
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dispersion forces acting between silica particles across water: influence of nanoscale roughness†
Valentina
Valmacco
a,
Magdalena
Elzbieciak-Wodka
b,
Céline
Besnard
c,
Plinio
Maroni
a,
Gregor
Trefalt
a and
Michal
Borkovec
*a
aDepartment of Inorganic and Analytical Chemistry, University of Geneva, Sciences II, Quai Ernest-Ansermet 30, 1205 Geneva, Switzerland. E-mail: michal.borkovec@unige.ch
bJerzy Haber Institute of Catalysis and Surface Chemistry, Polish Academy of Sciences, Niezapominajek 8, 30-239 Cracow, Poland
cLaboratory of Crystallography, University of Geneva, Quai Ernest Ansermet 24, 1211 Geneva 4, Switzerland
First published on 18th May 2016
Abstract
Force profiles between pairs of silica particles in concentrated aqueous solutions of a monovalent salt are measured using atomic force microscopy (AFM). Under such conditions, the double layer forces are negligible, and the profiles are dominated by van der Waals dispersion forces at larger distances. Heat treatment of the particles strongly influences the strength of dispersion forces. The dispersion force between the particles heated at 1200 °C was strongly attractive, and was characterized by a Hamaker constant of 2.4 × 10−21 J. This value is in good agreement with the current best theoretical estimate of the Hamaker constant for silica across water. For untreated particles, however, the dispersion force is much weaker and the Hamaker constant is 7 × 10−23 J. The Hamaker constant can be continuously tuned by adjusting the heating temperature between 1000 and 1200 °C. Such substantial variations of the Hamaker constant are caused by moderate differences in surface roughness on the nanoscale. The root mean square (RMS) of the roughness correlates inversely with the Hamaker constant, whereby the particles treated at 1200 °C have an RMS value of 0.63 nm, while the untreated particles have an RMS value of 2.5 nm. Other effects that could influence the Hamaker constant, such as changes in the degree of crystallinity, porosity, and shape of the particles, could be excluded.
Conceptual insightsDispersion forces are essential for controlling the stability of particle suspensions, synthesis of colloidal molecules, or fabrication of microelectromechanical devices. In the frequent situations, where silica substrates interact across aqueous solutions, the strength of dispersion forces was reported to vary by orders of magnitude. The present communication demonstrates that such substantial variations can be explained by varying degrees of nanoscale roughness. Such roughness effects could thus strongly influence suspension stability by weakening of dispersion interactions. As one often relies on dispersion forces to stabilize colloidal molecules, surface roughness could also be used to tune these forces accurately. As dispersion forces are further responsible for unwanted adhesion of surfaces in the fabrication of microelectromechanical devices, adjusting the surface roughness opens the possibility of minimizing such adhesion forces. |
Introduction
Dispersion forces act between any two types of bodies with dielectric discontinuities, and their effects can be substantial at distances of a few nanometers or smaller. They are the manifestation of the spontaneous fluctuations of the electromagnetic fields, and the respective theoretical framework was laid down about half a century ago by Lifshitz and co-workers.1–3 This theory generalized the earlier approach by van der Waals, which focused on non-retarded forces between non-conductive media at smaller separation distances, and the one of Casimir and Polder, which treated retarded forces acting between metallic conductors at larger distances. Dispersion forces are always attractive between the same media interacting across vacuum, gases, or liquids.2,4 They may occasionally become repulsive when two different media interact across a third one.5–7 The presence of ionic charges may reduce the magnitude of dispersion forces, even though this effect is relatively minor.1,4,8 Dispersion forces control particle aggregation, particle deposition, or flotation phenomena.1,2,9,10 More recently, they received renewed interest in the development of microelectromechanical and nanoscale systems, or low friction devices.3,5,11 Tuning dispersion forces is equally important in the fabrication of colloidal molecules.12,13Lifshitz theory represents an established framework for the calculation of dispersion forces based on the dielectric spectra of the materials in question.1,2,4,14 Some of these predictions turn out to be surprisingly accurate, especially for solids interacting across vacuum or air.4,15,16 Across liquids, however, substantial disparities between experimental and theoretical dispersion forces exist.8,17,18 The situation is particularly disturbing for the relevant case of silica (SiO2) interacting across water. For this system, some experiments suggest dispersion forces that are comparable to (or even stronger than) the ones predicted theoretically.18,19 For the same systems, other authors report dispersion forces that are one order of magnitude weaker.20,21 Some studies even remark the absence dispersion forces altogether.22–24 No satisfactory explanation of these discrepancies has been put forward so far. Given the widespread use of silica substrates in contact with water, a clarification of this discrepancy is imperative.
The present communication shows that the strength of dispersion forces acting between pairs of similar silica particles in aqueous solutions may indeed vary substantially. As will be demonstrated, this variation is mainly caused by different degrees of nanoscale surface roughness, which can be systematically vary through heat-treatment of the silica particles at temperatures in the range of 1000–1200 °C. Particle surfaces treated at 1200 °C become quite smooth, and dispersion forces are substantial, entirely in contrast to untreated particles.
Results and discussion
Forces between two individual silica particles with a diameter of about 5 μm were measured using the colloidal probe technique.25–27 The particles were attached to a tip-less cantilever and to a quartz substrate, and both are mounted in the AFM fluid cell. The scheme of the experiment is shown in Fig. 1a. Different series of experiments were carried out with particles heated for 2 hours at various temperatures in the range 1000–1200 °C and the results were compared with the untreated particles. The respective scanning electron microscopy (SEM) images are shown in Fig. 1b–d. The experiments were carried out in KCl electrolyte solutions adjusted to pH 4.0 as in an earlier study.20 Experimental details are given in the ESI.†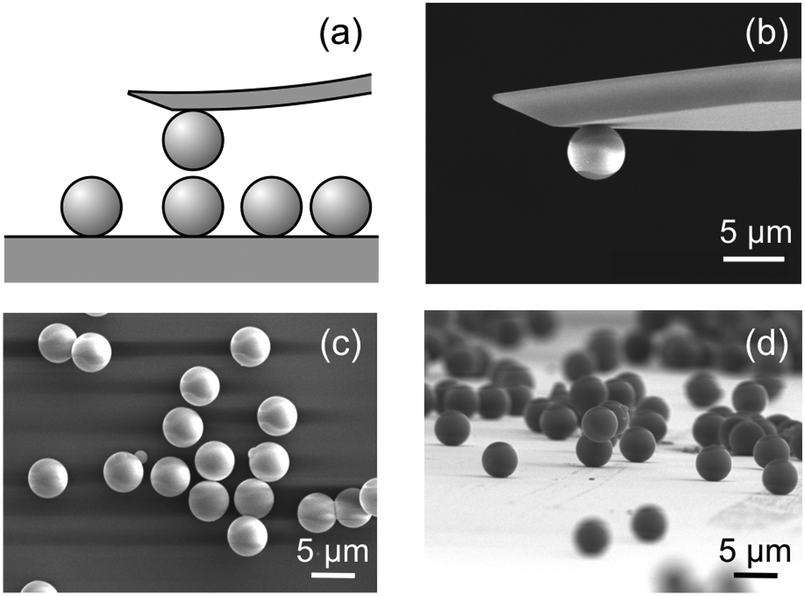 | ||
| Fig. 1 Force measurements between two individual silica particles of about 5 μm in diameter. (a) Scheme of the experiment. (b) SEM images of the cantilever with the attached colloidal particles. Particles deposited on the substrate. (c) Top view, and (d) side view. These images also illustrate the presence of few particle aggregates, but such particle aggregates were avoided in the force measurements. | ||
Let us first focus on forces acting between colloidal particles sintered at 1200 °C for different KCl concentrations (Fig. 2a). At larger separation distances, forces are always attractive. At lower salt concentrations and shorter distances, the forces become repulsive, while they remain attractive at higher salt concentrations. This behavior can be quantified by the classical theory of Derjaguin, Landau, Verwey, and Overbeek (DLVO). This theory assumes that the interactions between solids across aqueous solutions can be approximated as a superposition of attractive dispersion forces and repulsive double layer forces. We model this dependence of the force F on the separation distance h by means of the relation1
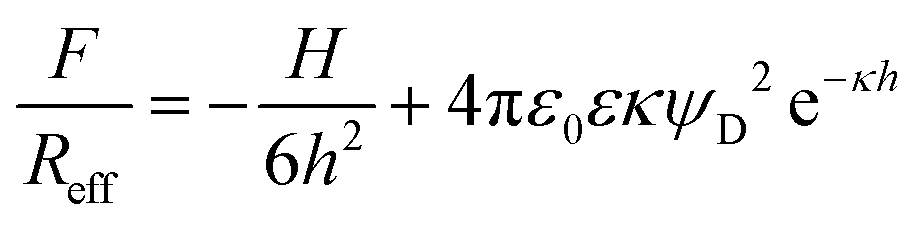 | (1) |
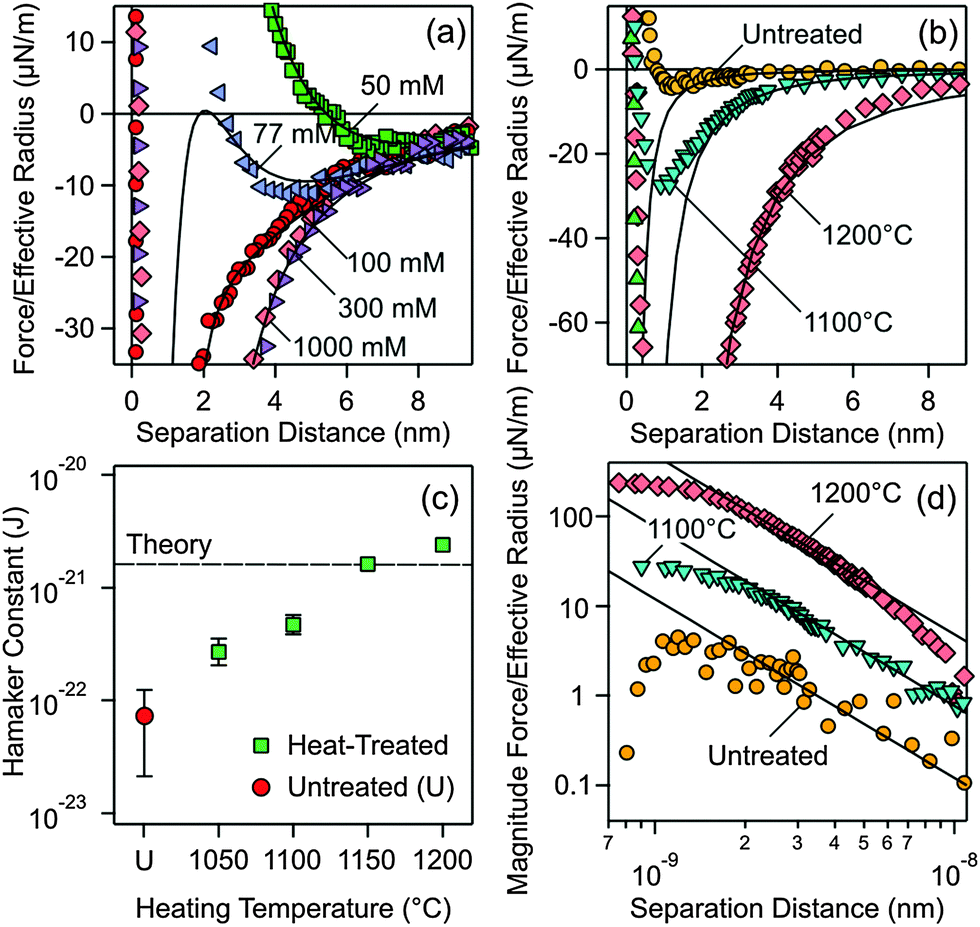 | ||
| Fig. 2 Measured force profiles between silica particles in aqueous KCl solutions of pH 4.0. Solid lines are best fits with DLVO theory. (a) Particles treated at 1200 °C measured at different salt concentrations. (b) Comparison of forces between the particles heated at different temperatures and the untreated ones in 1.0 M KCl solution, where double layer forces are fully screened. (c) Fitted Hamaker constants from experiments shown in (b) and additional ones at different temperatures. The dashed line in (c) represents the currently available best theoretical estimate by Ackler et al.14 (d) Doubly logarithmic representation of the magnitude of the attractive force. | ||
