
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancement of photochemical heterogeneous water oxidation by a manganese based soft oxometalate immobilized on a graphene oxide matrix
Santu
Das
,
Archismita
Misra
and
Soumyajit
Roy
*
EFAML, Material Science Centre, Department of Chemical Science, Indian Institute of Science Education and Research Kolkata, Mohanpur – 741246, India. E-mail: s.roy@iiserkol.ac.in
First published on 23rd November 2015
Abstract
Development of efficient and oxidatively stable molecular catalysts having abundant transition metals at the active site is an immediate challenge to synthetic chemists in order to photochemically split water into clean fuels oxygen and hydrogen to serve the ever-increasing energy demand. Herein we report a soft-oxometalate (SOM)-based heterogeneous photocatalytic system which effectively performs water oxidation giving oxygen. In the present work we placed a double sandwich type manganese-based polyoxometalate (POM), Na17[Mn6P3W24O94(H2O)2]·43H2O, on an electroactive graphene oxide matrix and synthesized a new SOM [Na17[Mn6P3W24O94(H2O)2]·43H2O@graphene oxide] 1 and performed water oxidation with it. The efficiency of photocatalytic water oxidation by SOM 1 is almost double than in the case of Na17[Mn6P3W24O94(H2O)2]·43H2O alone. The rationale behind this lies in the electron accepting nature of the graphene sheets which effectively relay the electrons generated in the water oxidation reaction, thus facilitating the forward reaction and increasing the oxygen yield. Variation of catalyst loading, pH-dependent and time-dependent experiments are performed to study their effects on photocatalytic water-splitting. The reaction kinetics is sigmoidal in nature, suggesting the heterogeneous nature of catalysis. The composite catalyst system is observed to be stable towards the reaction conditions.
Introduction
Water oxidation is one of the most promising routes towards the global goal of alternative energy.1–4 Many research groups have developed robust catalysts for efficient water oxidation.5–7 Recently chemists have been interested in developing molecular water oxidation catalysts by using cheap and abundant transition metals.6,8 Different chemical species are used as catalysts for that purpose, e.g. metal organic complexes,9–24 nanomaterials,25–29 and the recently developed polyoxometalates.6,8,30–42 Polyoxometalates (POM) show higher stability in an oxidizing environment compared to metal–organic complexes where organic ligands tend to get easily oxidized and thus offer better catalyst stability. Different routes of water splitting have been explored, such as chemical,9,10 electrochemical2,43–45 and photoelectrochemical methods.46–52 However, photochemical water oxidation seems to be the most facile and clean technique.6,13,53,54A recent challenge in photochemical water oxidation by polyoxometalates is to enhance the oxygen generation and increase the turnover number (TON) of the reaction.16 Up to now an iridium based complex has shown the maximum TON reported by Crabtree and Brudvig.55 We are interested in observing whether the reaction efficiency can be enhanced without changing the active center of the catalyst. It is known that POMs can easily be immobilized on the electroactive surface to form stable composite systems.56 So, we ask whether it is possible to employ a related composite system to perform water oxidation experiments.29,33,57
Recently, the Hill group developed a similar method using graphene modified electrodes and ruthenium based POM as active catalyst.57
The graphene modified electrodes show excellent catalytic activity and high stability toward the electrochemical water oxidation reaction at neutral pH. This work showed enhanced water oxidation reaction electrochemically.57 Here we ask whether it is possible to make a soft oxometalate56,58–61 based on polyoxometalate–graphene oxide to enhance the efficiency of photochemical water oxidation.
In our present work we use a manganese based polyoxometalate Na17[Mn6P3W24O94(H2O)2]·43H2O and synthesize a SOM 1 by immobilizing it on a graphene oxide surface [Na17[Mn6P3W24O94(H2O)2]·43H2O@graphene oxide] 1. This catalyst shows a turnover number of 22 at pH 8 for WO reaction. The SOM 1 dispersion is prepared by sonication. Formation of the composite is confirmed by Raman spectra, SEM images and EDX data. Finally we use this SOM as a photo-catalyst in water oxidation (Fig. 1). Interestingly, we observe that in the presence of the graphene oxide matrix the water oxidation activity of Mn–POM is almost doubled. A detailed account of synthesis and characterization of the composite catalyst and observations related to photochemical water oxidation studies are provided in the following sections.
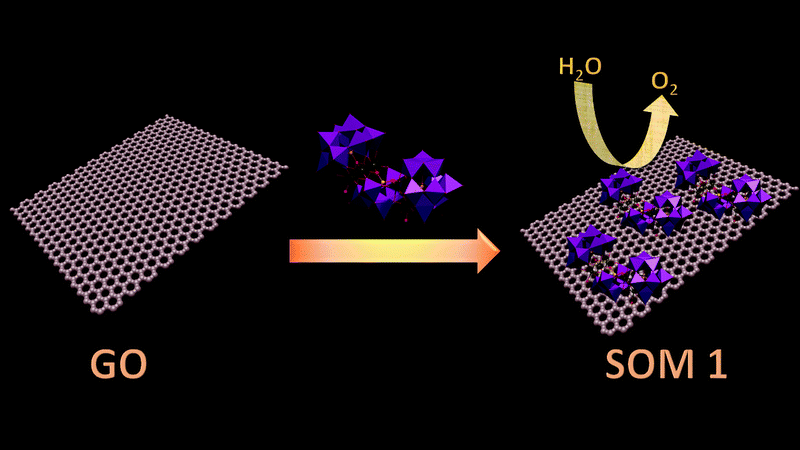 | ||
| Fig. 1 Schematic diagram of the overall process. | ||
Result and discussion
Formation of the SOM 1 composite based on graphene oxide
SOM 1 is prepared by following the classical route of immobilization of POM on an electroactive surface.56 In our present study we initially prepared graphene oxide dispersion in water. To this dispersion Na17[Mn6P3W24O94(H2O)2]·43H2O (Fig. 2) was added and the mixture was sonicated to finally get the composite SOM 1, which forms a stable dispersion. Composite formation was characterized by using Raman spectroscopy, SEM and EDX analysis.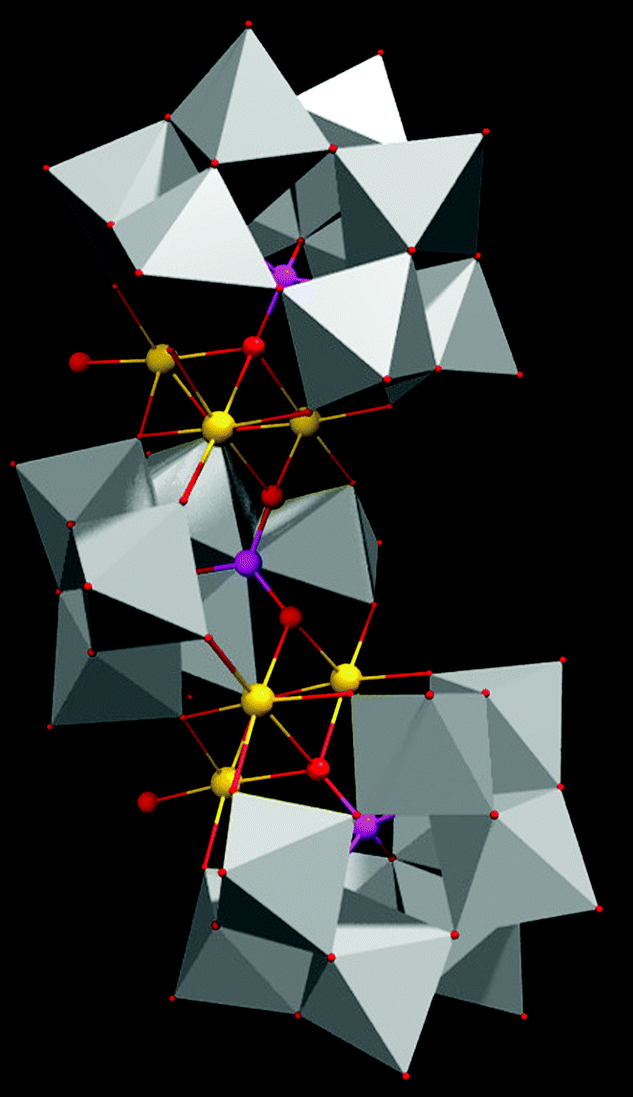 | ||
| Fig. 2 Single crystal structure of the POM constituent of SOM 1. | ||
Scanning electron microscopy (SEM): morphology of SOM 1
GO shows nanosheet type morphology. It is observed from scanning electron microscopy (Fig. 3a). The SOM 1 shows nanospheres embedded on graphene oxide layers (Fig. 3b). The white bright spot indicates the clustering of POM, suggesting that POM units are attached to the surface of GO by electrostatic interactions, as GO has an electron deficient surface (positively charged) and POM are large polyanions (negatively charged). This further indicates the formation of the composite in the reaction system. From the EDX data we can also infer that the molecular integrity of POM is intact in SOM 1. [Note: manganese, phosphorus and tungsten are present in the expected correct ratio of POM in SOM 1 (Fig. 3c).]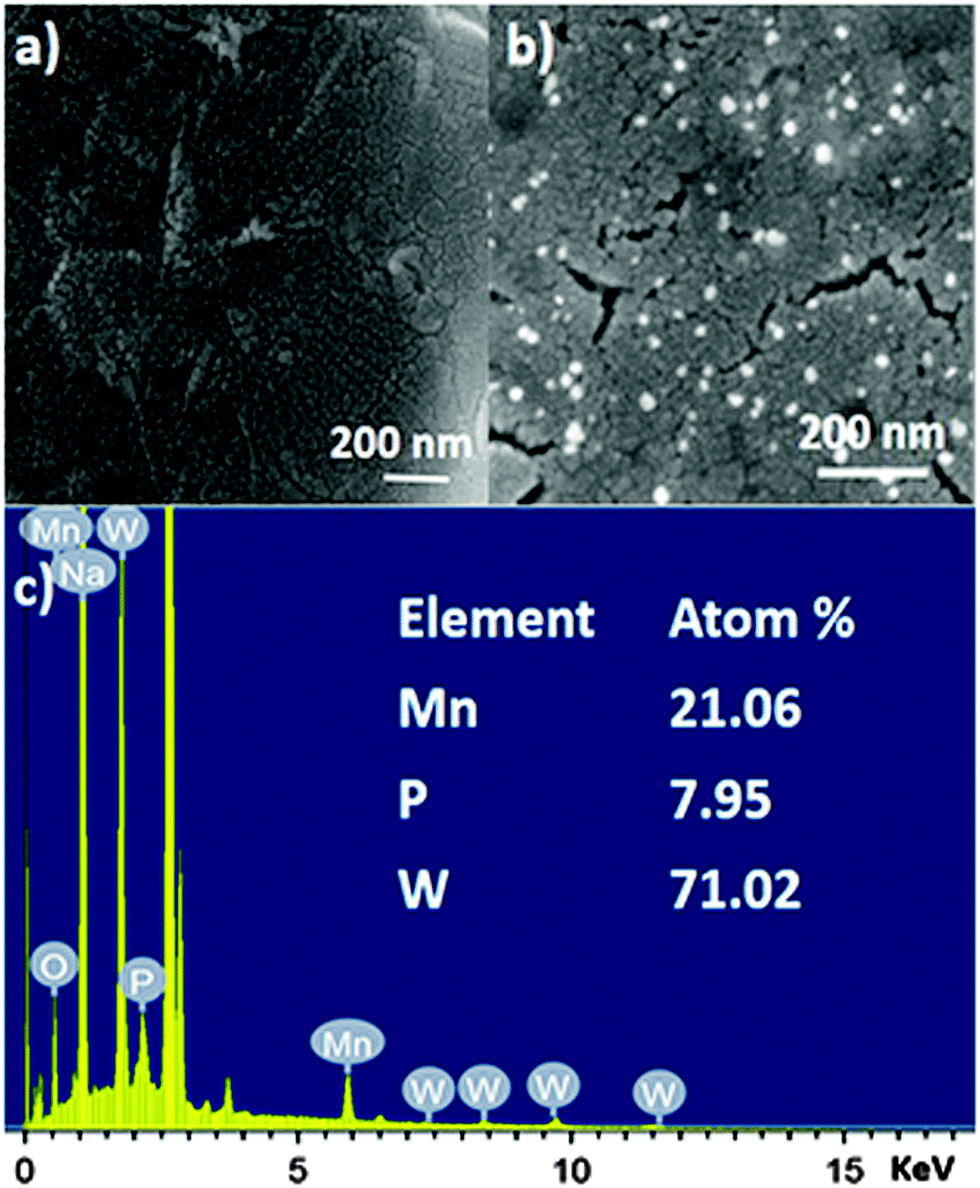 | ||
| Fig. 3 SEM images of (a) graphene oxide surface. (b) SOM 1, defined by aggregate nanospherical POMs immobilized on the graphene oxide layers. (c) EDX spectrum of SOM 1. | ||
HATR-IR spectroscopy and stability of POM in SOM 1
HATR-IR spectroscopy of SOM 1 and also HATR-IR spectroscopy of the POM constituent were performed. It was observed that a few broad bands were obtained in each case in the IR spectrum. This broadness is possibly due to the low concentration of the sample in the dispersion. Here we observed common peaks for POM and SOM 1 at 1637, 693, 569, 496 cm−1 respectively (Fig. 4a). Thus from the IR-spectrum we can conclude that the POM constituent remains stable and intact in the SOM 1 after composite formation and no catalyst is degraded at all. We further performed UV-VIS spectroscopy to check changes in the energy gap of the POM constituent after composite formation.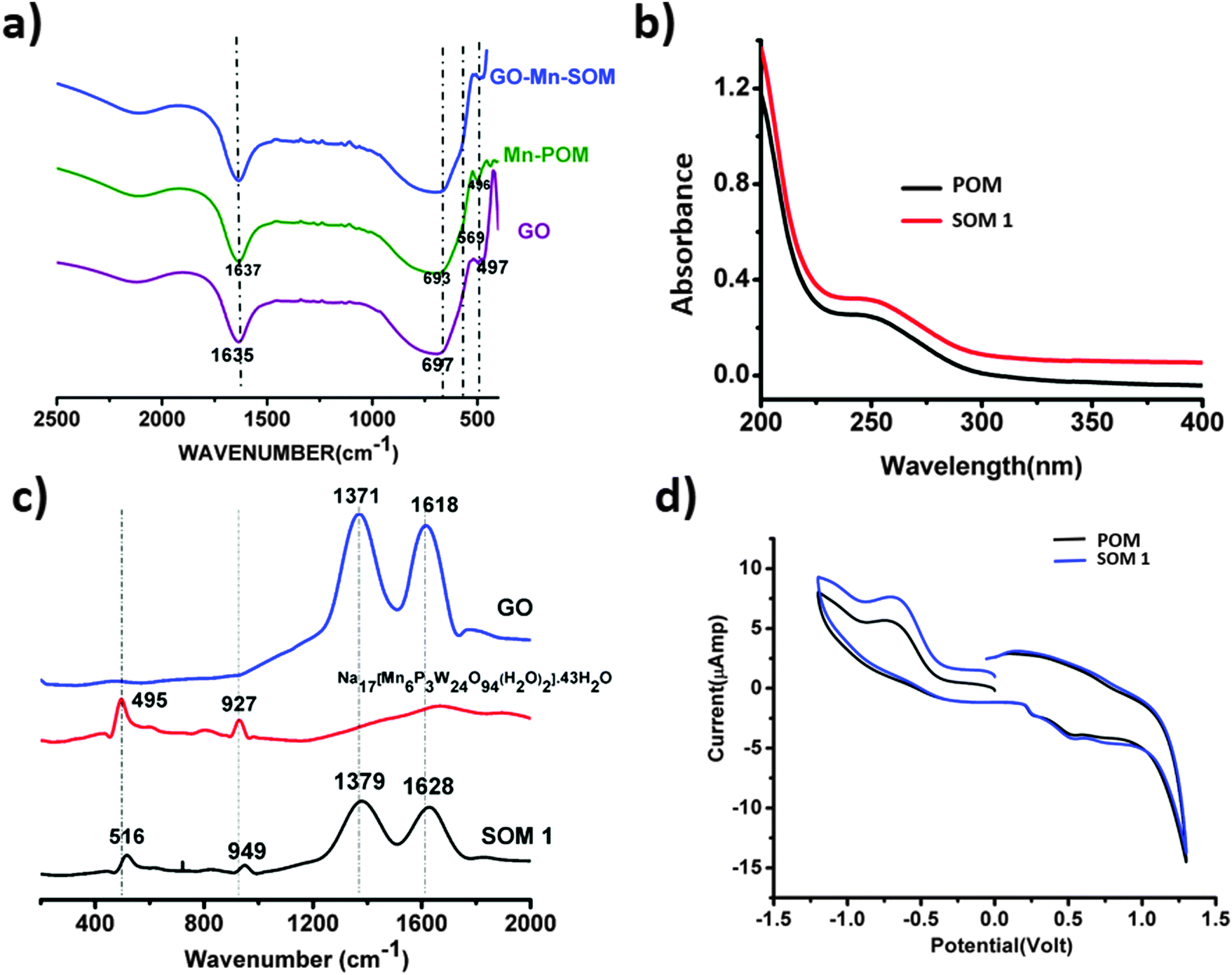 | ||
| Fig. 4 (a) HATR-IR spectroscopy of the POM constituent, GO, and SOM 1. (b) UV-VIS spectroscopy of the POM constituent of SOM 1 and SOM 1. (c) Raman spectra of the POM unit Na17[Mn6P3W24O94(H2O)2]·43H2O, graphene oxide and SOM 1. (d) Cyclic voltammogram of SOM 1 and the POM constituent of SOM 1. | ||
UV-VIS spectroscopy of the POM constituent and SOM 1
