
Naphthyl “capped” triazole-linked calix[4]arene hosts as fluorescent chemosensors towards Fe3+ and Hg2+: an experimental and DFT computational study†
Shofiur
Rahman
*a,
Yousif
Assiri
a,
Abdullah N.
Alodhayb
b,
Luc Y.
Beaulieu
b,
Ahmed K.
Oraby
a and
Paris E.
Georghiou
*a
aDepartment of Chemistry, Memorial University of Newfoundland, St. John's, Newfoundland and Labrador, Canada A1B3X7. E-mail: parisg@mun.ca; msrahman@mun.ca
bDepartment of Physics and Physical Oceanography, Memorial University of Newfoundland, St. John's, Newfoundland and Labrador, Canada A1B3X7. E-mail: lbeaulieu@mun.ca
First published on 6th November 2015
Abstract
Four new naphthyl “capped” 1,2,3-triazole-linked receptors 6a–d have been synthesized and characterized. Their binding properties towards different metal ions have been studied by 1H NMR and fluorescence spectroscopy and the association constants were analysed by a non-linear global fit method. The fluorescent spectral changes show that 6a–d are sensitive and selective towards Fe3+ and Hg2+ ions. DFT calculations of the binding modes of 6a–d with these metal ions are also presented.
1. Introduction
Many studies have been conducted on the use of triazole-functionalized calixarenes as highly selective and sensitive chemosensors.1a Calixarenes offer several advantages over other molecular systems and these advantages include their relatively easy functionalization at their lower- and upper-rims, the presence of hydrophobic cavities, and the presence of flexible core/scaffolds that can be modified for target substrate binding.1b The design and synthesis of new receptors for metal ions such as those of iron and mercury is of current ongoing interest in supramolecular chemistry due to the roles that these ions play in biological, environmental, and chemical processes.2–5Biologically, iron is the most essential transition-metal since it plays an essential role in carrying oxygen in the heme molecule. It acts also as a cofactor in enzymatic reactions of the mitochondrial respiratory chain. Either a deficiency, or an excess of Fe3+ ions can cause a variety of diseases such as anemia, liver and kidney damage, diabetes, heart disease and cancer.3 Chemosensors that can selectively detect Fe ions are rare and due to the importance of discrimination between Fe3+ and Fe2+ in the regulation of their biological function, the design of a fluorescent chemosensor capable of binding selectivity to Fe3+ ions is an important challenge, one that both Kim's4a and Rao's4b groups have recently tackled.
On the other hand, mercury is one of those elements that can accumulate in living tissues, thereby causing many harmful effects on human health. The toxicity of Hg2+ ions is related to the fact that biological ligands such as proteins, DNA, and enzymes can coordinate with these ions. Consequently, toxic levels of Hg2+ cause significant damage to the brain, kidney, stomach, and the Central Nervous System.5
The application of the Cu(I)-catalyzed 1,3-dipolar cycloaddition of an azide and a terminal alkyne (“CuAAC” or “click” reaction) to form 1,2,3-triazole-linked derivatives has provided a straightforward molecular linking strategy which has been adopted in a wide range of applications.1a,6 The properties of the 1,2,3-triazole moiety as a linker in multivalent derivatives can be exploited for the binding of cations, such as in the accelerated catalysis of the click reaction itself by the in situ formation of copper complexes. In calixarene chemistry, the 1,2,3-triazole moiety was first exploited for the functionalization of the calixarene scaffold by Ryu and Zhao in 2005.7 More relevant to this work has been the incorporation of the 1,2,3-triazole moiety onto calixarene and related8a–g,9 molecular scaffolds tailored for the selective binding of various metal cations. The determination of traces of these ions in clinical, medicinal, environmental and different industrial samples has been an important topic. Over the last two decades, the need has increased for the determination of extremely low concentrations of Fe3+ and Hg2+ in various samples. Therefore, the design and development of efficient ligands that may be used to selectively complex these metal ions are turning out to be extremely important. Herein, we report the synthesis, using a CuAAC methodology, and the properties of new 1,4-naphthyl-”capped” triazoyl-bridged calix[4]arene hosts 6a–6d as potential chemosensors for Fe3+ and Hg2+ ions. Santoyo-Gonzalez and co-workers had fist reported the use CuAAC reactions to form an anthracenyl- and two ferrocenyl-capped calix[4]arenes.9
2. Results and discussions
The 1,4-naphthyl-2,3-dialkoxy “capped” triazoyl-bridged hosts 6a–6d were synthesized by using the CuAAC methodology through the Cu(I)-catalyzed linking of the corresponding 1,3-bis(propargyl)-calix[4]arene and bis-azide derivatives (Scheme 1). The starting materials di-tert-butylcalix[4]arene 4a,10ap-tert-butylcalix[4]arene 4b,10bp-tert-butyl-25,27-di(O-propargyl)-calix[4]arene 5b10c–d were all synthesized according to the published literature procedures. A modified literature procedure, however was used to synthesize 5a.10e 1,4-bis(azidomethyl)-2,3-dialkoxynaphthalenes 3a–3b were synthesized via the corresponding 1,4-bis(bromomethyl)-2,3-dialkoxynaphthalenes 2a–2b.11 Reactions of 5a with 3a or 3b afforded 6a or 6b; and reactions of 5b with 3a or 3b afforded 6c or 6d, respectively.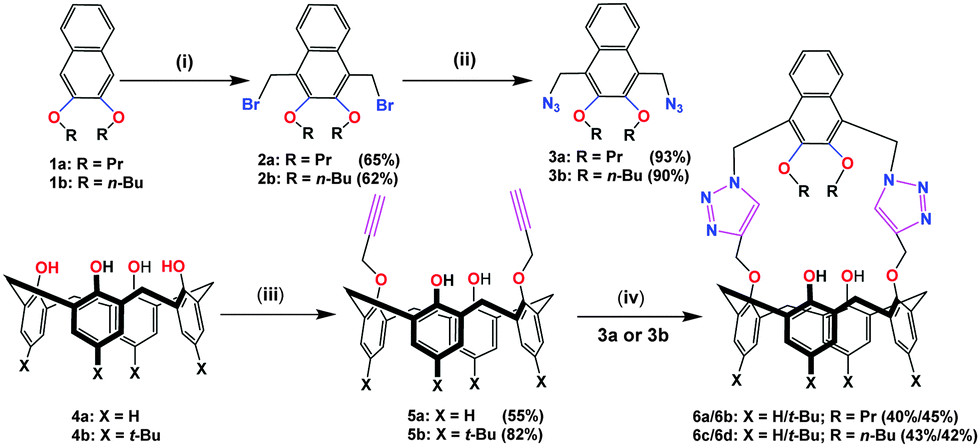 | ||
| Scheme 1 Synthesis of 6a–6d: (i) 1a (1b) + HBr 33 wt% solution in CH3COOH, rt, 72 h; (ii) 2a (2b) + NaN3 in DMF, 90 °C, 24 h; (iii) 4a (4b) + propargyl bromide, K2CO3, acetone, reflux, 24–48 h: (iv) 5a (5b) CuI/DIPEA in THF, 60 °C for 24 h. | ||
All of the synthesized compounds were characterized by 1H- and 13C-NMR and mass spectrometry. The 1H-NMR spectra in CDCl3 of macrocycles 6a–6d revealed new one-proton singlets at δ 7.58, 7.73, 7.52 and 7.65 ppm, respectively. These signals were assigned to the triazole protons of the macrocycles formed from the CuAAC reactions of the corresponding precursors. Two other two-proton singlets due to the two different –CH2– bridges which link the triazole units, were observed at δ 6.01 and 5.07 ppm for 6a; at δ 5.99 and 5.06 ppm for 6b; at δ 6.01 and 5.07 ppm for 6c, and at δ 5.93 and 5.04 ppm for 6d. The 1H-NMR spectra of 6a–d indicated that the calix[4]arene units are all in cone conformations since the chemical shifts of the calix[4]arene bridging –CH2– group protons, appeared as pairs of AB doublets: at δ 3.98 and 2.90 ppm (J = 13.5 Hz) for 6a; at δ 4.02 and 3.02 ppm (J = 12.0 Hz) for 6b; at δ 3.98 and 2.88 ppm (J = 15.0 Hz) for 6c; and at δ 4.00 and 2.99 ppm (J = 12.9 Hz) for 6d.
2.1 Complexation studies
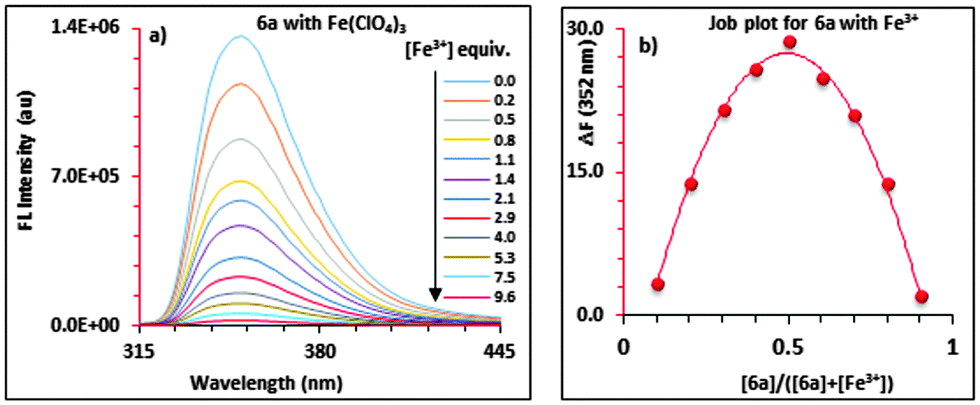
The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex formation between 6a and Fe3+ was also confirmed by a separate continuous variation method12 (Job) plot by fluorescence spectrometry. In the Job plot (Fig. 1b), a maximum fluorescence change was observed when the molar fraction of ionophore 6avs. Fe3+ was 0.5, indicative of 1:1 complex formation. Similar fluorescence titration behaviour and 1:1 binding stoichiometry were observed for all of the “capped” receptors 6a–6d tested with other metal ions. Fig. 2 shows a histogram comparing the Kassoc values for 6a (red), 6b (blue), 6c (purple) and 6d (yellow) with metal ions using a 1:1 global fit analysis of the entire fluorescence quenching spectra.13a,b The modified B–H method13c,d gave smaller values (Table S7 in the ESI†), but the limitations of the B–H have been well-addressed in the literature.13a Detailed titration data are summarized in the ESI† (Tables S6 and S7 and Fig. S18–S30).
:1 complex formation between 6a and Fe3+ was also confirmed by a separate continuous variation method12 (Job) plot by fluorescence spectrometry. In the Job plot (Fig. 1b), a maximum fluorescence change was observed when the molar fraction of ionophore 6avs. Fe3+ was 0.5, indicative of 1:1 complex formation. Similar fluorescence titration behaviour and 1:1 binding stoichiometry were observed for all of the “capped” receptors 6a–6d tested with other metal ions. Fig. 2 shows a histogram comparing the Kassoc values for 6a (red), 6b (blue), 6c (purple) and 6d (yellow) with metal ions using a 1:1 global fit analysis of the entire fluorescence quenching spectra.13a,b The modified B–H method13c,d gave smaller values (Table S7 in the ESI†), but the limitations of the B–H have been well-addressed in the literature.13a Detailed titration data are summarized in the ESI† (Tables S6 and S7 and Fig. S18–S30).
 | ||
| Fig. 1 (a) Fluorescence spectra of 6a from the titration of Fe(ClO4)3 (0–9.6 equivalents) to a fixed concentration (i.e. 1.50 × 10−5 M) of 6a, in 9:1, v:v CH3CN:CH3Cl. λex = 291 nm; (b) Job plot for 6a with Fe3+. | ||
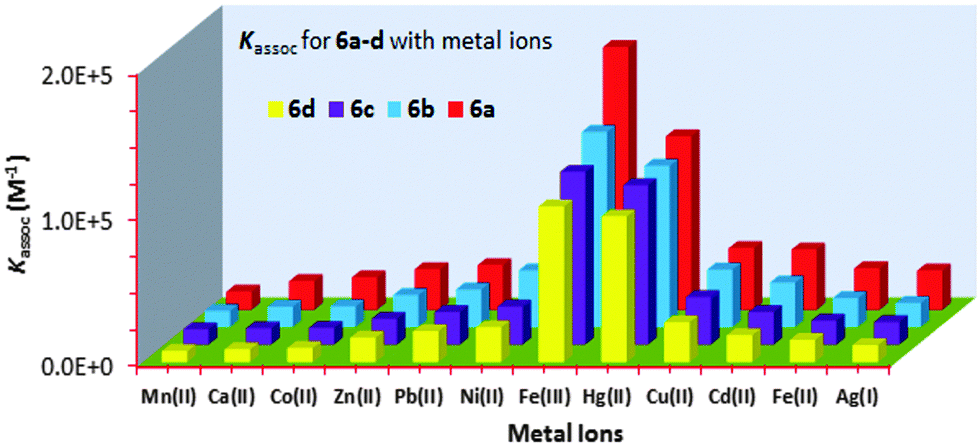 | ||
| Fig. 2 Histogram showing the Kassoc values for 6a (red), 6b (blue), 6c (purple) and 6d (yellow) with metal ions (Mn+). | ||
2.2 UV-vis and fluorescence spectral data
Spectroscopic experiments were conducted using 1 cm capped quartz fluorescence cuvettes. UV-vis spectra were recorded on an Agilent 8543 Diode Array Spectrophotometer. Data manipulations were conducted using software supplied by the manufacturer. Emission spectra were measured on a Photon Technology International (PTI) Quanta Master 6000 Spectrofluorometer equipped with a continuous xenon arc lamp as the excitation source.2.3 Fluorescence complexation studies for 6a–d with metal ions
To record the fluorescence spectra of 6a–d, stock solutions of 6a–d (∼1.50 × 10−5 M to ∼4.00 × 10−5 M), and the metal perchlorate salts (∼1.5 × 10−2 M) were prepared in 9:1 (v:v) CH3CN:CHCl3 solvent mixture. The receptors 6a–d displayed monomer emission at 331 nm at the 291 nm excitation wavelength. The fluorescence emissions were quenched upon the addition, in each case, of Mn2+, Ca2+, Co2+, Zn2+, Pb2+, Ni2+, Fe3+, Hg2+, Cu2+, Cd2+, Fe2+ or Ag+. The highest Kassoc values with the all of the receptors were obtained with Fe3+ and Hg2+. Fig. 2 shows the histogram for the Kassoc values determined for all of the complexes and it is clearly evident that the largest Kassoc values of 1.79 × 105 M−1, 1.33 × 105 M−1, 1.18 × 105 M−1 and 1.06 × 105 M−1 which were observed were for the binding between 6a–d and Fe3+, respectively.
2.4 Metal competitive experiments
To confirm the selectivity of 6a towards Fe3+ over the other metal ions (Mn+) competitive fluorescence experiments4b were carried out with 6a which were conducted by adding up to a molar equiv. of Fe3+ (Fig. S17, ESI†) to each respective solution in which the mole ratio of the various metal ions (Mn+ = 1.50 × 10−4 M) were kept constant at a 1:10 ratio with respect to 6a (1.50 × 10−5 M). The histogram in Fig. 3 shows the quenching by the addition of the Fe3+ indicating that 6a is more selective for Fe3+ over the other metal ions tested, with the exception of Hg2+ which showed very little change.
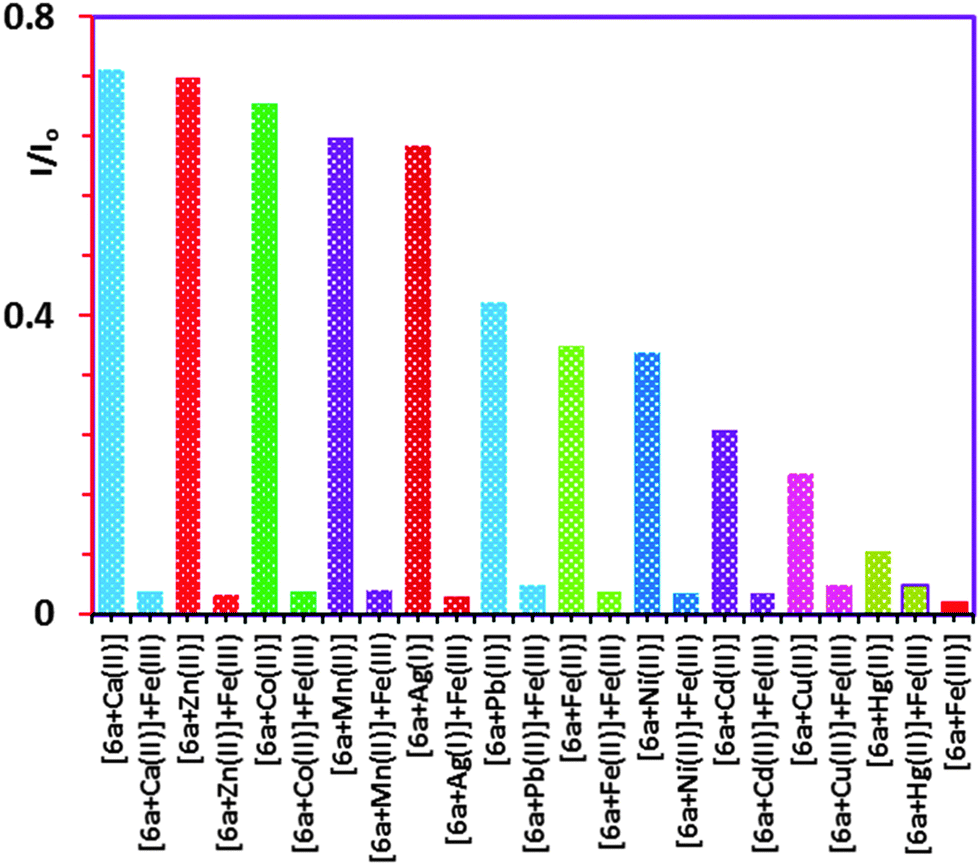 | ||
| Fig. 3 Histogram showing the metal competitive fluorescence quenching of 6a with Fe3+ in the presence of various metals ions (Mn+) in CH3CN:CHCl3 (9:1). I0 = fluorescence intensity of free 6a; I = fluorescence intensity of complex 6a with Mn+ ions; [6a] = 1.50 × 10−5 M; Mn+ = 1.5 × 10−4 M and Fe3+ = 1.50 × 10−4 M. | ||
During the same period that the present work was being completed we became aware of a study by W.-S. Chung and coworkers8g on the behaviour of Fe(ClO4)3 with a related calixarene-based ionic receptor. These authors demonstrated that Fe3+ was capable of oxidizing both the appended anthracenyl groups and also one or both of the free phenolic groups in their particular calixarene system. The oxidation resulted in the concomitant removal of the tert-butyl groups to form the corresponding mono- or bisquinonoidal calixarene derivatives, and also oxidation of their anthracenyl groups. It is therefore possible that reactions of Fe(ClO4)3 with 6a–d, based on Chung's work, that a similar occurrence could be involved with our system during the fluorescence study. Although this is a possibility in our case, the 1:1 Job plot behaviour shown in Fig. 1b makes this unlikely. At the very least, if oxidation was occurring instead of quenching involving a typical photoinduced electron transfer (PET) or heavy-metal effect,14 then the 1:1 stoichiometry would imply that 6a–d can act instead as sensitive chemodosimeters for Fe3+ (see references cited in ref. 8g).
2.5 1H-NMR complexation study of 6a with Hg(ClO4)2
Since 1H NMR spectra can potentially provide evidence for the site of binding of the metal ions and the receptors, titration experiments were conducted. To a solution of 6a for example, in 3:1 v/v CD2Cl2/CD3CN (1.98 × 10−3 M) in an NMR tube were added aliquots of 4.00 × 10−2 M Hg(ClO4)2 and in the same solvent. The 1H NMR spectra were recorded after each addition. The NMR probe temperature was maintained at 27 °C and the added [Hg2+] mole ratio was varied up to 2.0 equivalents (Fig. 4). The signals of the protons of 6a corresponding to the triazole-H (Hj, + Δδ = 0.15 ppm) and calix–OH (Hi, + Δδ = 0.09 ppm) protons were all found to shift downfield with Hg2+. The paramagnetism of Fe3+ prohibited an equivalent determination by 1H NMR. Furthermore, solubility and other considerations necessitated a change of the solvent system used for the NMR study compared with that used for the fluorescence studies.
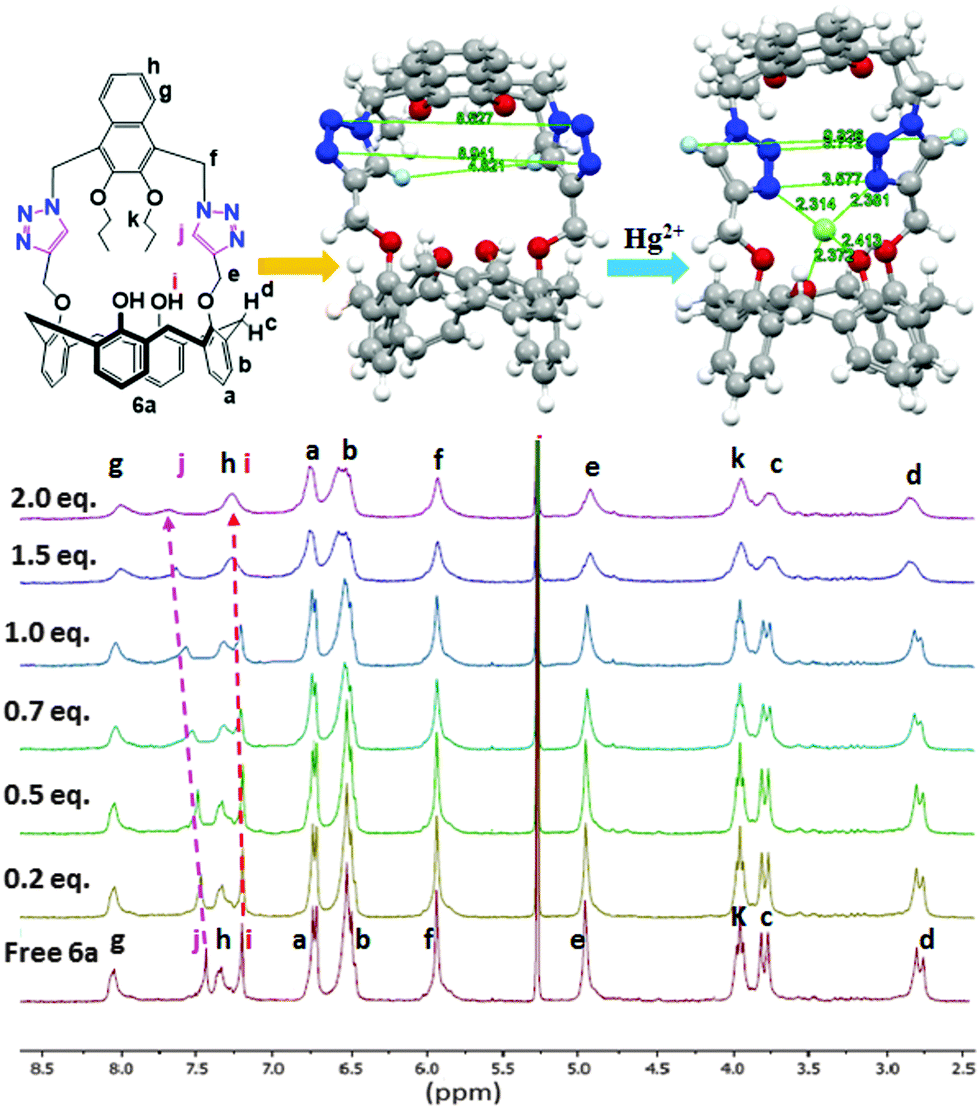 | ||
| Fig. 4 Top (left): ChemDraw structure of 6a. The geometry-optimized (ball-and-stick) structure of 6a (top centre) and complex 6a ⊃ Hg2+ (top right) using B3LYP/lanl2dz basis set. Colour code: Hg2+ = light green, triazole nitrogen = dark blue, triazole hydrogen = light blue and oxygen atom = red. Bottom: Partial 1H NMR (300 MHz) spectra of 6a (1.98 × 103 M) with Hg(ClO4)2 (0–2.0 equiv.) upon addition of 3:1, v/v CD2Cl2:CD3CN at 298 K. | ||
2.6 Computational studies
A computation study was carried out for each of the complexes formed from receptors 6a–6d with Fe3+ and Hg2+. The individual structures for all structures in the gas-phase were fully geometry-optimized using Gaussian 09 Revision D.0115 at the B3LYP level16 of DFT and the lanl2dz basis set.17,18a–c Significant distance changes were observed for the two triazole moieties in all of the receptors 6a–6d as their respective metal ion complexes, as can be seen for example, in Fig. 4 (top centre and right) for 6a with Hg2+ and in Table 1 for 6a with Fe3+ and Hg2+. The detailed computational results for 6a–d with Fe3+ and Hg2+ ions are summarized in the ESI† (Tables S1 to S5 and Fig. S1–S16).| Parameter | 6a | 6a⊃Fe3+ | 6a⊃Hg2+ |
|---|---|---|---|
| Distance (Å) | Distance (Å) | Distance (Å) | |
| N3–N24 | 8.941 | 3.024 | 3.577 |
| N4–N23 | 8.527 | 3.517 | 3.712 |
| H84–H112 | 4.821 | 8.548 | 9.326 |
| N3–O61 | 5.713 | 3.41 | 3.818 |
| N3–O74 | 6.39 | 3.901 | 4.2 |
| N24–O61 | 6.118 | 4.244 | 4.476 |
| N24–O74 | 4.829 | 2.898 | 3.307 |
| Mn+–N3 | — | 2.149 | 2.314 |
| Mn+–N24 | — | 2.272 | 2.381 |
| Mn+–O61 | — | 2.104 | 2.372 |
| Mn+–O74 | — | 2.126 | 2.413 |
The DFT B3LYP/lanl2dz basis set-calculated binding or interaction energies (BE) of the metal cation complexes of the receptors 6a–d formed between the metal cation (Mn+) and the free receptors 6a–d in the gas phase at 298 K are based on eqn (1). The results are summarized in Table S1 in the ESI.† For this system, the binding energy (BE) (where L = 6a–d, and Mn+ = metal ion) can be expressed as follows:
| BE = {(L:Mn+)complex − E(Lfree) − E(Mn+)} | (1) |
3. Experimental section
All reagents used for the syntheses of the calixarenes 3–6 and reagents used in the complexation studies were purchased from Sigma-Aldrich or AlfaAesar. 1H-NMR spectra were recorded at either 300 or 500 MHz as noted and for the 13C-NMR spectra at 75 MHz as noted.3.1 Reagents
All the metal ion salts were received from Sigma Aldrich in >99% purity. High-purity spectral grade CHCl3 and CH3CN was supplied by Sigma Aldrich or Cambridge Isotope Labs and were used as received.:20 hexane:ethyl acetate) to give a colourless oil (0.56 g, 80%, m.p. 123.2 °C). 1H NMR (300 MHz, CDCl3): δ = 7.98 (m, 2H, naphthyl-H), 7.53 (m, 2H, naphthyl-H), 4.86 (s, 4H, naphthyl-H), 4.05 (t, J = 6.6 Hz, 4H, –OCH2CH2CH3), 1.89 (m, 4H, –OCH2CH2CH3), 1.10 (t, J = 7.5 Hz, 6H, –O(CH2)2CH3). 13C-NMR (CDCl3, 75.46 MHz): δ = 150.4, 129.9, 126.1, 124.41, 124.2, 76.3, 45.2, 23.6, 10.6. APCI(+) MS (m/z, relative intensity) 353.4 [(M − 2N)+, 80].
:20 hexane:ethyl acetate) to give a colourless oil (0.64 g, 83%, m.p. 115.6 °C). 1H NMR (300 MHz, CDCl3): δ = 7.98 (m, 2H, naphthyl-H), 7.54 (m, 2H, naphthyl-H), 4.86 (s, 4H, naphthyl-H), 4.09 (t, J = 6.6 Hz, 4H, –OCH2(CH2)2CH3), 1.89 (m, 4H, OCH2CH2C2H5), 1.55 (m, 4H, –O(CH2)2CH2CH3), 1.02 (t, J = 7.5 Hz, 6H, O(CH2)3CH3); 13C-NMR (CDCl3, 75.5 MHz): δ = 151.4, 129.9, 126.1, 124.4, 124.2, 74.6, 45.2, 32.4, 19.4, 14.0. APCI(+) MS (m/z, relative intensity) 353.4 [(M − 2N)+], 100.
:10 dichloromethane:methanol) giving 6a (128 mg, 40%) as a colourless solid: mp. 129.1 °C (dec); 1H NMR (300 MHz, CDCl3): δ = 8.11 (m, 2H, naphthyl-H), 7.58 (s, 2H, triazoly-H), 7.398 (m, 2H, naphthyl-H), 7.15 (s, 4H, Ar-OH), 6.81 (d, J = 7.2 Hz, 4H, ArH), 6.01 (m, 8H, ArH), 6.01 (s, 4H, naphthyl-CH2-triazole), 5.07 (s, 4H, ArOCH2-triazole), 3.92 (m, 8H, ArCH2Ar AB system and –OCH2C2H5), 2.90 (d, J = 13.5 Hz, 4H, ArCH2Ar AB system), 1.83 (m, 4H, –OCH2CH2CH3), 1.02 (t, J = 7.2 Hz, 6H, –O(CH2)2CH3). 13C-NMR (CDCl3, 75.46 MHz): δ = 152.8, 151.1, 150.4, 143.3, 133.1, 129.6, 128.7, 128.3, 127.9, 126.8, 125.3, 124.3, 124, 123.7, 119.0, 76.1, 69.2, 44.8, 31.2, 23.6, 10.6; HRMS APPI(+) calcd for C52H50N6O6: 854.3792, found: 854.3794.
:10 dichloromethane:methanol) to give a colourless solid (140 mg, 45%, m.p. 145.5 °C). 1H NMR (300 MHz, CDCl3): δ = 8.06 (m, 2H, naphthyl-H); 7.73 (s, 2H, triazole-H); 7.38 (m, 2H, naphthyl-H), 6.92 (s, 4H, ArH), 6.83 (s, 2H, Ar-OH), 6.63 (s, 4H, ArH), 5.99 (s, 4H, naphthyl-CH2-triazole), 5.06 (s, 4H, ArOCH2-triazole), 4.02 (d, J = 12.0 Hz, 4H, ArCH2Ar, AB system), 4.02 (t, J = 6.0 Hz, naphthyl-OCH2C2H5), 3.02 (d, J = 12.0 Hz, 4H, ArCH2Ar, AB system), 1.82 (m, 4H, –OCH2CH2CH3), 1.27 (s, 18H, tBu) 1.02 (t, J = 9.0 Hz, 6H, –O(CH2)2CH3), 0.89 (s, 18H, tBu); 13C-NMR (CDCl3, 75.46 MHz): δ = 150.5, 150.3, 149.4, 147.0, 143.8, 141.5, 132.4, 129.6, 127.8, 125.5, 125.0, 124.1, 124.0, 123.6, 76.04, 69.3, 53.4, 44.9, 33.8, 31.7, 30.9, 24.0, 10.7; HRMS APPI(+) calcd for C68H82N6O6: 1078.6296, found: 1078.6236.
:10 dichloromethane:methanol) to give a colourless solid (130 mg, 43%, m.p. 153.4 °C). 1H NMR (300 MHz, CDCl3): δ = 8.09 (m, 2H, naphthyl-H), 7.52 (s, 2H, triazole-H), 7.38 (m, 2H, naphthyl-H), 7.10 (s, 2H, ArOH), 6.80 (d, J = 6.0 Hz, 4H, ArH), 6.55 (m, 8H, ArH), 6.01 (s, 4H, naph-CH2-triazole), 5.07 (s, 4H, ArOCH2-triazole), 4.04 (t, J = 6.0 Hz, 4H, –OCH2CH2CH2CH3), 3.94 (d, J = 15.0 Hz, 4H, ArCH2, AB system), 2.94 (d, J = 15.0 Hz, 4H, ArCH2, AB system), 1.76 (m, 4H, –O(CH2)2CH2CH3), 1.49 (m, 4H), 0.96 (t, J = 6.0 Hz, 6H, –O(CH2)3CH3). 13C-NMR (CDCl3, 75.46 MHz): δ = 152.8, 151.0, 143.2, 133.2, 129.6, 128.7, 128.3, 127.9, 126.9, 125.3, 124.3, 124.0, 123.8, 119.0, 74.5, 69.1, 44.9, 32.4, 31.2, 19.4, 14.0; HRMS APPI(+) calcd for C54H54N6O6: 882.4105, found: 882.4070.
:10 dichloromethane:methanol) to give a colourless solid (140 mg, 42%, m.p. 171.6 °C). 1H NMR (300 MHz, CDCl3): δ = 8.02 (m, 2H, naphthyl-H), 7.65 (s, 2H, triazole-H), 7.36 (m, 2H, naphthyl-H), 6.91 (s, 4H, ArH), 6.74 (s, 2H, ArOH), 5.98 (s, 4H, ArH), 5.93 (s, 4H, naphthyl-CH2-triazole), 5.05 (s, 4H, ArOCH2–), 4.05 (m, 4H, OCH2CH2CH2CH3), 4.00 (d, J = 12.9 Hz, 4H, ArCH2, AB system), 2.99 (d, J = 12.9 Hz, 4H, ArCH2Ar, AB system), 1.77 (m, 4H, –OCH2CH2CH2CH3), 1.49 (m, 4H, OCH2CH2CH2CH3), 1.27 (s, 18H, tBu), 0.95 (t, J = 14.4 Hz, 6H, –OCH2CH2CH2CH3), 0.86 (s, 18H, tBu); 13C-NMR (CDCl3, 75.46 MHz): δ = 150.6, 150.2, 149.4, 146.9, 143.7, 141.5, 132.4, 129.6, 127.8, 126.6, 125.4, 124.9, 124.1, 124.0, 123.6, 74.4, 69.2, 44.8, 33.8, 32.4, 31.7, 30.9, 29.7, 19.4, 14.0; HRMS APPI(+) calcd for C70H86N6O6: 1106.6609, found: 1106.6584.
4. Conclusions
Four new substituted-naphthyl ring “capped” 1,2,3-triazole-linked calix[4]arenes have been synthesized and characterized. Binding studies conducted by fluorescence quenching with twelve different metal ions showed that these new macrocyclic receptors were sensitive chemosensors for Fe3+ and Hg2+, in particular, for Fe3+ ions. The association, or binding constants, were determined using a 1:1 non-linear global analytical program developed by Thordarson and were compared with the values obtained with the classical Benesi–Hildebrand method. The site of the binding of the metal ions to the macrocyclic receptors was shown by a 1H NMR spectroscopic analysis and was confirmed by a detailed DFT computational analysis.
Acknowledgements
We thank the research support from the Research Development Corporation Newfoundland and Labrador, Vale, Memorial University of Newfoundland and the Ministry of Higher Education, Kingdom of Saudi Arabia for the Scholarships to Y. A. and A. N. A. The computational work has been assisted by the use of computing resources provided by WestGrid and Compute/Calcul Canada. We thank Dr Grigory Shamov, Westgrid/U. Manitoba for support.Notes and references
- For a recent review see: (a) M. Song, Z. Sun, C. Han, D. Tian, H. Li and J. S. Kim, Chem. – Asian J., 2014, 9, 2344–2357 CrossRef CAS PubMed; (b) W. Sliwa and M. Deska, ARKIVOC, 2011, 496–551 CAS.
- (a) S. Chen and P. Schopfer, Eur. J. Biochem., 1999, 260, 726–735 CrossRef CAS PubMed; (b) S. Hu, G. Wu, C. Xu, J. Dong and Q. Gao, J. Photochem. Photobiol., A, 2013, 270, 37–42 CrossRef CAS.
- (a) J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC; (b) D. Touati, Arch. Biochem. Biophys., 2000, 373, 1–6 CrossRef CAS PubMed; (c) B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS.
- (a) J. Cho, T. Pradhan, J. S. Kim and S. Kim, Org. Lett., 2013, 15, 4058–4061 CrossRef CAS PubMed; (b) R. K. Pathak, J. Dessingou, V. K. Hinge and C. P. Rao, Anal. Chem., 2013, 85, 3707–3714 CrossRef CAS PubMed.
- (a) M. D. P. De Costa and W. A. P. A. Jayasinghe, J. Photochem. Photobiol., A, 2004, 162, 591–598 CrossRef CAS; (b) B. R. Von, J. Appl. Toxicol., 1995, 15, 483–493 CrossRef; (c) H. H. Harris, I. J. Pickering and G. N. George, Science, 2003, 301, 1203 CrossRef CAS PubMed.
- (a) C. K. Hartmuth, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef; (b) Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848–2866 RSC; (c) A. H. El-Sagheera and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC; (d) C. Chu and R. Liu, Chem. Soc. Rev., 2011, 40, 2177–2188 RSC; (e) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC; (f) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed.
- E. H. Ryu and Y. Zhao, Org. Lett., 2005, 7, 1035–1037 CrossRef CAS PubMed.
- (a) K. D. Bhatt, H. S. Gupte, B. A. Makwana, D. J. Vyas, D. Maity and V. K. Jain, J. Fluoresc., 2012, 22, 1493–1500 CrossRef CAS PubMed; (b) X. L. Ni, Y. Wu, C. Redshaw and T. Yamato, Dalton Trans., 2014, 43, 12633–12638 RSC; (c) N. J. Wang, C. M. Sun and W. S. Chung, Tetrahedron, 2011, 67, 8131–8139 CrossRef CAS; (d) R. K. Pathak, V. K. Hinge, P. Mondal and C. P. Rao, Dalton Trans., 2012, 41(35), 10652–10660 RSC; (e) V. V. S. Mummidivarapu, V. K. Hinge, K. Tabbasum, R. G. Gomade and C. P. Rao, J. Org. Chem., 2013, 78, 3570–3576 CrossRef CAS PubMed; (f) R. K. Pathak, K. Tabbasum, A. Rai, D. Panda and C. P. Rao, Analyst, 2012, 137, 4069–4075 RSC; (g) Y.-J. Chen, S.-C. Yang, C.-C. Tsai, K.-H. Chuang, W.-L. Chu, V. Kovalev and W.-S. Chung, Chem. – Asian J., 2015, 10, 1025–1034 CrossRef CAS PubMed.
- J. Morales-Sanfrutos, M. Ortega-Muñoz, J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, J. Org. Chem., 2008, 73, 7768–7771 CrossRef CAS PubMed.
- (a) C. D. Gutsche, B. Dhawan, J. A. Levine, K. H. No and L. J. Bauer, Tetrahedron, 1983, 39, 409–426 CrossRef CAS; (b) C. D. Gutsche and M. Iqbal, Org. Synth., 1990, 68, 234 CrossRef CAS; (c) Z. Asfari, A. Bilyk, C. Bond, J. M. Harrowfield, G. A. Koutsantonis, N. Lengkeek, M. Mocerino, B. W. Skelton, A. N. Sobolev, S. Strano, J. Vicens and A. H. White, Org. Biomol. Chem., 2004, 2, 387–396 RSC; (d) R. K. Pathak, V. K. Hinge, M. Mondal and C. P. Rao, J. Org. Chem., 2011, 76, 10039–10049 CrossRef CAS PubMed; (e) W. Xu, J. J. Vittal and R. J. Puddephatt, Can. J. Chem., 1996, 74, 766–774 CrossRef CAS.
- A. H. Tran, D. O. Miller and P. E. Georghiou, J. Org. Chem., 2005, 70, 1115–1121 CrossRef CAS PubMed.
- P. Job, Ann. Chim., 1928, 9, 113–203 CAS.
- (a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC; (b) http://supramolecular.org ; (c) S. Bano, M. Ayaz, A. A. P. Khan and K. S. Siddiqi, J. Chem. Eng. Data, 2010, 55, 5759–5765 CrossRef CAS; (d) H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS . See the SI for the Kassoc determinations.
- K.-C. Chang, I.-H. Su, A. Senthilvelan and W.-S. Chung, Org. Lett., 2007, 9, 3363–3366 CrossRef CAS PubMed , and references cited therein.
- In order to determine whether the phenomena observed by Chung is a general one involving Fe(ClO4)3 and a calix[4]arene containing two free phenolic groups under the solvent conditions used for the fluorescence measurements, we subjected tetra-tert-butyl-1,3-di-propoxycalix[4]arene to titration experiments with up to 8-fold excesses of Hg(ClO4)2 and Fe(ClO4)3. Even after 72 h no changes in the chemical shifts of any of the protons could be detected. The presence of the anthryl-1,2,4-oxadiazole functional groups on Chung's calixarene scaffold appears to have a strong activating effect towards its oxidation with the Fe(ClO4)3 since Rao's group did not report any similar oxidations with their anthryl-triazole calixarene fluorescence studies.8d–f.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L; Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284–298 CrossRef; (c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details for the complexation and computational studies of 6a–d with Fe3+, and Hg2+ ions; 1H- and 13C-NMR and MS including HRMS spectra for all new compounds 3a–b and 6a–d. See DOI: 10.1039/c5nj01362c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |