
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, electrochemical and photophysical studies of the borondifluoride complex of a meta-linked biscurcuminoid†
Morgane
Rivoal
,
Elena
Zaborova
,
Gabriel
Canard
,
Anthony
D'Aléo
* and
Frédéric
Fages
Aix Marseille Université, CNRS, CINaM UMR 7325, Campus de Luminy, Case 913, 13288 Marseille, France. E-mail: daleo@cinam.univ-mrs.fr
First published on 8th December 2015
Abstract
The synthesis of the borondifluoride complex of a biscurcuminoid system is described, and its electrochemical and photophysical properties are compared to those of a series of monochromophoric models. The data show that the meta-linked biscurcuminoid exhibits enhanced optical properties in solution, such as high optical brightnesses obtained at one- and two-photon excitation (B1 = 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 and B2 = 313 GM, respectively). UV/visible absorption of solid-state particles formed in water solution revealed that the four investigated dyes are strongly aggregated and fluorescence spectroscopy showed that they are emissive in the NIR with fluorescence quantum yields spanning from 1.5 to 12.5%. In the case of the meta-linked biscurcuminoid, a high brightness is obtained using one- and two-photon excitation (B1 = 6952 M−1 cm−1 and B2 = 70 GM, respectively). Such red-shifted emission combined with a yet significant brightness in the solid-state is believed to arise from the choice of the meta-linkage which limits strong intermolecular packing.
000 M−1 cm−1 and B2 = 313 GM, respectively). UV/visible absorption of solid-state particles formed in water solution revealed that the four investigated dyes are strongly aggregated and fluorescence spectroscopy showed that they are emissive in the NIR with fluorescence quantum yields spanning from 1.5 to 12.5%. In the case of the meta-linked biscurcuminoid, a high brightness is obtained using one- and two-photon excitation (B1 = 6952 M−1 cm−1 and B2 = 70 GM, respectively). Such red-shifted emission combined with a yet significant brightness in the solid-state is believed to arise from the choice of the meta-linkage which limits strong intermolecular packing.
Introduction
The design of organic dyes with tailored optical properties is of utmost importance for applications in advanced fields such as bioimaging,1 photodynamic therapy,2 theranostics, display3 and telecommunication technologies,4 and photovoltaics.5–7 In that context, compounds displaying high molar absorption coefficients, large two-photon absorption cross sections, high luminescence quantum yields in solution and in the solid state, large Stokes shifts, high thermal and photochemical stability represent attractive candidates as fluorescent reporters. Especially, dyes that emit in the near infrared (NIR, wavelengths longer than 700 nm according to The International Commission on Illumination) using two-photon excitation in the NIR region are of great interest for use in cell imaging because both excitation and detection operate in the biological transparency window.Among the many classes of organic dyes, boron complexes, such as borondipyrromethene (BODIPY) compounds,8–11 are particularly attractive. In addition of the interesting optical properties in solution, many borondifluoride complexes (other than BODIPY) have also been shown to yield rather efficient photoluminescent behavior in the solid state.12 In particular, compounds deriving from acetylacetonate ligand have shown interesting properties such as high two-photon absorption cross sections,13,14 mechano-fluorochromic behaviors15,16 and efficient NIR emissions17,18 that led to their use for cells imaging19 or as sensors of volatile acid/base,20,21 fluorescent reporters for amyloid,22,23 optical sensors for anaerobic environment24 and electron donors in solar cells.25
Recently, we and others have shown that curcuminoid structures containing the borondifluoride unit represent efficient emitters that fulfill many of the previous requirements. However, our work also led to the conclusion that solid-state fluorescence of BF2 complexes of curcuminoids arose from highly stacked chromophores. Such interactions could explain the emission occurring in the NIR but inherently induce efficient face-to-face quenching, which limit the fluorescence quantum yield (Φf) to ca. 5%.17,18 These results led us to consider dye 4 (Chart 1) in which two curcuminoid subunits are covalently connected via a meta-phenylene linker. Indeed, this structural design has proven to be successful in the case of electroluminescent conjugated polymers such as poly(phenylenevinylene)s because the meta-linkage was observed to inhibit interchain interactions and excimer formation in the condensed phase, thereby ensuring brighter emission than in the para-linked counterparts.26 The introduction of three aliphatic long chains at the meta-phenylene bridge in 4 was anticipated to further limit solid-state interchromophoric interactions and to provide a higher solubility in organic solvents for the extended structure. As a result, compound 4 features a non symmetrical curcuminoid system, which required the investigation of the model compound 3 that possesses a non symmetrical structure as well. Compounds 1 and 2 served as models to estimate the electronic influence of the electron donor (D) peralkylated phloroglucinol ring in the photophysical investigation.
 | ||
| Chart 1 Molecular structures of the borondifluoride curcuminoid derivatives. | ||
We report herein the synthesis of compounds 1–4 and a study of their electrochemical and optical properties in organic solvents. The two-photon excited fluorescence (TPEF) properties of the four dyes were characterized in solution. We describe the preparation and fluorescence emission of organic nanoparticles of 1–4. Our approach gives a strategy toward the design of extended biscurcuminoids with improved emission ability in both the solution and the solid states.