We performed UV-VIS spectroscopy of SOM 1 and the POM constituent in water. For both POM and SOM 1 we got absorbance maxima at 250 nm (Fig. 4b). Thus it may be concluded that the band gap of the POM constituent does not change in the SOM 1 composite, which further proves the stability of the POM constituent in SOM 1 because if it was dissociated to other cluster units then there should have been a clear difference in the UV-VIS spectrum.Raman spectroscopy and the nature of SOM 1
We now want to show the effective formation of SOM. Raman spectra (Fig. 4c) of Na17[Mn6P3W24O94(H2O)2]·43H2O, graphene oxide and the SOM 1 composite were taken; for SOM 1 we observed 4 peaks at 516, 949, 1379, and 1628 cm−1 respectively. We assign these peaks as follows: 516 (νas,Mn–O), 949 (νW![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the other two peaks at 1379 and 1628 cm−1 for disorder and graphitic nature of graphene oxide respectively. These peaks are blue shifted compared to that of the spectrum of graphene oxide and Na17[Mn6P3W24O94(H2O)2]·43H2O alone.
O) and the other two peaks at 1379 and 1628 cm−1 for disorder and graphitic nature of graphene oxide respectively. These peaks are blue shifted compared to that of the spectrum of graphene oxide and Na17[Mn6P3W24O94(H2O)2]·43H2O alone.
The Raman spectrum of Na17[Mn6P3W24O94(H2O)2]·43H2O shows peaks at 495 and 927 cm−1 which can be attributed to the following modes: (νas,Mn–O), (νWO). The characteristic peaks at 1371 and 1618 cm−1 are on the other hand due to disorder and graphitic nature of graphene oxide respectively. This shift in the spectrum might indicate that there may be the possible presence of interaction between POM and the graphene oxide layer in SOM 1. In our system electrons are probably transferred from Na17[Mn6P3W24O94(H2O)2]·43H2O to graphene oxide.62,63 This was further explained by CV. We conclude from this shift that in SOM 1, graphene oxide may act as an electron acceptor and Na17[Mn6P3W24O94(H2O)2]·43H2O may act as an electron donor. The Raman spectrum also reveals that SOM 1 is not a physical mixture of the constituents graphene oxide and POM but an assembly of the two at the molecular level.
Cyclic voltammogram of the catalyst
To further monitor the stability of the POM constituent in SOM 1 we performed CV of SOM 1 and compared it with the CV of the POM constituent of SOM 1 (Fig. 4d) and it is clearly observed that both are identical, which indicates that the redox behavior of the POM constituent in SOM 1 remains unaltered and we can also conclude that the POM constituent is stable after composite formation. Also we observed that the peak current increased for SOM 1 which further indicates facile electron transport from the POM constituent to GO surface.Photochemical water splitting
Photochemical water splitting experiments were performed under a UV lamp (λmax = 373 nm) with the composite catalyst system. The composites were prepared as mentioned in the previous section. The oxygen evolution was monitored using a YSI optical sensor based dissolved oxygen meter and also by cyclic voltammetry. The maximum oxygen yield obtained was 19.2 μmol for 20% SOM 1 loading at pH 8 in phosphate buffer. The graphene oxide acts as an electron acceptor and traps the electrons released in the water oxidation reaction and facilitates electron transport as well.Confirmation of water oxidation and the effect of graphene oxide
In our present work SOM 1 absorbed light and was elevated to the excited state. This excited SOM 1 generated hole and electron pairs, and the holes oxidized water to oxygen in the presence of light. After photoillumination quantitative determination of evolved oxygen was performed by measuring the evolved oxygen (Fig. 5c) using a YSI optical sensor based dissolved oxygen meter. For further confirmation of evolution of oxygen cyclic voltammetry (Fig. 5a) was performed using samples after photoirradiation, where a sudden rise of current was observed near +1.2 V with respect to the Ag/AgCl reference electrode indicative of oxygen evolution from water. It implies oxidation of water.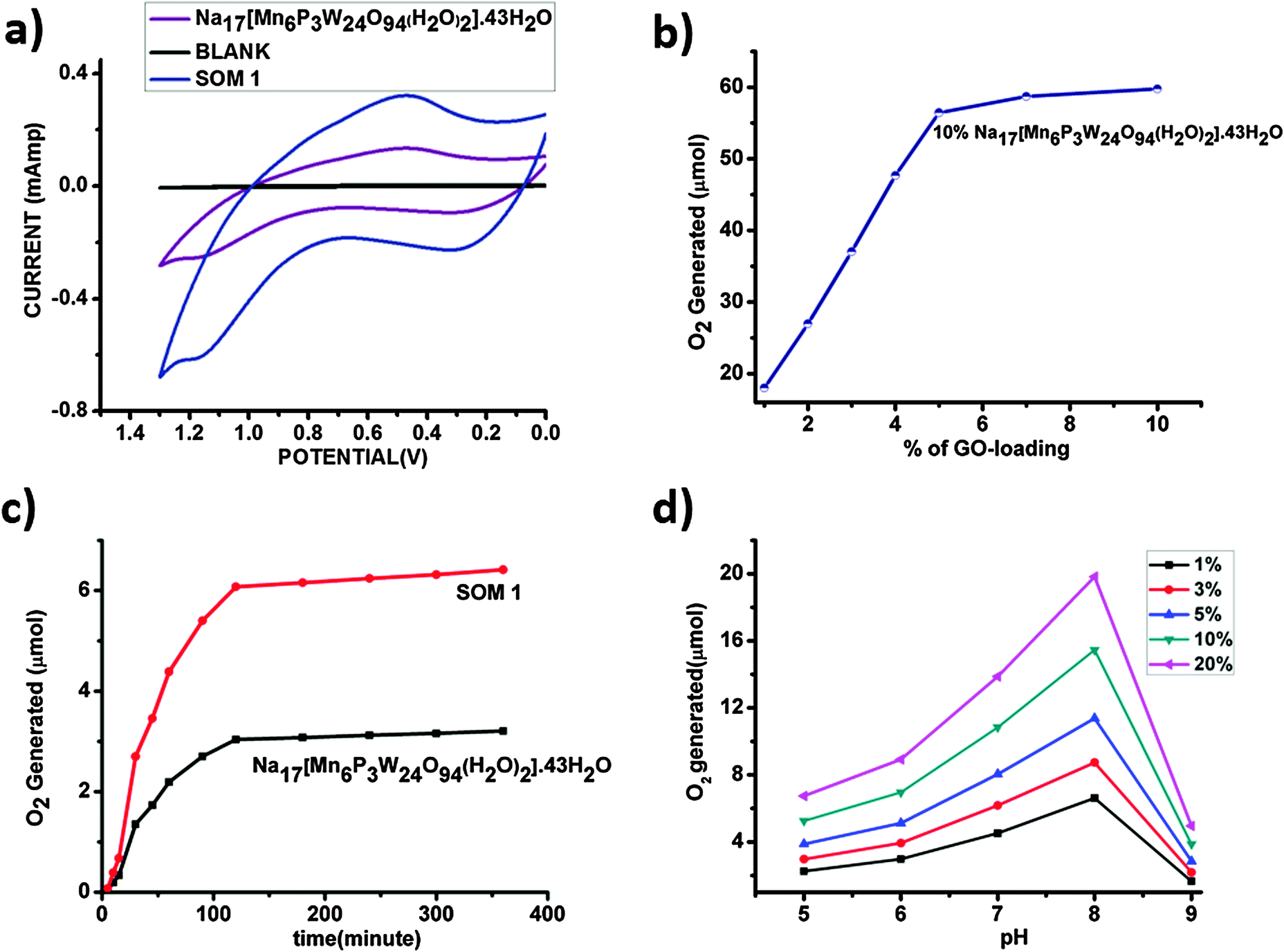 | ||
