
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiple di-leucines in the ATP7A copper transporter are required for retrograde trafficking to the trans-Golgi network†
Sha
Zhu
ab,
Vinit
Shanbhag
ab,
Victoria L.
Hodgkinson
ab and
Michael J.
Petris‡
*abc
aDepartment of Biochemistry, University of Missouri, Columbia, MO 65211, USA
bThe Christopher S. Bond Life Science Center, University of Missouri, 1201 Rollins St., Columbia, MO 65211, USA
cDepartment of Nutrition and Exercise Physiology, University of Missouri, Columbia, MO 65211, USA
First published on 15th June 2016
Abstract
The ATP7A protein is a ubiquitous copper-transporting P-type ATPase that is mutated in the lethal pediatric disorder of copper metabolism, Menkes disease. The steady-state location of ATP7A is within the trans-Golgi network (TGN), where it delivers copper to copper-dependent enzymes within the secretory pathway. However, ATP7A constantly cycles between the TGN and the plasma membrane, and in the presence of high copper concentrations, the exocytic arm of this cycling pathway is enhanced to promote a steady-state distribution of ATP7A to post-Golgi vesicles and the plasma membrane. A single di-leucine endocytic motif within the cytosolic carboxy tail of ATP7A (1487LL) was previously shown to be essential for TGN localization by functioning in retrieval from the plasma membrane, however, the requirement of other di-leucine signals in this region has not been fully investigated. While there has been some success in identifying sequence elements within ATP7A required for trafficking and catalysis, progress has been hampered by the instability of the ATP7A cDNA in high-copy plasmids during replication in Escherichia coli. In this study, we find that the use of DNA synthesis to generate silent mutations across the majority of both mouse and human ATP7A open reading frames was sufficient to stabilize these genes in high-copy plasmids, thus permitting the generation of full-length expression constructs. Using the stabilized mouse Atp7a construct, we identify a second di-leucine motif in the carboxy tail of ATP7A (1459LL) as essential for steady-state localization in the TGN by functioning in endosome-to-TGN trafficking. Taken together, these findings demonstrate that multiple di-leucine signals are required for recycling ATP7A from the plasma membrane to the TGN and illustrate the utility of large-scale codon reassignment as a simple and effective approach to circumvent cDNA instability in high-copy plasmids.
Significance to metallomicsThe regulated trafficking of the ATP7A copper transporter is one of the principle mechanisms for controlling cellular copper homeostasis. The current study solves the problem of human and mouse ATP7A cDNA instability in high copy plasmids of E. coli using methods that may be broadly applicable to other unstable genes. Furthermore, we identify a new di-leucine-based sorting signal in the carboxy-terminal region of ATP7A that regulates trafficking from endosomes to the Golgi. Our study sheds new light on the mechanisms by which mutations in the C-terminal region might give rise to ATP7A-related disorders of copper metabolism. |
Introduction
Copper is an essential enzymatic cofactor in all aerobic organisms. However, because copper is also potentially toxic, its accumulation within the cell must be strictly controlled. The copper transporting ATPase known as ATP7A is one of the major proteins in mammalian cells responsible for preventing the accumulation of copper to toxic levels. Under normal basal copper concentrations, the ATP7A protein is located predominantly in the trans-Golgi network (TGN),1,2 where it transports copper from the cytosol into secretory compartments for incorporation into nascent copper-requiring enzymes.3 However, under conditions of elevated copper concentrations, the ATP7A protein is sorted from the TGN into post-Golgi vesicles that fuse with the plasma membrane during copper egress from the cell.1 Like other resident proteins of the TGN, the ATP7A protein also constitutively cycles between the TGN and the plasma membrane in the absence of high copper concentrations.4The sorting of itinerant transmembrane proteins between the TGN and the plasma membrane has been shown to depend on the recognition of short linear signals in their cytoplasmic domains.5,6 Among the most well-characterized are the adaptor proteins, AP-1, AP-2, AP-3 and AP-4, which form components of membrane coats. These adaptor proteins are comprised of heterotetrameric subunits which bind directly to sorting signals within target proteins.7 AP-1, AP-2 and AP-3 are known to recognize both the “tyrosine-based”, YXXØ, and “dileucine-based”, (D/EXXXL[LI]), consensus motifs (where Y is tyrosine, D is aspartate, E is glutamate, L is leucine, I is isoleucine, X is any amino acid, and Ø is a bulky hydrophobic amino acid). Previous studies have shown that a single di-leucine (1487LL) in the cytosolic carboxy terminal region of ATP7A (hereafter designated LL3) is essential for maintaining a steady-state localization of ATP7A in the TGN.8,9 Mutation of this di-leucine results in the accumulation of ATP7A at the plasma membrane due to a reduction in endocytic retrieval of the protein to the TGN. Consistent with these findings, retrograde ATP7A trafficking from the plasma membrane to the TGN requires clathrin adaptor subunits AP-1 and AP-2.10,11 Interestingly, two additional di-leucines (which we have termed LL1 and LL2) are located upstream of the LL3 motif in the cytoplasmic tail of ATP7A (Fig. 1). Although these di-leucines do not fit the canonical D/EXXXL[LI] consensus, they are completely conserved among mammalian ATP7A proteins, raising the possibility that they may play a role ATP7A trafficking.
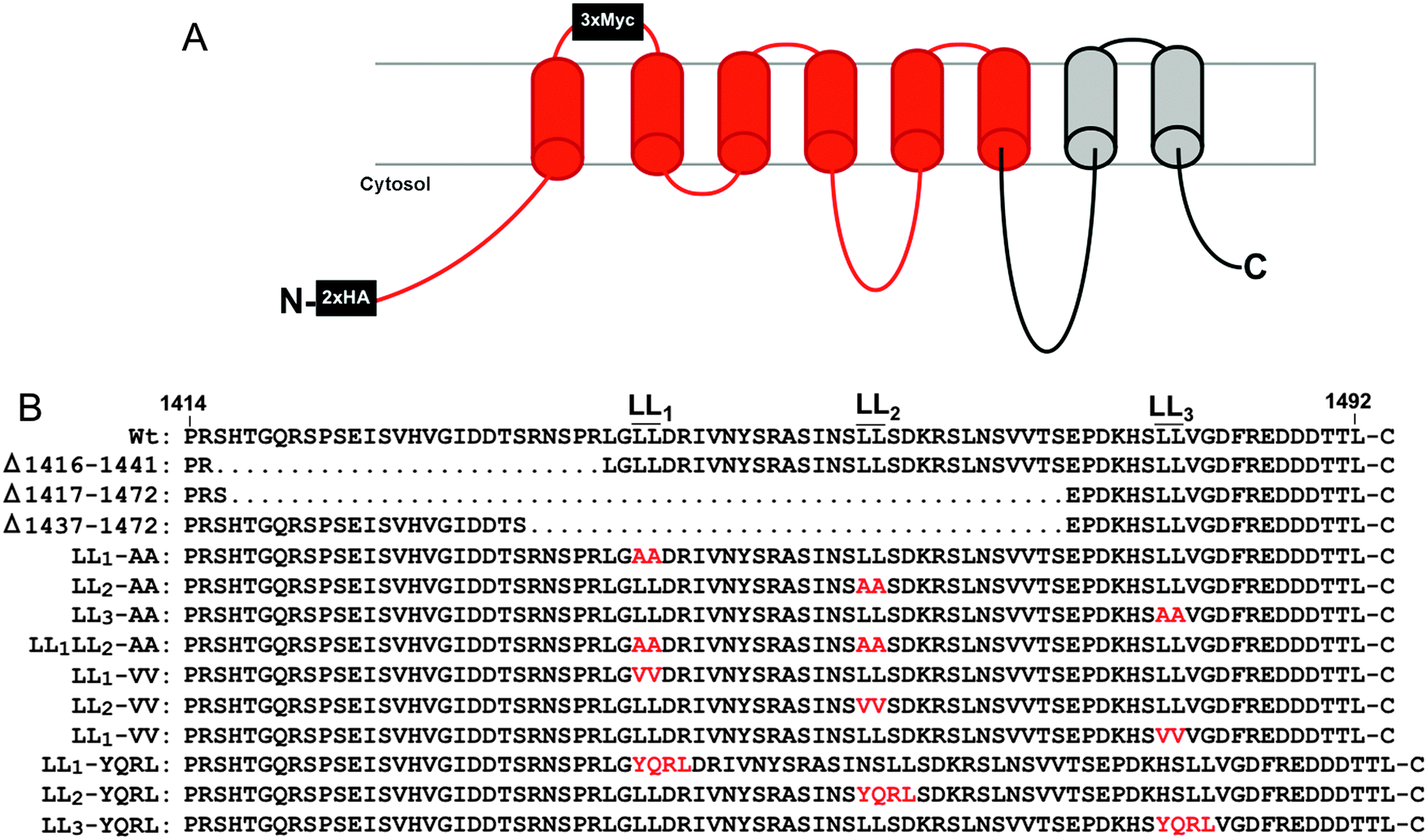 | ||
| Fig. 1 Topology of ATP7A-HA/Myc protein and mutations generated in this study. (A) The recombinant ATP7A protein with 2×HA tags inserted in the cytoplasmic amino terminus and 3×Myc tags in the first extracellular loop. Regions in red correspond to recoded DNA sequences with alternative codons used to stabilize the Atp7a cDNA. (B) The amino acid sequence of the carboxy terminal region is shown with the location of di-leucines LL1, LL2 and LL3. Amino acid substitutions are depicted in red. | ||
Several human diseases are caused by mutations in the ATP7A gene, the most well-characterized of which is Menkes disease (OMIM 309400), a pediatric disorder of copper deficiency.12 Others include Occipital Horn Syndrome (OMIM 304150), a disease that predominantly affects the connective tissue, and X-linked Spinal Muscular Atrophy type 3 (OMIM 300489), a motor neuropathy.12 Despite their rather disparate clinical manifestations, each of these disorders has been attributed to mutations that affect trafficking of the ATP7A protein, thus underscoring the complex relationships between ATP7A mutations and the extent to which specific tissues are affected.13,14
Despite the importance of ATP7A trafficking in copper metabolism, investigations into the underlying structure–function relationships of this protein have been hampered by instability of the ATP7A cDNA in high-copy plasmids during replication in Escherichia coli.15 The goal of the current study was two-fold: first, to solve the problem of ATP7A cDNA instability, and second, to address the importance of upstream di-leucines LL1 and LL2 in conferring steady-state localization of ATP7A in the TGN. In an attempt to solve the problem of ATP7A gene instability, we used commercial DNA synthesis to introduce silent mutations throughout much of the human and mouse ATP7A cDNA sequences, the majority of which were concentrated within the 3′ wobble positions of codons. This recoding of ATP7A cDNAs was found to stabilize propagation in high-copy plasmids. Using a combination of deletions and missense mutations, we demonstrate that LL1, LL2 and LL3 are each required for steady-state localization of ATP7A in the TGN. In contrast to LL3, which is required for ATP7A endocytosis from the plasma membrane, LL2 appears to be essential for trafficking of ATP7A from endosomes to the Golgi. As a whole, this study outlines a simple and rapid approach for overcoming problems of ATP7A cDNA instability with broad implications for other unstable genes in high-copy plasmids. Moreover, our findings provide new insights into disorders of copper metabolism caused by mutations in the carboxy terminal region of the ATP7A protein.
Methods
Reagents and plasmids
All reagents were from Sigma (St. Louis) unless otherwise noted. For the mouse Atp7a cDNA, commercial DNA synthesis using overlapping PCR was used to generate more than 1000 silent mutations within a 2.0 kb fragment flanked by AccIII/XbaI restriction sites, and a second 1.2 kb fragment flanked by XbaI/XhoI restriction sites (Life Technologies). Base substitutions were semi-random and in most cases involved the wobble base of each codon. Sequences with similarity to E. coli promoter elements or repeating elements were avoided. A third DNA fragment containing the last 1.4 kb of the native Atp7a open reading frame was generated by reverse transcription PCR using primers 5′-ACCATCTAGACTCGAGATGGCTCATAAGGTAAAGGTAGTGGTATTTGATAAGACTGG and 5′-AGATGCGGCCGCTTACAGTGTGGTGTCATCATCTTCCCGGAAGTCG. A complete open reading frame of Atp7a was then generated by successive subcloning each fragment into the high copy plasmid pQCXIP (Fig. S1, ESI†). Two hemagglutinin A (HA) epitope tags were then inserted after the start codon, and three Myc epitope tags were inserted into a PacI site immediately downstream of isoleucine-685 located in an exofacial loop between the first and second transmembrane domains to allow for anti-myc antibody uptake as a measure of ATP7A endocytosis.16 The final plasmid, pAtp7a-HA/Myc, was propagated in E. coli strain DH5α and found to be mutation free by DNA sequencing. An identical strategy was used to generate a stable version of the human ATP7A cDNA with the exception that silent codon reassignment was generated across the entire open reading frame (Fig. S2, ESI†). For the C-terminal mutations listed in Fig. 1, custom DNA synthesis (Life Technologies) was used to generate each mutation within a BamHI-NotI DNA fragment, which was then subcloned into the pAtp7a-HA/Myc plasmid replacing the corresponding wild type segment.Cell culture and transfection
HEK293T cells were maintained in Dulbecco's modified Eagle's medium (Life Technologies) containing 10% fetal bovine serum and 100 IU ml−1 penicillin and streptomycin (Life Technologies) in a 5% CO2 atmosphere at 37 °C. The above mutations were all stably expressed in HEK293T cells by transfection of the corresponding vectors using the FuGENE 6 transfection reagent (Promega) according to the manufacturer's instructions and selected for two weeks in 2 μg ml−1 puromycin (Life Technologies). The transfection efficiency of the pAtp7a-HA/Myc plasmid in HEK cells was approximately 40–50% after 24 hours using EndoFree plasmid preparations (Qiagen). Menkes disease patient fibroblasts, GM01981,17,18 were purchased from the Coriell Cell Repositories (http://www.ccr.coriell.org) and immortalized by expression of the SV40 large T antigen by transformation with a plasmid pSV3-neo (American Type Culture Collection).Immunoblots
Cells cultured in 6-well trays were scraped into ice-cold phosphate buffered saline (PBS) and pelleted by centrifugation at 800 × g. After three washes in ice-cold PBS, the cells were lysed for 20 minutes on ice in buffer containing 62.5 mM Tris-HCl (pH 7.4), 1% Triton X-100, 0.1% SDS, 1 mM EDTA, and Complete™ protease inhibitor cocktail (Roche). Samples were then centrifuged for 10 minutes at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g and 40 μg of protein supernatant was fractionated by 4–20% SDS-PAGE using a mini-PROTEAN 3 gel unit (Bio-rad) and then transferred to nitrocellulose membranes. Proteins were detected by probing with the indicated primary antibodies for 2 hours at room temperature, followed by the appropriate secondary antibody conjugated to horseradish peroxidase. Blots were developed using the SuperSignal West Pico Substrate according to the manufacturer's instructions (Pierce). Primary antibodies used were rabbit anti-ATP7A,19 rabbit anti-HA (Sigma), mouse β-actin antibody (Abcam), mouse anti-tyrosinase antibody (Santa Cruz Biotechnology). Molecular weight standards were SigmaMarker (Sigma) or PageRuler (ThermoFisher).
000 × g and 40 μg of protein supernatant was fractionated by 4–20% SDS-PAGE using a mini-PROTEAN 3 gel unit (Bio-rad) and then transferred to nitrocellulose membranes. Proteins were detected by probing with the indicated primary antibodies for 2 hours at room temperature, followed by the appropriate secondary antibody conjugated to horseradish peroxidase. Blots were developed using the SuperSignal West Pico Substrate according to the manufacturer's instructions (Pierce). Primary antibodies used were rabbit anti-ATP7A,19 rabbit anti-HA (Sigma), mouse β-actin antibody (Abcam), mouse anti-tyrosinase antibody (Santa Cruz Biotechnology). Molecular weight standards were SigmaMarker (Sigma) or PageRuler (ThermoFisher).
