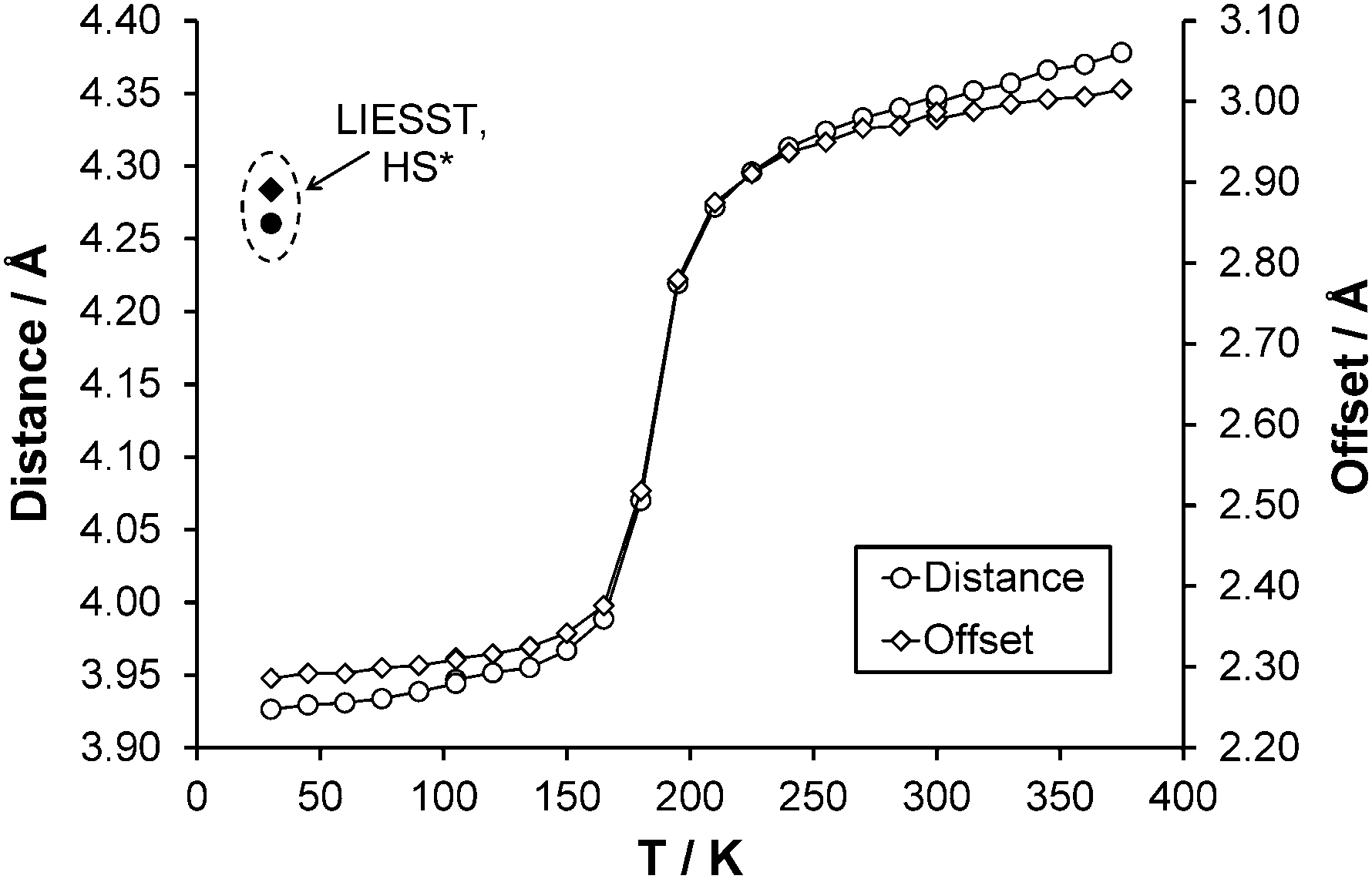
After the HS to LS transition upon cooling the interpenetration of adjacent pairs of molecules along approximately the ab direction increases, this results in a decrease of the centroid (N2, C2–C6) to centroid (N2#3, C2#3–C6#3, where #3 = 2 − x, 1 − y, 1 − z) distance from ∼4.3 Å with an offset of 3.0 Å (270 K) to ∼3.9 Å with an offset of 2.3 Å (30 K) (Fig. 6). Structures obtained upon flash freezing below the spin transition temperature are identical to those obtained with gradual cooling through the spin transition. No reduction in crystallinity was observed after the spin transition, probably due to the fact that the HS, LS and HS* crystal structures are isostructural and only very small changes occur as a result of the spin transition.
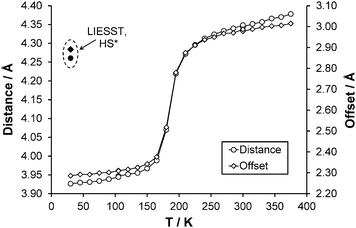 |
| | Fig. 6 Change in centroid (N2, C2–C6) to centroid (N2#3, C2#3–C6#3, where #3 = 2 − x, 1 − y, 1 − z) (○) distance and (◇) offset with temperature. ● and ◆ denote distance and offset observed respectively for the HS* structure at 30 K. Error bars are included but obscured by the data markers. | |
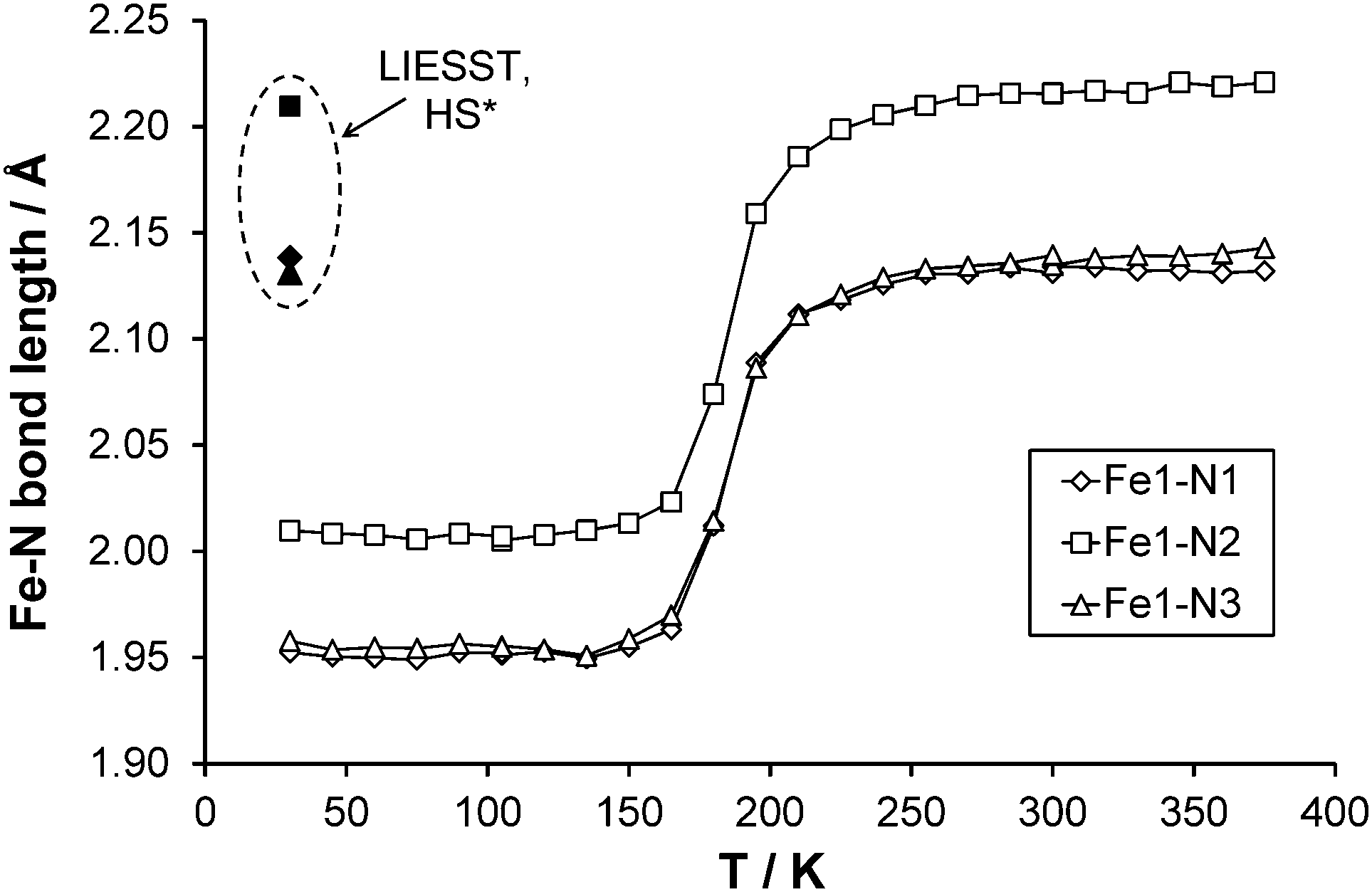
Full structure determinations are reported at 30, 120, 150, 165, 180, 210 and 270 K. Examining the three independent bond lengths around the Fe centre (Table 2 and Fig. 7) shows a marked decrease in all the Fe–N bond lengths upon cooling through the temperature range of the spin transition (datasets at 210–150 K). In the case of the unirradiated structure at 30 K and the 120 K structure; the Fe–N bond lengths, the distortion parameter Σ (∼49°) and Fe octahedron volume Vp (∼10 Å3) are essentially identical and Vp is consistent with values previously associated with the presence of a LS state in an FeN6 complex. At the other end of the temperature range the 270 K structure along with the ∼0.2 Å increase in the Fe–N bond lengths shows a significant increase in the value of Σ (∼70°) and Vp (∼13 Å3), supporting the formation of the HS state. For the structures obtained during the spin transition (165 K and 180 K) it can be seen that Σ and Vp also decrease markedly as the temperature decreases and suggest that the Fe centre is in between the HS and LS states as expected.
Table 2 Fe–N bond lengths for all structures, including the three 296 K structures collected in the DAC, along with the distortion parameter Σ and the volume of the Fe octahedron Vp
| |
30(2) K LIESST |
30(2) K |
120(2) K |
150(2) K |
165(2) K |
180(2) K |
210(2) K |
270(2) K |
Ambient |
1.4(2) kbar |
5.1(2) kbar |
|
Σ, the distortion parameter, is the sum of the absolute value of the deviation of all 12 cis N–Fe–N angles from 90°.
V
p is the volume of the Fe octahedron calculated in Olex2.22
|
| Fe1–N1 (Å) |
2.1384(13) |
1.952(2) |
1.953(2) |
1.955(1) |
1.964(2) |
2.012(2) |
2.112(2) |
2.131(2) |
2.127(5) |
2.128(3) |
2.006(3) |
| Fe1–N2 (Å) |
2.2096(13) |
2.010(2) |
2.007(2) |
2.013(1) |
2.023(1) |
2.074(2) |
2.185(1) |
2.215(2) |
2.217(6) |
2.202(4) |
2.063(4) |
| Fe1–N3 (Å) |
2.1312(13) |
1.958(2) |
1.955(2) |
1.958(1) |
1.970(1) |
2.015(3) |
2.112(1) |
2.134(2) |
2.129(8) |
2.111(6) |
1.991(6) |
|
Σ
(°) |
69.2(3) |
49.2(4) |
49.0(4) |
49.8(4) |
50.8(4) |
56.1(4) |
67.5(4) |
69.9(4) |
65.2(14) |
68.8(10) |
54.9(16) |
|
V
p
(Å3) |
12.957(5) |
10.080(5) |
10.053(5) |
10.104(4) |
10.255(5) |
10.964(5) |
12.564(5) |
12.947(6) |
12.96(2) |
12.74(2) |
10.76(2) |
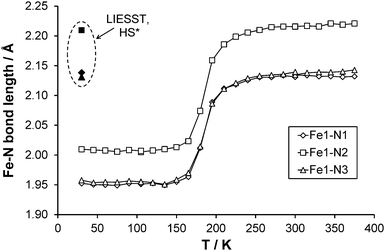 |
| | Fig. 7 Fe–N bond length changes as a function of temperature, LIESST values are contained within the dashed oval. Error bars are included but obscured by the data markers. | |
LIESST metastable HS* structure at 30 K
The reported value of TLIESST for A is ∼40 K.11 Given that previously reported magnetic data had established that at 30 K around 50% of the HS* state in a precipitated sample reverts back to the LS state after <1000 s and almost 100% relaxes within 4000 s,11 the laser was left on to continue irradiation throughout the data collection. Whether any residual ground state (LS) species remain even during continuous irradiation is not known for the crystals used in this study and the possibility of any resulting bias in the HS* structural parameters was not investigated.38 After irradiation there is a 4% increase in the unit cell volume at 30 K which would be anticipated with the formation of a metastable HS* state through LIESST. The HS* structure retains the monoclinic space group P21/n previously measured for the HS and LS ground state structures. As expected with the formation of the HS* state there is an increase of ∼0.2 Å in all of the Fe–N bond lengths upon irradiation at 30 K, ranging from 1.952(2) to 2.010(2) Å in the LS state structure to 2.1312(13) to 2.2096(13) Å in the HS* state structure. The range of Fe–N bond lengths seen in the HS* structure at 30 K align very closely with those observed in the HS structure at 270 K [2.131(2) to 2.215(2) Å]. In addition, upon the spin transition from LS to HS* at 30 K there is an increase in Σ from 49.2(4)° to 69.2(3)° and likewise an increase in Vp from ∼10 Å3 to ∼13 Å3, these values are in line with those observed in the HS state at 270 K [Σ = 69.9(4)° and Vp = 12.947(6) Å3]. Indeed the key structural features of the HS* structure at 30 K are very similar to those of the 270 K HS structure discussed previously and hence they are not examined in any further detail here.
LIESST metastable HS* structure relaxation at 30 K
The relaxation of the HS* state to the LS ground state was monitored crystallographically by irradiating a crystal at 30 K to induce the HS* state, switching the laser off and monitoring the evolution of unit cell parameters with time. This confirmed that at 30 K within ∼4000 s the metastable HS* state had reverted to the LS ground state structure (Fig. 8).11
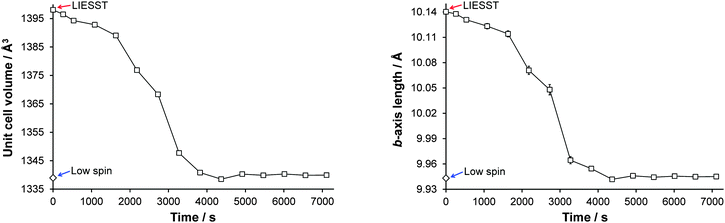 |
| | Fig. 8 Illustration of the change in (left) unit cell volume and (right) b-axis length as a function of time at 30 K as a result of relaxation of the LIESST HS* state. Note: Each data point is plotted at the time mid-point of its two consecutive omega runs, therefore an error between ±(535 to 538) s is associated with all data points except the 1st data point after turning the laser off (at 262 s) where the error is ±262 s. Plots showing a-axis, c-axis and β-angle relaxation are included in Fig. S1 (ESI†). | |
High pressure structure
Examining the change in Fe–N bond lengths between ambient and high pressure 5.1(2) kbar structures (Table 2) indicates that A undergoes a pressure induced spin transition. While a pressure induced spin transition can produce the same LS structure as that obtained from a thermal spin transition,39 this is not always the case.40 Indeed some compounds display a pressure induced spin transition where either no spin transition15 or only a partial spin transition41 is observed thermally. In the case of A, the application of pressure results in a decrease in the length of all of the cell axes and a reduction in the β-angle, this gives an ∼8% decrease in the cell volume between ambient pressure and 5.1(2) kbar (Table 3). The structure maintained the monoclinic P21/n crystal system and space group across the range of pressures measured, as was also observed in the variable temperature studies at ambient pressure. It is worth noting that the cell parameters for the pressure LS structures are compressed compared to the thermal LS cell parameters. The structural features associated with the pressure induced LS structure are similar to the thermal LS structure, although the bond lengths, distortion parameter Σ and Vp at 5.1(2) kbar are very slightly larger than those seen at 120 K. This indicates that the spin transition may not be fully complete at this pressure; however a partial spin transition has clearly occurred. It is highly likely that at slightly higher pressure the spin transition from HS to LS would be complete. Both the thermal and pressure induced spin transitions result in a darkening of the crystal colour which can clearly be seen between ambient pressure and 5.1(2) kbar (Fig. 9).
Table 3 Crystal data and refinement results for [Fe(abpt)2(NCS)2] polymorph A at 296 K in the DAC at ambient pressure, 1.4(2) kbar and 5.1(2) kbar
| |
Ambient |
1.4(2) kbar |
5.1(2) kbar |
| Empirical formula |
C26H20FeN14S2 |
C26H20FeN14S2 |
C26H20FeN14S2 |
| Formula weight |
648.53 |
648.53 |
648.53 |
|
λ (Å) |
0.56086 |
0.56086 |
0.56086 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/n |
P21/n |
|
a (Å) |
8.552(3) |
8.484(2) |
8.3440(15) |
|
b (Å) |
10.236(6) |
10.159(5) |
9.955(3) |
|
c (Å) |
16.444(6) |
16.253(5) |
15.960(3) |
|
β (°) |
93.978(11) |
93.894(12) |
93.414(7) |
|
V (Å3) |
1435.9(10) |
1397.7(9) |
1323.4(5) |
|
Z
|
2 |
2 |
2 |
| Temperature (K) |
296 |
296 |
296 |
|
D
c (Mg m−3) |
1.500 |
1.541 |
1.627 |
|
μ (mm−1) |
0.377 |
0.387 |
0.409 |
|
F(000) |
664 |
664 |
664 |
| Crystal size (mm3) |
0.17 × 0.15 × 0.10 |
0.17 × 0.15 × 0.10 |
0.17 × 0.15 × 0.10 |
|
θ range for data collection (°) |
3.92–41.00 |
3.96–39.47 |
4.03–39.48 |
| Ranges of h, k, l |
−10 ≤ h ≤ 10, −8 ≤ k ≤ 7, −20 ≤ l ≤ 20 |
−9 ≤ h ≤ 9, −6 ≤ k ≤ 6, −19 ≤ l ≤ 19 |
−9 ≤ h ≤ 9, −6 ≤ k ≤ 6, −19 ≤ l ≤ 19 |
| Refl. collected |
9085 |
5050 |
4597 |
|
R
int
|
0.0343 |
0.0317 |
0.0273 |
| Data/parameters |
1158/202 |
921/204 |
794/202 |
| Absorption coef. min/max |
0.939/0.963 |
0.937/0.962 |
0.812/0.928 |
| GooF (F2) |
1.130 |
1.055 |
1.052 |
| Final R1 [I > 2σ(I)] |
0.0430 |
0.0277 |
0.0229 |
| wR2 [I > 2σ(I)] |
0.0987 |
0.0561 |
0.0492 |
| Largest diff. peak/hole (e Å−3) |
0.19/−0.22 |
0.13/−0.12 |
0.12/−0.10 |
 |
| | Fig. 9 Photograph of a single crystal of A loaded in the DAC showing the colour change associated with the pressure induced spin transition: (left) ambient pressure, (right) 5.1(2) kbar. | |
Variable temperature UV-Vis transmission spectroscopy
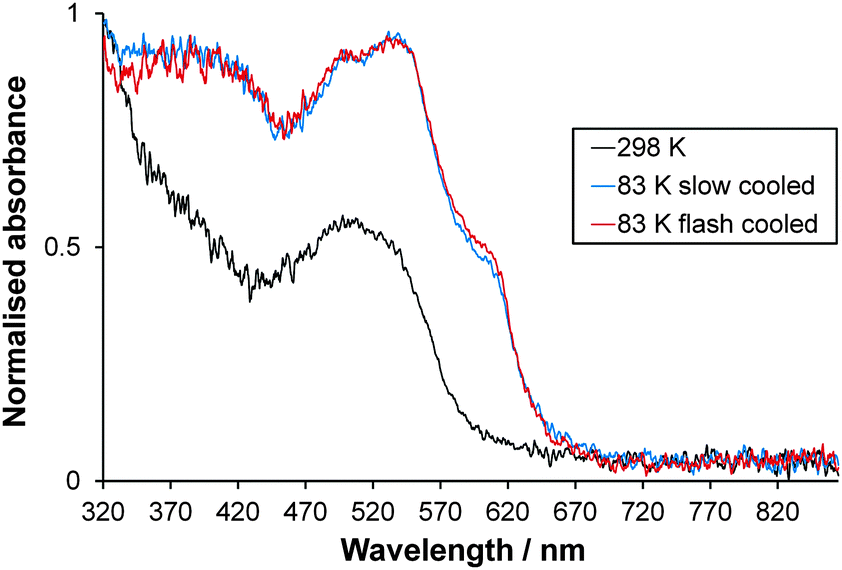
Single crystals of [Fe(abpt)2(NCS)2] polymorph A are red and dark red in the HS and LS states respectively. Normalised single crystal UV-Vis absorption spectra obtained for A in the HS state at 298 K and LS state at 83 K (the lowest temperature achievable with the Cryostream) are shown in Fig. 10. The intense colour in both spin states and possible residual HS fraction, γHS, remaining at low temperature make assignment of absorption bands to particular transitions challenging and beyond the scope of this study. The ligand field (d–d) transition bands could be partially obscured by low lying intense charge-transfer (CT) bands in the UV-Vis spectral region. At 298 K, the complex is expected to be purely HS.11,12 Whether the one featureless band centred at ∼505 nm, similar to the broad band at ∼500 nm observed for polymorph D at 300 K,13 can be attributed to the spin allowed, parity forbidden 5T2 → 5E transition2 is not yet known and computational studies would need to be carried out to draw further conclusions. The spectra at 83 K may incorporate a contribution from a small residual HS fraction. Two main overlapping bands are visible: one with two peaks at ∼535 nm and ∼496 nm and a shoulder at ∼605 nm and a second band below ∼450 nm. The completeness of the spin transition for this particular crystal is unknown; previously reported magnetic data suggest a residual γHS at low temperature of between 0% and 23% for a single crystal sample depending on the crystallisation method used11,12 but 4.4% for a precipitated microcrystalline sample.42 The crystal was repositioned in the beam between collection of HS and LS spectra due to contraction of the crystal mount during cooling; therefore, calculation of the % conversion is not possible. A similar spectrum is observed at 83 K either upon slow cooling (150 K h−1) or flash cooling (Fig. 10), indicating that thermal trapping of the HS state (TIESST)43 is not observed at 83 K.
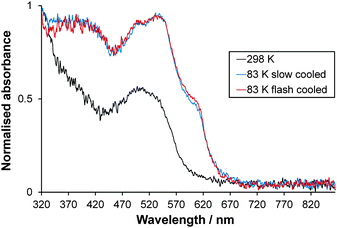 |
| | Fig. 10 Normalised single crystal UV-Vis absorption spectra collected at 298 K (HS) and 83 K (LS) for [Fe(abpt)2(NCS)2] polymorph A. The consistency in the spectrum at 83 K independent of cooling strategy is also shown. | |
Resonant ultrasound spectroscopy
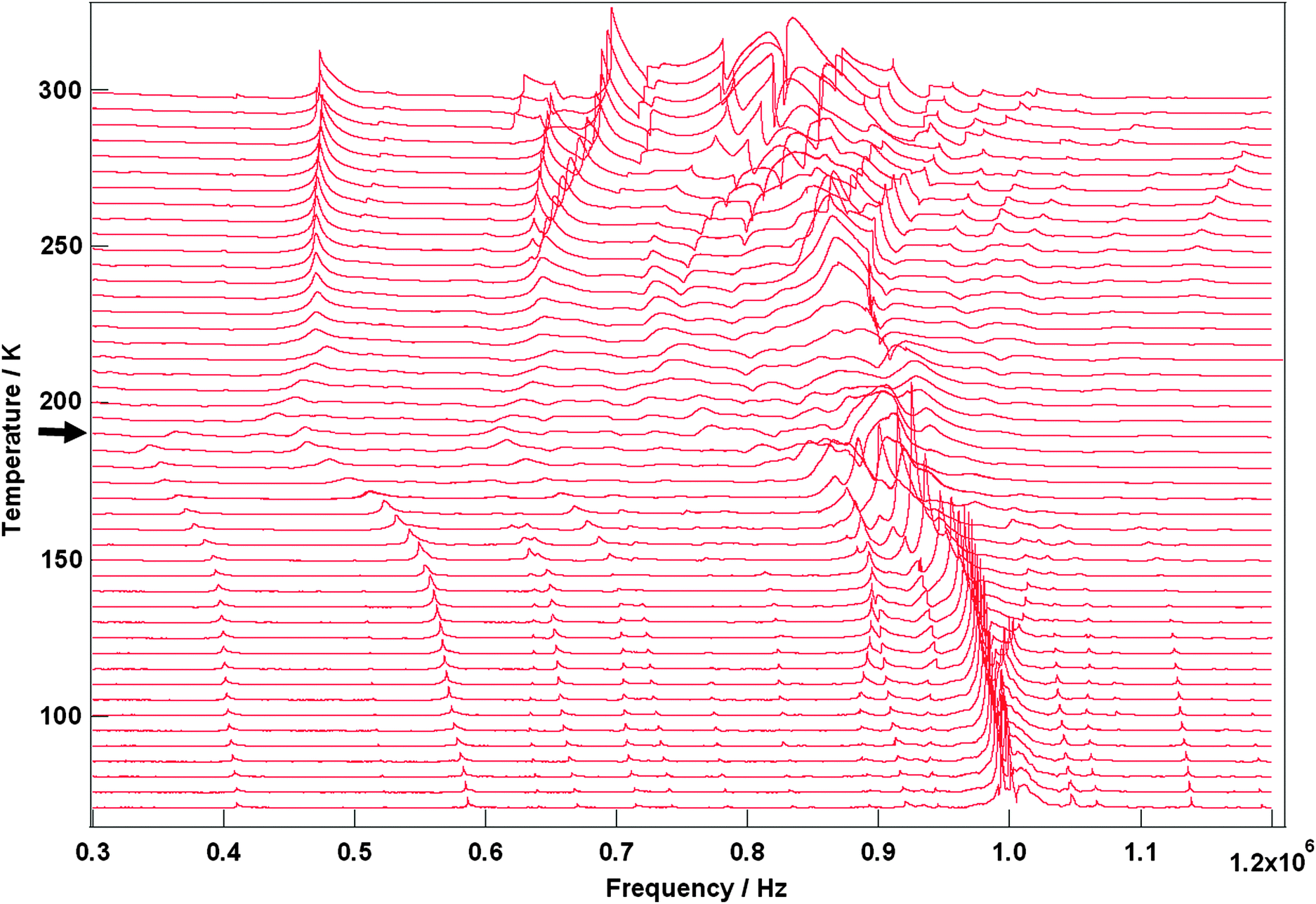
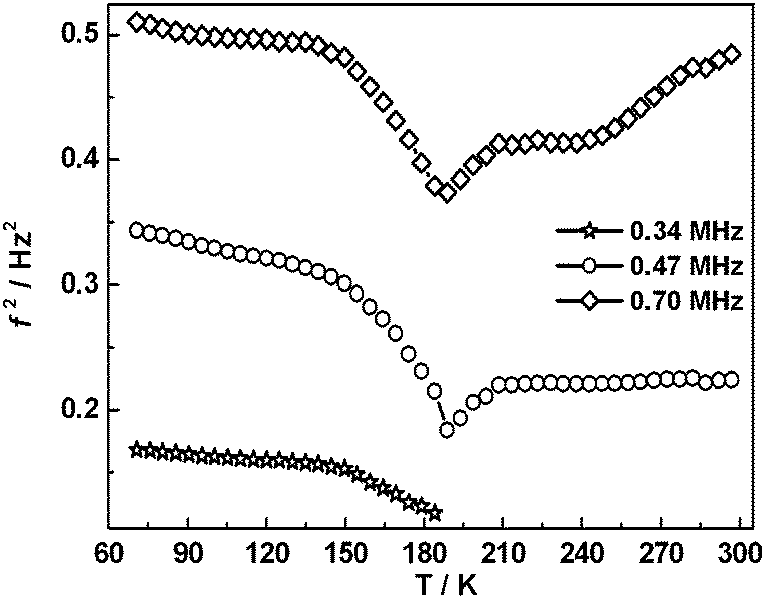
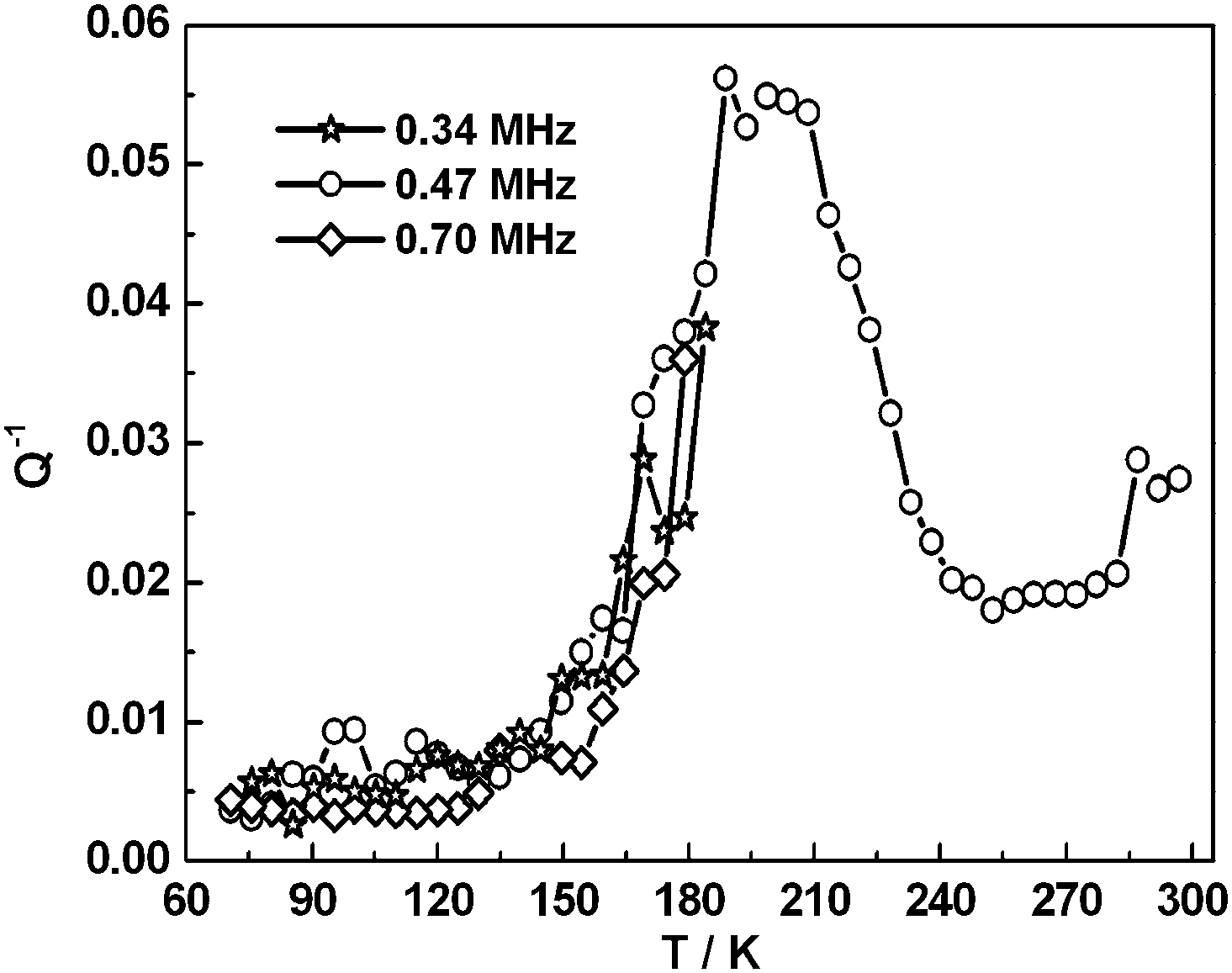
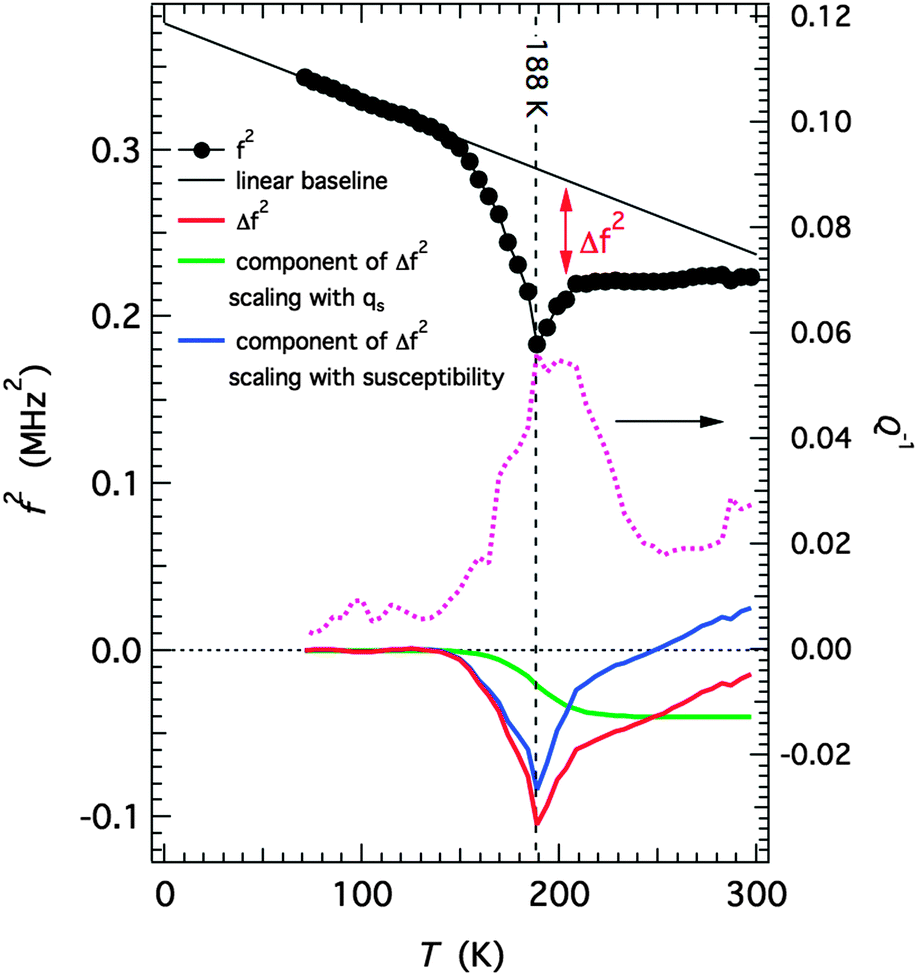
Selected RUS spectra from the single crystal collected during a heating sequence from 70–300 K are illustrated in Fig. 11. The y-axis is real amplitude from the voltmeter but the spectra have been offset in proportion to the temperature at which they were collected and the axis labelled as temperature. Peaks in the spectra which are dependent on temperature reflect the vibrational frequencies of the normal modes of the sample; the square of the frequency of each mode, f2, is proportional to the elastic constants associated with that mode. The inverse of the mechanical quality factor, Q−1, is a measure of acoustic dissipation (energy loss) in the sample.35 The raw spectra clearly show a change in trend of peak frequencies near ∼190 K, changes in line widths between ∼160 and ∼220 K, and relatively sharp peaks reappear in the low temperature region (Fig. 11). Data for f2 and Q−1 obtained from fitting selected peaks in the high temperature region are shown in Fig. 12 and 13. All the resonance peaks have the same form of elastic softening (reducing values of f2) as T1/2 is approached from both above and below, with a minimum at ∼190 K corresponding to the known spin crossover transition point T1/2 = 188 K12 for polymorph A (Fig. 12). This is a relaxational effect due to coupling of the spin state variation with strain, as seen also in the variation of lattice parameters with temperature. An applied stress in the mechanical resonance induces a strain which, in turn, results in a change in the spin state, giving rise to additional softening in the temperature interval of the spin crossover. Although one of the effects of changing spin configurations is a change in unit cell volume, the monoclinic symmetry means that there are also shear strains involved and the observed elastic softening is mainly through combinations of shear elastic constants. There is a significant increase in acoustic loss (increasing Q−1) in the vicinity of 200 K which appears to be frequency independent (Fig. 13). This is due to the strain/spin state relaxations occurring on the time scale of the applied stress, i.e. ∼10−6 s.
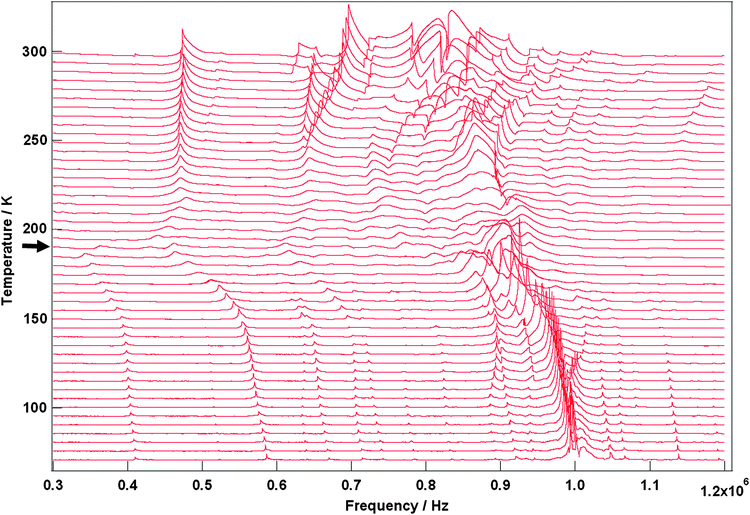 |
| | Fig. 11 Stack of RUS spectra as a function of frequency for [Fe(abpt)2(NCS)2] polymorph A from 70–300 K, the arrow at around ∼190 K indicates the spin crossover transition, T1/2 = 188 K.12 | |
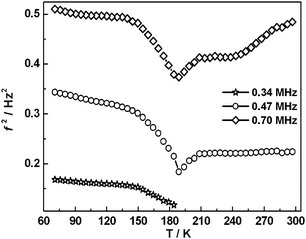 |
| | Fig. 12 Temperature dependency of resonance frequencies from fitting of selected peaks. f2 scales with combinations of shear elastic constants. The softening is due predominantly to coupling of spin state changes with changes in shear strain of the crystal. | |
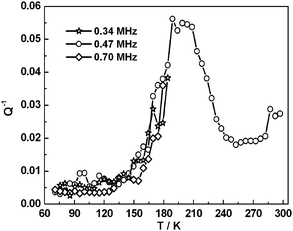 |
| | Fig. 13 Temperature dependency of Q−1 from fitting of selected resonance peaks. The increasing acoustic loss is due to dynamic coupling of strains with spin state relaxations on a time scale commensurate with the resonance frequencies. | |
A simple model for the strain relaxation behaviour
The strain coupling recognised both from the changes in lattice parameters and from the elastic anomalies is similar to that found in Co oxide phases for which a formal treatment has previously been set out.44,45 The spin state transition is first described in terms of an order parameter, qs, which takes on values between 0 and 1 to represent the proportion of atoms with low spin state. Variations in lattice parameters are represented in terms of coupling between qs and strains, ei, which in turn give rise to softening of the elastic constants, Cik. Relationships between the order parameter, strains and elastic constants derive from the change in free energy, G, due to the transition which, for a monoclinic crystal, may be expressed as| | 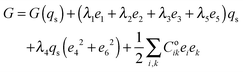 | (1) |
G(qs) describes the free energy change due to changes in spin state, excluding the contribution of lattice relaxation, and will be some form of partition function. The coupling coefficients, λi, describe the strength of coupling between the order parameter and strain, and the last term is the Hooke's law elastic energy. Coik represents the elastic constants of the crystal in its high spin state. The equilibrium condition, ∂G/∂e = 0, then gives| | | e1 ∝ e2 ∝ e3 ∝ e5 ∝ qs, | (2) |
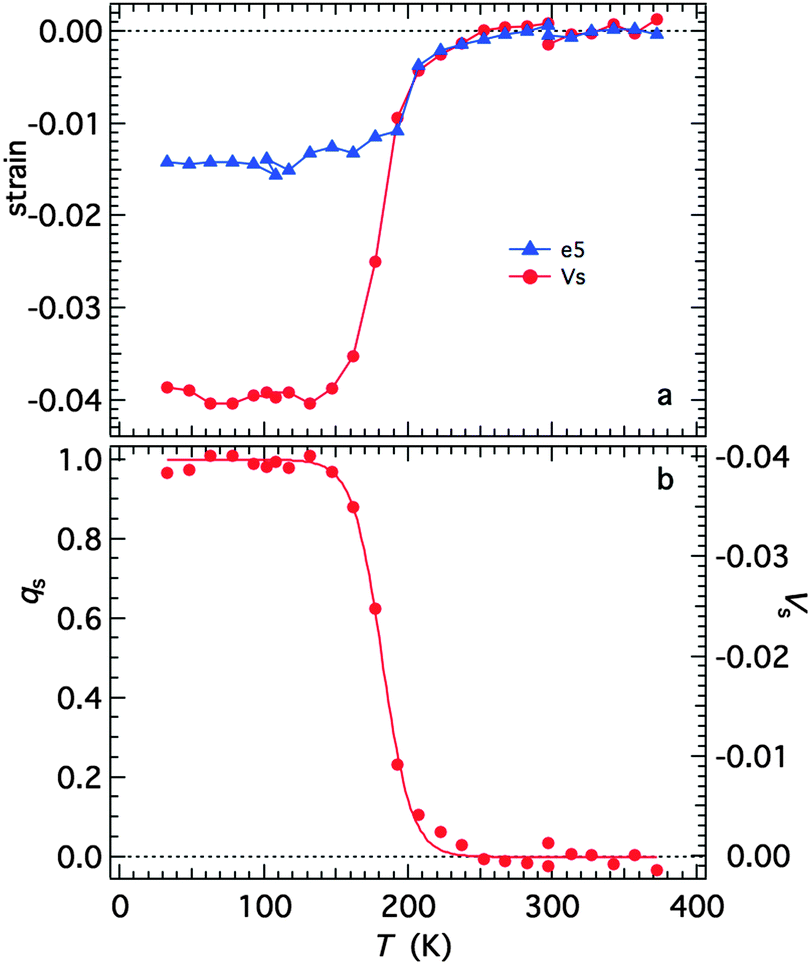
with proportionality constants depending on combinations of λi and Coik, and As shown in the supplementary material, values of the strains are defined with respect to the lattice parameters, ao, bo,… of the high spin state, giving e1 = Δa/ao, e2 = Δb/bo… Calculated strains obtained from the lattice parameter data are given in full in Fig. S3 (ESI†). From the observation that e1, e2 and e3 are approximately equal, it is evident that the negative unit cell volume strain of up to ∼4% accompanying the HS* (30 K)–LS (30 K) transition is nearly isotropic. There is however a significant shear strain, e5, with magnitude of up to ∼1.5%. All the strains show the same pattern of variation with temperature, consistent with eqn (2) and a spin transition model which includes the effects of lattice vibrations (e.g.Fig. 6 of Zimmerman and König).46Fig. 14a shows the shear strain e5 and the volume strain Vs = ΔV/Vo and the latter has been scaled to give the variation of qs in Fig. 14b. The variation of qs is well represented by a sigmoidal function| | 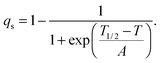 | (4) |
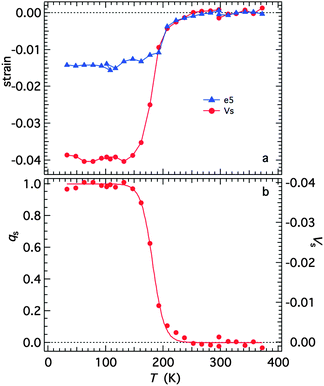 |
| | Fig. 14 (a) Spontaneous volume and shear strains determined from the lattice parameters, as set out in the ESI.† (b) Variation of the volume strain rescaled to values of between 0 and 1 as a representation of the order parameter. The curve is a fit of eqn (4) with T1/2 = 182 ± 1 K and A = 10.1 ± 0.6 K. | |
The fit parameters are T1/2 = 182 ± 1 K and A = 10.1 ± 0.6 K, and the transition occurs predominantly between limits of ∼130 and ∼250 K. Although the fit appears to produce a tightly constrained value of T1/2, it is actually highly dependent on a single data point at 192 K and a similar treatment of e5 gave T1/2 = 199 ± 2 K. The Fe–N bond lengths, Fe octahedron volume (Vp) and distortion parameter (Σ) are thought to be representative of the sample's spin state. Fitting a sigmoidal function to each of these parameters gives T1/2 values between 183 K and 185 K, see Fig. S4 (ESI†). The data are permissive of T1/2 = 188 K,12 reported previously.
The variation of elastic constants associated with changes in spin state can in principle be determined from eqn (1), by applying (after Slonczewski and Thomas,47 Zhang et al.).44
| | 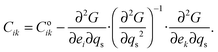 | (5) |
Strain coupling terms which are of the form
λqse will contribute to softening of the form
| | 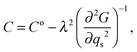 | (6) |
while terms of the form
λqse2 will give elastic constant variations of the form
| | 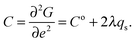 | (7) |
The difference, Δ
f2, between a simple linear baseline and the observed variations of
f2 from one of the resonances corresponds to
C–
C° and reaches a maximum softening of ∼35%. This is expected to be for shear elastic constants and made up of contributions of the form given by both
eqn (6) and (7) (including
C44,
C55,
C66). A breakdown into these two contributions is shown in
Fig. 15. The change due to the part proportional to
qs with
T1/2 taken as the temperature (188 K) where the softening is largest, was subtracted from Δ
f2 to give the remainder as the contribution proportional to the susceptibility. Softening of the latter is asymmetric about 188 K and this asymmetry is mirrored also in the acoustic loss,
Q−1. The maximum loss and the maximum value of (∂
2G/∂
qs2)
−1 approximately coincide, corresponding to the point at which a given applied stress induces the largest number of changes from one spin state to the other. The bulk modulus would be expected to follow
eqn (6) and should show a large softening, with the same form as the susceptibility component.
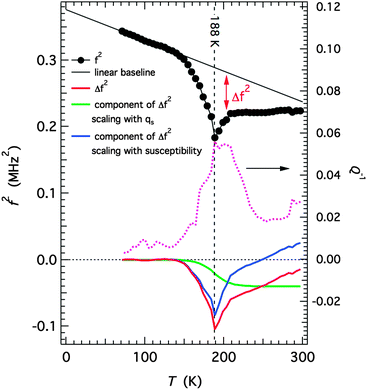 |
| | Fig. 15 Analysis of elastic softening through the thermal HS to LS spin transition. An estimate of the amount of softening, Δf2, for one of the resonance modes is given by the difference between a linear baseline and the observed values. This is then shown as being made up of two contributions, one which scales with qs (green curve) and the remainder which is assumed to scale with (∂2G/∂qs2)−1. This is only a semi-quantitative and non-unique solution but it fits with the pattern predicted on the basis of the model represented by eqn (1), (6) and (7). | |
Comparison of the four [Fe(abpt)2(NCS)2] polymorphs
As discussed earlier three of the known polymorphs undergo thermal spin crossover at ambient pressure, namely A, C and D. Sheu et al.12 reported that the value of the distortion parameter (Σ) and the volume of the Fe octahedron (Vp) for the HS and LS structures for all of these polymorphs are consistent with the expected values for an octahedral FeN6 geometry. These polymorphs all contain π–π interactions between uncoordinated and coordinated pyridyl rings of the abpt ligands on adjacent molecules. The shortest π–π separation (at room temperature) is different for each polymorph. For A, the centroid to centroid distance is ∼3.6 Å with an offset of ∼1.3–1.4 Å. For D, only the abpt ligands on one of the two independent Fe centres are involved in π–π interactions with a centroid to centroid distance of ∼3.8 Å and an offset of ∼2.0 Å. In the commensurate modulated structure of C at 130 K two of the four unique Fe centres have parallel or near parallel alignment for pairs of uncoordinated and coordinated pyridyl rings on adjacent abpt ligands with centroid to centroid distances of ∼3.8–4.1 Å and offsets of ∼1.7–2.0 Å. The presence of intermolecular interactions has often been linked to the occurrence of a spin transition and interestingly in polymorphs A and D the spin transition only occurs for Fe centres where the abpt ligands are involved in π–π interactions. In C the situation is complicated by the commensurate modulated structure prior to the spin transition, but at 130 K there is evidence of π–π interactions and at 25 K it is the Fe(II) centre with parallel alignment of pairs of rings on adjacent abpt ligands that is LS. In the case of B, which undergoes a thermal spin transition at high pressure, π–π interactions are also present between the uncoordinated and coordinated pyridyl rings on adjacent molecules (centroid to centroid distances of ∼3.7 Å and an offset of ∼1.1 Å at room temperature). Similar to the Fe centres in C and D which do not undergo spin crossover at ambient pressure, in B the abpt pyridyl group not coordinated to the Fe centre is twisted out of the plane of the rest of the abpt ligand.12
All three of the polymorphs that undergo a thermal spin transition at ambient pressure also display LIESST. In the case of C the LIESST causes the formation of a commensurate modulated phase in which the c-axis is tripled,12,13 while for D upon irradiation at 20 K two photoinduced metastable HS states can be formed, one the standard LIESST structure and the second with linkage isomerism of the NCS group.14 In the case of A, reported here, the LIESST structure formed was not found to be associated with any modulation or linkage isomerism and at 30 K was consistent with the HS structure above the thermal spin transition.
Conclusions
In summary, the spin crossover behaviour of [Fe(abpt)2(NCS)2] polymorph A has been structurally characterised. The integrity of the single crystal was maintained throughout the transition enabling structural information to be obtained. This showed that the Fe–N bond lengths change gradually and the molecules distort slightly over the temperature range of the spin transition. π–π interactions exist between adjacent abpt ligands in the crystal allowing potential communication between Fe(II) centres,12 this could explain the moderately cooperative nature of the spin transition in A. In addition to characterising the HS and LS structures of A, a LIESST HS* structure was obtained at 30 K which showed a 4% increase in unit cell volume occurred upon irradiation at that temperature. The structural parameters associated with the HS* structure were found to be consistent with those in the 270 K HS structure. It was necessary to irradiate the sample continuously throughout the data collection as the LIESST state relaxes back to the LS ground state within ∼4000 s, i.e. during the timeframe of the experiment. It has also been demonstrated that A undergoes a pressure induced spin transition resulting in a similar LS structure to that obtained at low temperature. The results of variable temperature UV-Vis spectroscopy and RUS experiments, which show obvious anomalies of both elastic moduli and acoustic dissipation at about 190 K, further confirm the thermal spin crossover transition and demonstrate strong spin–lattice coupling.
Acknowledgements
H. E. M. is grateful to Professor Jonathan W. Steed (Durham University) for useful discussions and Dr Michael R. Probert (Newcastle University) for advice on high pressure data collection. She is also grateful to the EPSRC and Durham University for financial support. W. L. acknowledges funding support from the Natural Science Foundation of China (Grant no. 21571072 and 61138006) and the Opening Project of the Key Laboratory of Cryogenics in the Technical Institute of Physics and Chemistry, Chinese Academy of Sciences. RUS facilities have been established and maintained in Cambridge through grants from the Natural Environment Research Council and the Engineering and Physical Sciences Research Council of Great Britain to M. A. C., which are gratefully acknowledged (NE/B505738/1, NE/F17081/1, EP/I036079/1).
References
- P. Gütlich, V. Ksenofontov and A. B. Gaspar, Coord. Chem. Rev., 2005, 249, 1811–1829 CrossRef.
-
A. Hauser, Light-Induced Spin Crossover and the High-Spin→Low-Spin Relaxation. Spin Crossover in Transition Metal Compounds II, Top. Curr. Chem., Springer-Verlag, Berlin, Heidelberg, New York, 2004, vol. 234, pp. 155–198 Search PubMed.
- P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342–391 CrossRef.
- M. Nihei, T. Shiga, Y. Maeda and H. Oshio, Coord. Chem. Rev., 2007, 251, 2606–2621 CrossRef.
- A. B. Gaspar, M. C. Muñoz, V. Niel and J. A. Real, Inorg. Chem., 2001, 40, 9–10 CrossRef CAS.
- P. Nielsen, H. Toftlund, A. D. Bond, J. F. Boas, J. R. Pilbrow, G. R. Hanson, C. Noble, M. J. Riley, S. M. Neville, B. Moubaraki and K. S. Murray, Inorg. Chem., 2009, 48, 7033–7047 CrossRef CAS.
-
Spin Crossover in Transition Metal Compounds I, II and III, ed. P. Gütlich and H. A. Goodwin, Springer-Verlag, Berlin, Heidelberg, New York, 2004 Search PubMed.
-
Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Ltd, Chichester, UK, 2013 Search PubMed.
- M. Griffin, S. Shakespeare, H. J. Shepherd, C. J. Harding, J.-F. Létard, C. Desplanches, A. E. Goeta, J. A. K. Howard, A. K. Powell, V. Mereacre, Y. Garcia, A. D. Naik, H. Müller-Bunz and G. G. Morgan, Angew. Chem., Int. Ed., 2011, 50, 896–900 CrossRef CAS.
- J. Tao, R.-J. Wei, R.-B. Huang and L.-S. Zheng, Chem. Soc. Rev., 2012, 41, 703–737 RSC.
- N. Moliner, M. C. Muñoz, S. Létard, J. F. Létard, X. Solans, R. Burriel, M. Castro, O. Kahn and J. A. Real, Inorg. Chim. Acta, 1999, 291, 279–288 CrossRef CAS.
- C.-F. Sheu, S.-M. Chen, S.-C. Wang, G.-H. Lee, Y.-H. Liu and Y. Wang, Chem. Commun., 2009, 7512–7514 RSC.
- C.-H. Shih, C.-F. Sheu, K. Kato, K. Sugimoto, J. Kim, Y. Wang and M. Takata, Dalton Trans., 2010, 39, 9794–9800 RSC.
- C.-F. Sheu, C.-H. Shih, K. Sugimoto, B.-M. Cheng, M. Takata and Y. Wang, Chem. Commun., 2012, 48, 5715–5717 RSC.
- A. B. Gaspar, M. C. Muñoz, N. Moliner, V. Ksenofontov, G. Levchenko, P. Gütlich and J. A. Real, Monatsh. Chem., 2003, 134, 285–294 CrossRef CAS.
- J.-F. Létard, P. Guionneau, L. Rabardel, J. A. K. Howard, A. E. Goeta, D. Chasseau and O. Kahn, Inorg. Chem., 1998, 37, 4432–4441 CrossRef.
- A. E. Goeta, L. K. Thompson, C. L. Sheppard, S. S. Tandon, C. W. Lehmann, J. Cosier, C. Webster and J. A. K. Howard, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1243–1246 CAS.
-
Bruker, SMART-NT, Data Collection Software, Version 5.63, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 2000 Search PubMed.
-
Bruker, SAINT-NT, Data Reduction Software Ver. 6.14, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 2000 Search PubMed.
-
Bruker, SADABS, Bruker AXS area detector scaling and absorption correction, Bruker Analytical X-ray Instruments Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. More, G. Odou and J. Lefebvre, Acta Crystallogr., Sect. B: Struct. Sci., 1987, 43, 398–405 CrossRef.
- T. Fukami, S. Akahoshi, K. Hukuda and T. Yagi, J. Phys. Soc. Jpn., 1987, 56, 2223–2224 CrossRef CAS.
- P. N. Martinho, B. Gildea, M. M. Harris, T. Lemma, A. D. Naik, H. Müller-Bunz, T. E. Keyes, Y. Garcia and G. G. Morgan, Angew. Chem., Int. Ed., 2012, 51, 12597–12601 CrossRef CAS.
-
M. R. Probert, SMARTreduce, Durham University, UK, 2009 Search PubMed.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46, 2774–2780 CrossRef CAS.
- M. R. Probert, J. A. Coome, A. E. Goeta and J. A. K. Howard, Acta Crystallogr., Sect. A: Found. Crystallogr., 2011, 67(suppl.), C528 Search PubMed.
- M. R. Probert, C. M. Robertson, J. A. Coome, J. A. K. Howard, B. C. Michell and A. E. Goeta, J. Appl. Crystallogr., 2010, 43, 1415–1418 CAS.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
-
Bruker, APEX2, Data Collection Software, Version 1.08, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 2004 Search PubMed.
-
Bruker, SAINT + Integration Engine, Data Reduction Software, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 2007 Search PubMed.
-
S. Parsons, ECLIPSE, University of Edinburgh, Edinburgh, UK, 2009 Search PubMed.
-
OriginLab, OriginPro 8, OriginLab, Northampton, MA, USA Search PubMed.
-
A. Migliori and J. L. Sarrao, Resonant Ultrasound Spectroscopy: Applications to Physics, Material Measurements and Nondestructive Evaluation, Wiley, New York, 1997 Search PubMed.
- R. E. A. McKnight, M. A. Carpenter, T. W. Darling, A. Buckley and P. A. Taylor, Am. Mineral., 2007, 92, 1665–1672 CrossRef CAS.
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC.
- V. Legrand, S. Pillet, H.-P. Weber, M. Souhassou, J.-F. Létard, P. Guionneau and C. Lecomte, J. Appl. Crystallogr., 2007, 40, 1076–1088 CAS.
- H. J. Shepherd, S. Bonnet, P. Guionneau, S. Bedoui, G. Garbarino, W. Nicolazzi, A. Bousseksou and G. Molnár, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 145401 CrossRef.
- V. Legrand, S. Pechev, J.-F. Létard and P. Guionneau, Phys. Chem. Chem. Phys., 2013, 15, 13872–13880 RSC.
- H. J. Shepherd, P. Rosa, L. Vendier, N. Casati, J.-F. Létard, A. Bousseksou, P. Guionneau and G. Molnár, Phys. Chem. Chem. Phys., 2012, 14, 5265–5271 RSC.
- N. Moliner, M. C. Muñoz, P. J. van Koningsbruggen and J. A. Real, Inorg. Chim. Acta, 1998, 274, 1–6 CrossRef CAS.
- M. Marchivie, P. Guionneau, J. F. Létard, D. Chasseau and J. A. K. Howard, J. Phys. Chem. Solids, 2004, 65, 17–23 CrossRef CAS.
- Z. Zhang, J. Koppensteiner, W. Schranz and M. A. Carpenter, Am. Mineral., 2012, 97, 399–406 CrossRef CAS.
- Z. Zhang, J. Koppensteiner, W. Schranz, D. Prabhakaran and M. A. Carpenter, J. Phys.: Condens. Matter, 2011, 23, 145401 CrossRef.
- R. Zimmermann and E. König, J. Phys. Chem. Solids, 1977, 38, 779–788 CrossRef CAS.
- J. C. Slonczewski and H. Thomas, Phys. Rev. B: Condens. Matter Mater. Phys., 1970, 1, 3599–3608 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: LIESST relaxation plots, change in C2–H2⋯N4#1 (#1 = −x + 1, −y + 1, −z + 1) distance plot, additional crystal data not in main paper, tables of hydrogen bonding distances and strain analysis. CCDC 1422236–1422246. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj02359a |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a