| Fig. 5 (a) Cyclic voltammogram of the water oxidation study by the POM Na17[Mn6P3W24O94(H2O)2]·43H2O and SOM 1. (b) Extent of oxygen liberated with variation in graphene oxide-loading with Na17[Mn6P3W24O94(H2O)2]·43H2O concentration kept constant. (c) Comparative oxygen evolution by the POM Na17[Mn6P3W24O94(H2O)2]·43H2O only and SOM 1. (d) Variation of oxygen yield with pH. | ||
We observe that the extent of oxygen evolution is almost doubled in the case of the SOM 1 composite catalyst as compared to water oxidation by Na17[Mn6P3W24O94(H2O)2]·43H2O alone. Using POM alone the maximum amount of oxygen liberated is almost 3.2 μmol for 0.071 μmol loading of the catalyst, with a TON of around 46, whereas in the case of the SOM 1 composite catalyst system the amount of O2 evolved is almost 6.5 μmol for 0.071 μmol loading of the catalyst, with a TON of 92, which is roughly double than that of the POM alone. We thus investigate the role of graphene oxide in water oxidation.
In the next set of experiments, Na17[Mn6P3W24O94(H2O)2]·43H2O loading was kept constant (10 mg/10 ml in all the catalyst dispersions) and the graphene oxide concentration was increased (Fig. 5b). Here we observe a similar type of sigmoidal curves and the maximum O2 generation is almost 58 μmol for 10% SOM 1 loading. The comparative studies clearly show that the increase of graphene oxide loading has a prominent effect on water oxidation. We observed two different aspects: (i) up to a certain loading of graphene oxide (5 mg) O2 evolution increases to a maximum of 58 μmol and (ii) thereafter the increase of graphene oxide loading has no further effect on O2 evolution. We explain this as follows. With the increase in graphene oxide loading the extent of electron relay facilitated by graphene oxide increases, thereby increasing the effective O2 evolution. However beyond a certain threshold of graphene oxide loading the active POM concentration in SOM 1 gets diluted. Hence the evolution of O2 does not increase anymore.
Needless to say, graphene oxide invariably enhances the water oxidation activity. In SOM 1 we also infer that graphene oxide most likely (1) provides an enhanced active catalytic surface area and (2) facilitates electron transport and thereby enhances water oxidation effectively. Also one of the prominent reasons for the enhancement of water oxidation by using the GO matrix may be due to the increase in the effective surface area of the catalyst. In the case of SOM 1, the hydrodynamic radius is around 300 nm (from dynamic light scattering experiments) as compared to single SOM having a hydrodynamic radius of 130 nm. We have calculated the surface area by assuming the catalyst materials to form nanospheres in dispersion and for a spherical surface we calculated the area by the following equation: surface area = 4πRh2. Now we address the problem of how water oxidation is affected with variation in pH and loading of SOM 1 dispersion in the next section.
pH dependent study
pH dependent water oxidation study reveals some interesting results (Fig. 5d). We observe that with the increase in pH the amount of evolved oxygen increases gradually. At pH 8 we observe the maximum yield. On further increasing the pH, oxygen evolution decreases abruptly and this may be attributed to the degradation of clusters at higher pH. This observation may be explained by the shift of equilibrium involved with oxygen evolution to the right with the increase in pH.| H2O → O2 + 4H+ + 4e− |
Catalyst loading variation studies
In this set of experiments, the graphene oxide concentration is kept constant (1 mg/10 ml in all the catalyst dispersions) and the SOM 1 loading is increased to observe the change in water oxidation. It is observed that with increasing POM loading oxygen evolution increases for a given pH. The nature of the oxygen evolution curve with catalyst loading reveals that initially with increasing catalyst loading oxygen evolution increases rapidly, but after exceeding a certain loading of POM on SOM 1 catalyst (Fig. 6), the rate of enhancement of oxygen evolution in the reaction decreases to some extent. This may be due to the stability factor of the dispersion. More precisely oxygen evolution decreases when phase separation is observed and when we cross the dispersion stability window. This decrease in oxygen evolution is also due to the decrease in the active surface area of the catalyst.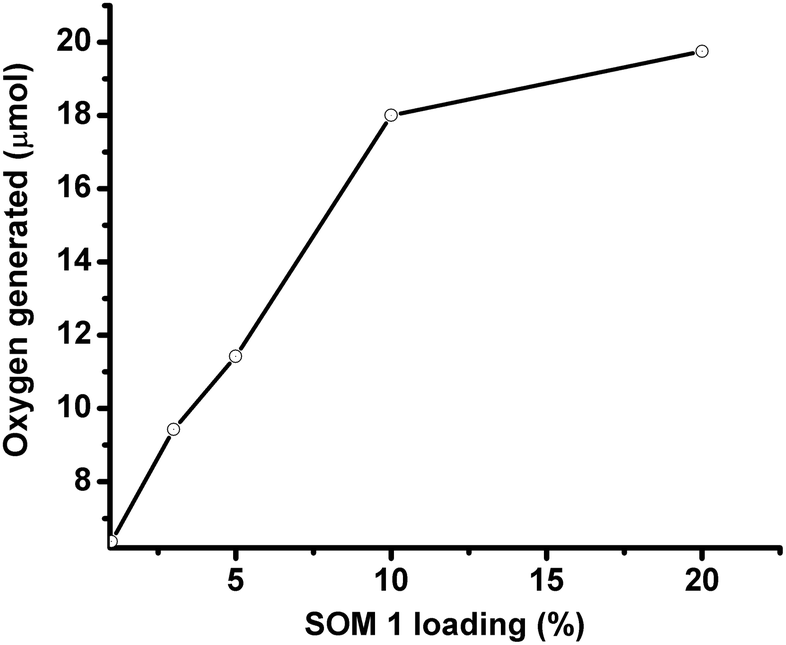 | ||
| Fig. 6 Variation of SOM 1 loading at pH 7. | ||
Time depended studies of oxygen evolution reaction
Time dependent water oxidation experiments show general sigmoidal kinetic patterns typical of heterogeneous catalysis reactions, where up to a certain limit of time oxygen evolution increases and reaches a plateau (Fig. 7a). There is an induction period of reaction which may be due to light absorption limitation. For excitation of SOM 1 it needs to cross a minimum energy barrier, which is attained after some time and therefore initially there is no reaction. When SOM 1 possesses minimum energy for excitation water oxidation starts (Fig. 7b). As water is taken in excess in the reaction, the reaction rate only depends on the intensity of light and not on the amount of water present in the reaction medium. At early times of the reaction, i.e., at low light intensity (up to 9.32 mW cm−2) there is no O2 evolution. However, beyond a threshold light intensity (9.32 mW cm−2) O2 evolution begins. The induction period (before threshold light intensity) probably simply reflects the time for O2 product equilibration before the analysis of evolved O2. It increases in a sigmoidal fashion suggesting co-operative photo-activation of the SOM sites for water oxidation. However with the increase in energy density oxygen evolution reaches saturation. Hence in other words it might be said that water oxidation reaction requires a threshold energy density to begin with, then increases in a sigmoidal fashion implying co-operative photo-activation of the SOM sites, finally reaching saturation with energy density. Thus it implies that the water oxidation reaction is topped off after a certain time. The maximum TOF of the reaction is 0.75 min−1 which is comparatively less than that of the recently developed photochemical water oxidation using cobalt based POM. This difference in TOF may be due to the involvement of different redox couples in the reaction. Here we observe that the maximum amount of O2 generated is almost 19 μmol for 20% of SOM 1 loading. Now we ask whether SOM 1 is stable in the course of the reaction. To determine its stability we measured the Raman spectrum of SOM 1 before and after the completion of the reaction. A detailed account of this study is provided in the next section.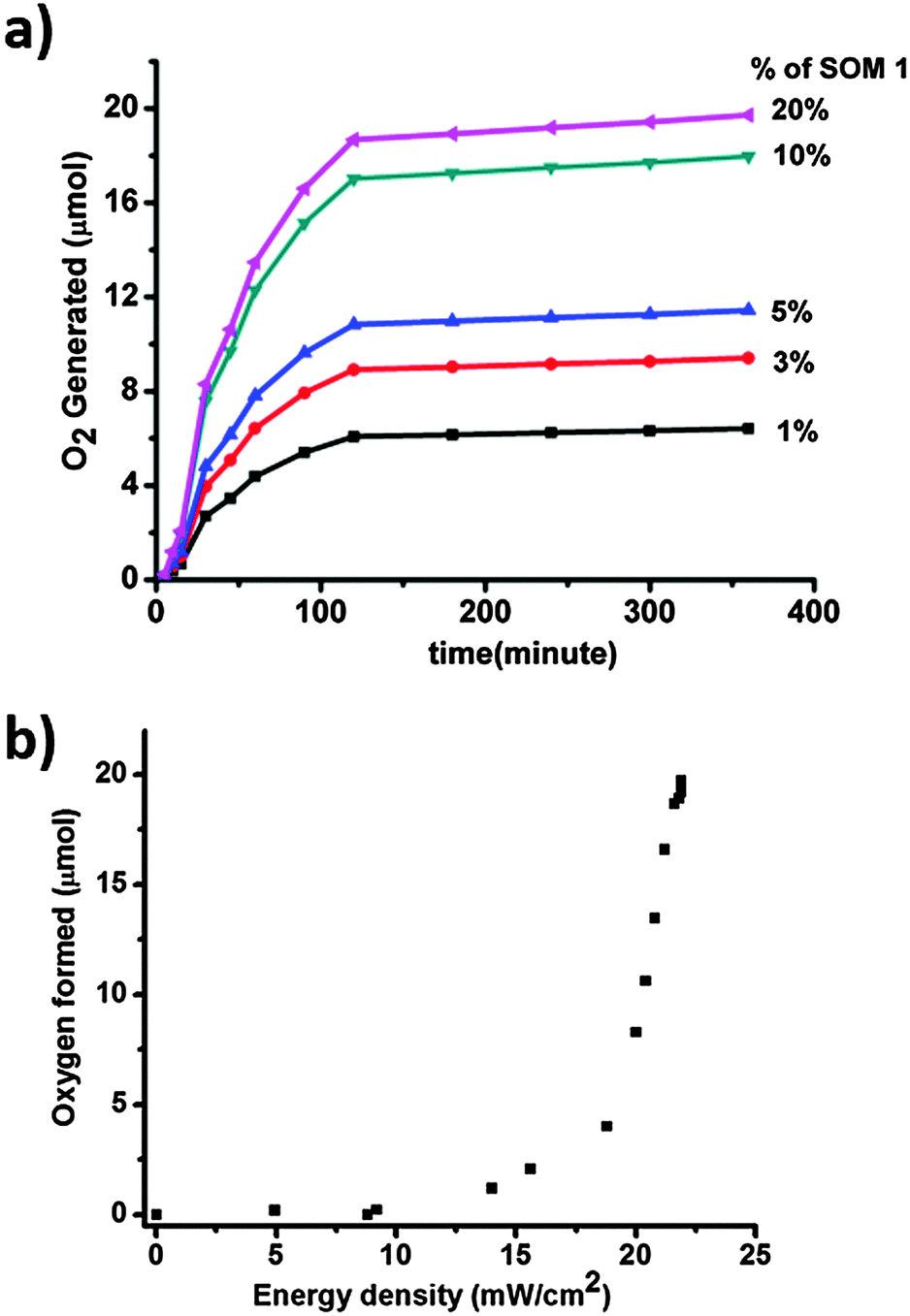 | ||
| Fig. 7 (a) Concentration and time dependent water oxidation study. (b) Plot of energy density of the photoreactor with oxygen formed. | ||
Stability of the composite SOM catalyst
Raman spectroscopic investigations were performed on the SOM 1 composite catalyst before and after the reaction. The spectra were observed to be identical (Fig. 8a). We also performed the HATR-IR (Fig. 8b) and UV-VIS (Fig. 8c) spectroscopy of the SOM 1 catalyst after the reaction. We also performed cyclic voltammetry (Fig. 8d) with the post reaction dispersion and in all the cases we observed identical spectra compared with the spectra obtained with the dispersion SOM 1 before the reaction. So we can possibly comprehend that the composite catalyst system remains intact during the water splitting reaction. Thus the reported catalyst system is stable under the water oxidation conditions that are used in this study. The POM constituent does not dissociate to form MnO2 or some other fragment. Therefore during photochemical water oxidation reaction it is reasonable to believe that no MnO2 is generated under reaction conditions which can possibly oxidize water. Only the POM constituent is clearly responsible for the water oxidation reaction. More experiments and analyses are needed to pin-point the active species, excited species lifetime and other deeper mechanistic details which will be performed by us in the future. We also took SEM images (Fig. 9) of the post reaction composite catalyst and observed almost a similar kind of morphology as in the images taken before the commencement of the reaction.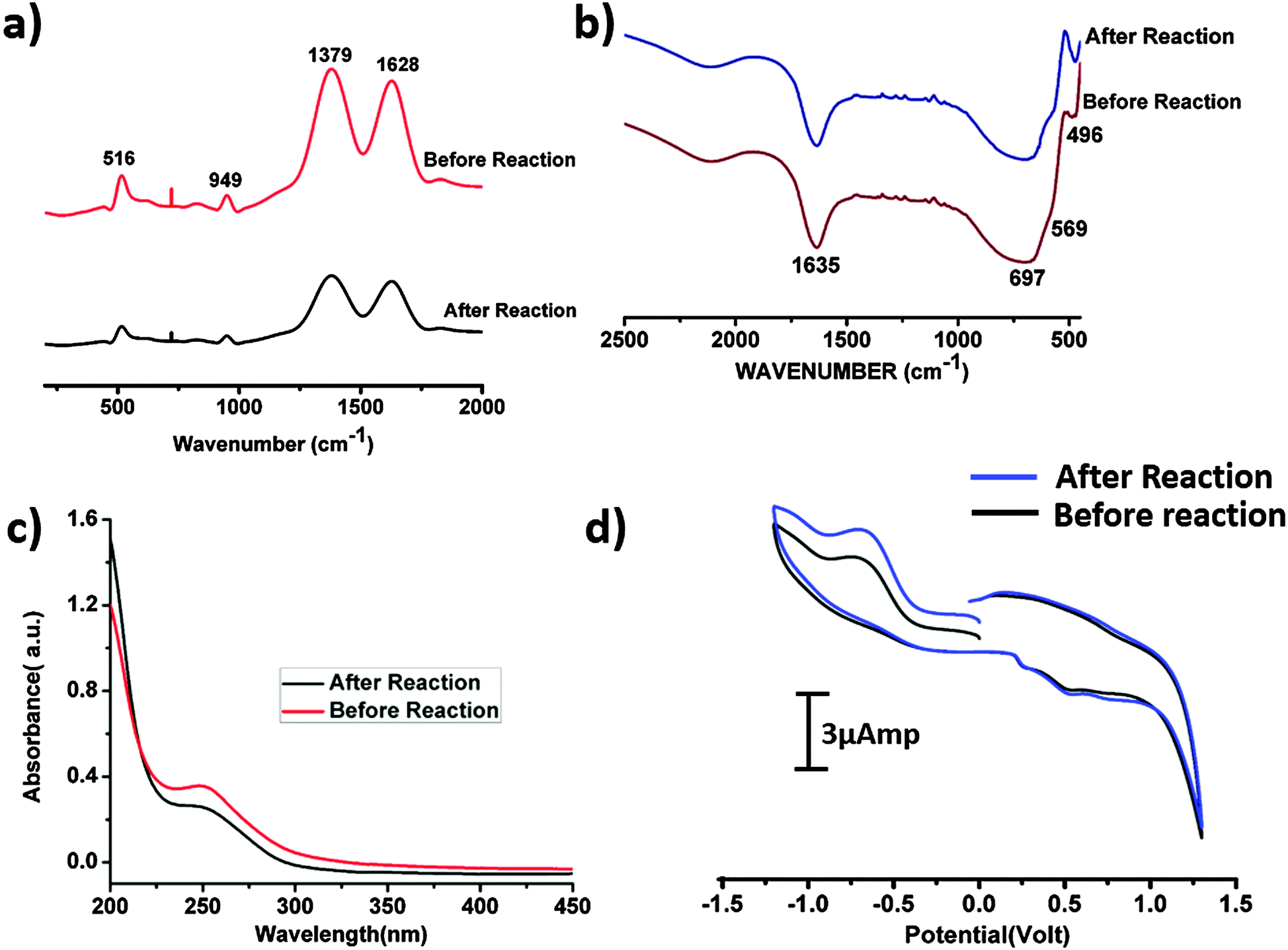 | ||
| Fig. 8 (a) Raman spectrum of the SOM 1 catalyst before and after the reaction. (b) HATR-IR spectrum of the SOM 1 catalyst before and after the reaction. (c) UV-VIS spectrum of the SOM 1 catalyst before and after the reaction. (d) Cyclic voltammogram of the catalyst before and after the reaction. | ||
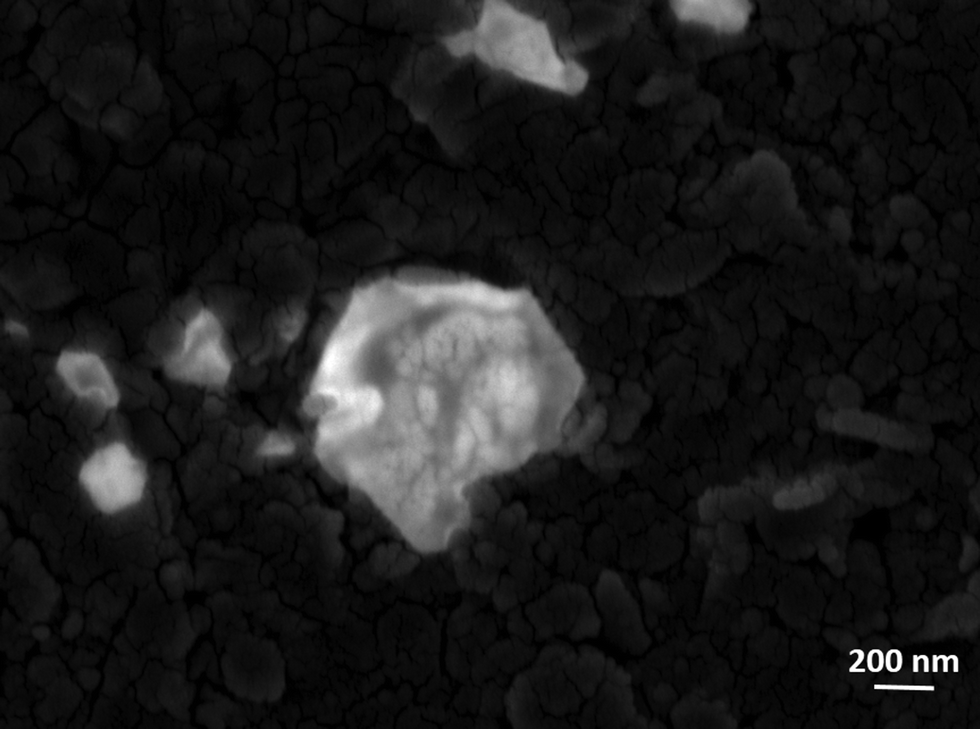 | ||
| Fig. 9 SEM image of the post reaction SOM 1. | ||
Catalytic recyclability of SOM 1
As the catalyst is stable after the reaction, we can effectively reuse this catalyst for further catalytic cycles. For this purpose we checked the recyclability up to 10 catalytic cycles and we observed that each time an equal amount oxygen is evolved in the catalytic cycle (Fig. 10). Therefore the catalyst is completely reusable.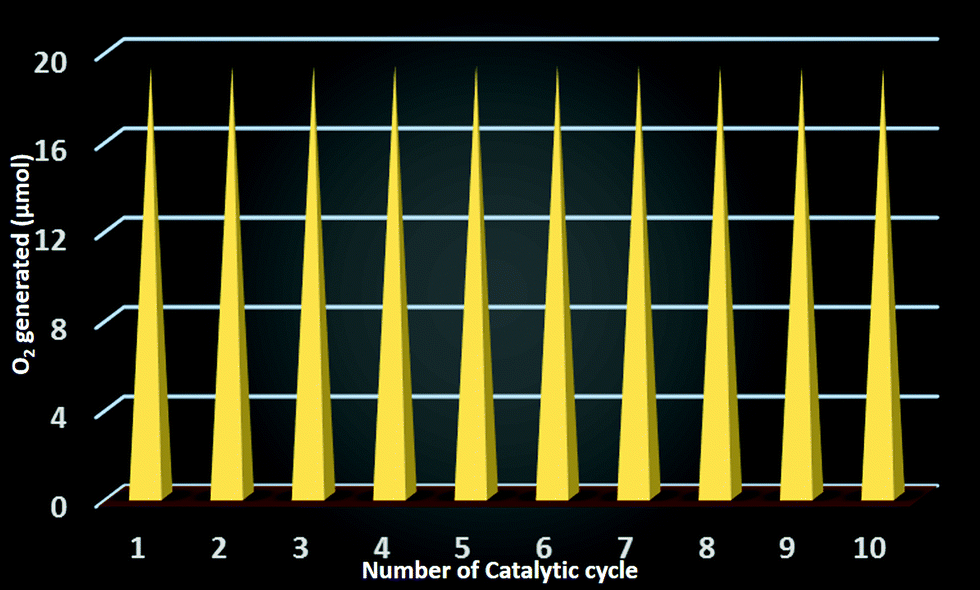 | ||
| Fig. 10 Catalytic recyclability of the composite catalyst SOM 1. | ||
Mechanism of evolution of O2 from water
Photochemical water oxidation with polyoxometalates generally takes place in the presence of an additional photosensitizer and a sacrificial electron acceptor. In our present work, the photocatalytic heterogeneous reaction possibly follows a completely different pathway. Here we need not add any photosensitizer and sacrificial electron acceptor. SOM 1 itself may be absorbing light and going to the excited state which oxidizes water to oxygen (Fig. 11), but elucidation of the actual photophysical mechanism will require additional studies. To further prove that the excited species oxidized water we added catechol in the reaction and observed that water oxidation ceased under these conditions. This may be due to the fact that catechol oxidation is more favorable compared to water oxidation and therefore water oxidation does not take place in the presence of catechol. The graphene oxide sheets are expected to act as electron acceptor platforms for the electrons generated in the water oxidation process64,65 and also enhance the surface area of the POM constituent of SOM 1.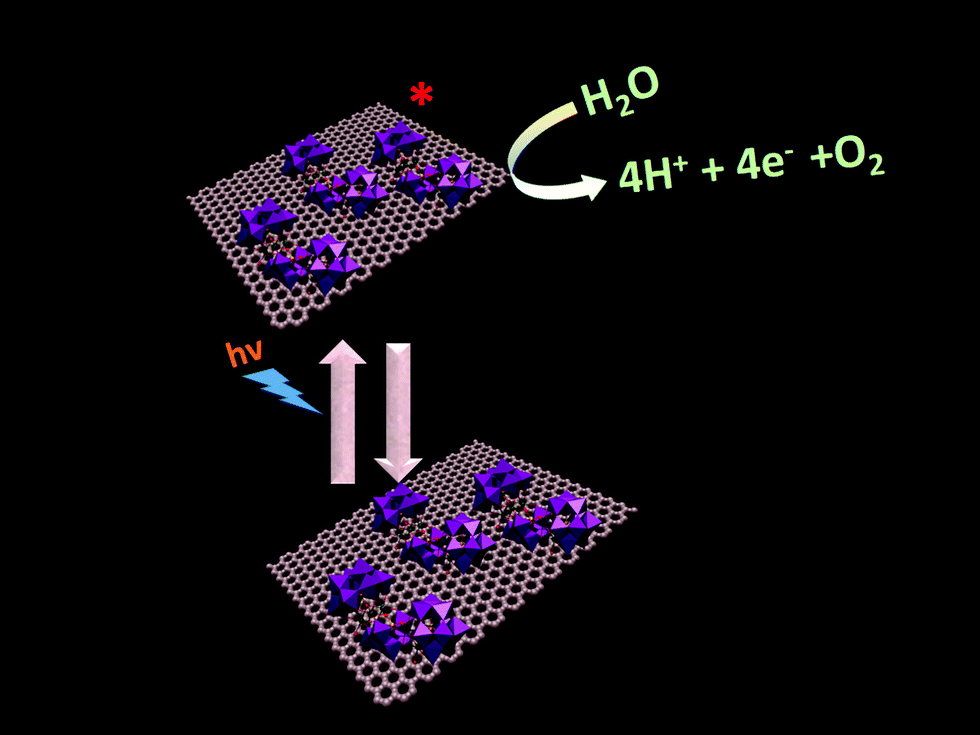 | ||
| Fig. 11 Pathway of photochemical water oxidation. | ||
Conclusion
To summarize, we have demonstrated the water oxidation by Mn–polyoxometalate (POM) based soft-oxometalate (SOM 1) dispersion and the efficiency is almost doubled by immobilizing Mn–POM on an electroactive graphene oxide matrix. The catalyst system acts as a water oxidizing agent to generate oxygen under photochemical conditions. The graphene oxide layers possibly act as electron acceptors and surface area enhancers and facilitate water oxidation by SOM 1. Thereafter we describe the effect of catalyst loading and pH on photocatalytic water-splitting. From the kinetics of the reaction we show the operation of heterogeneous mode of catalysis. After demonstrating the stability of the catalyst in the course of the water splitting reaction we have proposed the plausible pathway of the catalyst action. Further work is in progress in our laboratory in order to design more SOM based water splitting catalysts.Experimental procedure
Materials and reagents
All the materials were purchased from commercially available sources and used without further purification. All the glass apparatus were first boiled in an acid bath, then in water and finally rinsed with acetone. All the glass apparatus were properly dried in a hot air oven overnight. Doubly distilled deionized water was used to carry out all the experiments.Synthesis of graphene oxide
Graphene oxide was synthesized by the improved Hummers' method.66–68 Hummers' method69 (KMnO4, NaNO3, H2SO4) is the most common method used for preparing graphene oxide. A recent methodology study has modified the process to some extent and improved the efficiency of the oxidation process and this modified Hummers' method68 was employed here to synthesize graphene oxide for our experiments.Concentrated H2SO4 (69 ml) was added to a mixture of graphite flakes (3.0 g, 1 wt equiv.) and NaNO3 (1.5 g, 0.5 wt equiv.), and the mixture was cooled using an ice bath to 0 °C. KMnO4 (9.0 g, 3 wt equiv.) was added slowly to keep the reaction temperature below 20 °C as KMnO4 addition is exothermic. The reaction was warmed to 35 °C and stirred for 7 h. Additional KMnO4 (9.0 g, 3 wt equiv.) was added in one portion, and the reaction was stirred for 12 h at 35 °C. The reaction mixture was cooled to room temperature and poured into ice with 30% H2O2 (3 ml). The mixture was then purified following the usual protocol of sifting, filtering, centrifugation, decanting with multiple washes followed by a final vacuum drying to give 4.0 g of solid product.
Synthesis of SOM 1
1 mg of graphene oxide was added into 10 ml of water and 2 ml of ethylene glycol was added to it for better separation of the graphene oxide sheets. Then it was sonicated for 3 hours at room temperature to prepare graphene oxide dispersion. After that, 10 mg of Na17[Mn6P3W24O94(H2O)2]·43H2O was added and the dispersion was sonicated for 3 more hours. The stability of the dispersion was checked and it was found to be stable.Photocatalytic water splitting
Photocatalytic water splitting reactions were performed as follows. In the composite dispersion for water oxidation experiment buffer solution of pH 7 was added. The reaction mixture was then sealed and N2 gas was purged for 3 hours to get rid of the trace amount of oxygen in it. Then the reaction mixture was kept in a photoreactor under UV-light (energy density of the photoreactor is −19.5 mW cm−2 with λmax = 373 nm) for 2 hours. After irradiation we measured the amount of evolved oxygen in the reaction by using YSI optical sensor based dissolved oxygen meter standardized by using degassed double distilled water. Evolution of oxygen in the reaction was further investigated by performing cyclic voltammetry using the irradiated samples. In cyclic voltammetry we observed a sharp rise of the current–voltage curve near +1.2 V, which is typically indicative of O2 generation by water splitting.pH dependent water splitting
This experiment was performed by following the previous procedure using different buffer solutions in the pH range of 5 to 9. Measurement of oxygen evolution was carried out by a similar method mentioned earlier.Characterization techniques
Acknowledgements
SR gratefully acknowledges the grants from IISER-Kolkata, India, DST-fast track and BRNS-DAE grant.References
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- M. Jacobson, W. Colella and D. Golden, Science, 2005, 308, 1901–1905 CrossRef CAS PubMed.
- L. Hammarström and S. Hammes-Schiffer, Acc. Chem. Res., 2009, 42, 1859–1860 CrossRef PubMed.
- L. S. Dennis and G. H. Hetterscheid, Eur. J. Inorg. Chem., 2014, 568–790 Search PubMed.
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17632–17635 CrossRef CAS PubMed.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore and D. Gust, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed.
- G. Zhu, E. N. Glass, C. Zhao, H. Lv, J. W. Vickers, Y. V. Geletii, D. G. Musaev, J. Song and C. L. Hill, Dalton Trans., 2012, 41, 13043–13049 RSC.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- L. Francas, X. Sala, E. Escudero-Adán, J. Benet-Buchholz, L. Escriche and A. Llobet, Inorg. Chem., 2011, 50, 2771–2781 CrossRef CAS PubMed.
- I. López, M. Z. Ertem, S. Maji, J. Benet-Buchholz, A. Keidel, U. Kuhlmann, P. Hildebrandt, C. J. Cramer, V. S. Batista and A. Llobet, Angew. Chem., 2014, 126, 209–213 CrossRef.
- J. K. Hurst, Coord. Chem. Rev., 2005, 249, 313–328 CrossRef CAS.
- S. Roeser, P. Farràs, F. Bozoglian, M. Martínez-Belmonte, J. Benet-Buchholz and A. Llobet, ChemSusChem, 2011, 4, 197–207 CrossRef CAS PubMed.
- J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730–8731 CrossRef CAS PubMed.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446–11449 CrossRef CAS PubMed.
- R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802–12803 CrossRef CAS PubMed.
- J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. Murakami Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed.
- N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210–217 CrossRef CAS PubMed.
- J. T. Muckerman, D. E. Polyansky, T. Wada, K. Tanaka and E. Fujita, Inorg. Chem., 2008, 47, 1787–1802 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- D. M. Robinson, Y. B. Go, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2010, 132, 11467–11469 CrossRef CAS PubMed.
- A. Indra, P. W. Menezes, I. Zaharieva, E. Baktash, J. Pfrommer, M. Schwarze, H. Dau and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 13206–13210 CrossRef CAS PubMed.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed.
- A. K. Vannucci, L. Alibabaei, M. D. Losego, J. J. Concepcion, B. Kalanyan, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20918–20922 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841–1844 CrossRef CAS PubMed.
- T. Zidki, L. Zhang, V. Shafirovich and S. V. Lymar, J. Am. Chem. Soc., 2012, 134, 14275–14278 CrossRef CAS PubMed.
- R. D. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- M. Quintana, A. M. López, S. Rapino, F. M. Toma, M. Iurlo, M. Carraro, A. Sartorel, C. Maccato, X. Ke and C. Bittencourt, ACS Nano, 2012, 7, 811–817 CrossRef PubMed.
- A. Sartorel, M. Carraro, G. Scorrano, R. D. Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed.
- Y. V. Geletii, Z. Huang, Y. Hou, D. G. Musaev, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 7522–7523 CrossRef CAS PubMed.
- C. Besson, Z. Huang, Y. V. Geletii, S. Lense, K. I. Hardcastle, D. G. Musaev, T. Lian, A. Proust and C. L. Hill, Chem. Commun., 2010, 46, 2784–2786 RSC.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch and T. Da Ros, Nat. Chem., 2010, 2, 826–831 CrossRef CAS PubMed.
- M. Murakami, D. Hong, T. Suenobu, S. Yamaguchi, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11605–11613 CrossRef CAS PubMed.
- G. Zhu, Y. V. Geletii, P. Kögerler, H. Schilder, J. Song, S. Lense, C. Zhao, K. I. Hardcastle, D. G. Musaev and C. L. Hill, Dalton Trans., 2012, 41, 2084–2090 RSC.
- S. Tanaka, M. Annaka and K. Sakai, Chem. Commun., 2012, 48, 1653–1655 RSC.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- J. Soriano-López, S. Goberna-Ferrón, L. Vigara, J. J. Carbó, J. M. Poblet and J. R. N. Galán-Mascarós, Inorg. Chem., 2013, 52, 4753–4755 CrossRef PubMed.
- S. Goberna-Ferrón, L. Vigara, J. n. Soriano-López and J. R. N. Galán-Mascarós, Inorg. Chem., 2012, 51, 11707–11715 CrossRef PubMed.
- J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110–14118 CrossRef CAS PubMed.
- R. Al-Oweini, A. Sartorel, B. S. Bassil, M. Natali, S. Berardi, F. Scandola, U. Kortz and M. Bonchio, Angew. Chem., Int. Ed., 2014, 53, 11182–11185 CrossRef CAS PubMed.
- J. M. Sumliner, H. Lv, J. Fielden, Y. V. Geletii and C. L. Hill, Eur. J. Inorg. Chem., 2014, 635–644 CrossRef CAS.
- S. Chatterjee, K. Sengupta, S. Dey and A. Dey, Inorg. Chem., 2013, 52, 14168–14177 CrossRef CAS PubMed.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- S. Dey, B. Mondal and A. Dey, Phys. Chem. Chem. Phys., 2014, 16, 12221–12227 RSC.
- D. K. Zhong and D. R. Gamelin, J. Am. Chem. Soc., 2010, 132, 4202–4207 CrossRef CAS PubMed.
- S. Hoang, S. Guo, N. T. Hahn, A. J. Bard and C. B. Mullins, Nano Lett., 2011, 12, 26–32 CrossRef PubMed.
- W. J. Jo, J. W. Jang, K. j. Kong, H. J. Kang, J. Y. Kim, H. Jun, K. Parmar and J. S. Lee, Angew. Chem., Int. Ed., 2012, 51, 3147–3151 CrossRef CAS PubMed.
- Y. H. Ng, A. Iwase, A. Kudo and R. Amal, J. Phys. Chem. Lett., 2010, 1, 2607–2612 CrossRef CAS.
- M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2012, 134, 8332–8335 CrossRef CAS PubMed.
- G. Wang, Y. Ling and Y. Li, Nanoscale, 2012, 4, 6682–6691 RSC.
- Y. Hou, F. Zuo, A. Dagg and P. Feng, Angew. Chem., 2013, 125, 1286–1290 CrossRef.
- Z. Huang, Z. Luo, Y. V. Geletii, J. W. Vickers, Q. Yin, D. Wu, Y. Hou, Y. Ding, J. Song and D. G. Musaev, J. Am. Chem. Soc., 2011, 133, 2068–2071 CrossRef CAS PubMed.
- W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–92755 CrossRef CAS PubMed.
- J. M. Thomsen, D. L. Huang, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2015, 44, 12452–12472 RSC.
- S. Roy, M. C. Mourad and M. T. Rijneveld-Ockers, Langmuir, 2007, 23, 399–401 CrossRef CAS PubMed.
- S.-X. Guo, Y. Liu, C.-Y. Lee, A. M. Bond, J. Zhang, Y. V. Geletii and C. L. Hill, Energy Environ. Sci., 2013, 6, 2654–2663 CAS.
- S. Roy, CrystEngComm, 2014, 16, 4667–4676 RSC.
- B. Roy, M. Arya, P. Thomas, J. K. Jürgschat, K. Venkata Rao, A. Banerjee, C. Malla Reddy and S. Roy, Langmuir, 2013, 29, 14733–14742 CrossRef CAS PubMed.
- S. Roy, Comments Inorg. Chem., 2011, 32, 113–126 CrossRef CAS.
- S. Roy, H. J. Meeldijk, A. V. Petukhov, M. Versluijs and F. Soulimani, Dalton Trans., 2008, 2861–2865 RSC.
- P. D. Tran, S. K. Batabyal, S. S. Pramana, J. Barber, L. H. Wong and S. C. J. Loo, Nanoscale, 2012, 4, 3875–3878 RSC.
- H. Li, S. Pang, X. Feng, K. Müllen and C. Bubeck, Chem. Commun., 2010, 46, 6243–6245 RSC.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- S.-S. Li, K.-H. Tu, C.-C. Lin, C.-W. Chen and M. Chhowalla, ACS Nano, 2010, 4, 3169–3174 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- W. Hummers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |