DOI:
10.1039/C5NJ02287H
(Paper)
New J. Chem., 2016,
40, 1640-1648
Schiff base tailed silatranes for the fabrication of functionalized silica based magnetic nano-cores possessing active sites for the adsorption of copper ions†
Received
(in Montpellier, France)
28th August 2015
, Accepted 8th December 2015
First published on 11th December 2015
Abstract
One pot fabrication of functionalized magnetite nanoparticles using Schiff base tethered silatranes as modifiers is reported. In this work a new Schiff base functionalized silane and the corresponding silatrane were synthesized by the condensation of 2-hydroxy-4-methoxybenzophenone with 3-aminopropyltriethoxysilane and -silatrane, respectively. The latter was purified by convenient recrystallization. Thus, the silatrane, which was characterized by elemental analysis, spectroscopic techniques and single crystal X-ray diffraction, was used as a modifier to functionalize magnetite nano-cores to introduce active lone-pair donor systems on their surface. The Schiff base functionalized magnetite nano-particles were characterized by Powder X-Ray Diffraction (XRD), Fourier Transform Infrared Spectroscopy (FT-IR), Scanning Electron Microscopy (SEM), Transmission Electron Microscopy (TEM) and Vibrating Sample Magnetometry (VSM). These studies revealed superparamagnetic nanoparticles of polydispersed spherical shape. Although functionalization of the nano-cores introduces some organic moieties onto the surface, some properties of the nano-particles have not been altered significantly, i.e. their magnetic nature, spherical shape and XRD patterns were retained even after the modification. Moreover, Langmuir adsorption isotherm measurement revealed that the Schiff base functionalized nanoparticles possess better adsorption capacity for copper ions (0.207 mmol g−1) as compared to magnetite nanocores coated with silica (0.141 mmol g−1). The Schiff base functionalized magnetic nanomaterial can be used for the adsorption of copper ions without interferences because the adsorption remained unaffected in the presence of other metal ions up to certain limits.
1. Introduction
Tailed siloxy derivatives have attracted much attention in the past few years due to their potential to functionalize various solid surfaces via Si–O covalent bonds.1,2 These functionalized surfaces have a wide range of applications in the field of catalysis,3 chemosensing,4 atomic force microscopy,5 fabrication of monolithic columns,6 enzyme immobilization,7 molecular imprinting,8 material science,9 and separation science.10 Engaged in the synthesis of tailed silatranes,11–13 we examined the potential of dye functionalized silatranes as useful precursors for the modification of silica nano-cores by azo- and azomethine functionalities.14 It was investigated that silatranes may act as better precursors relative to their trialkoxysilane analogues because they can be purified in a convenient manner (by recrystallization), and their comparatively slow immobilization results in the uniform coating of surfaces.15–17 Although the dye coated silica nanoparticles offered high adsorption capacity for copper ions, some problems were encountered during the adsorption studies. Major problems were associated with the isolation of silica nanoparticles as they often blocked the pores of various filters because of their size, which made the filtration process slow and laborious. Therefore, it is important to engineer the adsorbent in such a way that it can be isolated easily from the solution. In the present study, we selected magnetic nano-cores instead of silica nano-cores to functionalize a solid surface using silatrane as a precursor and used the particles for the adsorption of copper ions.
The magnetic nano-cores of magnetite nanoparticles (MNPs) exhibit a high surface-to-volume ratio resulting in a high adsorption capacity for metal ions.18–22 These particles have been widely used as inorganic supports in biology and medicine such as in protein and enzyme immobilization,23 immunoassay,24 RNA and DNA purification,25 magnetic resonance imaging (MRI),26 cell isolation and target drug.27 They have attracted particular interest in separation science and heterogeneous catalysis, because they can be easily isolated from the reaction mixture using an external magnetic field.28–31 They can be prepared by different chemical methods like co-precipitation, hydrothermal, microemulsion, electrochemical deposition, sonochemical, and thermal decomposition.32–36 Co-precipitation is one of the most efficient and simple methods for the synthesis of MNPs.37
Direct modification of magnetite nano-particles (Fe3O4) with organic functionalities is difficult due to certain problems like: (1) the anisotropic dipolar interaction between magnetic nanoparticles leads to their agglomeration to form large clusters losing some specific properties associated with the individual domains; (2) bare iron oxide nanoparticles may undergo rapid biodegradation under certain conditions; and (3) they possess few hydroxyl groups on the surface for derivatization.38 Therefore, coating of MNPs is required with some suitable coating materials such as surfactants, polymers, gold, silica etc.39,40 Amongst these, silica coating is more favourable because it is chemically stable, nontoxic, biocompatible and possesses a high surface area.41 Moreover, it reduces agglomeration of MNPs as it acts as a spacer between the magnetic dipoles and enhances repulsive forces between negatively charged silica coated surfaces. Furthermore, the presence of silanol groups on the surface of silica provides a platform for grafting of a variety of surface modifiers. Silica encapsulation of MNPs can be achieved by sol–gel processing42 and micro emulsion.43
Till date silica coated MNPs have been modified by some tailed alkoxysilanes like aminopropyl- and mercaptopropyltriethoxysilane (MPTES).44 In few reports, the amino functionalized MNPs have been further modified to Schiff base functionalized MNPs by their treatment with carbonyl compounds and used for catalytic as well as metal adsorption purposes.45,46 Herein, we elucidate the one-pot surface modification of MNPs to afford Schiff base functionalized silica magnetite nanoparticles (SB-SiO2@MNPs), their characterization and application. The newly fabricated SB-SiO2@MNPs showed remarkable adsorption capacity for copper ions. Previously, the adsorption of copper ions has been reported on the solid surfaces functionalized with a Schiff base,47 a surfactant,48 dithiazone,49 and triethoxysilanes.19,50 It is advantageous to use a Schiff base functionalized material as an adsorbent over silica sorbents as it eliminates pre- and post-derivatization steps, possesses excellent stability of MNPs, facile separation of the sorbent using an external magnet and easy functionalization of the surface in one step only. To the best of our knowledge, this is the first report wherein magnetically recoverable particles are functionalized in one pot using a Schiff base containing tailed silatrane as a modifier.
2. Experimental
2.1. Materials
Toluene, hexane and diethyl ether were dried by heating over sodium with benzophenone and dichloromethane over phosphorus pentaoxide before use and stored over molecular sieves for 24 h under a nitrogen atmosphere. Absolute ethanol with 99.9% purity (0.05% methanol, 0.01% iso-propyl alcohol, and 0.1% water) was used as such. Ferric sulphate (Acros), ferrous sulphate (Acros), ammonia (Sd-fine), tetraethoxysilane (Acros), 2-hydroxy-4-methoxybenzophenone (Aldrich), 3-aminopropyl(triethoxy)silane (Aldrich), triethanolamine (Merck), trisodium citrate (Merck), ethanol absolute (Merck), and CaCl2 (Fisher Scientific) were used as such without any purification. 3-Aminopropylsilatrane was synthesized from 3-aminopropyltriethoxysilane and triethanolamine as reported in the literature.51
2.2. Physical measurements
Infrared spectra were routinely obtained on a Thermo scientific NICOLET IS50 FT-IR and a Perkin Elmer RX-I FT IR spectrophotometer. Mass spectral measurements (ESI source with capillary voltage 2500 V) were carried out on a VG Analytical (70-S) spectrometer. C, H, N elemental microanalyses were obtained on a FLASH-2000 organic elemental analyzer. The solution NMR spectra were recorded at 25 °C on a Bruker Avance II FT NMR (AL 400 MHz) spectrometer (1H, 13C) and on a Bruker DPX 400 spectrometer (1H, 13C, 29Si). Chemical shifts in ppm are reported relative to tetramethylsilane (TMS). Single crystal X-ray structure analyses were carried out on a Bruker X8 APEX2 CCD diffractometer (1) and on a Stoe IPDS-2T diffractometer (2) using Mo Kα-radiation (λ = 0.71073 Å). The structures were solved by direct methods (SHELXS-97) and refined with full-matrix least-squares method (refinement of F2 against all reflections with SHELXL-97 for 2 and SHELXL-2014 for 1). Even though the structure of 1 in modification 1 does not exhibit any disorder, the crystal had very poor diffraction power. Thus, in order to save variables (to perform a final refinement with an observed data/parameter ratio >5), the phenyl groups were refined as idealised benzene rings (refinement code AFIX 66). Furthermore, for better bond length precision each set of chemically corresponding bonds of the two crystallographically independent molecules in the asymmetric unit was treated with the same distance restraint (SADI 0.005 for non-H atom bonds, SADI 0.01 for the OH groups). The crystal of the 2nd modification of this compound, 1a, exhibited satisfactory diffraction power, but the molecule (in this case only one molecule of 1 forms the asymmetric unit) exhibits severely disordered ethoxy groups of the Si(OEt)3 moiety. Thus, each O–CH2–CH3 moiety was refined in two positions and for each set the site occupancies refined to different ratios [0.81(1),0.19(1); 0.77(1),0.23(1); 0.40(1),0.60(1)]. The sets of Si–O, O–C and C–C bonds for each set of two alternative OEt moieties were restrained to same distances (SADI 0.005). Furthermore, SIMU 0.01 0.01 and DELU 0.01 0.01 restraints were used for each O–CH2–CH3 chain to produce reasonable thermal displacement ellipsoids. In the structure of 2 the CH2–CH2 bridges of the silatrane cage are disordered in a mutual manner (alternative direction of the propeller tilt of the cage), thus the whole disorder of the N(CH2CH2)3 cage was refined with one set of site occupancy variables [0.828(2),0.172(2)]. Same distance restraints (SADI 0.001) were applied to corresponding C–O, C–C and C–N bonds. Same thermal displacement parameters (EADP) were refined for corresponding C and N atoms. Furthermore, this structure contains water as a solvent of crystallization. The site occupancy of the water molecule was refined to a value close to 15% and was then fixed to 15% in the final refinement. The hydrogen atoms of this water molecule were refined with fixed O–H distances (0.92 Å) and a fixed H–H separation of 1.5 Å to give a reasonable H–O–H angle. A vibrating sample magnetometer (EV-9, Microsense, ADE) was utilized for obtaining the magnetization curves. The morphology, size and shape of magnetic silica nanoparticles were investigated by Scanning Electron Microscopy (SEM) using a Tescan Mira 3 FESEM instrument and Transmission Electron Microscopy (TEM) at 80 kV using a Hitachi H-7500 instrument. Powder X-ray Diffraction (XRD) data were collected on a PANalytical's X'Pert PRO diffractometer using Cu-Kα radiation (λ = 1.541 Å) in 2θ range from 20° to 80°. Electronic spectral measurements were carried out on an ESICO 23750 double beam spectrophotometer in the range 200–700 nm. Brunauer–Emmett–Teller (BET) surface areas of the nanoparticles were determined by N2 adsorption/desorption on a Quantachrome instrument.
2.3. Syntheses
2.3.1 Schiff base functionalized silane 1; (2-((E)-(3-(triethoxysilyl)propylimino)(phenyl)methyl)-5-methoxyphenol).
In a 500 mL round bottom flask fitted with a dropping funnel and a Dean-Stark-trap, a solution of 2-hydroxy-4-methoxybenzophenone (39.9 g, 175 mmol) in toluene (200 mL) was heated to reflux. Then, 3-aminopropyltriethoxysilane (38.7 g, 175 mmol) was slowly added via a dropping funnel and the contents were heated under reflux until the completion of water formation (as indicated by the water collected in the trap). Thereafter, the solvent was removed under vacuum to afford the product as yellow oil. Upon storage of the oil at room temperature (for 2 weeks), it transformed into a yellow sticky solid. For X-ray structure analysis some small crystalline needles were manually extracted from this solid.
Yield: quantitative. (According to 29Si NMR spectroscopy, this product contains ca. 5 mol% of the corresponding disiloxane [(imine)-(CH2)3–Si(OEt)2]2O, indicated by a signal at −53.5 ppm. Because of this impurity melting point determination and elemental analyses have not been performed.) 1H NMR (400 MHz, CDCl3): δ (ppm) 0.65 (t, 2H, SiCH2, J = 8.0 Hz), 1.20 (t, 9H, Me, J = 6.8 Hz), 1.77 (m, 2H, CCH2C), 3.26 (m, 2H, NCH2C), 3.78 (q, 6H, O–CH2, J = 6.8 Hz) + (s, 3H, OMe), 6.07 (d, 1H9, J = 8.8 HZ), 6.38 (s, 1H11), 6.60 (d, 1H8, J = 8.8 Hz), 7.21–7.48 (m, 5H, phenyl), 16.51 (s, 1H, OH). 13C NMR (100.6 MHz, CDCl3): δ (ppm) 8.0 (SiCH2), 18.3 (O–CH2), 24.2 (CCH2C), 51.3 (CCH2N), 55.2 (OCH3), 58.4 (OCH2), 102.1 (C11), 105.5 (C9), 112.2 (C7), 127.5 (C3,5), 128.7 (C2,6), 129.3 (C4), 132.9 (C8), 132.9 (C1), 164.5 (C12), 172.0 (C10), 173.2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). 29Si NMR (CDCl3, 79.5 MHz): δ (ppm) −49.5.
N). 29Si NMR (CDCl3, 79.5 MHz): δ (ppm) −49.5.
2.3.2 Schiff base functionalized silatrane 2; (2-((E)-(3-(silatranyl)propylimino)(phenyl)methyl)-5-methoxyphenol).
In a 100 mL round bottom flask equipped with a Dean-Stark-trap, 3-aminopropylsilatrane (0.50 g, 2.15 mmol) was dissolved in toluene (10 mL). The equimolar amount of 2-hydroxy-4-methoxybenzophenone (dissolved in toluene, 10 mL) was added drop wise using a syringe. The contents were heated under reflux and water produced during reaction (forming an azeotrope with toluene) was collected in the Dean-Stark-trap. The contents of the reaction flask were cooled to room temperature and toluene was removed under vacuum to afford a solid, which was washed with diethyl ether and dried under reduced pressure.
Yield (0.73 g, 78%). M.p.: 120-122 °C. Elemental analysis: anal. calcd for C23H30N2O5Si: Found: C, 62.18; H, 6.90; N, 6.28 requires C, 62.42; H, 6.83; N, 6.33. IR (KBr pellet, νmax cm−1): 578 m (νSi ← N), 716 s, 758 s (νsSiO), 836 m, 872 w (νC–N), 907 m (νsNC3), 933 m (νC–C), 968 w, 1014 s (νasNC3), 1095 vs. (νasSiO), 1124 vs. (νC–O), 1163 m (τCH2O), 1216 m (ωCH2O), 1272 m (ωCH2N), 1340 w, 1444 s (δCH3C), 1586 vs. (νCN), 2872.4 s, 2929 (νsCH2), 3493 s (νOH). 1H NMR (400 MHz, CDCl3, Me4Si): δ (ppm) 0.27 (m, 2H, SiCH2), 1.86 (m, 2H, CCH2C), 2.67 (t, 6H, NCH2C, J = 5.8 Hz), 3.14 (t, 2H, CCH2N, J = 7.7 Hz), 3.60 (t, 6H, OCH2, J = 5.8 Hz), 3.70 (s, 3H, OCH3), 5.99 (d, 1H9, J = 2.52), 6.29 (d, 1H11, J = 2.52), 6.55 (d, 1H8, J = 9.08), 7.25 (m, 2H2,6), 7.48 (m, 3H3,4,5) 16.41 (s, 1H, OH). 13C NMR (100.6 MHz, CDCl3): δ (ppm) 13.2 (SiCH2), 26.4 (CCH2C), 51.5 (CCH2N), 54.9 (CH2N), 57.8 (OCH3), 68.7 (OCH2), 102.6 (C11), 105.0 (C9), 111.2 (C7), 127.7 (C3,5), 128.4 (C2,6), 129.2 (C4), 132.2 (C8), 133.1(C1), 165.0 (C12), 172.5 (C10), 175.5 (CN). MS: m/z (relative abundance (%), assignment): 174 (3.48, Si(OCH2CH2)3N+), 433 (100, M + H+), 465 (2.79, M + Na+).
A suitable crystal for X-ray crystallography of compound 2 was obtained by recrystallization at room temperature from DCM/hexane mixtures.
2.3.3 Magnetite nanoparticles (MNPs).
Magnetite (Fe3O4) nanoparticles were synthesized according to a previously reported co-precipitation technique.52 In a typical synthesis, ferric sulphate (6.00 g, 14.3 mmol) and ferrous sulphate heptahydrate (4.20 g, 15.1 mmol) salts were dissolved in double de-ionized water (250 mL) and stirred at 60 °C under nitrogen to afford a yellow-orange solution. Then, 25% NH3 solution (12 mL) was added drop wise and vigorous stirring was continued for an hour. The crystalline black precipitate of Fe3O4 nanoparticles was isolated simply via an external magnetic force, washed thoroughly with water and ethanol several times until the solution reached neutral pH. To stabilize the nanoparticles, the “magnetic mud” was redispersed in 200 mL of a solution of trisodium citrate (0.5 M) and stirred for an hour at 80 °C. Thereafter, the Fe3O4 nanoparticles were harvested using an external magnet and washed with acetone to remove the excessive citrate groups. The nanoparticles obtained were dried under vacuum at 50 °C.
2.3.4 Schiff base functionalized silica magnetite nanoparticles (SB-SiO2@MNPs).
The modification of MNPs with silica and Schiff base functional groups was carried out in one pot. First, 0.34 g of magnetite nanoparticles was dispersed in 60 mL of absolute ethanol (99.9%) via sonication under inert atmosphere to form a suspension. Then, under vigorous stirring at room temperature, 1.5 mL of tetraethoxysilane and 2 mL of 25% ammonia solution were added to the suspension of magnetic nanoparticles. Vigorous stirring was continued for 3 h followed by the addition of an absolute ethanolic solution of 0.40 g of Schiff base silatrane 2 (modifier). Stirring of the suspension under inert atmosphere was continued overnight. The resulting product was separated using an external magnet and washed with ethanol in order to remove the excess modifier. The modified nanoparticles were dried under vacuum at room temperature. For reference, silica coated magnetite nanoparticles (SiO2@MNPs) were synthesized via a similar procedure without adding Schiff base silatrane 2.
2.4 Adsorption studies
SiO2@MNPs and SB-SiO2@MNPs were used for the adsorption of copper(II) ions. The adsorption experiment was performed at room temperature by stirring 30 mg of functionalized and non-functionalized silica coated magnetite nanoparticles for 25 min with 5 mL of copper chloride solution (in ethanol) of different concentrations (0.787 mmol L−1 to 2.36 mmol L−1) to reach the equilibrium. After stirring, nanoparticles were separated magnetically, and the remaining concentration of copper ions in the solution was determined by UV spectrophotometry at λ = 270 nm.
The adsorption amount (Qe, mmol g−1) was calculated by knowing the values of concentration of copper ions before and after adsorption according to eqn (1)
where
C0 is the initial concentration of Cu
2+ ions (mmol L
−1),
Ce is the concentration of Cu
2+ ions at adsorption equilibrium (mmol L
−1),
V is the volume of CuCl
2 solution (0.005 L), and
M is the weight of the adsorbent,
i.e., SiO
2@MNPs or SB-SiO
2@MNPs (0.030 g). The adsorption capacity of both types of MNPs was calculated using the Langmuir equation, the linear form of the isotherm is given by
eqn (2).
| | | Ce/Qe = Ce/Qm + 1/(KaQm) | (2) |
where
Ce (mmol L
−1) and
Qe (mmol g
−1) are the Cu
2+ ion concentration and adsorption amount at adsorption equilibrium.
Qm (mmol g
−1) and
Ka (L mmol
−1) are the theoretical maximum adsorption capacity and the Langmuir equilibrium constant related to theoretical maximum adsorption capacity and energy of adsorption, respectively.
The interference studies were done for the adsorption of copper ions in the presence of other metal ions. In this operation, different amounts of various metal ions ranging from 1 μg mL−1 to 150 μg mL−1 were added to 5 mL of copper ion solution (1.57 mM). The adsorption amount of copper ions was calculated following the same procedure mentioned above.
3. Results and discussion
3.1 Syntheses
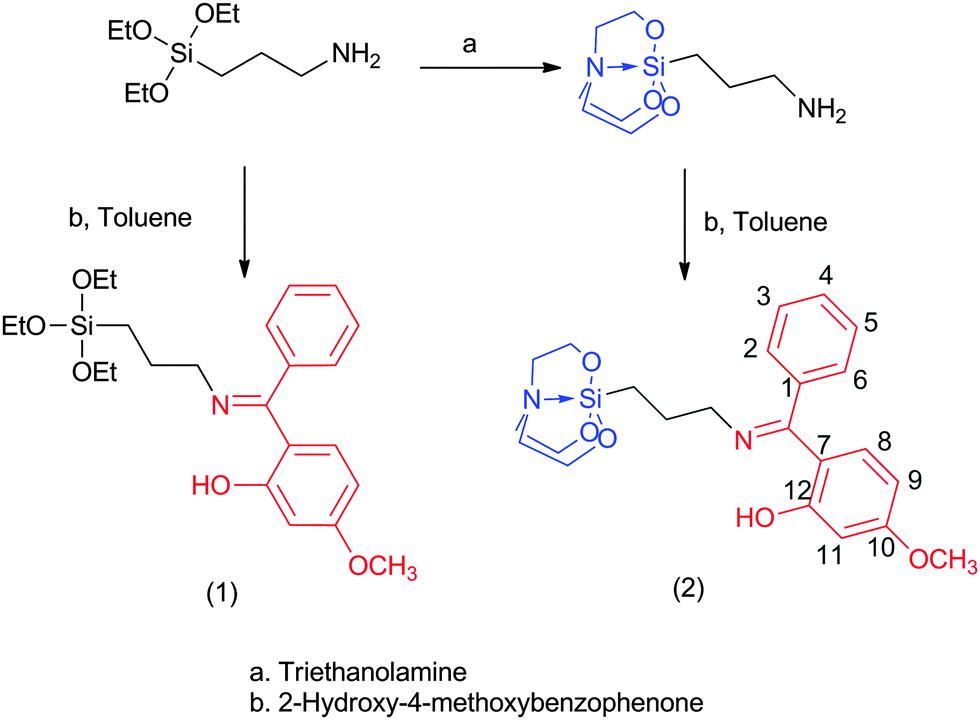
The reaction of 3-aminopropyltriethoxysilane with 2-hydroxy-4-methoxybenzophenone yielded the corresponding Schiff base silane (1) as an oil, which solidified after two weeks. This compound is very well soluble in various organic solvents, and therefore purification by recrystallization (in order to remove some byproducts, which had formed during the condensation reaction) was not successful. In comparison, the reaction of 2-hydroxy-4-methoxybenzophenone with 3-aminopropyltriethoxysilatrane yielded the corresponding Schiff base silatranes in solid form (Scheme 1), which could easily be separated from the supernatant. Therefore, we decided to use 2 as a modifier of the silica coated magnetite nanoparticles as it can be isolated in pure form.
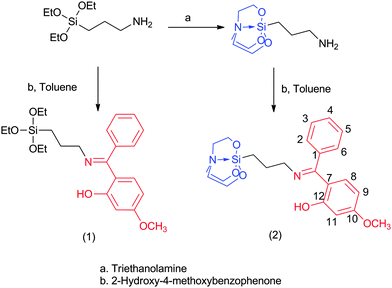 |
| | Scheme 1 Synthetic route for Schiff base-triethoxysilane (1) and -silatrane (2). | |
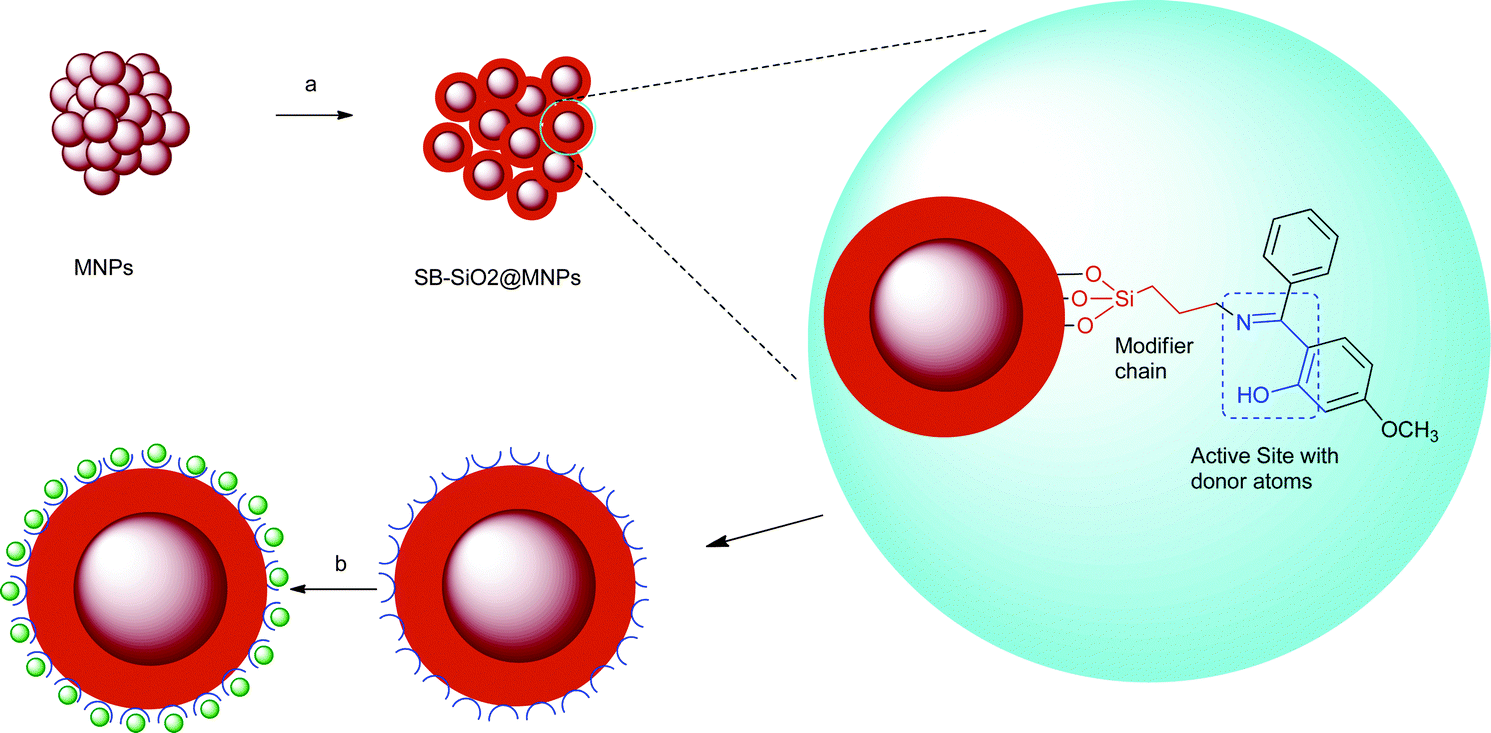
In the fabrication of SB-SiO2@MNPs, magnetic nanocores were isolated in the first step and then treated with citrate solution to reduce agglomeration of the nanocores. In the next step, TEOS was added to the citrate coated MNPs, which formed a silica layer composed of an O–Si–O polymeric network on the surface of MNPs (SiO2@MNPs).53 This step generated a large number of silanol groups and facilitated the silanization of magnetic nanocores with the modifier. The modifier, SB–Si(OCH2CH2)3N, contains a silatranyl moiety, which hydrolyzes under basic conditions with the formation of triethanolamine (N(CH2CH2OH)3) and SB–Si(OH)3. The latter undergoes condensation polymerization with silica via silanol groups to form Si–O bonds without affecting the Schiff base moiety. Further polymerization in the presence of a modifier and TEOS distributed the azomethine groups in the silica network to form SB-SiO2@MNPs, which facilitated the adsorption of Cu(II) ions (Scheme 2).
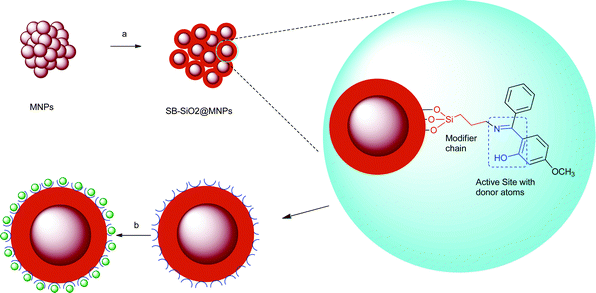 |
| | Scheme 2 Illustration for the fabrication of SB-SiO2@MNPs and adsorption of copper ions; (a) TEOS, 2, ethanol, 25% ammonia, stirring; (b) copper ions. | |
3.2 Characterization of compounds
Silatrane 2 exhibits a band in the IR region 1600–1620 cm−1 indicating the formation of the Schiff base derivative of 3-aminopropylsilatrane. The appearance of the absorption band at 3493 cm−1 in compound 2 indicated the presence of the hydroxyl group. The absorption bands observed in the regions of 1086–1095 cm−1 and 577–579 cm−1 can be assigned to the stretching vibration of Si–O and Si–N, respectively. 1H NMR spectra of 2 having triplets in regions 2.69–2.78 and 3.65–3.75 ppm due to the silatrane NCH2 and OCH2 groups, respectively, supported retention of the silatrane motif. Mass spectra of compound 2 exhibit corresponding molecular ion peaks in addition to the peak at m/z = 174 due to the cleavage of a Si–CH2 bond.
3.3. Single crystal X-ray crystallography
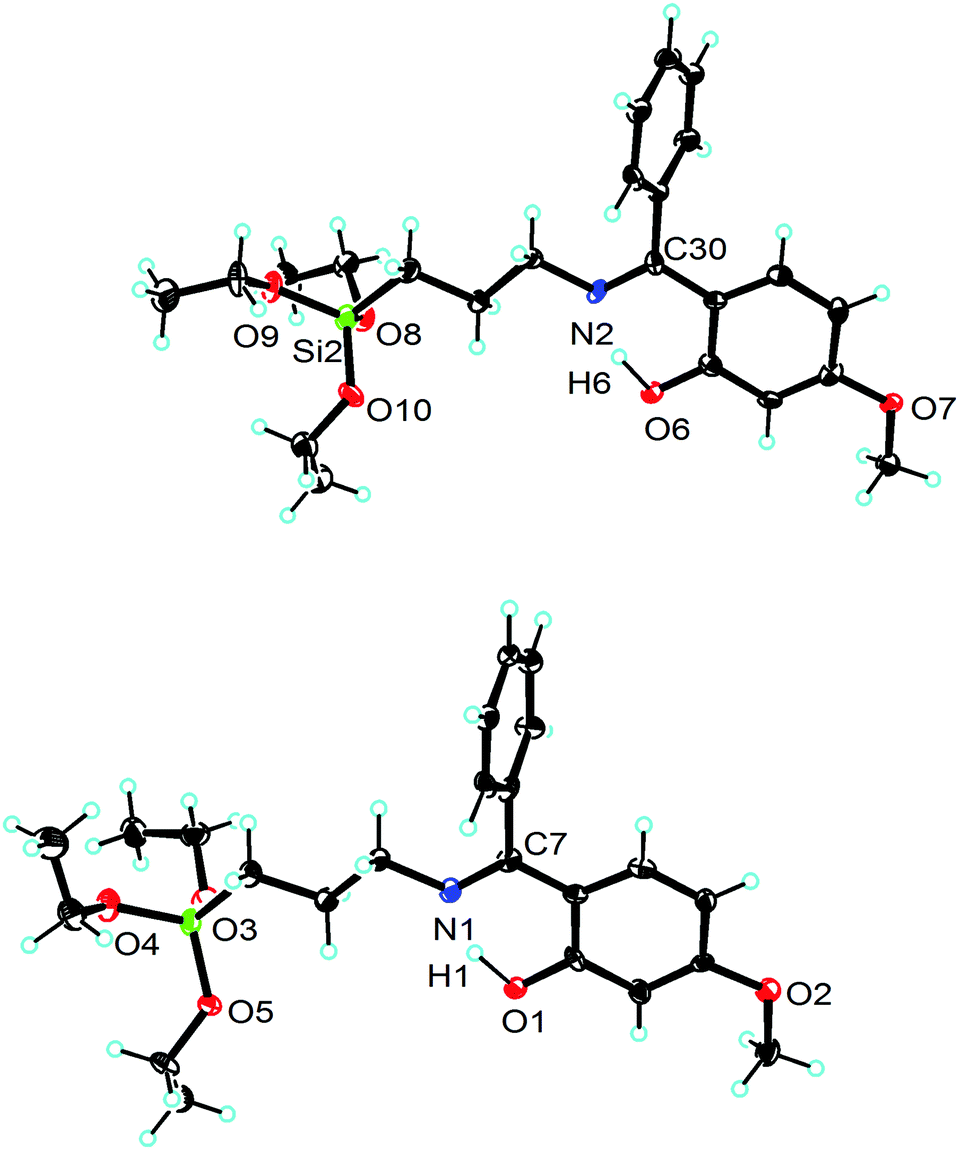
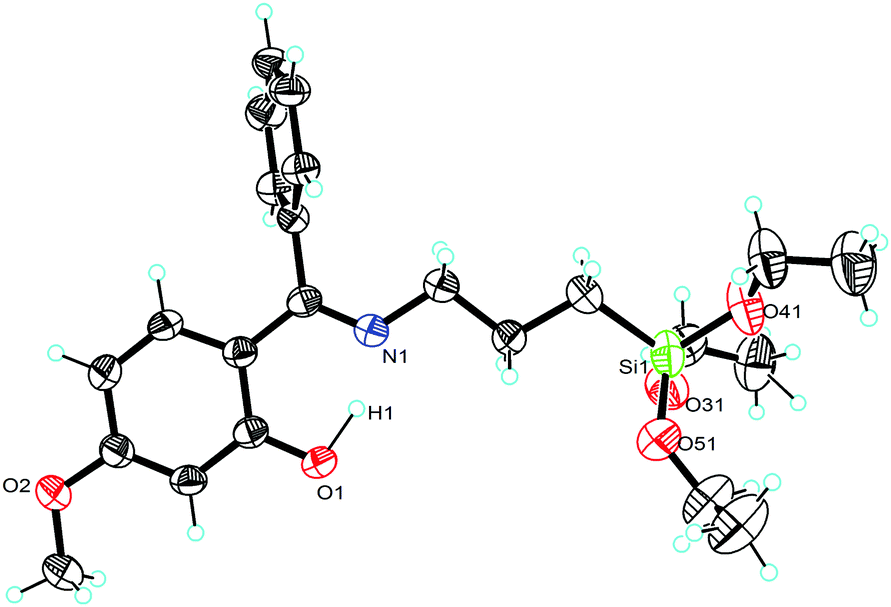
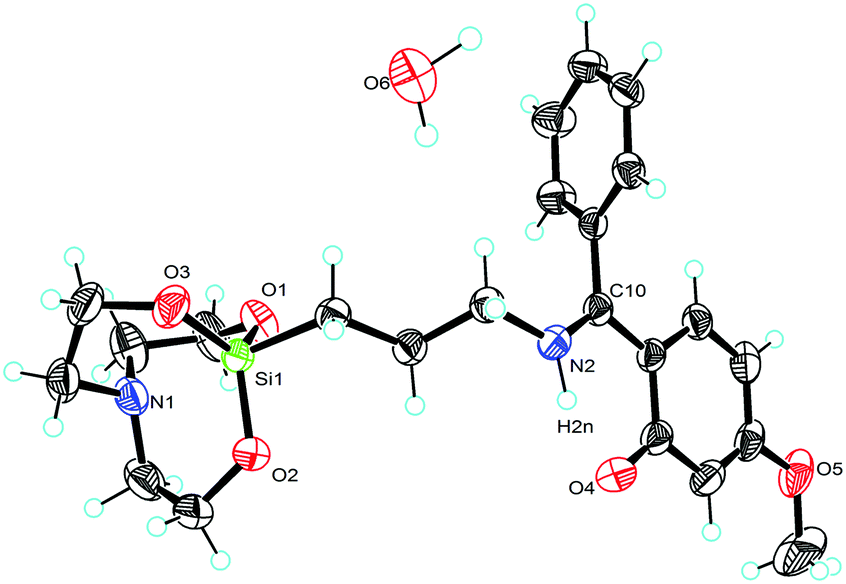
Molecular structures of compounds 1 and 2 with selected bond angles and bond lengths are shown in Fig. 1–3 along with atomic numbering scheme. X-ray diffraction data sets for compound 1 were collected from two different crystals which turned out to be two different modifications (reported as 1 and 1a). X-ray crystallographic parameters are given in Table 1. The silatrane propeller of 2 is two-fold disordered in ratio 0.828(2)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.172(2). In case of the orientation with lower occupancy, a water molecule could be situated next to the silatrane moiety, and an electron density peak found in this cavity was thus refined as a water molecule. Its occupancy was refined to 0.142, therefore it was then set to 0.15 for the final refinement.
:0.172(2). In case of the orientation with lower occupancy, a water molecule could be situated next to the silatrane moiety, and an electron density peak found in this cavity was thus refined as a water molecule. Its occupancy was refined to 0.142, therefore it was then set to 0.15 for the final refinement.
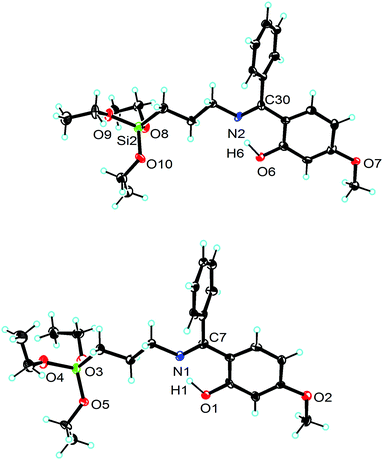 |
| | Fig. 1 Ortep view of 1 (ellipsoids are shown at the 40% probability level). The asymmetric unit consists of two independent molecules. Selected bond lengths [Å] and angles [deg]: molecule 1: Si(1)–O(3) 1.628(3), Si(1)–O(4) 1.627(3), Si(1)–O(5) 1.627(3), O(5)–Si–O(3) 107.7(3), O(5)–Si–O(4) 113.1(3), O(3)–Si–O(4) 105.8(3); molecule 2: Si(2)–O(8) 1.623(3), Si(2)–O(9) 1.631(3), Si(2)–O(10) 1.629(3), O(8)–Si(2)–O(10) 107.9(3), O(8)–Si(2)–O(9) 106.4(3), O(10)–Si(2)–O(9) 112.6(3). | |
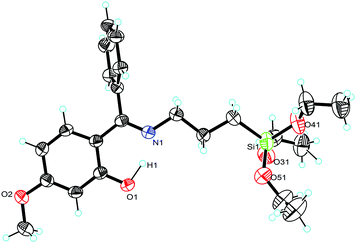 |
| | Fig. 2 Molecular structure of 1a (ellipsoids are shown at the 40% probability level). The asymmetric unit consists of one molecule, its Si(OEt)3 group exhibits severe disorder, only one part is depicted for clarity. Selected bond lengths [Å]: Si(1)–O(31) 1.626(2), Si(1)–O(41) 1.621(3), Si(1)–O(51) 1.621(4). | |
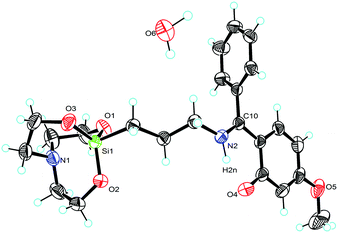 |
| | Fig. 3 Molecular structure of 2 (ellipsoids are shown at the 40% probability level). Selected bond lengths [Å] and angles [deg]: N(1)–Si(1) 2.161(6), O(1)–Si(1) 1.6652(12), O(2)–Si(1) 1.6648(12), O(3)–Si(1) 1.6659(11), O(2)–Si–O(1) 117.53(7), O(2)–Si–O(3) 117.26(7), O(1)–Si–O(3) 120.87(7), N(1)–Si(1)–C(7) 178.07(8). | |
Table 1 Parameters of X-ray diffraction data collection and structure refinement of 1 (two modifications) and 2
| Parameters |
1
|
1a
|
2
|
| Empirical formula |
C23H33NO5Si |
C23H33NO5Si |
C23 H30 N2O5Si |
| Formula weight |
431.59 |
431.59 |
455.28 |
|
T (K) |
93(2) |
233(2) |
150(2) |
|
λ (Å) |
0.71073 |
0.71073 |
0.71073 |
| Crystal system, space group |
Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Triclinic, P |
Monoclinic, P21/c |
| Unit cell dimensions |
|
|
|
|
a (Å) |
7.9600(10) |
7.8815(12) |
10.7352(6) |
|
b (Å) |
13.588(2) |
13.2589(8) |
13.3873(3) |
|
c (Å) |
22.433(3) |
13.5466(6) |
16.2758(11) |
|
α (°) |
87.035(6) |
117.517(2) |
90 |
|
β (°) |
83.056(5) |
103.013(3) |
100.095(4) |
|
γ (°) |
73.204(5) |
93.115(3) |
90 |
|
V (Å3) |
2305.5(5) |
1202.5(2) |
2302.9(2) |
|
Z
|
4 |
2 |
4 |
|
ρ
calc. (Mg m−3) |
1.243 |
1.192 |
1.284 |
|
μ
Mokμα (mm−1) |
0.135 |
0.135 |
0.139 |
|
F(000) |
928 |
464 |
950 |
| Crystal size (mm) |
0.25 × 0.06 × 0.02 |
0.25 × 0.10 × 0.08 |
0.30 × 0.16 × 0.13 |
| Theta range for data collection (deg) |
2.4 to 25.0 |
2.7 to 25.3 |
2.5 to 27.0 |
| Limiting indices |
−9 ≤ h ≤ 5 |
−9 ≤ h ≤ 9 |
−13 ≤ h ≤ 13 |
| −16 ≤ k ≤ 16 |
−15 ≤ k ≤ 15 |
−15 ≤ k ≤ 17 |
| −26 ≤ l ≤ 26 |
−16≤ l ≤ 15 |
−20 ≤ l ≤ 20 |
| Reflections collected |
16270 |
12341 |
22920 |
| Unique, R(int) |
7979, 0.1183 |
4181, 0.0244 |
5020, 0.0313 |
|
θ
max (°)/completeness (%) |
25.0/98.2 |
25.0/98.5 |
27.0/99.9 |
| Absorption correction |
None |
Multi-scan |
Integration |
| Max. and min. transmission |
n/a |
0.9242 and 0.9897 |
0.9935 and 0.9579 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
7979/66/523 |
4181/99/359 |
5020/10/317 |
| GoF on F2 |
0.965 |
1.012 |
1.037 |
|
R[I > 2σ(I)] |
R
1 = 0.0729, wR2 = 0.1412 |
R
1 = 0.0438, wR2 = 0.1119 |
R
1 = 0.0397, wR2 = 0.0969 |
|
R (all data) |
R
1 = 0.2606, wR2 = 0.2150 |
R
1 = 0.0720, wR2 = 0.1304 |
R
1 = 0.0507, wR2 = 0.1029 |
| Largest diff. peak and hole (e Å−3) |
0.376 and −0.307 |
0.166 and −0.207 |
0.317 and −0.273 |
3.4 Characterization of SB-SiO2@MNPs
3.4.1 Powder X-ray diffraction studies.
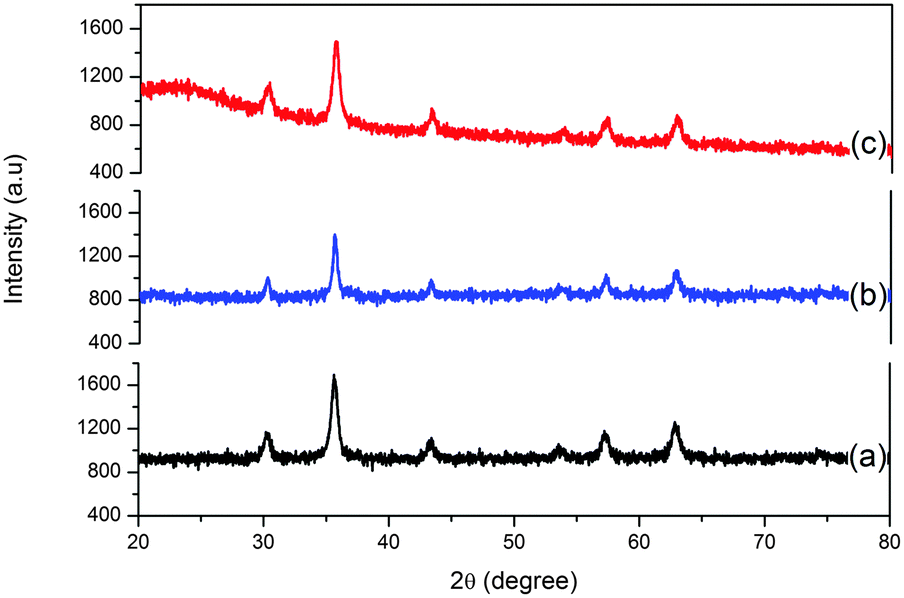
Powder XRD patterns of MNPs, SiO2@MNPs and SB-SiO2@MNPs depicted in Fig. 4 are similar to the pattern reported in Joint Committee on Powder Diffraction Standards [JCPDS, ref. 19-629]. Bragg's diffraction peaks at 30.0, 35.4, 43.0, 53.4, 56.9, and 62.5° refer to (2 2 0), (3 1 1), (4 0 0), (4 2 2), (5 1 1) and (4 4 0) crystal planes of cubic spinel Fe3O4, respectively. The absence of extra peaks also indicated the high phase purity of crystalline nanoparticles. The average crystal size of Fe3O4 cores was obtained by the Debye Scherrer formula (Dhkl = Kλl/βcosθ), where D is the size of the axis parallel to the (hkl) plane, K is a constant with a typical value of 0.89 for spherical particles, λ is the wavelength of radiation, β is full width at half maxima 40 in radians and θ is the position of the diffraction maximum of the peak with highest intensity from the XRD pattern of the MNPs. The size of MNPs was found to be 21 nm and it is increased to 27.5 nm and 29 nm in SiO2@MNPs and SB-SiO2@MNPs, respectively. The basically identical peaks in the X-ray diffraction patterns after modification of MNPs with silica and Schiff base-silatrane indicated that the crystalline nature of the particles persists even after modification. However, a less intense band was observed in the region 20–30° in the XRD curve of SiO2@MNPs, which became broad and more intense in SB-SiO2@MNPs. This suggested the introduction of an amorphous shell around the nanocores due to their functionalization with Schiff base silyl chains. The observed XRD patterns are also supported by literature reports.48
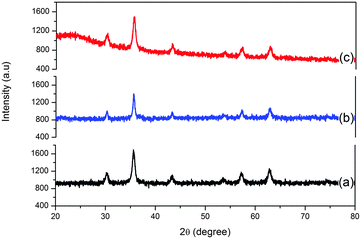 |
| | Fig. 4 Powdered XRD curves of (a) Fe3O4, (b) SiO2@ Fe3O4 and (c) SB-SiO2@ Fe3O4. | |
3.4.2 IR studies.
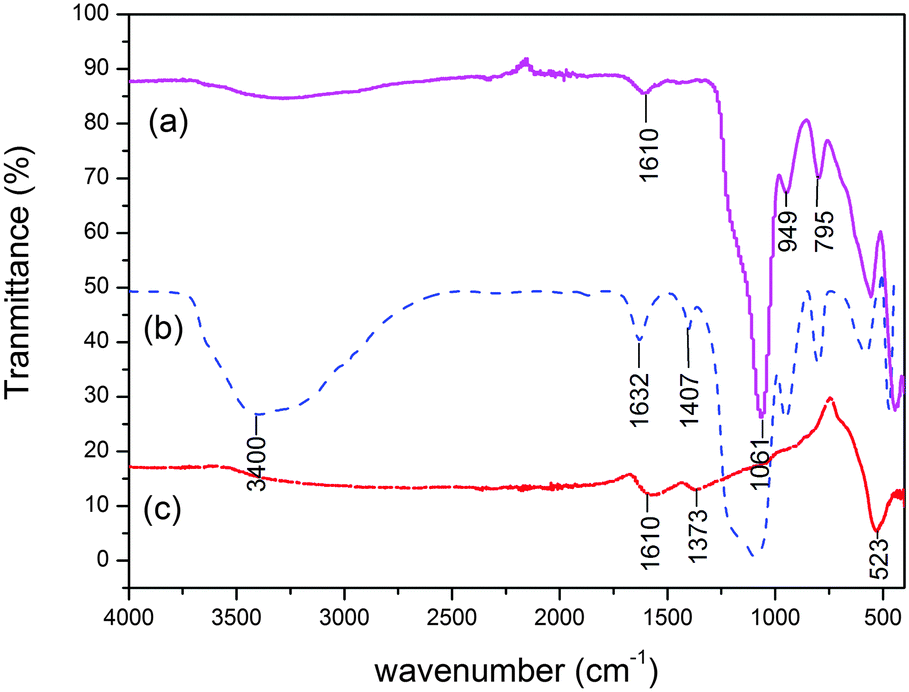
The IR spectra of citrate treated MNPs, SiO2@MNPs and SB-SiO2@MNPs are shown in Fig. 5. It shows a characteristic peak of Fe–O stretching vibration at 523 cm−1 in citrate treated MNPs. This peak is prominent in all spectra confirming the presence of a Fe3O4 core in all samples. Two additional absorptions at 1610 cm−1 and 1373 cm−1 can be assigned to the stretching of a COO–Fe bond, which indicated grafting of citrate onto the surface of MNPs. The IR spectrum of SiO2@MNPs showed a strong band at 1061 cm−1 due to Si–O–Si. Two weak bands at 795 cm−1 and 949 cm−1 revealed Si–O–H stretching and –OH vibrations, respectively, on the surface of MNPs. The stretching vibration frequency of Si–O–Fe (at around 970 cm−1) overlapped with Fe–O stretching. In the IR spectrum of SB-SiO2@MNPs, a broad band at 3400 cm−1 was observed in addition to the above mentioned peaks, which can be ascribed to –OH stretching vibration. A band observed at 1632 cm−1 is attributed to the stretching frequency of the CN moiety introduced by the silatranyl Schiff base modifier, which corroborates the functionalization of MNPs with silica and Schiff base functionality.
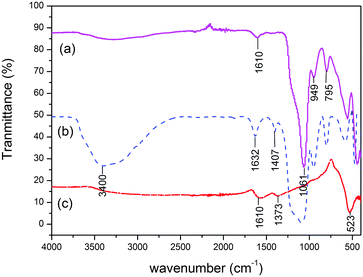 |
| | Fig. 5 FT-IR spectra of (a) SiO2@MNPs, (b) SB-SiO2@MNPs and (c) sodium citrate treated MNPs. | |
3.4.3 TEM, SEM and BET analysis.
The surface morphology of the magnetite nanoparticles before and after surface modification was investigated by SEM and TEM studies (Fig. 6). The SEM micrograph of the MNPs and SB-SiO2@MNPs gave considerable information about the surface topology of the materials. The surface of SB-SiO2@MNPs became rough relative to SiO2@MNPs due to the deposition of silyl chains around the magnetic nanocores. However, aggregation of nanoparticles was reduced after functionalization because the formation of a silica layer around magnetic nanocores dispersed the particles apart from each other due to reduction in the magnetic interactions. In addition, pure silica aggregates were not seen in the micrographs suggesting the silanization of the modifier only on the surface of magnetic nanocores. Besides SEM, TEM micrograph of both SiO2@MNPs and SB-SiO2@MNPs revealed roughly spherical shape of the particles with an average size of 25–33 nm. In the micrographs of SB-SiO2@MNPs, a coating of amorphous silica can be clearly seen around the bare nanocore suggesting the functionalization of MNPs. To get the information about the surface area of the fabricated material, BET analysis was performed. According to the BET data, the surface area of MNPs and SB-SiO2@MNPs was found to be 119 m2 g−1 and 42.1 m2 g−1, respectively.
 |
| | Fig. 6 (a and b) SEM images of MNPs and SiO2@MNPs; (c–e) TEM images of MNPs, SiO2@MNPs, and SB-SiO2@MNPs; (f) TEM image of SB-SiO2@MNPs showing the coating of SiO2 on MNP cores of the SiO2@MNP specimen. | |
3.4.4 VSM studies.
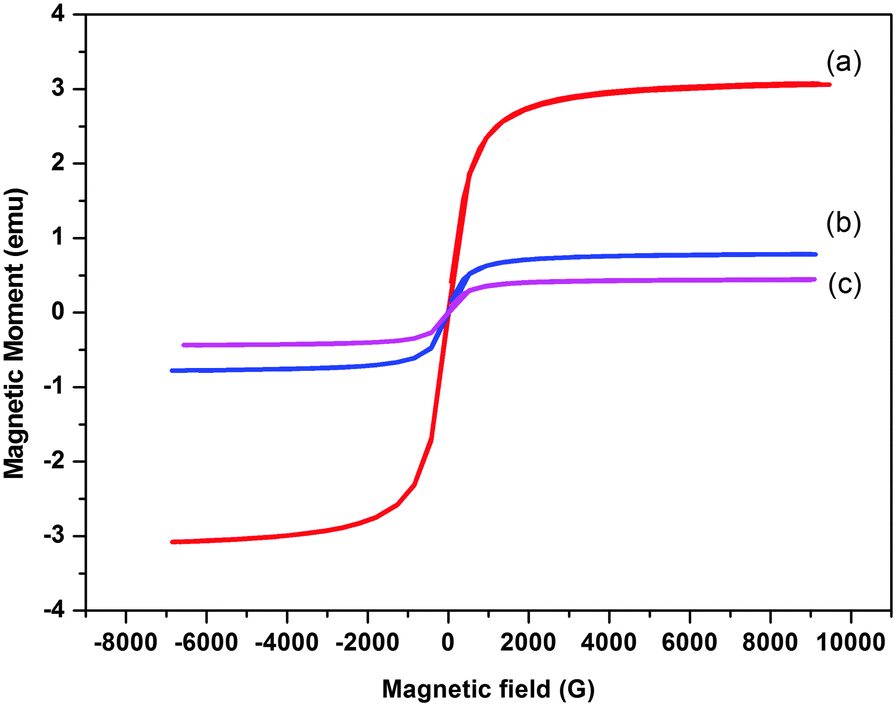
Magnetization behavior of MNPs, SiO2@MNPs and SB-SiO2@MNPs was studied by applying a magnetic field as depicted in Fig. 7. The saturation magnetization of citrate coated MNPs were found to be less than in bulk magnetite due to citrate coating. The curves showed decrease in values of saturation magnetization (Ms) from MNPs (55 emu g−1) to SiO2@MNPs (28.7 emu g−1) to SB-SiO2@MNPs (18.1 emu g−1). This phenomenon confirmed the coating with silica and with Schiff base functionalized silica on the surface of MNPs. Furthermore, the presence of sufficient magnetization in the modified MNPs offers advantage of effective separation from solutions via an external magnetic field. Moreover, MNPs are superparamagnetic in nature because coercivity and remanence are negligible as both magnetization as well as demagnetization curves pass through the origin.
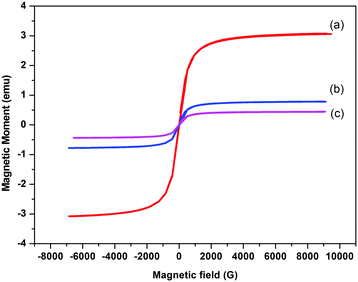 |
| | Fig. 7 Magnetization loops of (a) MNPs, (b) SiO2@MNPs and (c) SB-SiO2@MNPs. | |
3.5 Adsorption studies
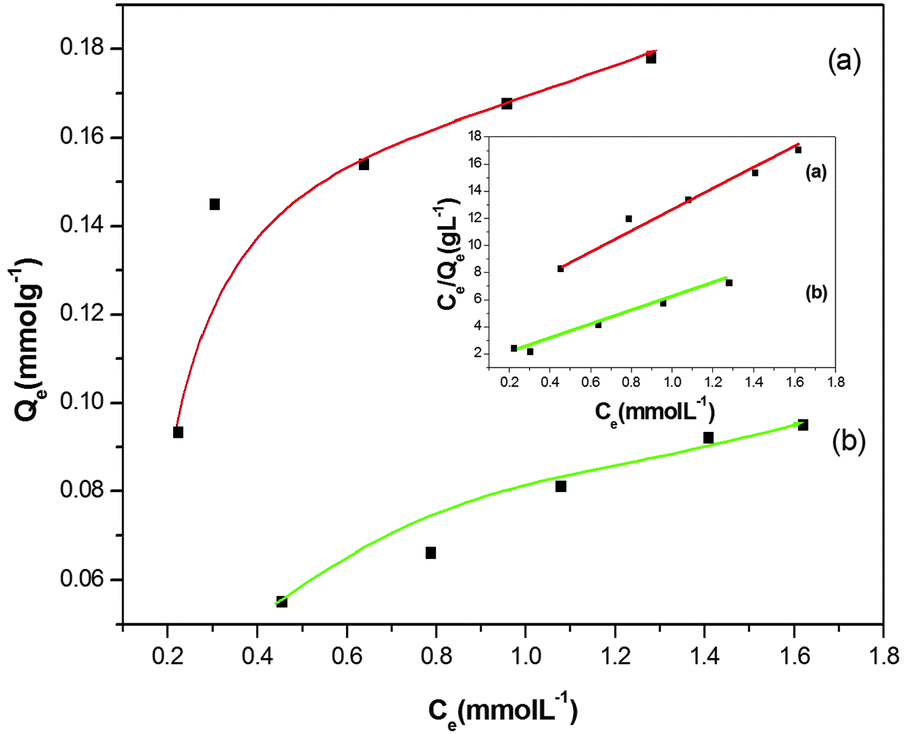
The newly fabricated SiO2@MNPs and SB-SiO2@MNPs were used to study the adsorption of copper ions. SiO2@MNPs showed the adsorption of Cu2+ ions due to the presence of active silanol groups on the surface whereas SB-SiO2@MNPs adsorb Cu2+ ions due to the presence of Schiff base functionality. When SB functionalized MNPs were stirred with copper(II) chloride solutions, an azomethine system captures the copper ions more efficiently than SiO2@MNPs. The adsorption kinetics was studied by varying the stirring time. It was observed that equilibrium is attained after 25 min and thereafter the adsorption remains constant. Therefore, the stirring time was set to 25 min for further studies. Fig. 8 illustrates equilibrium isotherms for the adsorption of Cu(II) by the SB functionalized magnetite nanoparticles at 30 °C. The plot of Ce/Qevs. Ce yielded a straight line, revealing that the adsorption of Cu(II) ions on the functionalized magnetic nanoparticles follows the Langmuir adsorption isotherm. The value of Ka and Qm is found to be 0.469 L mmol−1 (0.075 L mmg−1) and 0.207 mmol g−1 (13.17 mmg−1), respectively, calculated from the slope and intercept equal to 1/Qm and 1/KaQm. Similarly, Ka and Qm values calculated from the Langmuir plot of copper ion adsorption using SiO2@MNPs were found to be 1.27 L mol−1 (0.020 L mmg−1) and 0.141 mmol g−1 (8.9 mmg−1). It is clear that the adsorption capacity of SB-SiO2@MNPs is almost double as compared to SiO2@MNPs. Despite having less surface area relative to SiO2@MNPs, SB-SiO2@MNPs showed better adsorption of copper ions, which may be attributed to the presence of potentially good coordinating Schiff base sites. The Scatchard plot of Qe/Cevs. Qe is a straight line and it revealed that a single type of adsorption sites is involved when SB-SiO2@MNPs were used as an adsorbent and the contribution of silanol groups is minimal or negligible.
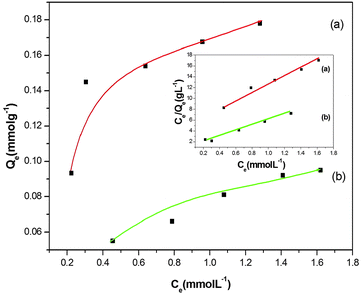 |
| | Fig. 8 Langmuir adsorption isotherm for the adsorption studies of copper ions on (a) SB-SiO2@MNPs and (b) SiO2@MNPs. | |
3.6 Interference studies
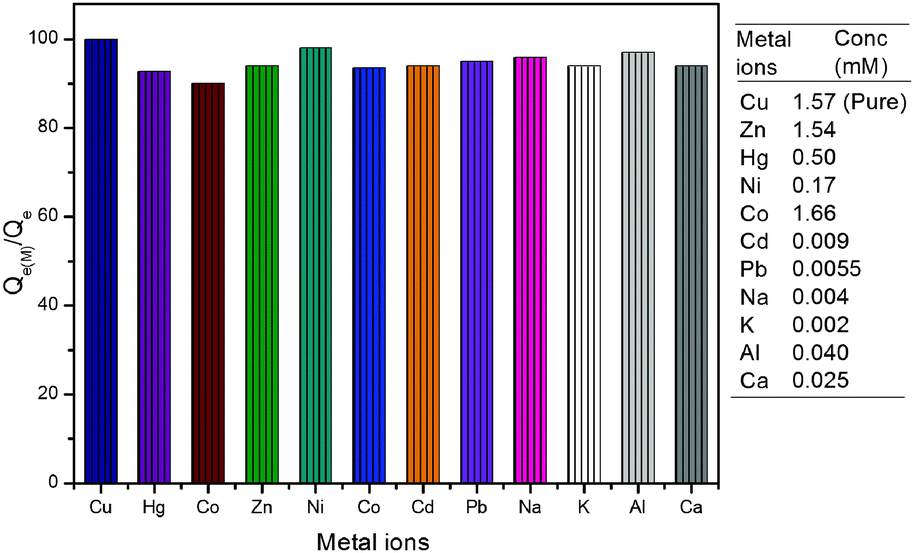
The interference of coexisting metal ions on the adsorption of copper ions (1.57 mM) was investigated. It was observed that the adsorption capacity of material for copper ions remained unaffected up to certain amounts of other metal ions. Interestingly, some of the metals like Zn2+, Hg2+, Ni2+ and Co2+ did not interfere with the adsorption of copper ions even at higher concentrations (up to 100 μg mL−1). However, interference due to other metal ions like Cd2+, Pb2+, Na+, K+, Al3+ and Ca2+ was observed above 10 μg mL−1. The value of Qe(M) for the adsorption of copper ions in the presence of other metal ions was calculated and plotted as a percent of Qe (calculated in the absence of metal ions). A bar graph for the adsorption of copper ions in terms of Qe(M)/Qe percent is given in Fig. 9. It clearly shows that Qe for the adsorption of copper ions remained above 90% even in the presence of Zn2+ (1.54 mM), Hg2+ (0.50 mM), Ni2+ (0.17 mM), Co2+ (1.66 mM), Cd2+ (0.009 mM), Pb2+ (0.006 mM), Na+ (0.004 mM), K+ (0.002 mM), Al3+ (0.040 mM) and Ca2+ (0.025 mM). It indicates that copper ions may be adsorbed effectively and selectively in the presence of other metal ions up to certain limits.
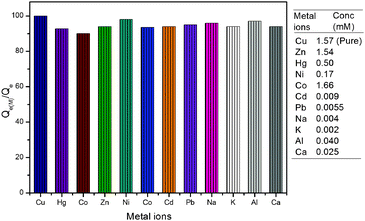 |
| | Fig. 9 A bar graph showing adsorption of copper ions in the presence of other metal ions (concentration of added metal ions is given in brackets along with the metal ion in the graph; the first column represents copper ion adsorption in the absence of other metal ions and taken as 100%). | |
4. Conclusion
In this work, we have successfully fabricated silica based magnetic nanoparticles with newly synthesized tailed silatrane possessing a Schiff base moiety. Functionalization of magnetic nanoparticles with the Schiff base reduced the aggregation of magnetic nano-cores without altering their shape. Modified Schiff base magnetic nanoparticles possessed superparamagnetic behaviour and are decorated with active sites for the adsorption of metal toxins. The adsorption capacity of magnetic nanoparticles was not as high as monoliths, which may be attributed to surface coating of functional moieties. In case of monoliths and other mesoporous materials, metal ions can penetrate deep into the adsorbent due to high porosity of the structure. But in this case, coordinating sites are present only on the surface of magnetic nanoparticles, therefore, adsorption equilibrium is attained in a very short period. This was also validated by the Langmuir adsorption isotherm, which revealed that the adsorption of copper ions is purely surface adsorption. In conclusion, silatranes can be used as modifiers in one pot fabrication of magnetite nano-particles.
Acknowledgements
The authors are thankful to UGC, New Delhi [No. F. 20-1/2012 (BSR)/20-9(12)/2012 (BSR)] and DST, New Delhi [Regd. No. CS-099/2012] for providing financial support.
References
- I. I. Slowing, J. L. V. Escoto, B. G. Trewyn and V. S. Y. Lin, J. Mater. Chem., 2010, 20, 7924–7937 RSC.
- J. W. Park, Y. J. Park and C. H. Jun, Chem. Commun., 2011, 47, 4860–4871 RSC.
- M. P. Conley, C. Copéret and C. Thieuleux, ACS Catal., 2014, 4, 1458–1469 CrossRef CAS.
- E. Rampazzo, E. Brasola, S. Marcuz, F. Mancin, P. Tecilla and U. J. Tonellato, Mater. Chem., 2005, 15, 2687–2696 RSC.
- Y. L. Lyubchenko, L. S. Shlyakhtenko and T. Ando, Methods, 2011, 54, 274–283 CrossRef CAS PubMed.
- J. Ou, Z. Liu, H. Wang, H. Lin, J. Dong and H. Zou, Electrophoresis, 2015, 36, 62–75 CrossRef CAS PubMed.
- M. Hartmann and X. Kostrov, Chem. Soc. Rev., 2013, 42, 6277–6289 RSC.
- Y. Lv, T. Tan and F. Svec, Biotechnol. Adv., 2013, 31, 1172–1186 CrossRef CAS PubMed.
- L. Treccani, T. Y. Klein, F. Meder, K. Pardun and K. Rezwan, Acta Biomater., 2013, 9, 7115–7150 CrossRef CAS PubMed.
- J. H. Chang, S. Y. Lee and J. Lee, Curr. Org. Chem., 2013, 17, 1014–1022 CrossRef CAS.
- R. Singh, R. Mutneja, V. Kaur, J. Wagler and E. Kroke, J. Organomet. Chem., 2013, 724, 186–191 CrossRef CAS.
- J. K. Puri, R. Singh and V. K. Chahal, Chem. Soc. Rev., 2011, 40, 1791–1840 RSC.
- J. K. Puri, R. Singh, V. K. Chahal, R. P. Sharma, J. Wagler and E. Kroke, J. Organomet. Chem., 2011, 696, 1341–1348 CrossRef CAS.
- R. Mutneja, R. Singh, V. Kaur, J. Wagler and E. Kroke, Dyes Pigm., 2014, 108, 41–49 CrossRef CAS.
- L. S. Shlyakhtenkoa, A. A. Gallc, A. Filonovd, Z. Cerovaca, A. Lushnikova and Y. L. Lyubchenko, Ultramicroscopy, 2003, 97, 279–287 CrossRef.
- B. J. Brennan, A. E. Keirstead, P. A. Liddell, S. A. Vail, T. A. Moore, A. L. Moore and D. Gust, Nanotechnology, 2009, 20, 505203 CrossRef PubMed.
- K. W Huang, C. W. Hsieh, H. C. Kan, M. L. Hsieh, S. Hsieh, L. K. Chau, T. E. Cheng and W. T. Lin, Sens. Actuators, B, 2012, 163, 207–215 CrossRef.
- J. H. Jung, J. H. Lee and S. Shinkai, Chem. Soc. Rev., 2011, 40, 4464–4474 RSC.
- J. Wang, S. Zheng, Y. Shao, J. Liu, Z. Xu and D. Zhu, J. Colloid Interface Sci., 2010, 349, 293–299 CrossRef CAS PubMed.
- J. G. Pastora, E. Bringas and I. Ortiz, Chem. Eng. J., 2014, 256, 187–204 CrossRef.
- P. I. Girginova, A. L. D. da-Silva, C. B. Lopes, P. Figueira, M. Otero, V. S. Amaral, E. Pereira and T. Trindade, J. Colloid Interface Sci., 2010, 345, 234–240 CrossRef CAS PubMed.
- F. Ge, M. M. Li, H. Ye and B. X. J. Zhao, Hazard. Mater., 2012, 211–212, 366–372 CrossRef CAS PubMed.
- M. Caoa, Z. Lia, J. Wanga, W. Gea, T. Yuea, R. Lib, V. L. Colvinc and W. W. Yu, Trends Food Sci. Technol., 2012, 27, 47–56 CrossRef.
- I. Safarik and M. Safarikova, BioMagn. Res. Technol., 2004, 2, 7–24 CrossRef PubMed.
- J. He, M. Huang, D. Wang, Z. Zhang and G. J. Li, J. Pharm. Biomed. Anal., 2014, 101, 84–101 CrossRef CAS PubMed.
- L. Li, W. Jiang, K. Luo, H. Song, F. Lan, Y. Wu and Z. Gu, Theranostics, 2013, 3, 595–615 CrossRef PubMed.
- V. Biju, Chem. Soc. Rev., 2014, 43, 737–962 RSC.
-
R. K. Gautam, S. K. Sharma and M. C. Chattopadhyaya, Heavy Metals In Water: Presence, Removal and Safety, Royal Society of Chemistry, Cambridge, 2014 Search PubMed.
- I. Ali, Chem. Rev., 2012, 112, 5073–5091 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2014, 114, 6949–6985 CrossRef CAS PubMed.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J. M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- S. Ge, X. Shi, K. Sun, C. Li, C. Uher, J. R. Baker, J. M. M. B. Holl and B. G. Orr, J. Phys. Chem. C, 2009, 113, 13593–13599 CAS.
- C. Okoli, M. S. Dominguez, M. Boutonnet, S. Jaras, C. Civera, C. Solans and G. R. Kuttuva, Langmiur, 2012, 28, 8479–8485 CrossRef CAS PubMed.
- P. P. Goswami, H. A. Choudhury, A. Chakma and V. S. Moholkar, Ind. Eng. Chem. Res., 2013, 52, 17848–17855 CrossRef CAS.
- Y. C. Han, H. G. Cha, C. W. Kim, Y. H. Kim and Y. S. Kang, J. Phys. Chem. C, 2007, 111, 6275–6280 CAS.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- H. Lu, A. E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef PubMed.
- I. Y. Goon, L. M. H. Lai, M. Lim, P. Munroe, J. J. Gooding and R. Amal, Chem. Mater., 2009, 21, 673–681 CrossRef CAS.
- H. Bagheri, A. Afkhami, M. S. Tehrani and H. Khoshsafar, Talanta, 2012, 97, 87–93 CrossRef CAS PubMed.
- Y. H. Deng, C. C. Wang, J. H. Hu, W. L. Yang and S. K. Fu, Colloids Surf., A, 2005, 262, 87–93 CrossRef CAS.
- D. Yang, J. Hu and S. Fu, J. Phys. Chem. C, 2009, 113, 7646–7651 CAS.
- S. Santra, R. Tapec, N. Theodoropoulou, J. Dobson, A. Hebard and W. Tan, Langmuir, 2001, 17, 2900–2906 CrossRef CAS.
- L. M. Rossi, N. J. S. coasta, F. P. Silva and R. Wojceiszak, Green Chem., 2014, 16, 2906–3380 RSC.
- M. Mohammadikish, M. M. Farahani and S. Mahdavi, J. Magn. Magn. Mater., 2014, 354, 317–323 CrossRef CAS.
- A. Afkhami, R. Moosavi, T. Madrakian, H. Keypour, A. R. Aktij and M. M. Monsef, Electroanalysis, 2014, 26, 786–795 CrossRef CAS.
- S. Moradinasab and M. Behzad, Desalin. Water Treat., 2014 DOI:10.1080/19443994.2014.991760.
- A. E. Karatapanis, Y. Fiamegos and C. D. Stalikas, Talanta, 2011, 84, 834–839 CrossRef CAS PubMed.
- G. Cheng, M. He, H. Peng and B. Hu, Talanta, 2012, 88, 507–515 CrossRef CAS PubMed.
- C. Huang and B. Hu, Spectrochim. Acta, Part B, 2008, 63, 437–444 CrossRef.
- R. Singh, J. K. Puri, R. P. Sharma, V. K. Chahal and P. Venugopalan, J. Organomet. Chem., 2010, 695, 183–188 CrossRef CAS.
- R. K. Sharma, S. Dutta and S. Sharma, Dalton Trans., 2015, 44, 1303–1316 RSC.
- J. Zhang, S. Zhai, S. Li, Z. Xiao, Y. Song, Q. An and G. Tian, Chem. Eng. J., 2013, 215–216, 461–471 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a