Immunofluorescence microscopy
Cells were cultured for 24 h in 6-well trays on sterile poly-L-lysine-coated glass coverslips, washed twice with 1 ml of ice-cold PBS, and then fixed for 10 minutes at room temperature using 4% paraformaldehyde in PBS. Cells were then permeabilized with 0.1% Triton X-100 in PBS for 8 minutes, blocked overnight with 1% casein in PBS, and then probed with either rabbit anti-HA antibodies (Sigma) or antibodies against the TGN marker p230 (BD Biosciences). After three washes in PBS, cells were probed with secondary antibodies Alexa 488-conjugated anti-rabbit IgG or Alexa 594-conjugated anti-mouse IgG (Life Technologies). Cells were then incubated with 4′,6-diamidino-2-phenylindole (DAPI) to stain nuclei, washed three times with PBS and mounted with MOWIOL (Sigma). Cells were imaged using a Leica DMRE fluorescence microscope or Leica SP8 MP spectral scanning confocal microscope.Myc-antibody uptake assay
Endocytosis of the Myc-tagged ATP7A protein was determined by assaying the uptake of anti-Myc antibodies added to the culture media of cells grown on glass coverslips using a modified assay described previously.4 Internalization of anti-Myc antibodies bound to the exofacial Myc-tag protein provides an indirect assessment of ATP7A cycling via the plasma membrane. Cells were incubated for 4 hours at 37 °C in media containing 10 μg ml−1 anti-Myc antibodies (9E10; Developmental Studies Hybridoma Bank). Cells were then washed three times with ice cold PBS. To reduce surface-bound antibody, cells were washed three times for 2 minutes in ice-cold acidic buffer (100 mM glycine, 20 mM magnesium acetate, 50 mM potassium chloride, pH 2.2). Cells were then fixed with 4% PFA in PBS, permeabilized with 0.1% Triton X-100 in PBS and blocked overnight with 1% casein in PBS. Anti-myc antibodies were then detected using Alexa 488- or Alexa 594-conjugated anti-mouse IgG. Where indicated, the total cellular pool of ATP7A protein was detected by incubating the same cells with rabbit anti-HA antibodies (Sigma). Early endosomes were labeled using rabbit antibodies against EEA1 (Abcam).Tyrosinase assays
The cell in situ tyrosinase assay was conducted as previously described20 by assaying the production of brown DOPA-chrome from the oxidation of 3,4-dihydroxy-L-phenyl-alanine (L-DOPA). Menkes patient GM01981 cells were cultured for 16 hours on poly-L-lysine coated coverslips prior to transient transfection with the pTYR plasmid (a kind gift from Ann Hubbard, Johns Hopkins) with or without the pATP7A-HA/Myc plasmid using the FuGENE 6 transfection reagent (Roche), according to the manufacturer's instructions. Cells were allowed to recover for 48 hours in growth medium, washed twice in PBS and then fixed for 30 s in acetone/methanol (1:1) at −20 °C. Coverslips were then incubated for 1 hour at 37 °C in 0.15% (w/v) L-DOPA dissolved in 0.1 M phosphate buffer (pH 6.8) to allow pigmented dopachrome formation. Coverslips were mounted on slides and analyzed by bright field microscopy. The colorimetric in gel tyrosinase assay was performed as previously described.3
Results
Construction of stable human and mouse ATP7A expression vectors in high-copy plasmids
Instability of the human ATP7A cDNA in high-copy plasmids in E. coli is a well-documented problem for mutagenesis studies of this copper transporter.15,21 We considered the possibility that the mouse Atp7a cDNA might be stably propagated in high-copy plasmids. In an attempt to make a contiguous construct of the entire 4.6 kb open reading frame of the mouse Atp7a cDNA, partial EST clones spanning the entire coding sequence were purchased. However, clones spanning the first 2000 bp downstream of the transcription start site were all found to have numerous deletions and rearrangements. These observations suggested that, like the human ATP7A cDNA, the mouse Atp7a cDNA contains sequences that are deleterious in E. coli, and that these sequences reside within the first 2000 bp. In an attempt to overcome this problem, we used commercial DNA synthesis to recode the first 2.9 kb of the mouse Atp7a open reading frame using silent mutations (Fig. S1, ESI†). These silent nucleotide substitutions were semi-random and were typically placed within the wobble position of each codon, except where such a change would generate a region with similarity to E. coli promoter elements such as an AT-rich region or Pribnow box.22 The recoded DNA fragments were then used to generate a full-length mouse Atp7a open reading frame by subcloning into the high-copy mammalian expression plasmid, pQCXIP. Two HA epitope tags were placed at the 5′ end of the Atp7a open reading frame, and three Myc tags were inserted into a region corresponding to the first extracellular loop to enable endocytosis to be detected by the uptake of extracellular Myc-antibodies bound to the mouse ATP7A protein (Fig. 1A).4 The final construct, the pAtp7a-HA/Myc, was confirmed to be stably propagated in E. coli using both restriction digest analysis and DNA sequencing of the Atp7a open reading frame. The same approach was applied to generate a stably propagated human ATP7A cDNA, although in this case silent mutations were introduced throughout the entire open reading frame (Fig. S2, ESI†).To assess whether the mouse ATP7A protein was expressed from the recoded construct and whether the HA and Myc tags interfered with the expected localization and trafficking, we stably transfected the pAtp7a-HA/Myc plasmid into HEK293T cells. Immunofluorescence microscopy using anti-HA antibodies revealed that the recombinant ATP7A protein was localized in the juxtanuclear region, and found to considerably overlap with the TGN marker, p230 (Fig. 2A). Copper-regulated trafficking of the recombinant ATP7A protein was not affected by the location of the tags because the addition of elevated copper concentrations to the growth medium shifted the steady-state distribution of ATP7A towards the cell periphery (Fig. 2A). We then tested if the TGN pool of ATP7A-HA/Myc protein in basal medium constitutively cycled via the plasma membrane using a method previously developed for human ATP7A.4 Because the Myc-tag was inserted into an exofacial loop that is exposed to the extracellular medium whenever ATP7A is at the plasma membrane, by incubating transfected cells with the anti-Myc antibody and allowing internalization to occur at 37 °C, the subsequent detection of these anti-Myc antibodies in fixed and permeabilized cells using fluorescent secondary antibodies provides an indirect measure of ATP7A protein that has cycled via the plasma membrane.4 As shown in Fig. 2B, the incubation of cells for 4 hours in basal medium containing anti-Myc antibodies resulted in the accumulation of these antibodies within a juxtanuclear region that completely overlapped with the total pool of ATP7A-HA/Myc protein that was labeled with anti-HA antibodies after fixing and permeabilizing cells. Anti-Myc antibodies were only detected in cells that expressed the ATP7A-HA/Myc protein, indicating that the internalization of anti-Myc antibodies was specific to endocytosis of the ATP7A-HA/Myc protein. Collectively, these findings demonstrate that the ATP7A-HA/Myc protein correctly localizes to the TGN, undergoes trafficking from this location in response to elevated copper concentrations, and constitutively cycles via the plasma membrane, as shown previously for the human ATP7A protein.16
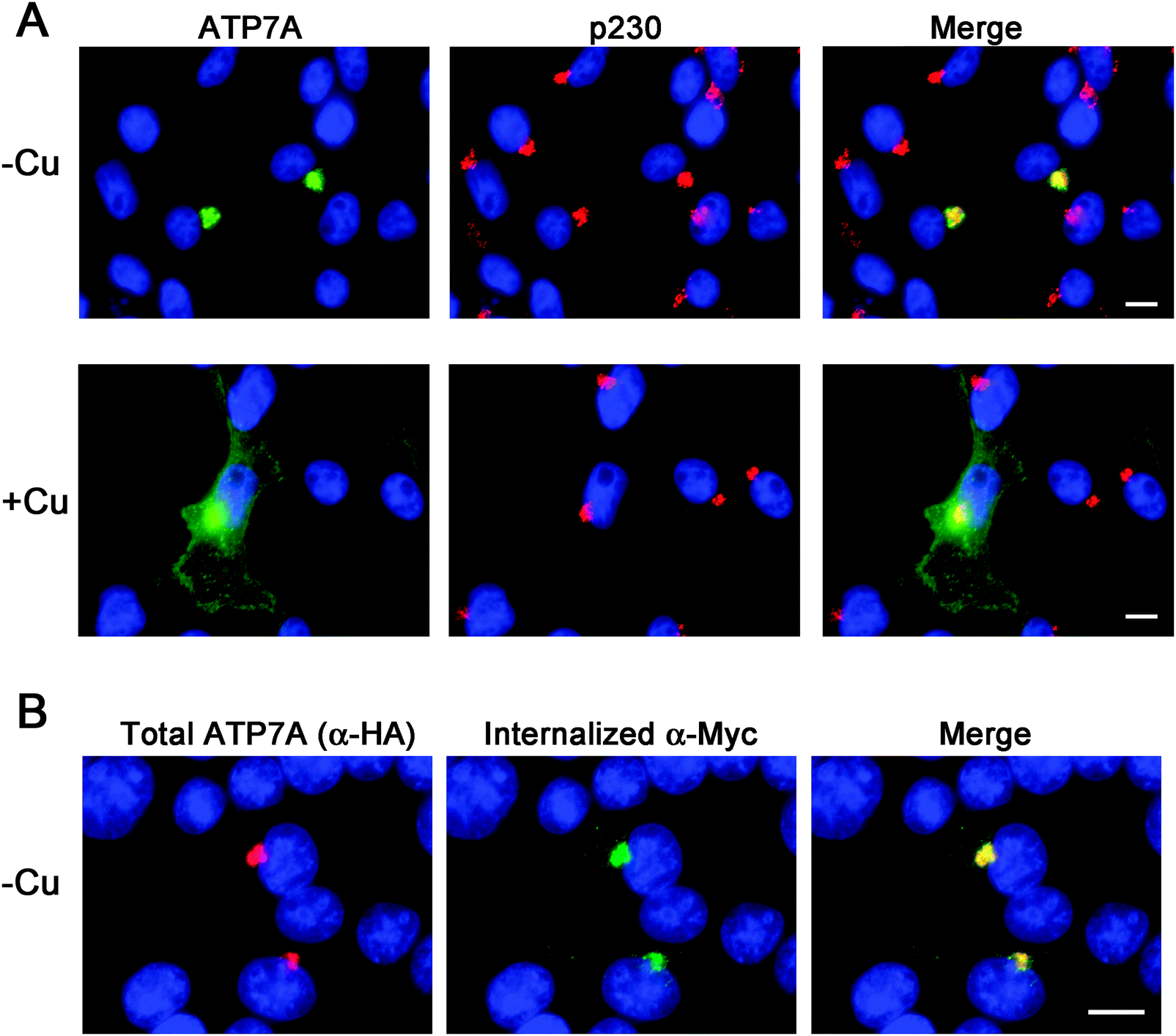 | ||
| Fig. 2 Immunofluorescence analysis of the steady-state localization and trafficking of ATP7A-HA/Myc protein. (A) HEK293T cells stably expressing the ATP7A-HA/Myc protein cultured in either basal medium (−Cu) or for 2 hours in medium containing 50 μM CuCl2 (+Cu) were probed with HA antibodies to label ATP7A-HA–Myc protein (green) and antibodies against the TGN marker protein p230 (red). (B) HEK293T cells stably expressing the ATP7A-HA/Myc protein were incubated in basal medium containing anti-Myc antibodies for 4 hours to allow internalization of the antibodies via ATP7A endocytosis. After fixing and permeabilization of cells, the internalized anti-Myc antibodies were then detected using Alexa 488 goat anti-mouse IgG (green). The total pool of ATP7A-HA–Myc protein was detected in the same cells using rabbit anti-HA antibodies followed by Alexa 594 goat anti-rabbit IgG (red). Nuclei were counterstained with DAPI (blue). Scale bars = 10 μm. | ||
To verify whether the ATP7A-HA/Myc protein is a functional copper transporter, we tested whether it could activate tyrosinase in Menkes disease fibroblasts lacking endogenous ATP7A protein. Tyrosinase is a cuproenzyme that must receive its copper cofactor via ATP7A-dependent copper transport into the secretory pathway.23 As expected, transfection of Menkes fibroblasts with the tyrosinase expression plasmid alone failed to result in detectable tyrosinase activity, however, co-expression of the pAtp7a-HA/Myc plasmid in these cells resulted in abundant tyrosinase activity (Fig. 3A and B), confirming that the ATP7A-HA/Myc protein was indeed a functional copper transporter.
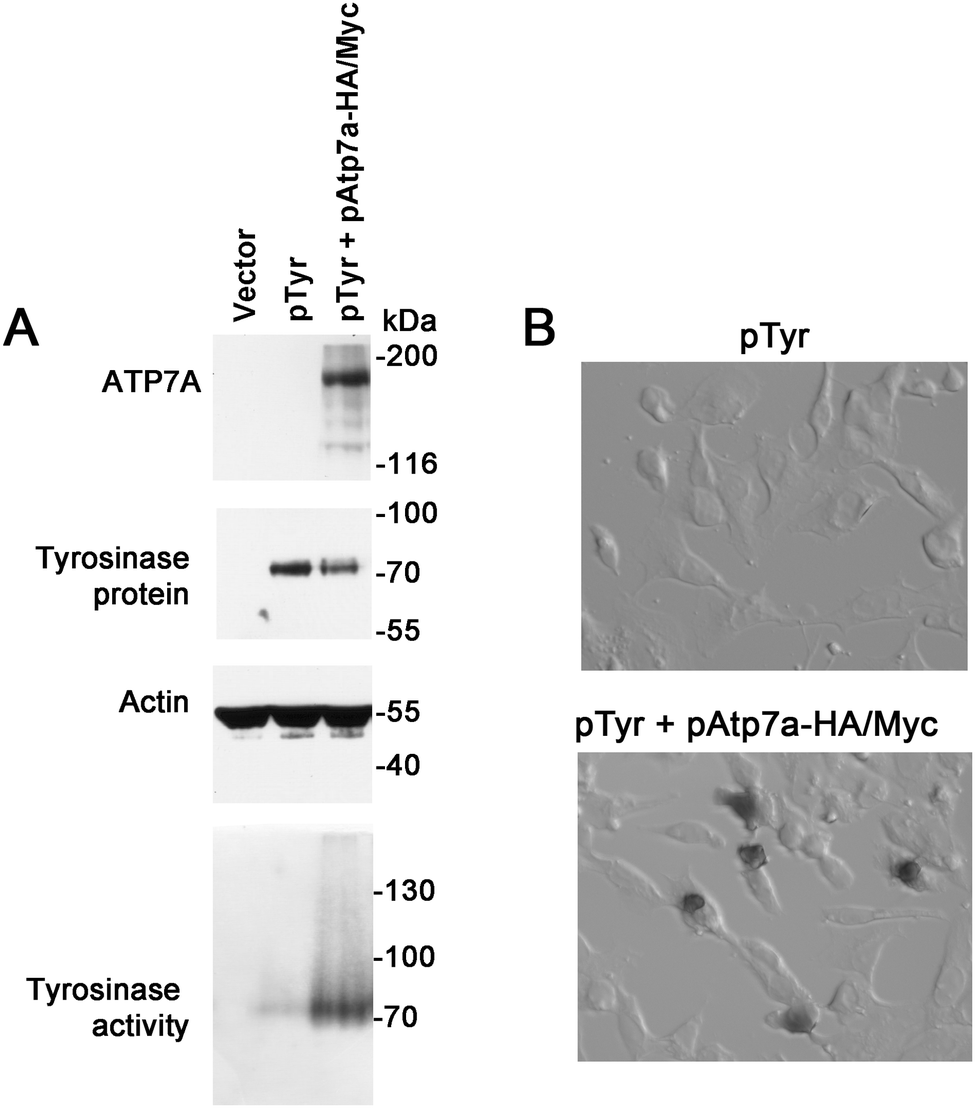 | ||
| Fig. 3 Functional analysis of the ATP7A-HA/Myc protein. (A) Menkes patient GM01981 cells were transiently transfected with empty pQCXIP vector only (lane 1), tyrosinase expression construct (pTyr) (lane 2), or co-transfected with pTyr and pAtp7a-HA–Myc plasmids (lane 3) and cultured in basal medium for 48 hours. Lysates were prepared and used for immunoblot analysis of ATP7A and tyrosinase. Actin was detected as a loading control. Tyrosinase activity was determined using a colorimetric in-gel assay of the oxidation of L-DOPA substrate (bottom panel). (B) GM01981 cells were transiently transfected with the indicated plasmids, fixed with acetone/methanol and then incubated with the L-DOPA substrate to detect tyrosinase activity in situ. | ||
LL1 and LL2 are required for TGN localization of the ATP7A protein via different mechanisms
Having generated a stably propagated Atp7a cDNA in a high-copy plasmid vector, we returned to the question of whether di-leucines LL1 and LL2 in the carboxy terminal region of ATP7A might be necessary for maintaining its steady-state localization in the TGN. A series of deletion mutations were generated in the carboxy terminal region of the pAtp7a-HA/Myc plasmid and stably transfected into HEK293T cells (Fig. 1). The positive control, LL3-AA, resulted in the accumulation of ATP7A at the plasma membrane (Fig. 4E), consistent with the importance of this di-leucine in ATP7A endocytosis.8,21 Deletions Δ1417–1472 and Δ1437–1472 which encompassed both LL1 and LL2, resulted in the marked accumulation of ATP7A protein at the plasma membrane, with little if any of the protein detected in the juxtanuclear region (Fig. 4C and D). In contrast, the Δ1416–1441 deletion, which did not encompass the LL1 and LL2 sequences, did not affect the localization of ATP7A (Fig. 4B). These finding suggest that sequences located upstream of LL3 that encompass both LL1 and LL2 are essential for steady state localization of ATP7A in the TGN.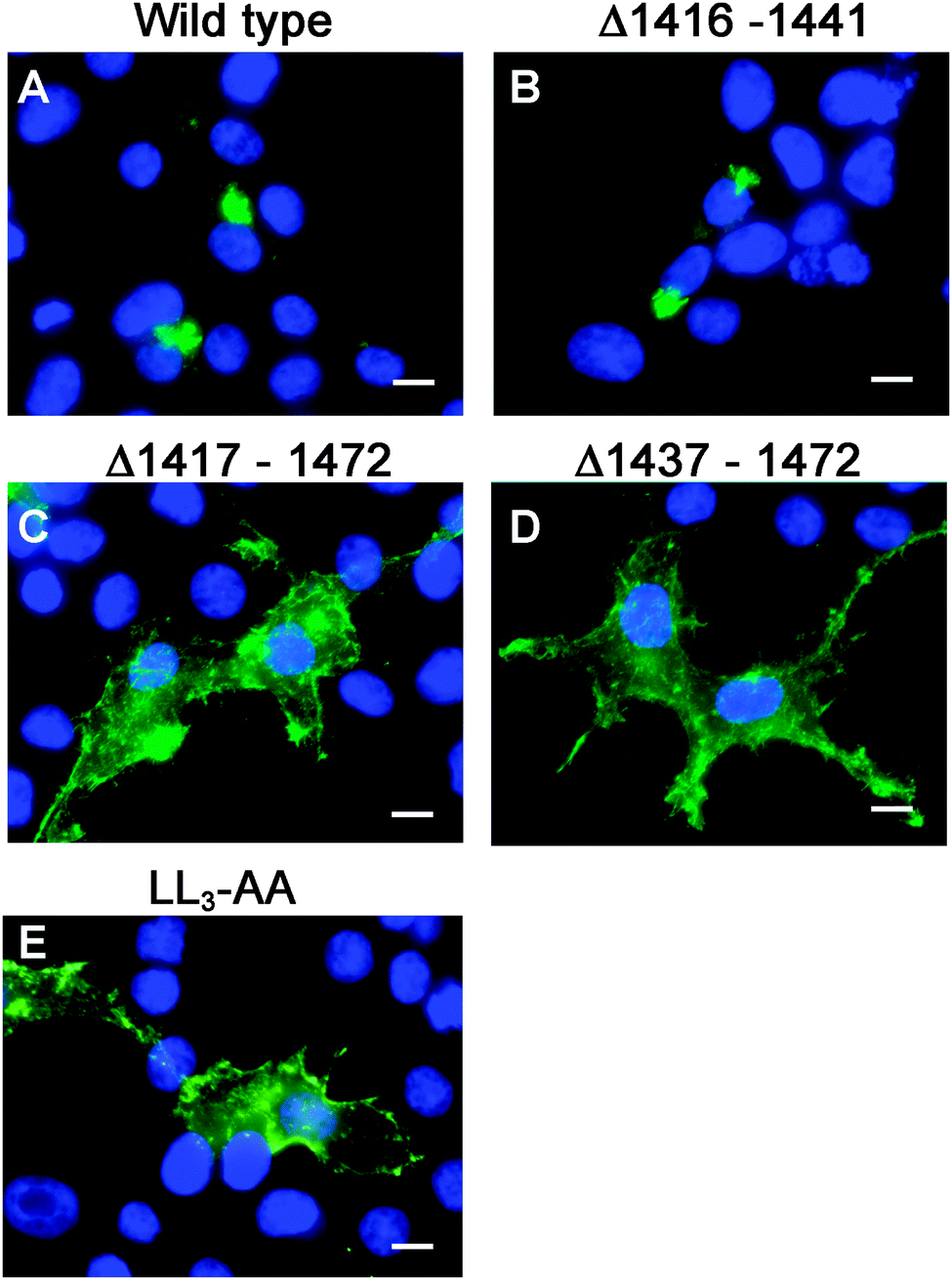 | ||
| Fig. 4 Deletion analysis of the carboxyl terminal region of ATP7A. Immunofluorescence microscopy was used to assess the intracellular localization of wild type ATP7A-HA/Myc protein (A), or mutants Δ1416–1441 (B), Δ1417–1472 (C), Δ1437–1472 (D), or LL3-AA (E) using anti-HA antibody followed by Alexa 488 anti-rabbit IgG (green). Note that the Δ1416–1441 mutation had no effect on TGN localization of ATP7A, while Δ1417–1472 and Δ1437–1472 encompassing both LL1 and LL2 resulted in the accumulation of ATP7A at the plasma membrane similar to that of LL3-AA. Nuclei were counterstained with DAPI (blue). Scale bars = 10 μm. | ||
To directly test the importance of LL1 and LL2, in ATP7A localization, both di-leucines were mutated to di-alanine in the pAtp7a-HA/Myc plasmid and stably expressed in HEK293T cells. Although there was some labeling of LL1-AA and LL2-AA mutant proteins in the juxtanuclear region, there was significant accumulation of both mutant proteins in cytoplasmic puncta and the plasma membrane (Fig. 5B and C). A combined mutation of both LL1-AA and LL2-AA produced a more extensive dispersion of ATP7A with little, if any, in the TGN (Fig. 5D), indicating an additive effect of these mutations. These findings suggest that both LL1 and LL2 are essential for normal localization of ATP7A in the TGN.
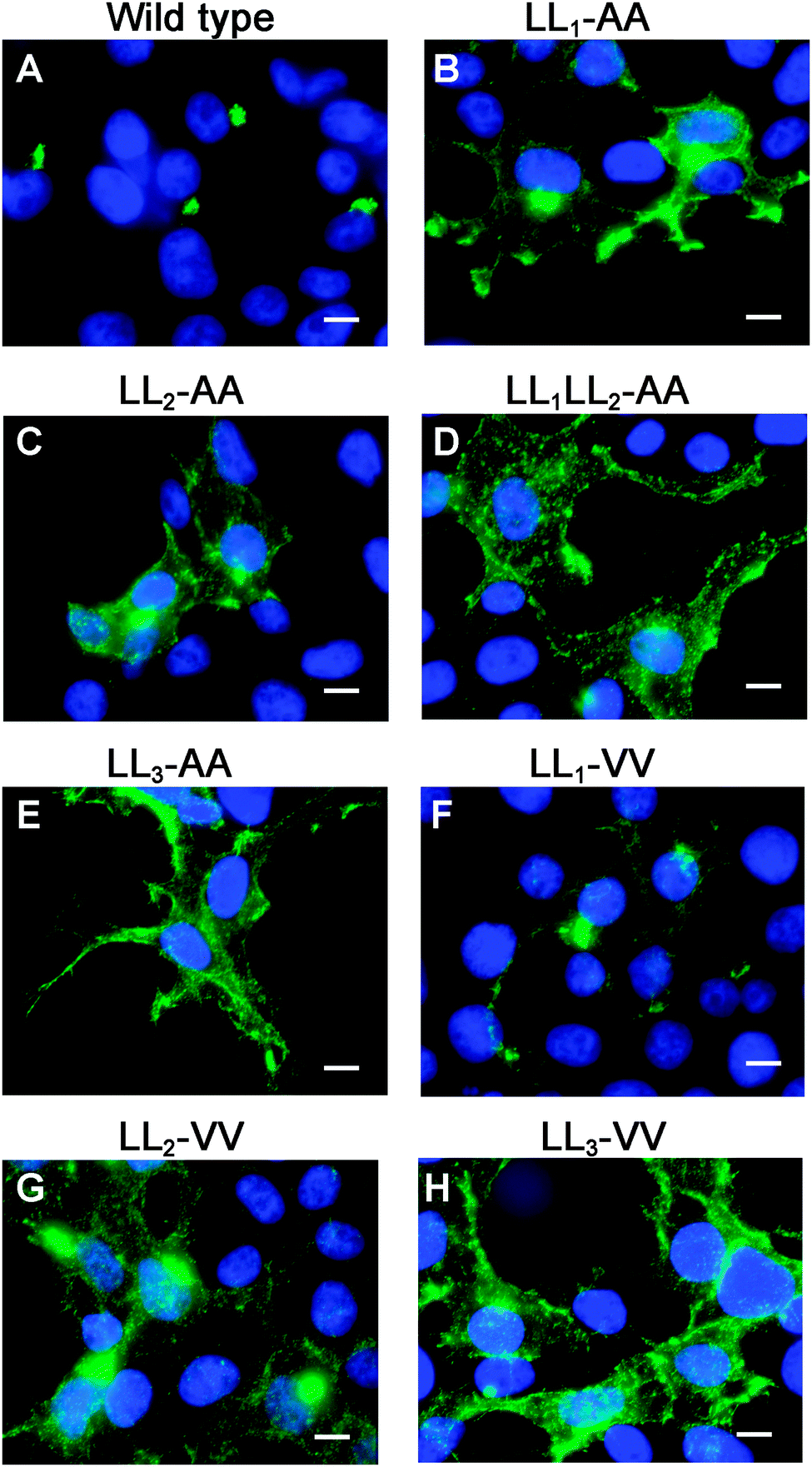 | ||
| Fig. 5 LL1 and LL2 are required for TGN localization of the ATP7A protein via different mechanisms. Immunofluorescence microscopy with anti-HA antibodies (green) was used to assess the steady-state localization of ATP7A protein in basal medium in stably transfected HEK293T cells expressing wild type ATP7A or the indicated di-leucine mutants. Note that mutation of individual LL1 and LL2 to di-alanine resulted in the accumulation of mutants at the plasma membrane with partial staining in the juxtanuclear region (B and C), whereas a double mutation of both LL1 and LL2 to di-alanine produced a complete mislocalization of ATP7A at the cell surface (D), similar to LL3-AA (E). Note that the LL1-VV substitution resulted in a predominantly normal intracellular localization of ATP7A in the Golgi (F). In contrast, the LL2-VV and LL3-VV mutations resulted in plasma membrane accumulation of ATP7A (G and H). Nuclei were counterstained with DAPI (blue). Scale bars = 10 μm. | ||
To generate mutations that more closely preserve size and hydrophobicity of the di-leucine sequences, we engineered di-valine substitutions into LL1, LL2 and LL3. Surprisingly, the LL1-VV mutation resulted in a normal localization of ATP7A in the TGN suggesting that these di-leucines can be replaced by amino acids that conserve hydrophobicity (Fig. 5F). In contrast, replacement of either LL2 or LL3 with di-valine failed to correct mislocalization of ATP7A (Fig. 5G and H). These findings suggest that it is not hydrophobicity per se that determines the importance of LL2 and LL3 for steady-state localization in the TGN, and suggests that LL2, like LL3, may function as a sorting signal.
Because di-leucine sorting signals are known to bind to the same adaptor subunits as tyrosine-based sorting signals, we reasoned that if LL2 and LL3 are bona fide sorting signals, they should be replaceable with a known tyrosine-based endocytosis motif. Thus, we investigated whether a well-characterized tyrosine-based endocytosis motif of the protein TGN38, YQRL,24–26 could functionally replace LL1, LL2 or LL3. As shown in Fig. 6, replacement of LL1 with YQRL failed to prevent accumulation of ATP7A at the plasma membrane. In contrast, we found that the YQRL sequence could functionally replace both LL2 and LL3 by correctly localizing ATP7A in the perinuclear region. These findings suggest that both LL2 and LL3 are required for maintaining a TGN localization of ATP7A by functioning as di-leucine sorting signals with analogous functions to tyrosine-based signals.
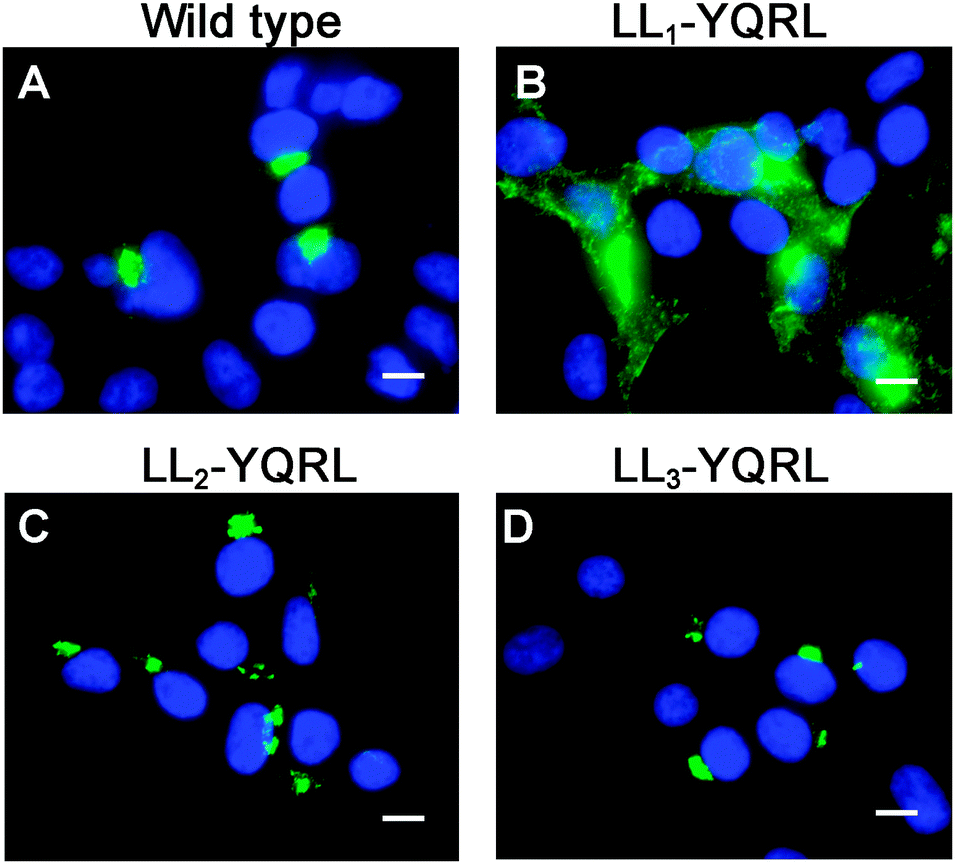 | ||
| Fig. 6 The tyrosine-based endocytosis motif, YQRL, can functionally replace LL2 and LL3, but not LL1. Immunofluorescence microscopy was used to detect the steady-state localization of ATP7A in which LL1, LL2 and LL3 were replaced by the tyrosine-based motif, YQRL. Note that the YQRL sequence could not functionally replace LL1 (B), whereas this motif conferred normal localization in the juxtanuclear Golgi region when substituted for LL2 (C) and LL3 (D). Nuclei were counterstained with DAPI (blue). Scale bars = 10 μm. | ||
LL2 is required for retrieval of ATP7A from endosomes to Golgi
Our observations are consistent with LL2 function as a sorting signal necessary for TGN localization of ATP7A. We determined whether LL2 is required for retrograde trafficking of ATP7A from the plasma membrane to the Golgi, by testing whether LL2 is necessary for the ATP7A-dependent internalization of anti-Myc antibodies from the culture medium. Anti-Myc antibodies were added to the culture medium of Myc-tagged wild type ATP7A protein and permitted to accumulate for 4 hours. As expected these antibodies accumulated within a tight perinuclear region indicating that the TGN pool of ATP7A protein cycles via the plasma membrane (Fig. 7A–C). In contrast, anti-Myc antibodies added to the culture medium of the LL3 mutant protein failed to become internalized and remained largely associated with the plasma membrane, confirming the defect in endocytosis for this mutant (Fig. 7G–I). Anti-Myc antibodies added to the media of cells expressing the LL2 mutant failed to accumulate in the tight perinuclear location as seen for wild type ATP7A, and instead were internalized to cytoplasmic endosomes that partially overlapped with the early endosome marker, EEA1 (Fig. 7D–F). Taken together, these data suggest that the LL2 motif is essential for steady state localization of ATP7A in the TGN by retrieving ATP7A from endosomes to the Golgi.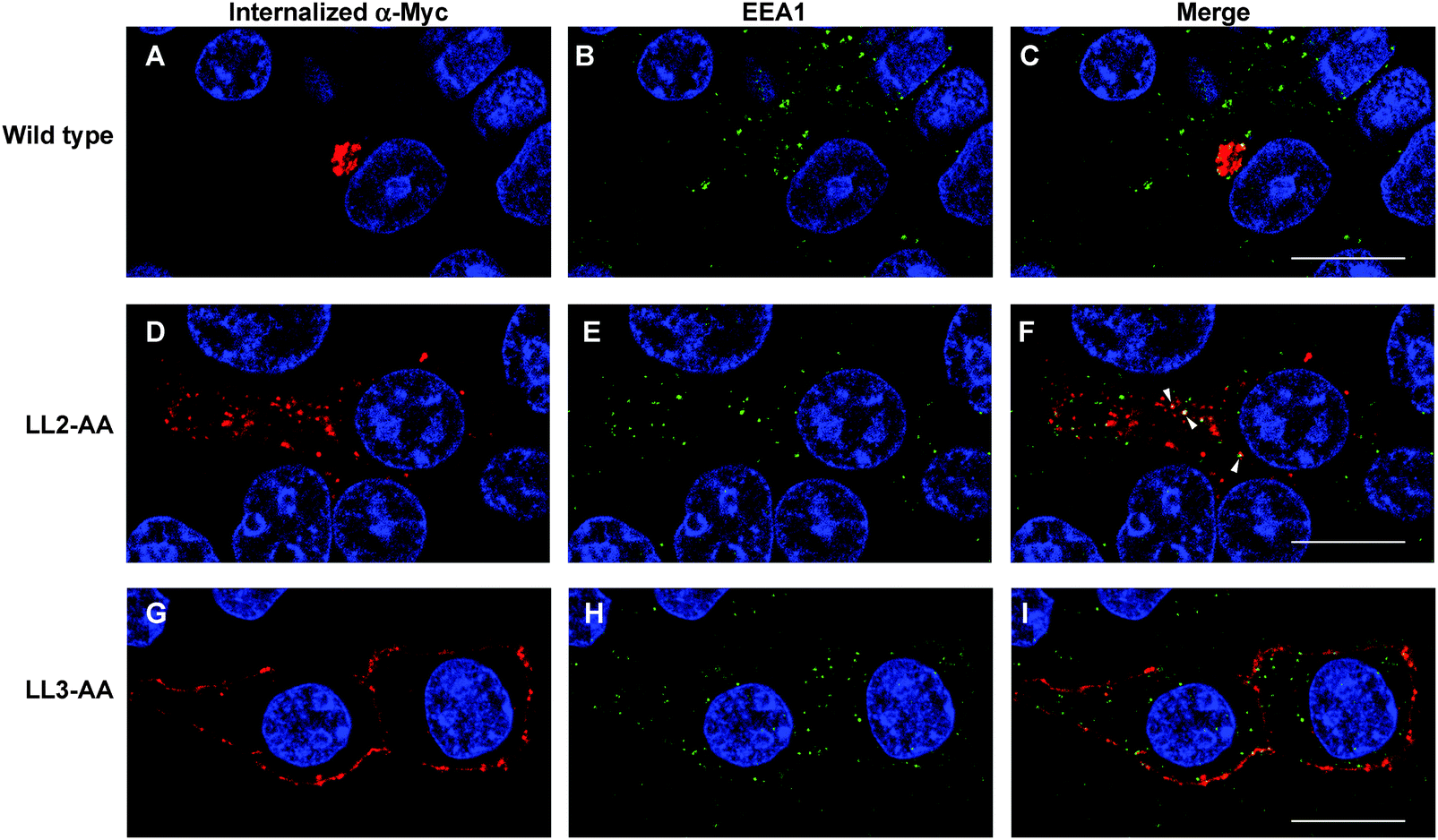 | ||
| Fig. 7 LL2 is required for retrieval of ATP7A from endosomes to Golgi. HEK293T cells stably expressing wild type or ATP7A mutants were incubated in basal medium containing anti-Myc antibodies for 4 hours. The intracellular distribution of anti-Myc antibody was detected using a Leica SP8 MP spectral scanning confocal microscope after labeling fixed cells with anti-mouse antibodies conjugated to Alexa 594 (A, D, G; red). Early endosomes were labeled with rabbit antibodies against EEA1 followed anti-rabbit antibodies conjugated to Alexa 488 (B, E, H; green). Areas of colocalization are indicated in the merged panels (arrowheads). Nuclei were counterstained with DAPI (blue). Scale bars = 10 μm. | ||
Discussion
The results of this study demonstrate that the introduction of silent mutations throughout much of the coding region of the mouse and human ATP7A cDNAs is a simple and effective approach to disrupt regions of instability that lead to the accumulation of mutations in high-copy plasmids within E. coli. Early attempts to stably propagate the ATP7A cDNA in high-copy plasmids required reducing the temperature of the culture medium to 30 °C.21 However, this approach was only marginally successful as many clones were still found to contain mutations, and complete sequencing of multiple clones was required to identify those lacking mutations. Other approaches have utilized very low-copy plasmids that use the pSC101 origin of replication to enable the stable propagation of the ATP7A cDNA15 or other unstable cDNAs,27 however, such plasmids require large culture volumes and lack many desirable features available in high-copy commercial plasmids. In general, the reasons for DNA instability in high-copy plasmids are not well defined, and may be caused by different factors including long repeats, AT-rich sequences, unusual secondary/tertiary structures, or cryptic promoters recognized by the bacterial transcriptional machinery that result in the generation of toxic proteins. Approaches to solve the problem of DNA instability, which typically involve deletional mutagenesis to identify and then remove or alter problematic DNA elements, are both labor-intensive and time consuming.28,29 The silent recoding of an unstable open reading frame described herein is, to our knowledge, the first application of commercial DNA synthesis to solve the problem of cDNA instability, and may be broadly applicable to other similarly unstable cDNAs.The availability of a stabilized Atp7a construct enabled us to clarify whether or not LL3 is the sole di-leucine necessary for TGN localization of ATP7A or whether upstream di-leucines LL1 and LL2 are also required. Previous studies have demonstrated that the carboxy-terminal region of ATP7A is able to autonomously mediate endocytosis of the plasma membrane protein, CD8.9 Interestingly, mutation of LL3, but not LL1 or LL2, was found to disrupt this endocytic function of the carboxy terminal region of ATP7A when tethered to CD8, suggesting that of the three di-leucines, only LL3 is necessary for endocytosis of ATP7A. Our studies expand upon these observations by demonstrating that LL1 and LL2 are necessary for TGN localization of ATP7A, albeit by different mechanisms. Substitution of LL1 with di-valine, but not di-alanine, did not disrupt localization of ATP7A in the TGN, suggesting LL1 is not a bona fide di-leucine targeting motif, but rather that hydrophobicity is important at this location. Consistent with this concept was the finding that the tyrosine-based sorting motif, YQRL, could not functionally substitute for LL1. In contrast, the finding that neither di-alanine nor di-valine could substitute for the loss of LL2 and LL3 in maintaining the steady-state localization of ATP7A in the TGN indicated that hydrophobicity per se is not essential at this location. That the tyrosine-based sorting signal, YQRL, could functionally replace LL2 supports the concept that this di-leucine is a bona fide sorting motif. In contrast to wild type ATP7A which internalized anti-Myc antibodies to the TGN, and the LL3-AA mutant which failed to internalize anti-Myc antibodies, the LL2-AA mutation resulted in the accumulation of anti-Myc antibodies within endosomes. The partial co-localization with the EEA1 protein suggests the LL2 motif is required, in part, for trafficking via early endosomes. The nature of other ATP7A-containing endosomal compartments is the subject of ongoing studies. Taken together, these observations suggest that LL2 is a sorting motif that functions in retrograde trafficking of ATP7A from endosomes to the Golgi.
Consistent with these observations, LL3 conforms to the canonical D/EXXXL[LI] endocytosis motif in which an acidic amino acid lies in the −4 position relative to the first leucine, which facilitates electrostatic interactions with specific basic residues within the adaptor subunits.7,11 It is currently unknown whether the LL2 sequence also interacts with adaptor subunits. The LL2 sequence lacks an acidic residue in the −4 position and thus does not conform to the canonical D/EXXXL[LI] motif, however, studies have shown that an acidic residue is not critical in this location for adaptor protein interactions.5 X-ray crystallography studies suggest that residues in the −4 position of di-leucine sorting signals interact with a positively charged hydrophilic patch within the adaptor subunits.30 It is interesting to note that a serine located upstream of LL2 is known to be phosphorylated based on proteomic analyses,31 thus, it is tempting to speculate that the addition of this negative charge may facilitate LL2 binding to adaptor subunits. Further studies will be required to delineate the mechanism by which LL2 and surrounding residues facilitate ATP7A trafficking.
Conclusions
The identification of a second targeting di-leucine in the ATP7A carboxy-terminal region raises important new questions as to how mutations affecting this region might impact ATP7A-related diseases. To date, no ATP7A mutations have been described in humans or animals that solely affect the LL2 or LL3 di-leucines, thus one can only speculate to what extent mutation of these sequences would affect copper metabolism in vivo. It has recently emerged that mutations affecting general trafficking pathways may give rise to multisystem defects with an underlying disturbance in copper metabolism. This concept is illustrated by the MEDNIK syndrome (acronym for mental retardation, enteropathy, deafness, neuropathy, ichthyosis, keratodermia), which is caused by mutations in the AP-1 adaptor subunit, AP1S1. MEDNIK patients exhibit hypocupremia and hypoceruloplasminemia, which are postulated to arise, in part, from defective AP-1 dependent trafficking of ATP7A or the homologous copper transporter, ATP7B.32 Similarly, Hermansky–Pudlak syndrome is caused by mutations affecting the BLOC-1 complex, which is required for ATP7A trafficking to melanosomes,23 and accordingly, ATP7A trafficking defects are postulated to contribute to hypopigmentation and potentially other systemic defects in Hermansky–Pudlak patients. It will be important for future studies to elucidate whether mutations in BLOC-1, AP1S1 or other trafficking components differentially affect ATP7A sorting via the LL2 and LL3 motifs in specific tissues, which may explain how such mutations give rise to disorders with distinct copper phenotypes. The approaches and findings presented in this study will have far reaching implications for these efforts.Acknowledgements
We thank Ann Hubbard for kindly providing the tyrosinase expression plasmid. We thank all members of our laboratory for their support and helpful comments. This work was supported by funding from the National Institutes of Health (DK093386 and CA190265).References
- M. J. Petris, J. F. B. Mercer, J. G. Culvenor, P. Lockhart, P. A. Gleeson and J. Camakaris, EMBO J., 1996, 15, 6084–6095 CAS.
- Y. Yamaguchi, M. E. Heiny, M. Suzuki and J. D. Gitlin, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14030–14035 CrossRef CAS.
- M. J. Petris, D. Strausak and J. F. B. Mercer, Hum. Mol. Genet., 2000, 9, 2845–2851 CrossRef CAS PubMed.
- M. J. Petris and J. F. Mercer, Hum. Mol. Genet., 1999, 8, 2107–2115 CrossRef CAS PubMed.
- J. S. Bonifacino and L. M. Traub, Annu. Rev. Biochem., 2003, 72, 395–447 CrossRef CAS PubMed.
- L. M. Traub, Nat. Rev. Mol. Cell Biol., 2009, 10, 583–596 CrossRef CAS PubMed.
- R. Mattera, M. Boehm, R. Chaudhuri, Y. Prabhu and J. S. Bonifacino, J. Biol. Chem., 2011, 286, 2022–2030 CrossRef CAS PubMed.
- M. J. Petris, J. Camakaris, M. Greenough, S. LaFontaine and J. F. Mercer, Hum. Mol. Genet., 1998, 7, 2063–2071 CrossRef CAS PubMed.
- M. J. Francis, E. E. Jones, E. R. Levy, R. L. Martin, S. Ponnambalam and A. P. Monaco, J. Cell Sci., 1999, 112(Pt 11), 1721–1732 CAS.
- Z. G. Holloway, A. Velayos-Baeza, G. J. Howell, C. Levecque, S. Ponnambalam, E. Sztul and A. P. Monaco, Mol. Biol. Cell, 2013, 24, 1735–1748 CrossRef CAS PubMed , S1731–1738.
- L. Yi and S. G. Kaler, Hum. Mol. Genet., 2015, 24, 2411–2425 CrossRef CAS PubMed.
- S. G. Kaler, Nat. Rev. Neurol., 2011, 7, 15–29 CrossRef CAS PubMed.
- L. Yi, A. Donsante, M. L. Kennerson, J. F. Mercer, J. Y. Garbern and S. G. Kaler, Hum. Mol. Genet., 2012, 21, 1794–1807 CrossRef CAS PubMed.
- S. L. Dagenais, A. N. Adam, J. W. Innis and T. W. Glover, Am. J. Hum. Genet., 2001, 69, 420–427 CrossRef CAS PubMed.
- S. L. Fontaine, S. D. Firth, P. J. Lockhart, J. A. Paynter and J. F. Mercer, Plasmid, 1998, 39, 245–251 CrossRef CAS PubMed.
- M. J. Petris and J. F. B. Mercer, Hum. Mol. Genet., 1999, 8, 2107–21175 CrossRef CAS PubMed.
- S. Das, B. Levinson, S. Whitney, C. Vulpe, S. Packman and J. Gitschier, Am. J. Hum. Genet., 1994, 55, 883–889 CAS.
- Y. Yamaguchi, M. E. Heiny, M. Suzuki and J. D. Gitlin, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14030–14035 CrossRef CAS.
- Y. Wang, S. Zhu, G. A. Weisman, J. D. Gitlin and M. J. Petris, PLoS One, 2012, 7, e43039 CrossRef CAS PubMed.
- M. J. Petris, D. Strausak and J. F. Mercer, Hum. Mol. Genet., 2000, 9, 2845–2851 CrossRef CAS PubMed.
- M. J. Francis, E. E. Jones, E. R. Levy, S. Ponnambalam, J. Chelly and A. P. Monaco, Hum. Mol. Genet., 1998, 7, 1245–1252 CrossRef CAS PubMed.
- E. G. Minkley and D. Pribnow, J. Mol. Biol., 1973, 77, 255–277 CrossRef CAS PubMed.
- S. R. Setty, D. Tenza, E. V. Sviderskaya, D. C. Bennett, G. Raposo and M. S. Marks, Nature, 2008, 454, 1142–1146 CrossRef CAS PubMed.
- J. Zhan, L. Ge, J. Shen, K. Wang and S. Zheng, Biochem. Biophys. Res. Commun., 2004, 313, 1053–1057 CrossRef CAS PubMed.
- K. Bos, C. Wraight and K. K. Stanley, EMBO J., 1993, 12, 2219–2228 CAS.
- I. Rapoport, Y. C. Chen, P. Cupers, S. E. Shoelson and T. Kirchhausen, EMBO J., 1998, 17, 2148–2155 CrossRef PubMed.
- R. J. Gregory, S. H. Cheng, D. P. Rich, J. Marshall, S. Paul, K. Hehir, L. Ostedgaard, K. W. Klinger, M. J. Welsh and A. E. Smith, Nature, 1990, 347, 382–386 CrossRef CAS PubMed.
- S. Y. Pu, R. H. Wu, C. C. Yang, T. M. Jao, M. H. Tsai, J. C. Wang, H. M. Lin, Y. S. Chao and A. Yueh, J. Virol., 2011, 85, 2927–2941 CrossRef CAS PubMed.
- A. C. Boyd, F. Popp, U. Michaelis, H. Davidson, H. Davidson-Smith, A. Doherty, G. McLachlan, D. J. Porteous and S. Seeber, J. Gene Med., 1999, 1, 312–321 CrossRef CAS PubMed.
- B. T. Kelly, A. J. McCoy, K. Spate, S. E. Miller, P. R. Evans, S. Honing and D. J. Owen, Nature, 2008, 456, 976–979 CrossRef CAS PubMed.
- N. A. Veldhuis, V. A. Valova, A. P. Gaeth, N. Palstra, K. M. Hannan, B. J. Michell, L. E. Kelly, I. Jennings, B. E. Kemp, R. B. Pearson, P. J. Robinson and J. Camakaris, Int. J. Biochem. Cell Biol., 2009, 41, 2403–2412 CrossRef CAS PubMed.
- D. Martinelli, L. Travaglini, C. A. Drouin, I. Ceballos-Picot, T. Rizza, E. Bertini, R. Carrozzo, S. Petrini, P. de Lonlay, M. El Hachem, L. Hubert, A. Montpetit, G. Torre and C. Dionisi-Vici, Brain, 2013, 136, 872–881 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mt00093b |
| ‡ 540d Life Sciences Center, 1201 Rollins St., University of Missouri, Columbia, MO 65211, USA. E-mail: petrism@missouri.edu; Fax: 573-884-2537; Tel: 573-882-9685 |
| This journal is © The Royal Society of Chemistry 2016 |