Results and discussion
Synthesis
The synthetic routes toward 1–4 are outlined in Scheme 1. The borondifluoride complexes 1 and 2 were prepared using a previously reported procedure.27 Syntheses of the unsymmetrical BF2 complexes 3 and 4 required first of all the preparation of the hemicurcuminoid 5. Similarly to what has been published,28 this intermediate was prepared by a Knoevenagel reaction using an excess of acetylacetone (acac/aldehyde 3:1), which provided the compound 5 in a reasonable yield of 60%. Then, the reaction of the intermediate 5 with one equivalent of 2,4,6-trimethoxybenzaldehyde afforded the ligand Lig 3. Lig 4 was prepared in a yield of 34% by reacting two equivalents of 5 with 1,3,5-tris(n-octyloxy)benzaldehyde.
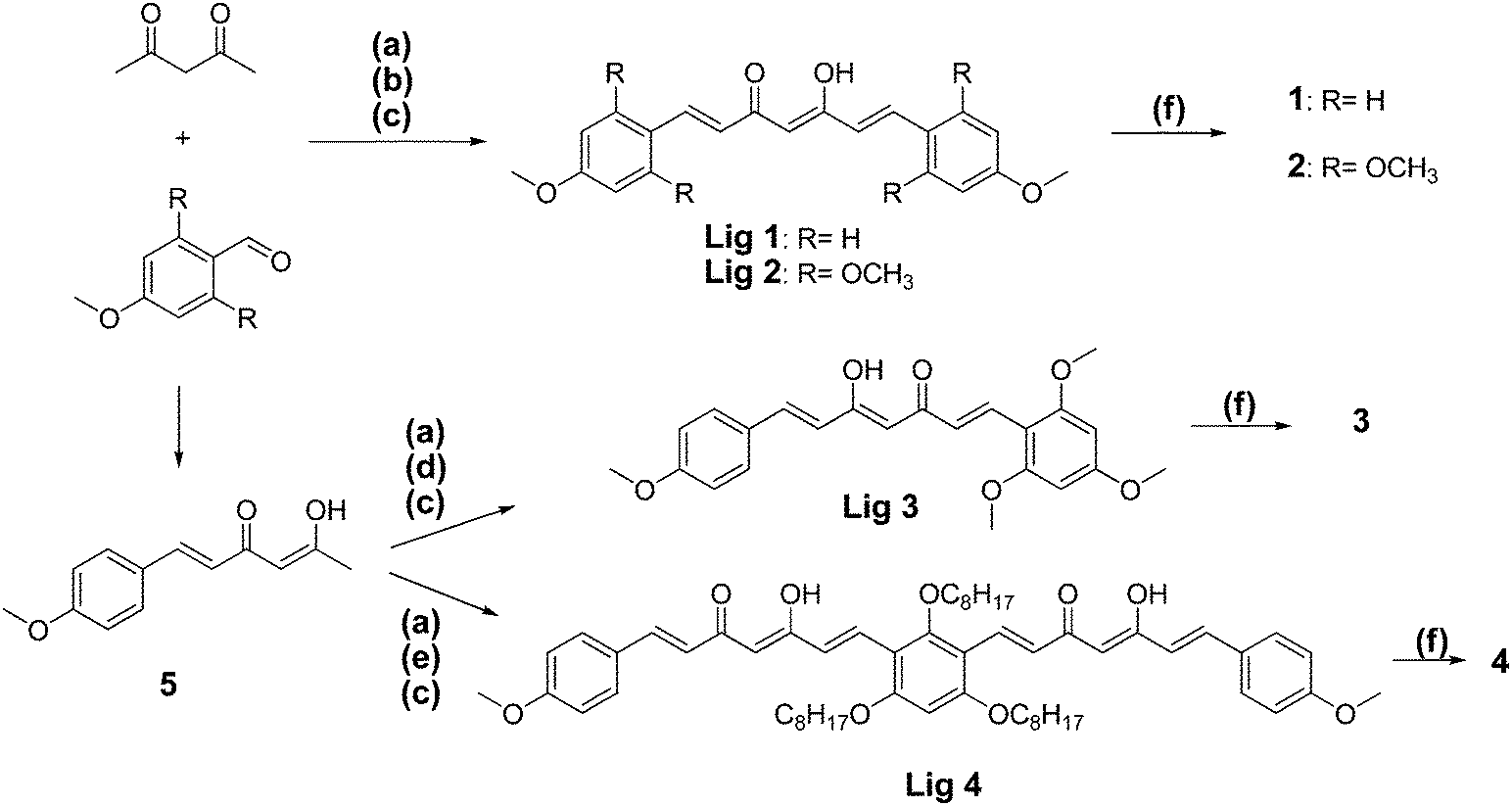 | ||
| Scheme 1 Syntheses of the borondifluoride curcuminoid derivatives 1, 2, 3 and 4. (a) B2O3, ethylacetate, 60 °C; (b) anisaldehyde, tri-n-butylborane, ethylacetate, 60 °C; (c) n-butylamine, ethylacetate, 80 °C, then HCl, 60 °C; (d) 2,4,6-trimethoxybenzaldehyde, tri-n-butylborane, ethylacetate, 60 °C; (e) 2,4,6-tris(octyloxy)isophthalaldehyde, tri-n-butylborane, ethylacetate, 60 °C; (f) BF3·Et2O, DCM, reflux. | ||
Complexation to boron difluoride was performed by reacting the ligands with a slight excess of the boron trifluoride etherate in dichloromethane solution (DCM). The dyes were purified by crystallization or, when necessary, by column chromatography. Dyes 1–4 were obtained as highly colored solids and characterized by 1H- and 19F-NMR spectroscopies and by high resolution mass spectrometry (HRMS).