Two parameters can be extracted from such fits, namely the diffuse layer potential ψD and the Hamaker constant H. The latter parameter was obtained from the force profile measured at 1.0 M, and one finds H = (2.4 ± 0.5) × 10−21 J. For the profiles measured at lower salt concentration, the Hamaker constant was fixed and the diffuse layer potential was adjusted. This value is about −11 mV for 50 mM salt solution, decreases in magnitude with the increasing salt concentration, and vanishes for higher salt concentrations (Table S1, ESI†). The potential is negative due to the dissociation of silanol groups.9
The observed force profiles can be indeed well described by DLVO theory, including the minimum at larger distances and lower salt concentrations, which is characteristic for the superposition of dispersion and double layer forces. The other typical feature is that the double layer contribution becomes negligible at higher salt concentrations (>250 mM) and under these conditions the force profile is determined by dispersion forces only. The fact that forces between silica surfaces across water can be well described by DLVO theory may seem surprising, especially since some authors even claimed that dispersion forces are absent in such systems.22–24
The reason for this unexpected behavior becomes obvious when one compares the force profiles for heat-treated particles with the untreated ones in 1.0 M KCl solution. In this medium, only dispersion forces contribute and the force profiles will not be influenced by different surface charge densities, which might have been induced by the different heat-treatments used. Major differences in the strength of the dispersion force can be evidenced (Fig. 2b). The data were fitted at larger distances with the expression for the non-retarded dispersion force (first term in eqn (1)). Fig. 2d shows the magnitude of the force versus separation in a double logarithmic representation, where the non-retarded dispersion force law appears as a straight line. The force profile decays more rapidly at larger distances. This effect is especially prominent for the 1200 °C data and is probably caused by retardation effects.1 At shorter distances, the forces become repulsive. This short-ranged repulsion has a range of about 0.3 nm and is probably due to the hydration force or the overlapping of hairy layers of polysilicilic acid.9,20,28–30 The different heat-treatment procedures influence this short-ranged component of the force somewhat, as can be inferred from different strengths of this component. However, changes in surface hydrophobicity were reported not to influence such short-ranged forces too strongly.31 In the intermediate range of about 2–8 nm, however, the data follow well the power-law, which is characteristic of the non-retarded dispersion force. This dependence clearly confirms that dispersion forces are being measured. Additional contributions from hydration or hydrophobic forces would result in an exponential dependence.30
One observes that the strength of the measured dispersion forces decreases strongly with decreasing heating temperature, and they become extremely weak for the untreated particles. Given our excellent force resolution of about 0.9 pN, such weak forces are readily measurable. Additional force profiles are given in the ESI† (Fig. S1 and S2). From the measured force profiles, the respective Hamaker constants are extracted. Their dependence on the heating temperature is given in Fig. 2c (see also Table S2, ESI†). The Hamaker constant decreases strongly with decreasing heating temperature, from (2.4 ± 0.1) × 10−21 J for the particles treated at 1200 °C to (7 ± 5) × 10−23 J for the untreated particles. These substantial differences reflect the widely varying Hamaker constants for the same system reported in the literature.18,19,21 Fielden et al.19 measured the largest Hamaker constant of 1.5 × 10−20 J, Sivan and coworkers18 2.2 × 10−21 J, while Wang et al.21 2.5 × 10−22 J. All these previous measurements were carried out using a colloidal silica probe against a flat silicon wafer. The description of the sample preparation by Fielden et al.19 suggests that a thin, native silica layer was used, and therefore one indeed expects a larger Hamaker constant due to the nearby silicon phase with a high refractive index. Sivan and coworkers18 and Wang et al.21 grow a thicker silica layer, and therefore the underlying silicon will contribute less. As a consequence, lower Hamaker constants are reported. These difficulties underline the advantage of the present sphere–sphere geometry, where a fully symmetric system is automatically realized. The reproducibility of the measured forces involving different pairs of particles, which typically is around 20%, represents a measure of deviations from the ideal symmetry. In an asymmetric setting, the data analysis would be much more demanding. The low Hamaker constant reported by Wang et al.21 is in line with the present value observed for the heat-treatment at 1050 °C. The very small Hamaker constant for the untreated particles rationalizes the absence of dispersion forces reported by some authors.22–24 Such weak dispersion forces could have simply remained undetected given an inferior force resolution in the previous studies to the one achieved here.
Forces between the same silica particles heated at 1050 °C were investigated earlier.20 These authors found a Hamaker constant of 3.3 × 10−22 J, which should be compared to the value of 2.7 × 10−22 J reported here. The reason for this discrepancy is that the earlier study introduced a shift of 0.85 nm in the plane of origin of the dispersion force. This shift leads to a somewhat larger Hamaker constant, but such shifts cannot explain the large variations in the Hamaker constant observed here.
Measurements carried out with particles heated at the highest temperatures, namely 1150 °C and 1200 °C, lead to Hamaker constants that are well comparable to theoretical estimates. The currently most reliable theoretical value for silica across water is 1.6 × 10−21 J, which was calculated using accurate dielectric spectra over a wide frequency range.14 This value should be compared to the presently measured value of (2.4 ± 0.1) × 10−21 J. Given the uncertainties in the force measurements and spectral calculations, the agreement between experimental and theoretical values is satisfactory. The difficulties in obtaining an accurate theoretical estimate are illustrated by earlier calculations based on simplified spectra, which span a rather wide range, and typically yield substantially larger values (5 ± 3) × 10−21 J.14,32
After having established the strong variation of the Hamaker constant with the heating temperature, let us discuss the underlying mechanism leading to these variations. As the first hypothesis to explain the variation of the Hamaker constant, we have considered a possible transformation in the crystallinity of the silica particles. X-ray diffraction spectra were recorded for the untreated and heat-treated particles (Fig. S3, ESI†). All spectra feature a broad peak at diffraction angles of 21–22°, which is typical for amorphous silica. Detailed X-ray diffraction and NMR studies demonstrate that the size of the crystalline subunits is a few nm only.33 A minor shift of the position of this peak to smaller angles with heating temperature was also consistent with an earlier study.34 Therefore, the silica remains amorphous throughout, and the variation of the Hamaker constant cannot be explained in this way. In any case, one does not expect large variations of the Hamaker constants for different silica allotropes (e.g., quartz, cristobalite, tridymite).32
The second hypothesis concerning the variation of the Hamaker constant focuses on possible variations in particle porosity (i.e., volume fraction of voids). Suppose that the untreated particles are very porous, and that they shrink during heat treatment. Thereby, their porosity might decrease, which would modify the dielectric constant and, as a consequence, the Hamaker constant. To address this question, the size distributions of the untreated and heat-treated particles were measured using SEM. The average particle radii are reported in Fig. 3a, while further information on the size distributions is given in the ESI† (Table S2 and Fig. S4). One observes that the particles shrink somewhat during the heat treatment, but their final size is independent of the heating temperature within experimental error. Therefore, variations in particle porosity cannot explain the observed trend.
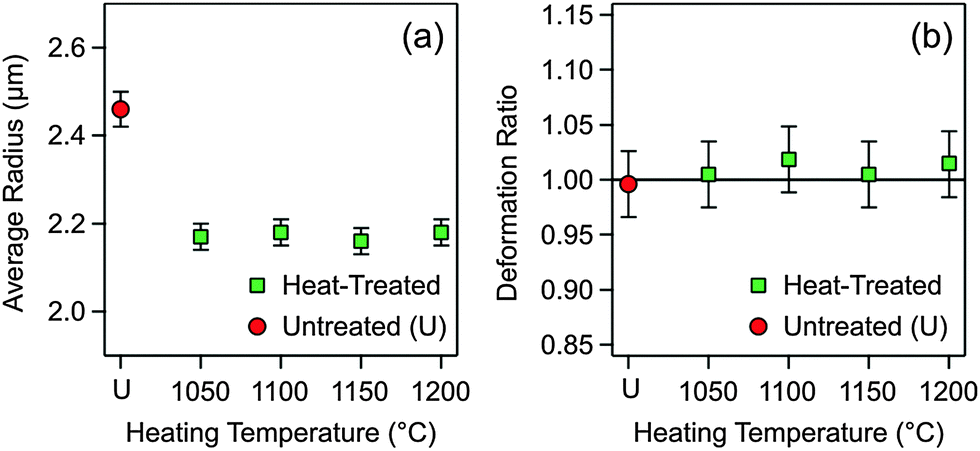 | ||
| Fig. 3 SEM characterization of the heat-treated particles at different temperatures and comparison with the untreated ones. (a) Average particle radius and (b) deformation ratio. This ratio is the quotient of the averages of the normal and lateral extension of the particles with respect to the surface, and is unity for a perfect sphere. | ||
The third hypothesis to consider is a change in the shape of the heated particles. When the particle surface would flatten during the heat treatment, one would have a larger effective radius Reff, and one would obtain an incorrect Hamaker constant. We have therefore imaged the tilted samples of the untreated and heat-treated silica particles. The SEM images shown in Fig. 1b and d illustrate that no deviations from the spherical particle shape for the heat-treated particles take place. This observation was quantified by comparing the ratio of the average radii in the normal and parallel direction to the substrate. Indeed, no deformation takes place within experimental error up to heating temperatures of 1200 °C (Fig. 3b). However, the silica particles melt at temperatures above 1300 °C, and thereby they deform completely. Images of the deformed particles are provided in the ESI† (Fig. S5 and S6).
The final and in our view the correct hypothesis focuses on surface roughness. The AFM images shown in Fig. 4 indicate substantial variations in the amplitude and correlation length of the surface roughness. The root mean square (RMS) roughness is plotted for the untreated and heat-treated particles in Fig. 4a, and one observes that the particle surface becomes smoother with increasing heating temperature. One actually obtains an excellent correlation between the Hamaker constant and the RMS roughness (Fig. 4b). The value for the untreated particles is an outlier. The RMS roughness is actually comparable to the ones treated at 1050 °C, but for the particles heated at higher temperatures the Hamaker constant is considerably larger. Recall that the untreated particles shrink somewhat when heated at 1050 °C, but heating at higher temperatures results in the same shrinkage (Fig. 3a). This initial shrinkage could actually induce a larger Hamaker constant due to a decrease in porosity. Another possibility for the differences between the untreated and heat-treated particles could be due to volatilization of residual ammonium ions originating from the particle synthesis.
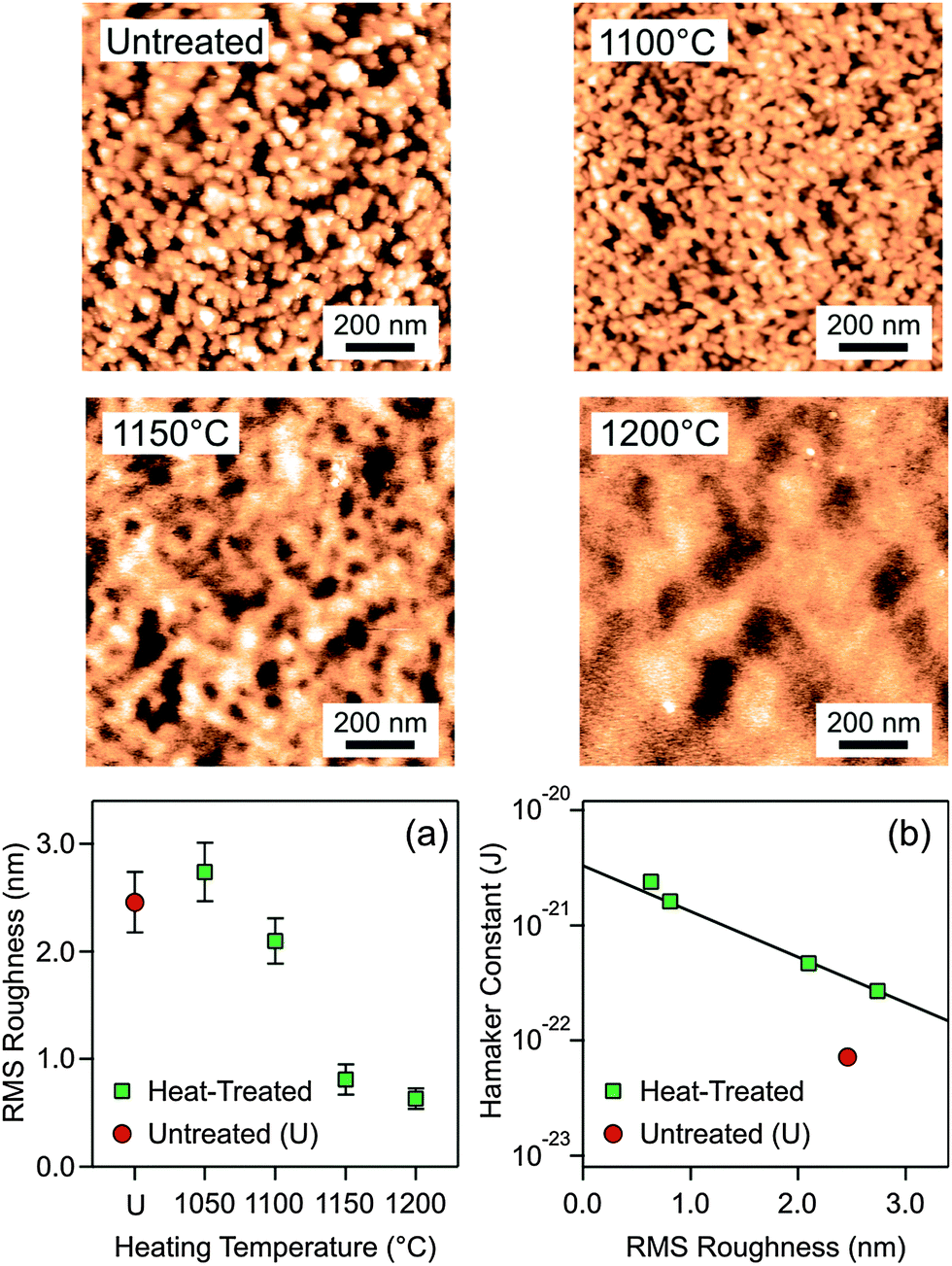 | ||
| Fig. 4 Roughness determination using AFM imaging. (a) RMS roughness versus the heat-treated particles at different temperatures and comparison with the untreated ones. (b) Hamaker constant versus RMS roughness. The line serves to guide the eye only. Representative AFM images of the particle surfaces are shown above. | ||
The effect of roughness on the dispersion forces has been investigated theoretically in various ways. One approach relies on the Derjaguin approximation, and averaging the effect of roughness over a distribution of pillars.8,35 This approach predicts that roughness does substantially reduce the strength of dispersion forces. This effect can be simply understood since intervening voids within the rough surface remain present at contact. These studies further suggest that the height–height correlation length has little influence on the strength of the dispersion forces. Our data support this aspect as well. The height–height correlation length is substantially larger for the particles heated at 1200 °C than the ones at 1150 °C as can be seen by comparing the characteristic size of the surface corrugations in the respective AFM-images (Fig. 4). We have confirmed this trend by quantitative image analysis (Table S2 and Fig. S5, ESI†). Nevertheless, the observed Hamaker constants remain similar. There is also a minor difference in the correlation length between the untreated sample and the one heated at 1100 °C, and this difference probably reflects the modification in the particle porosity mentioned above. However, the Derjaguin approximation may also break down, and the effects of deformation or non-additivity may have to be considered.35,36 Recently, the elastic deformation of the spikes in the height profile was found to be relevant, but this effect is likely to be important near the contact region only.37
Conclusions
The present direct force measurements demonstrate that modest variations in nanoscale roughness may substantially modify the strength of dispersion forces acting between silica particles across water. In the present case, we control the surface roughness by heat-treatment. In other systems, differences in surface roughness may originate from different particle synthesis protocols. These observations rationalize why widely different strengths of dispersion forces for silica particles across water were found18–21 and why the absence of dispersion forces was reported by others.22–24 In the latter situations, the dispersion forces were presumably so weak that they were impossible to detect using the experimental setup used at that time.The varying strength of dispersion forces through surface roughness could have major implications in various research areas. Let us mention three examples. First, colloidal suspensions of silica particles are known to be surprisingly stable in many situations,9 and this stability could be related to the weakening of dispersion interactions. Second, the design of colloidal molecules often relies on dispersion forces to stabilize the resulting clusters, and when such molecules contain silica particles, their surface roughness could be used to tune these forces precisely.13 Finally, during the fabrication of microelectromechanical devices, dispersion forces are responsible for unwanted adhesion of surfaces,11 and controlling the surface roughness would again open the possibility of tuning these forces precisely.
Acknowledgements
This research was supported by the Swiss National Science Foundation through grants 140327 and 159874 and University of Geneva.References
- W. B. Russel, D. A. Saville and W. R. Schowalter, Colloidal Dispersions, Cambridge University Press, Cambridge, 1989 Search PubMed.
- M. Elimelech, J. Gregory, X. Jia and R. A. Williams, Particle Deposition and Aggregation: Measurement, Modeling, and Simulation, Butterworth-Heinemann Ltd, Oxford, 1995 Search PubMed.
- G. L. Klimchitskaya, U. Mohideen and V. M. Mostepanenko, Rev. Mod. Phys., 2009, 81, 1827–1885 CrossRef.
- M. A. Bevan and D. C. Prieve, Langmuir, 1999, 15, 7925–7936 CrossRef CAS.
- J. N. Munday, F. Capasso and V. A. Parsegian, Nature, 2009, 457, 170–173 CrossRef CAS PubMed.
- R. F. Tabor, R. Manica, D. Y. C. Chan, F. Grieser and R. R. Dagastine, Phys. Rev. Lett., 2011, 106, 064501 CrossRef PubMed.
- A. Milling, P. Mulvaney and I. Larson, J. Colloid Interface Sci., 1996, 180, 460–465 CrossRef CAS.
- M. Elzbieciak-Wodka, M. Popescu, F. J. Montes Ruiz-Cabello, G. Trefalt, P. Maroni and M. Borkovec, J. Chem. Phys., 2014, 140, 104906 CrossRef PubMed.
- M. Kobayashi, F. Juillerat, P. Galletto, P. Bowen and M. Borkovec, Langmuir, 2005, 21, 5761–5769 CrossRef CAS PubMed.
- N. Tufenkji and M. Elimelech, Langmuir, 2005, 21, 841–852 CrossRef CAS PubMed.
- F. W. Delrio, M. P. De Boer, J. A. Knapp, E. D. Reedy, P. J. Clews and M. L. Dunn, Nat. Mater., 2005, 4, 629–634 CrossRef CAS PubMed.
- S. M. Gatica, M. W. Cole and D. Velegol, Nano Lett., 2005, 5, 169–173 CrossRef CAS PubMed.
- G. R. Yi, V. N. Manoharan, E. Michel, M. T. Elsesser, S. M. Yang and D. J. Pine, Adv. Mater., 2004, 16, 1204–1208 CrossRef CAS.
- H. D. Ackler, R. H. French and Y. M. Chiang, J. Colloid Interface Sci., 1996, 179, 460–469 CrossRef CAS.
- S. K. Lamoreaux, Phys. Rev. Lett., 1997, 78, 5–8 CrossRef CAS.
- R. S. Decca, E. Fischbach, G. L. Klimchitskaya, D. E. Krause, D. Lopez and V. M. Mostepanenko, Phys. Rev. D: Part. Fields, 2003, 68, 116003 CrossRef.
- M. Finessi, P. Sinha, I. Szilagyi, I. Popa, P. Maroni and M. Borkovec, J. Phys. Chem. B, 2011, 115, 9098–9105 CrossRef CAS PubMed.
- M. Dishon, O. Zohar and U. Sivan, Langmuir, 2009, 25, 2831–2836 CrossRef CAS PubMed.
- M. L. Fielden, R. A. Hayes and J. Ralston, Phys. Chem. Chem. Phys., 2000, 2, 2623–2628 RSC.
- V. Valmacco, G. Trefalt, P. Maroni and M. Borkovec, Phys. Chem. Chem. Phys., 2015, 17, 16553–16559 RSC.
- Y. H. Wang, L. G. Wang, M. A. Hampton and A. V. Nguyen, J. Phys. Chem. C, 2013, 117, 2113–2120 CAS.
- M. Giesbers, J. M. Kleijn and M. A. Cohen Stuart, J. Colloid Interface Sci., 2002, 248, 88–95 CrossRef CAS PubMed.
- R. F. Considine and C. J. Drummond, Langmuir, 2001, 17, 7777–7783 CrossRef CAS.
- S. Rentsch, R. Pericet-Camara, G. Papastavrou and M. Borkovec, Phys. Chem. Chem. Phys., 2006, 8, 2531–2538 RSC.
- W. A. Ducker, T. J. Senden and R. M. Pashley, Langmuir, 1992, 8, 1831–1836 CrossRef CAS.
- H. J. Butt, Biophys. J., 1991, 60, 1438–1444 CrossRef CAS PubMed.
- M. Borkovec, I. Szilagyi, I. Popa, M. Finessi, P. Sinha, P. Maroni and G. Papastavrou, Adv. Colloid Interface Sci., 2012, 179–182, 85–98 CrossRef CAS PubMed.
- G. Vigil, Z. H. Xu, S. Steinberg and J. Israelachvili, J. Colloid Interface Sci., 1994, 165, 367–385 CrossRef CAS.
- J. J. Valle-Delgado, J. A. Molina-Bolivar, F. Galisteo-Gonzalez, M. J. Galvez-Ruiz, A. Feiler and M. W. Rutland, J. Chem. Phys., 2005, 123, 034708 CrossRef CAS PubMed.
- S. H. Donaldson, A. Royne, K. Kristiansen, M. V. Rapp, S. Das, M. A. Gebbie, D. W. Lee, P. Stock, M. Valtiner and J. Israelachvili, Langmuir, 2015, 31, 2051–2064 CrossRef CAS PubMed.
- A. Grabbe and R. G. Horn, J. Colloid Interface Sci., 1993, 157, 375–383 CrossRef CAS.
- L. Bergstrom, Adv. Colloid Interface Sci., 1997, 70, 125–169 CrossRef CAS.
- D. D. Kragten, J. M. Fedeyko, K. R. Sawant, J. D. Rimer, D. G. Vlachos, R. F. Lobo and M. Tsapatsis, J. Phys. Chem. B, 2003, 107, 10006–10016 CrossRef CAS.
- J. R. Martinez, S. Palomares-Sanchez, G. Ortega-Zarzosa, F. Ruiz and Y. Chumakov, Mater. Lett., 2006, 60, 3526–3529 CrossRef CAS.
- V. B. Svetovoy and G. Palasantzas, Adv. Colloid Interface Sci., 2015, 216, 1–19 CrossRef CAS PubMed.
- G. Bimonte, T. Emig, R. L. Jaffe and M. Kardar, EPL, 2012, 97, 50001 CrossRef.
- D. F. Parsons, R. B. Walsh and V. S. J. Craig, J. Chem. Phys., 2014, 140, 164701 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental techniques, tables, and figures with additional force profiles, X-ray powder spectra, size distributions, and SEM images. See DOI: 10.1039/c6nh00070c |
| This journal is © The Royal Society of Chemistry 2016 |