Electrochemistry
The electrochemical properties of dyes 1–4 were investigated in DCM solution containing 0.1 M of (n-Bu)4NPF6. The cyclic voltammograms (CV) are given in Fig. 1 and Fig. S1 (ESI†) and the oxidation and reduction half-wave potential values (E1/2vs. ferrocene/ferrocenium) are collected in Table 1. | ||
| Fig. 1 Cyclic voltammogram of the bis borondifluoride complex 4 in DCM solution containing 0.1 M [(nBu4N)PF6] (scan rate of 100 mV s−1). | ||
The four complexes exhibit one-electron oxidation and reduction waves that can be attributed to the donor alkoxy-substituted phenyl (D) end-groups and the acceptor (A) dioxaborine ring, respectively. The first oxidation potential decreases with the strength of the D units, depending on the number of appended methoxy groups (Table 1). In contrast, increasing the donor strength induces a cathodic shift of the reduction potential but overall, the electrochemical gap is decreased by ca. 0.2 V.
The CV of 2 (Fig. S1, ESI†) clearly shows two successive and well separated one-electron oxidation waves that reflect the electronic communication between trimethoxyphenyl end-groups through the curcuminoid backbone. It can be noted that the first oxidation and reduction potential values of the unsymmetrical complex 3 are intermediate between those of the symmetrical complexes 1 and 2.
The first oxidation and reduction potential values obtained for 4 are closer to those recorded for 1 than for 3. Furthermore, the HOMO–LUMO electrochemical gap is also very close for both compounds 1 and 4. These observations suggest that the first oxidation in 4 is likely centered on the p-methoxy aryl groups. The increase of the oxidation potential in 4 relative to 3 could be due to presence the second curcuminoid moiety. In addition, since both reduction and oxidation processes of 4 involve the same number of electrons, we assume that methoxyphenyl end-groups and dioxaborine rings in 4 are simultaneously oxidized and reduced, respectively, at the same potential. This effect is in agreement with the reduced conjugation between meta-linked chromophores.
Photophysical study in solution
The electronic absorption spectra of 1–4 were recorded in DCM solutions (Fig. 2a) and the spectroscopic data are reported in Table 2. The spectra consist mainly of one intense transition band at low energy (450–550 nm) attributed to a strongly allowed π–π* transition. A second electronic transition band of much lower intensity appears as a shoulder at higher energy (<400 nm, vide infra). The spectra of compounds 1–3 display identical shape of the absorption profiles, they only differ by the position of the bands. The increase of donor strength from 1 to 2 induces a red-shift of the electronic transitions. As expected, the unsymmetrical dye 3 absorbs at a wavelength intermediate between those recorded for 1 and 2. In contrast, compound 4 exhibits a low-energy absorption band with very different shape, full width at half maximum, and intensity. The molar absorption coefficient determined for 4 is twice as high as that of the three other dyes. The more complex shape and the larger Stoke shift of the absorption band is likely to stem from intramolecular excitonic coupling between the two curcuminoid chromophores. This excitonic coupling explains the low value of the optical gap determined for 4. | ||
Fig. 2 (a) Overlaid electronic absorption spectra and (b) overlaid corrected normalized fluorescence emission spectra (conc. ≈ 10−6 M. λexc at the absorption maximum) of 1 (—), 2 ( ), 3 ( ), 3 ( ) and 4 ( ) and 4 ( ) recorded in DCM at room temperature. ) recorded in DCM at room temperature. | ||
| Compound | UV-vis | Fluorescence | TPEF | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ abs | ε max | λ em | ΔνST | Φ f | B 1 | τ f | k f | k nr | λ max 2 | σ TPA | B 2 | |
| a Absorption maximum wavelengths λabs (nm); molar absorption coefficients at maximum εmax (M−1 cm−1); fluorescence maximum wavelengths λem (nm); Stokes shifts ΔνST (cm−1); fluorescence quantum yields Φf; brightness B = Φf × ε (M−1 cm−1); fluorescence lifetimes τf (ns); radiative kf (108 s−1) and nonradiative knr = (1 − Φf)/τf(108 s−1) rate constants; two-photon absorption maximum λmax2 (nm); two-photon cross section σTPA (GM); two-photon brightness B2 = Φf × σTPA (GM). b Pseudo-Stokes shift determined using the maximum absorption. c A biexponential decay was found. d Not determined due to high scattering of light with those particles. | ||||||||||||
| 1 (DCM) | 488 | 75480 |
538 | 1904 | 0.44 | 33211 |
1.30 | 3.4 | 4.3 | 770 | 155 | 68 |
| 2 (DCM) | 524 | 84860 |
566 | 1416 | 0.52 | 44127 |
1.72 | 3.0 | 2.8 | 810 | 248 | 129 |
| 3 (DCM) | 511 | 74510 |
561 | 1957 | 0.46 | 34275 |
1.74 | 2.6 | 3.1 | 790 | 208 | 96 |
| 4 (DCM) | 529 | 143140 |
574 | 1421 | 0.61 | 87315 |
1.69 | 3.6 | 2.3 | 800 | 513 | 313 |
| 1 (water) | 451 | 34170 |
710 | 8088b | 0.06 | 2050 | 6.44 | 0.09 | 1.5 | 780 | 210 | 13 |
| 2 (water) | 489 | 27380 |
717 | 6503b | 0.035 | 783 | 1.32 | — | — | —d | —d | —d |
| 2.91c | ||||||||||||
| 3 (water) | 407 | 29500 |
711 | 10505b |
0.015 | 443 | 2.13 | — | — | —d | —d | —d |
| 6.01c | ||||||||||||
| 4 (water) | 468 | 55620 |
692 | 6917b | 0.125 | 6952 | 1.89 | — | — | 850 | 560 | 70 |
| 6.64c | ||||||||||||
The complexes 1–4 are fluorescent in the visible region (540–575 nm) upon excitation into the low-energy transition band and exhibit fluorescence quantum yields ranging from 44 to 61% in DCM. In agreement with electronic absorption data, an increase of the donor strength causes a red-shift of the fluorescence emission from 538 nm (1) to 574 nm (4). It is worth noting that the highest value of Φf (61%) is obtained for complex 4, giving a high brightness value of ca. 87000 M−1 cm−1.
The solvent dependence of absorption and emission properties was studied for the four borondifluoride complexes (Tables S1 and S2, ESI†). Both electronic UV/visible absorption and fluorescence emission spectra undergo a bathochromic shift and lose their vibronic structures when solvent polarity increases (Fig. S3–S6, ESI†). These features show that the Franck–Condon excited state S1 is more polar in nature than the ground state. Furthermore, the positive slope of the Lippert–Mataga plots reveals that, in those systems, the relaxation occurs toward a solvent-equilibrated charge transfer (CT) singlet excited state along with an increase of dipole moment with respect to the ground state (Fig. S2, ESI†).29
The fluorescence intensity of compounds 1–3 continuously increases with increasing solvent polarity. Both solvatochromism and the negative solvatokinetic behavior suggest the formation of an emissive CT state. The fact that the fluorescence lifetimes also increase with solvent polarity leads to constant values of the radiative rate constants kf for the three dyes (1–3). Concomitantly, the nonradiative rate constants knr decreases in polar solvents. The case of 4 is different. Upon increasing solvent polarity, the fluorescence quantum yield first increases (negative solvatokinetics) then decreases (positive solvatokinetics), a maximum being observed. The positive solvatokinetic behavior of 4 in polar solvent suggests an enhanced population of a highly polar CT state prone to a strong nonradiative deactivation. At this stage, the peculiar photophysical behavior of 4 remains unclear.
Two-photon excited fluorescence emission and excitation spectra of 1–4 were recorded in the 700–1000 nm wavelength range using a femtosecond Ti-Sapphire pulsed laser source, according to the experimental protocol described by Webb et al. using coumarin-307 and rhodamine B as references.30 The observation of a quadratic dependence of the fluorescence intensity versus incident laser power at several wavelengths unambiguously confirmed that the origin of the fluorescence emission can be assigned to a TPA process in DCM solution (Fig. S7, ESI†). In the experimental laser power range used for these measurements, we checked that no saturation or photodegradation occurred. The two-photon excitation spectra of 1, 2, 3 and 4 in DCM are shown in Fig. 3 and Fig. S9 (ESI†) and the corresponding data are reported in Table 2. As reported elsewhere,141 has a TPA cross section (σTPA) of 155 GM at 770 nm while dye 2, containing stronger donating groups, has its maximum red-shifted to 810 nm with a larger σTPA value of ca. 248 GM. It is interesting to note that 3 presents an intermediate σTPA value of 208 GM at 790 nm. For dye 4, as already observed for the molar absorption coefficient, the two-photon cross section is slightly more than twice (i.e. 513 GM) the value determined for 3. This gives a two-photon brightness of 313 GM, that is much higher than those obtained with the model borondifluoride complexes 1–3.
 | ||
Fig. 3 Two-photon excitation ( , higher x-coordinate and , higher x-coordinate and  , right y-coordinate) with their error bars, OPA spectra (—, lower x-coordinate and —, left y-coordinate) in DCM: (a) dye 3 and (b) dye 4. , right y-coordinate) with their error bars, OPA spectra (—, lower x-coordinate and —, left y-coordinate) in DCM: (a) dye 3 and (b) dye 4. | ||
As noticed previously for curcumin31 and borondifluoride complexes of curcuminoids,14 the TPA band of the four complexes does not match the one-photon transition as it is strongly blue-shifted, corresponding to the two-photon allowed transition at higher energy mentioned previously. The lowest-energy transitions of 1–4 are not completely TPA forbidden: σTPA values of ca. 10–15, 30–35, 25–30 and 100 GM were measured for 1, 2, 3 and 4, respectively (Fig. 3 and Fig. S8, ESI†). Such situation is often encountered for D–A–D chromophores32–35 and, in the case of free curcumin, Hernández et al. have shown that the S0–S2 transition was more TPA-allowed than the S0–S1 transition. However, a much weaker contribution from the S0–S1 transition was still observed.31
Solid-state optical properties
We prepared solid-state particles, P-2–P-4, by quickly adding a concentrated THF solution of the dyes 2–4 into water according to the classical fast precipitation method.36 The so-obtained suspensions enabled the measurement of the UV/visible absorption and fluorescence spectra of the aggregated molecules. We reported the preparation of P-1 in a previous publication14 and the resulting spectroscopic data are recalled here for the sake of comparison. The shape of the electronic absorption spectra of P-1–P-4 is strongly affected relative to the solution spectra showing that Davydov splitting is occurring within such dyes. The absorption profiles of P-1, P-2 and P-4 (Fig. 4a, b, and d, respectively) are complex, broad, and their maximum presents a hypsochromic shift (ca. 30–60 nm). These observations show that excitonic interactions prevail in the condensed phase, which could be correlated with single-crystal X-ray diffraction data in the case of 1. Such complex structure is in line with Davydov splitting into high- and low-energy transitions. The absorption spectrum of P-3 displays a very different effect (Fig. 4c). The low-energy transition is considerably blue-shifted (of ca. 96 nm) and has a narrower shape as compared to those of the above mentioned particles P-1, P-2 and P-4. These spectroscopic features are the signature of H-aggregation in the solid state for compound 3. Noticeably, P-4, also containing unsymmetrical curcuminoid chromophores, does not adopt such arrangement.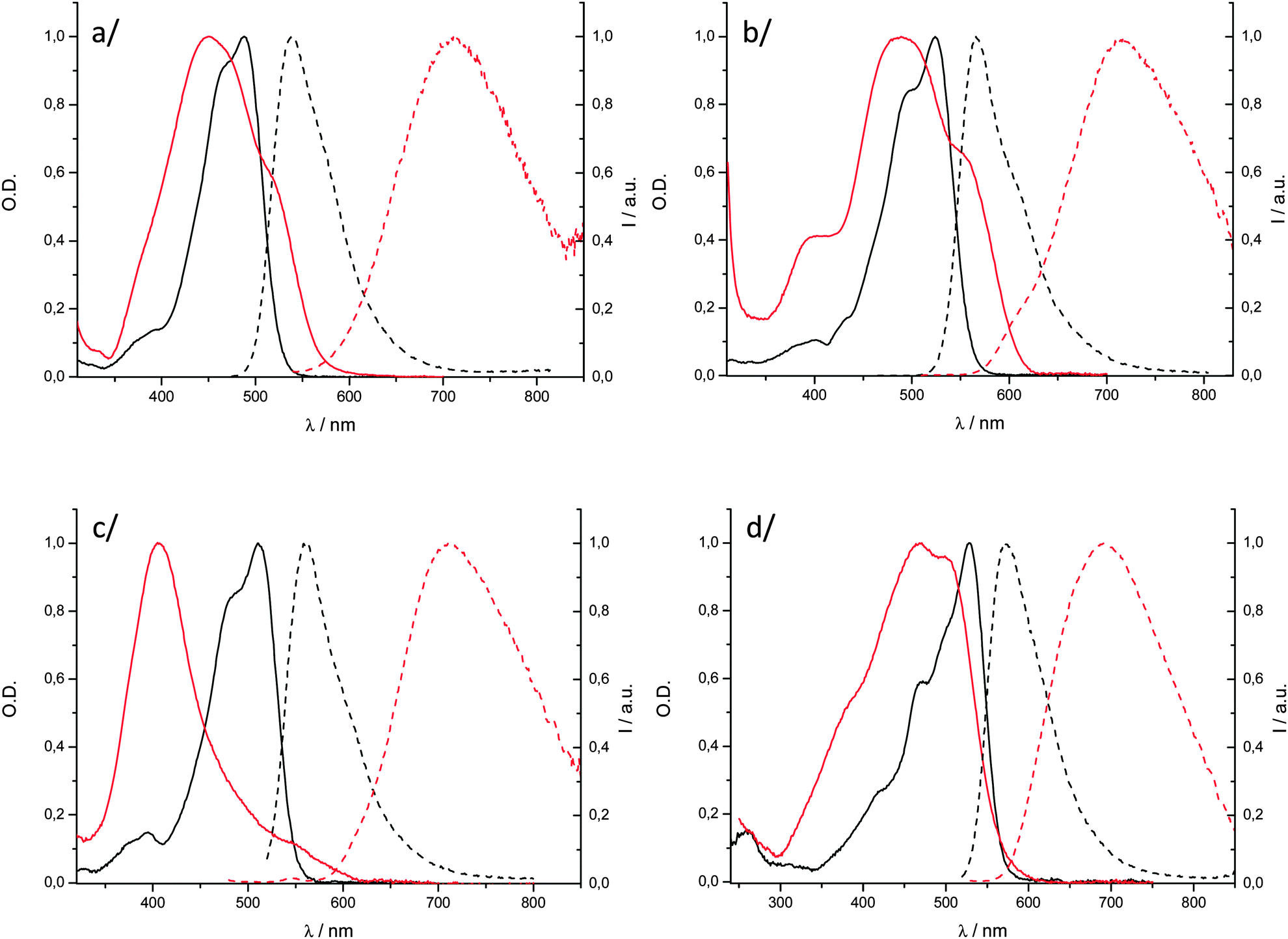 | ||
| Fig. 4 Overlaid absorption (solid lines) and emission (dashed lines) spectra of DCM solutions (black) and particles in water (red) for (a) 1; (b) 2; (c) 3 and (d) 4. | ||
Particles P-2–P-4 fluoresce in the NIR, from 692 to 717 nm (Fig. 4, Table 2). The spectra are considerably red-shifted (superior to 95 nm) as compared to those obtained in solution in the highly polar acetonitrile solvent (Fig. S3–S6, ESI†). Such long emission wavelengths can be attributed to chromophore packing in the solid state, as shown by X-ray diffraction for P-1 and by electronic absorption spectroscopy for the others (Fig. 4). The fluorescence quantum yields of P-1–P-3 are found in the range of 1.5–6.0% which are significant values for aggregated organic dyes emitting in the NIR. Not surprisingly, the H-aggregated compound 3 affords the lowest value of 1.5%. Remarkably, the Φf of dye 4 reaches a substantial value of 12.5% in the solid state, which makes P-4 a very bright solid-state NIR fluorophore (brightness at 700 nm of 6952 M−1 cm−1). Actually, a rather large part of emitted photons by the four compounds have energies below 700 nm because the particle emission spectra are broad. However, it can be highlighted that, when only photons at longer wavelength than 700 nm are integrated, dye 4 remains the most luminescent borondifluoride complex of the series with a NIR luminescence quantum yield of ca. 6.5% while the other three dyes have quantum yields of 4.0, 1.5 and 1.0%, respectively.
In addition, two-photon properties of P4 could be measured and compared to the previously reported P1 particles. Noticeably, the P2 and P3 particles could not be measured due to the much higher light scattering in those samples which precluded the obtention of reliable data. As observed in DCM solution and for P1 in water, the two-photon maximum of P4 does not overlap the maximum of the one photon absorption S0–S1 transition (Fig. 5) but it better matches the S0–S2 one (vide supra). This maximum is located at 850 nm with a two-photon cross section of ca. 560 GM. Such two-photon cross section value is 2.5 times higher than that of P1 which, associated to the higher fluorescence quantum yield of P4, results in a much higher two-photon brightness for P4 (more than 5 times greater than the one of P1).
 | ||
Fig. 5 Two-photon excitation ( , higher x-coordinate and , higher x-coordinate and  , right y-coordinate) with their error bars, OPA spectra (—, lower x-coordinate and —, left y-coordinate) of P4 in water. , right y-coordinate) with their error bars, OPA spectra (—, lower x-coordinate and —, left y-coordinate) of P4 in water. | ||
Conclusion
We have described the synthesis of three borondifluoride complexes of curcuminoid derivatives, 2–4. Compound 4 contains two curcuminoid subunits connected via a meta-position of an aryl linkage, and as such it represents a first example of a new class of extended biscurcuminoids. The electrochemical investigations show the interruption of conjugation in 4, the two chromophoric subunits behaving independently. However, the absorption spectrum reveals that an intramolecular excitonic interaction exists in solution, showing that the two chromophoric units are not optically independent. Compound 4 displays a high fluorescence quantum yield, and a good value of the two-photon absorption cross section, which makes it an attractive fluorophore. Compounds 2–4 are fluorescent in the solid state with wavelengths reaching the NIR. Like 1–3, compound 4 experience intermolecular interactions in the condensed phase. However, it exhibits the higher value of fluorescence quantum yield within the series investigated, which may result from the meta-linkage that limits strong π–π stacking with respect to the monochromophoric analogues. Indeed, the unsymmetrical model compound 3 forms H-aggregates, which illustrates the strong propensity of monochromophoric curcuminoids to form tightly packed solid-state arrangements. These findings should help in the design of new fluorophores with improved NIR emitting properties in solution and in the solid state for imaging applications.Experimental
Material and steady state spectroscopy
| A1 = (F1)(S1)/λ2 | (1) |
| Φfx/Φfr = [ODr(λ)/ODx(λ)][Ix/Ir][nx/nr]2 | (2) |
Lifetime measurements were carried out on a HORIBA Jobin Yvon IBH FluoroLog-3 spectrofluorimeter that was adapted for time-correlated single-photon counting. For these measurements, pulsed LEDs with an appropriate wavelength were used. Emission was monitored perpendicular to the excitation pulse and spectroscopic selection was achieved by a passage through the spectrograph. A thermoelectrically cooled single-photon-detection module (HORIBA Jobin Yvon IBH, TBX-04-D) incorporating a fast-rise-time photomultiplier tube, a wide-bandwidth preamplifier, and a picosecond-constant fraction discriminator was used as the detector. Signals were acquired using an IBH DataStation Hub photon counting module and data analyses were performed by using the commercially available DAS 6 decay-analysis software package from HORIBA Jobin Yvon IBH; the reported τ values are given with an estimated uncertainty of about 10%.
Cyclic voltammetric (CV) data were acquired using a BAS 100 Potentiostat (Bioanalytical Systems) and a PC computer containing BAS100W software (v2.3). A three-electrode system with a Pt working electrode (diameter 1.6 mm), a Pt counter electrode and an Ag/AgCl (with 3 M NaCl filling solution) reference electrode was used. [(nBu)4N]PF6 (0.1 M in dichloromethane) served as an inert electrolyte. Cyclic voltammograms were recorded at a scan rate of 100 mV s−1. Ferrocene was used as internal standard.39
Synthetic procedure
Dye 1, its ligand (Lig 1),27 the ligand of 2 (Lig 2),40 2,4,6-tris(octyloxy)isophthalaldehyde41 and (1E,4Z)-5-hydroxy-1-(4-methoxyphenyl)hexa-1,4-dien-3-one (Lig 5)28 were prepared as previously reported.General procedure for borondifluoride complexes
In a 50 mL round bottom flask, the ligand (1 mol eq.) was solubilized in dichloromethane (20 mL) before boron trifluoride etherate (1.1 mol eq.) was added. The reaction mixture was refluxed overnight. After cooling to room temperature, the solvent was evaporated and the resulting solid was suspended in diethyl ether. The precipitate was filtered off yielding the pure complex except for 2. For the latter dye, the crude complex was purified by column chromatography using silica gel with a mixture of cyclohexane and dichloromethane (1/4).Acknowledgements
The authors want to thank Dr Wladimir Marine for his help in setting up the two-photon excitation set-up.References
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- A. B. Ormond and H. S. Freeman, Materials, 2013, 6, 817–840 CrossRef CAS.
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed.
- P.-A. Bouit, G. Wetzel, G. R. Berginc, B. Loiseaux, L. C. Toupet, P. Feneyrou, Y. Bretonnière, K. Kamada, O. Maury and C. Andraud, Chem. Mater., 2007, 19, 5325–5335 CrossRef CAS.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- A. Zitzler-Kunkel, M. R. Lenze, N. M. Kronenberg, A.-M. Krause, M. Stolte, K. Meerholz and F. Würthner, Chem. Mater., 2014, 26, 4856–4866 CrossRef CAS.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496–501 RSC.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- A. D'Aléo and F. Fages, Disp. Imaging, 2014, 2, 149–174 Search PubMed.
- E. Cogné-Laage, J.-F. Allemand, O. Ruel, J.-B. Baudin, V. Croquette, M. Blanchard-Desce and L. Jullien, Chem. – Eur. J., 2004, 10, 1445–1455 CrossRef PubMed.
- A. D'Aleo, A. Felouat, V. Heresanu, A. Ranguis, D. Chaudanson, A. Karapetyan, M. Giorgi and F. Fages, J. Mater. Chem. C, 2014, 2, 5208–5215 RSC.
- N. D. Nguyen, G. Zhang, J. Lu, A. E. Sherman and C. L. Fraser, J. Mater. Chem., 2011, 21, 8409–8415 RSC.
- G. Zhang, J. P. Singer, S. E. Kooi, R. E. Evans, E. L. Thomas and C. L. Fraser, J. Mater. Chem., 2011, 21, 8295–8299 RSC.
- A. D'Aléo, D. Gachet, V. Heresanu, M. Giorgi and F. Fages, Chem. – Eur. J., 2012, 18, 12764–12772 CrossRef PubMed.
- A. D'Aléo, V. Heresanu, M. Giorgi, B. Le Guennic, D. Jacquemin and F. Fages, J. Phys. Chem. C, 2014, 118, 11906–11918 Search PubMed.
- G. Bai, C. Yu, C. Cheng, E. Hao, Y. Wei, X. Mu and L. Jiao, Org. Biomol. Chem., 2014, 12, 1618–1626 CAS.
- X. Cheng, D. Li, Z. Zhang, H. Zhang and Y. Wang, Org. Lett., 2014, 16, 880–883 CrossRef CAS PubMed.
- J. Hu, Z. He, Z. Wang, X. Li, J. You and G. Gao, Tetrahedron Lett., 2013, 54, 4167–4170 CrossRef CAS.
- C. Ran, X. Xu, S. B. Raymond, B. J. Ferrara, K. Neal, B. J. Bacskai, Z. Medarova and A. Moore, J. Am. Chem. Soc., 2009, 131, 15257–15261 CrossRef CAS PubMed.
- X. Zhang, Y. Tian, Z. Li, X. Tian, H. Sun, H. Liu, A. Moore and C. Ran, J. Am. Chem. Soc., 2013, 135, 16397–16409 CrossRef CAS PubMed.
- A. Pfister, G. Zhang, J. Zareno, A. F. Horwitz and C. L. Fraser, ACS Nano, 2008, 2, 1252–1258 CrossRef CAS PubMed.
- S. Chambon, A. D'Aléo, C. Baffert, G. Wantz and F. Fages, Chem. Commun., 2013, 49, 3555–3557 RSC.
- A. M. Sarker, E. E. Gürel, M. Zheng, P. M. Lahti and F. E. Karasz, Macromolecules, 2001, 34, 5897–5901 CrossRef CAS.
- A. Felouat, A. D'Aléo and F. Fages, J. Org. Chem., 2013, 78, 4446–4455 CrossRef CAS PubMed.
- G. Mann, L. Beyer and A. Arrieta, Z. Chem., 1987, 27, 172–173 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4031 CrossRef PubMed.
- C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481–491 CrossRef CAS.
- J. A. Tiburcio-Moreno, J. J. Alvarado-Gil, C. Diaz, L. Echevarria and F. E. Hernández, Chem. Phys. Lett., 2013, 583, 160–164 CrossRef CAS.
- H. Hu, O. V. Przhonska, F. Terenziani, A. Painelli, D. Fishman, T. R. Ensley, M. Reichert, S. Webster, J. L. Bricks, A. D. Kachkovski, D. J. Hagan and E. W. Van Stryland, Phys. Chem. Chem. Phys., 2013, 15, 7666–7678 RSC.
- W. M. McClain, Acc. Chem. Res., 1974, 7, 129–135 CrossRef CAS.
- F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742–15755 CrossRef CAS PubMed.
- Q. Bellier, N. S. Makarov, P.-A. Bouit, S. Rigaut, K. Kamada, P. Feneyrou, G. Berginc, O. Maury, J. W. Perry and C. Andraud, Phys. Chem. Chem. Phys., 2012, 14, 15299–15307 RSC.
- H. Kawai, H. S. Nalwa, H. Oikawa, S. Okada, H. Matsuda, N. Minami, A. Kakuda, K. Ono, A. Mukoh and H. Nakanishi, Jpn. J. Appl. Phys., 1992, 31, L1132–L1134 CrossRef.
- C. A. Parker, Photoluminescence of Solutions, Elsevier Publishing, Amsterdam, 1969 Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer, NewYork (NY), 2006 Search PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- Y. Zuo, J. Huang, B. Zhou, S. Wang, W. Shao, C. Zhu, L. Lin, G. Wen, H. Wang, J. Du and X. Bu, Eur. J. Med. Chem., 2012, 55, 346–357 CrossRef CAS PubMed.
- S. B. Bharate, R. Mudududdla, R. Sharma and R. A. Vishwakarma, Tetrahedron Lett., 2013, 54, 2913–2915 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nj00925a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |