
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mass spectrometry of B. subtilis CopZ: Cu(I)-binding and interactions with bacillithiol†
Kristine L.
Kay
a,
Chris J.
Hamilton
b and
Nick E.
Le Brun
*a
aCentre for Molecular and Structural Biochemistry, School of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: n.le-brun@uea.ac.uk; Fax: +44 1603 592003; Tel: +44 1603 592699
bSchool of Pharmacy, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK
First published on 13th May 2016
Abstract
CopZ from Bacillus subtilis is a well-studied member of the highly conserved family of Atx1-like copper chaperones. It was previously shown via solution and crystallographic studies to undergo Cu(I)-mediated dimerisation, where the CopZ dimer can bind between one and four Cu(I) ions. However, these studies could not provide information about the changing distribution of species at increasing Cu(I) levels. To address this, electrospray ionisation mass spectrometry using soft ionisation was applied to CopZ under native conditions. Data revealed folded, monomeric CopZ in apo- and Cu(I)-bound forms, along with Cu(I)-bound dimeric forms of CopZ at higher Cu(I) loading. Cu4(CopZ)2 was the major dimeric species at loadings >1 Cu(I)/CopZ, indicating the cooperative formation of the tetranuclear Cu(I)-bound species. As the principal low molecular weight thiol in B. subtilis, bacillithiol (BSH) may play a role in copper homeostasis. Mass spectrometry showed that increasing BSH led to a reduction in Cu(I)-bound dimeric forms, and the formation of S-bacillithiolated apo-CopZ and BSH adducts of Cu(I)-bound forms of CopZ, where BSH likely acts as a Cu(I) ligand. These data, along with the high affinity of BSH for Cu(I), determined here to be β2(BSH) = ∼4 × 1017 M−2, are consistent with a role for BSH alongside CopZ in buffering cellular Cu(I) levels. Here, mass spectrometry provides a high resolution overview of CopZ–Cu(I) speciation that cannot be obtained from less discriminating solution-phase methods, thus illustrating the potential for the wider application of this technique to studies of metal–protein interactions.
Significance to metallomicsThe application of mass spectrometry under native conditions to studies of Cu(I)-binding to the copper chaperone CopZ provides a high resolution overview of the evolving range of Cu(I)-bound CopZ species formed at increasing Cu(I) concentrations, providing information about how the chaperone likely functions in copper-buffering in the cytoplasm. The data also demonstrate the interplay between CopZ, Cu(I) and bacillithiol, the recently discovered low molecular weight thiol of the Bacillus cytoplasm, revealing bacillithiolation of apo-CopZ and likely complex formation with Cu–CopZ, consistent with a joint role in copper metabolism in the cell. |
Introduction
Mass spectrometry (MS) of folded proteins employs soft ionisation (electrospray ionisation, ESI) together with broad mass detection (time of flight, TOF) to study proteins in their native, folded states at, or close to, physiological pH.1–3 Although MS has been successfully applied to a number of metalloproteins, most notably metallothioneins,4,5 the copper chaperone Cox17,6 and the zinc sensor SmtB,7 it has great potential for much wider utility, particularly for understanding metal–protein interactions because, under appropriate conditions, these are preserved in the gas phase. To further investigate the potential of MS for studies of metalloproteins, we sought a protein that has been well characterized by solution spectroscopy and crystallographic methods, so that results from MS could be validated in terms of the nature of species detected. A further requirement was that important questions remain that are not easily answered by traditional spectroscopic and crystallographic techniques. The copper chaperone CopZ from Bacillus subtilis was selected.Copper is essential for a wide range of cellular processes, including respiration and oxidative stress response.8,9 However, the physico-chemical properties that make copper so useful also mean that it is potentially extremely toxic. To counter this, organisms have evolved complex machineries for trafficking copper from the point of uptake into the cell to copper-dependent enzymes.10–12 In the reducing environment of the cell cytoplasm, copper is found in the +1 oxidation state and it is this form that is transported by a network of chaperone proteins. These minimise the risk of deleterious binding or redox processes, thereby controlling copper toxicity, by binding Cu(I) with extremely high affinity (K ∼ 1017–1018 M−1). Movement of Cu(I) along the transport pathway occurs only via highly specific protein–protein interactions that enable facile metal transfer, ensuring that levels of ‘free’ copper in the cell are vanishingly small.13
One of the best-studied and most prevalent copper trafficking pathways involves a copper chaperone and a P-type ATPase transporter,14 which function in transmembrane Cu(I)-transport. In humans, the chaperone Hah1 (also called Atox1) delivers Cu(I) to two P-type ATPase transporters, the Menkes and Wilson proteins,15 while in yeast the chaperone Atx1 delivers Cu(I) to the transporter Ccc2.16 In the Gram-positive bacterium Bacillus subtilis, as in many prokaryotes, the copper efflux system comprises a copper chaperone (CopZ) and a P-type ATPase (CopA).17 The Atx1-like CopZ chaperones are small (∼70 residues) proteins that adopt a characteristic ferredoxin βαββαβ-fold in which the antiparallel β strands form a β-sheet on which the two α-helices are superimposed.18 A MXCXXC Cu(I)-binding motif, in which the two Cys residues provide thiolate coordination to the metal, is situated on a flexible solvent-exposed loop at the beginning of the first α-helix.19 Coordination of the Cu(I) is typically trigonal, with the third ligand provided either by a low molecular weight thiol (LMWT) ligand, or by a second chaperone molecule.20 In the case of B. subtilis CopZ, this copper-dependent dimeric structure has been shown to be capable of accommodating between one and four Cu(I) ions, and a high resolution structure of the latter species, Cu4(CopZ)2, has been reported.21B. subtilis lacking CopZ contains only one third of the copper of wild-type cells indicating that, in addition to a role in copper trafficking, CopZ also functions as a cytoplasmic store/buffer for Cu(I).22 Higher order Cu(I)-bound forms of CopZ may contribute to this role. Although much is known about CopZ, important gaps in our knowledge remain. Chief amongst these is the lack of information about the distribution of CopZ species as Cu(I)-loading increases, which cannot be determined from spectroscopic methods because they are insufficiently discriminating between closely related species.
Another important open question concerns the possible interplay between CopZ and bacillithiol, the recently discovered low molecular weight thiol (LMWT) of B. subtilis, and other low G + C Gram-positive bacteria (Firmicutes).23 BSH is the α-anomeric glycoside of L-cysteinyl-D-glucosamine with L-malic acid and so, like glutathione (GSH) (found in eukaryotes and most Gram negative bacteria) and mycothiol (MSH) (found in the actinomycetes), it is a derivative of cysteine. LMWTs, in addition to functioning in maintaining redox balance, also function to protect Cys residues in proteins from over-oxidation through the formation of a mixed disulfide with the protein Cys thiol.24 This confers a protective effect, by preventing irreversible oxidation to sulfinic or sulfonic acid forms. Formation of mixed disulfides with GSH, a process called S-glutathionylation, not only protects but also serves regulatory functions.25 S-Bacillithiolation has been observed in B. subtilis in response to oxidative stress26 and a similar picture of protective and signalling/regulatory functions is emerging.27
Because of the natural affinity of thiols for several transition metals, it has been proposed that the LMWT pool may also play a role in metal ion trafficking. Recently, it was shown that BSH functions in Zn(II) buffering in B. subtilis,28 and the high affinity with which BSH binds Zn(II) suggests a possible cooperative role between this LMWT and other cellular metal trafficking protein networks. Previous in vitro studies of the influence on Cu(I)-binding to CopZ of LMWT such as dithiothreitol (DTT), GSH and Cys revealed an effect, but the spectroscopic techniques employed did not offer detailed speciation information.10,29
Here, we report MS studies of Cu(I)-binding to B. subtilis CopZ and the influence of LMWTs, including BSH. The data demonstrate copper-mediated dimerisation of CopZ and the formation of all the species predicted from previous spectroscopic and structural studies. Changes in species distribution as Cu(I) load increases revealed the cooperative formation of a Cu4(CopZ)2 species. LMWTs, particularly at high thiol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CopZ ratios, were observed to inhibit the formation of higher order Cu(I)-bound forms of dimeric CopZ, and the presence of BSH led to S-bacillithiolation of apo-CopZ and the formation of CopZ–Cu–BSH adducts. Along with the determined affinity of BSH for Cu(I), the data provide insight into the capability of BSH for buffering cellular Cu(I).
:CopZ ratios, were observed to inhibit the formation of higher order Cu(I)-bound forms of dimeric CopZ, and the presence of BSH led to S-bacillithiolation of apo-CopZ and the formation of CopZ–Cu–BSH adducts. Along with the determined affinity of BSH for Cu(I), the data provide insight into the capability of BSH for buffering cellular Cu(I).
Methods
Purification of CopZ
CopZ was prepared essentially as previously described29 with the following modifications: E. coli JM109/pMKNC6 cultures were grown to an OD600 of 0.4–0.6 prior to the addition of IPTG; DNAse I and RNAse A (Sigma) were added to a final concentration of 6 μg mL−1; to remove contaminating nucleic acids, following anion exchange and prior to gel filtration chromatography, ammonium sulfate was added to a final concentration of 3 M and the protein solution loaded onto a 15 mL Phenyl-Sepharose HIC column (GE Healthcare), previously equilibrated with 50 mL of 100 mM HEPES, 100 mM NaCl, 3 M NH4SO4, 15 mM DTT, pH 7.0. The column was washed with 250 mL binding buffer before applying a 100 mL gradient of 3–0 M ammonium sulfate, with CopZ eluting at approximately 2.4 M ammonium sulfate. CopZ-containing fractions (determined by SDS-PAGE) were buffer exchanged into 100 mM HEPES, 100 mM NaCl, pH 7.0 and concentrated to <5 mL using a spin concentrator (Vivaspin) at 8000 × g and 4 °C. The protein solution was passed through a 0.45 μm filter (Sartorius) and DTT added to a final concentration of 15 mM before applying to the Sephacryl S-100 gel filtration column.Spectroscopic methods
UV-visible absorbance spectra were recorded on a Jasco V-550 spectrophotometer. Near-UV CD spectra were recorded using a Jasco J-810 spectropolarimeter with a slit width of 2 nm. Intensity is expressed as Δε in units of M−1 cm−1. Prior to all Cu(I) addition experiments, CopZ was reduced with excess DTT under anaerobic conditions and buffer exchanged to remove DTT. Protein concentrations were calculated using an extinction coefficient, ε276nm, of 1450 M−1 cm−1.29 Additions of LMWTs were made anaerobically. Additions of Cu(I) to CopZ were made using a microsyringe (Hamilton) to a 1 cm pathlength septum-sealed cell.30Determination of Cu(I)–BSH binding affinity
BSH was synthesized as described previously,31 and dissolved under anaerobic conditions using deoxygenated LC-MS grade water (HiPerSolv, VWR). The affinity of BSH for Cu(I) was determined through competition experiments with bathocuproine disulfonate (BCS), a Cu(I) chelator which forms a 2:1 complex, [Cu(BCS)2]3−, that exhibits an absorption band at 483 nm with ε483nm = 13300 M−1 cm−1.32 2.5 μL additions of 60 mM BSH were made in an anaerobic glove box to a 8.95 μM Cu(BCS)2 solution in 100 mM MOPS, 100 mM NaCl, pH 7.5, containing 2 μM dithionite or ascorbate to scavenge trace oxygen and ensure copper existed solely as reduced Cu(I). After each BSH addition, samples were left to equilibrate for 10 min before spectra were recorded using a septum sealed cuvette. The A483nm values were used to calculate changes in concentration of [Cu(BCS)2]3− (as copper was transferred to Cu(BSH)2), from which the concentrations of Cu(BSH)2, free BSH and free BCS could be determined. The affinity of BSH for Cu(I) was calculated using eqn (1) and (2),33 and assuming that BSH forms a 2:1 complex with Cu(I), as would be expected for a monothiol low molecular weight species.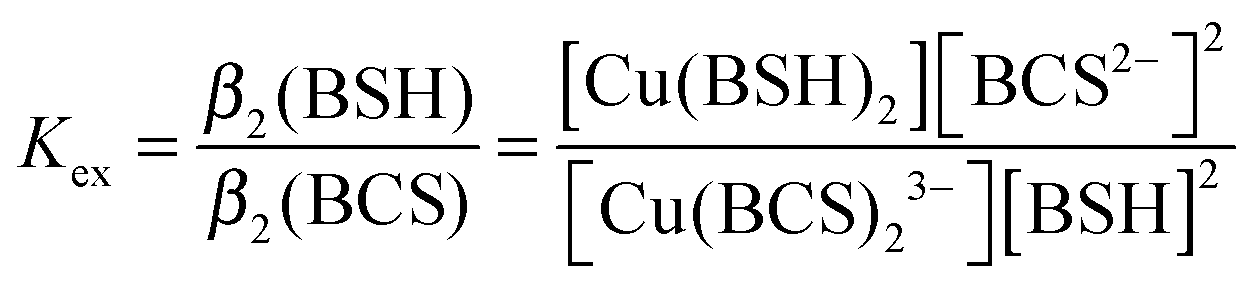 | (1) |
| β2(BSH) = Kexβ2(BCS) | (2) |
β2 = 19.8).32 The acid–base properties of BSH indicate that Cu(I)-binding is likely to be pH-dependent;34 and so the value determined here is an apparent β2 formation constant at pH 7.5.
Electrospray ionisation mass spectrometry
Mass spectrometry samples were prepared by first adding 15 mM DTT (Formedium) and removing reductant by passage down a G25 Sephadex column (PD10, GE Healthcare) in an anaerobic glovebox (Faircrest Engineering, O2 concentration <2 ppm) using 20 mM ammonium acetate, pH 7.4 (Sigma) as the elution buffer. The protein sample was diluted with 20 mM ammonium acetate to a working sample concentration of 15 μM. To prepare Cu(I)-bound CopZ samples, a deoxygenated solution of Cu(I)Cl prepared in 100 mM HCl and 1 M NaCl was added to anaerobic, reduced CopZ using a microsyringe (Hamilton) in an anaerobic glovebox. Unbound Cu(I) was removed by passage of the sample down a G25 Sephadex column (PD10, GE Healthcare) equilibrated with 20 mM ammonium acetate, pH 7.4. Thiol experiments were carried out using GSH (Sigma), DTT (Formedium) or BSH. Protein solutions were prepared using aliquots of 1.0 Cu(I) per CopZ, prepared as above, where thiol solution was added to yield ratios of 5, 10, or 25 thiol per protein.Mass spectra were acquired using a Bruker micrOTOF-QIII electrospray ionisation (ESI) time-of-flight (TOF) mass spectrometer (Bruker Daltonics, Coventry, UK), in positive ion mode. The ESI-TOF was calibrated in the m/z range 300–2200 using ESI-L Low Concentration Tuning Mix (Agilent Technologies, San Diego, CA). Native protein samples were introduced to the ESI source via a syringe pump (Cole-Parmer) at 5 μL min−1, and data acquired for 3 min, with ion scans between 50–3000 m/z. MS acquisition was controlled using Bruker oTOF Control software, with parameters as follows: dry gas flow 4 L min−1, nebuliser gas pressure 0.4 bar, dry gas 130 °C, capillary voltage 4500 V, offset 500 V, isCID energy 35 eV, collision RF 2200 Vpp, collision cell energy 10 eV. LC-MS experiments were performed using an UltiMate 3000 HPLC system (Dionex, Sunnyvale, CA, USA). A 1 μL injection volume of protein in 2% acetonitrile was applied to a ProSwift® reversed phase RP-1S column (4.6 × 50 mm; Dionex) at 25 °C. Gradient elution was performed at a flow rate of 200 μL min−1 using solvents A (0.1% formic acid) and B (acetonitrile, 0.1% formic acid), with the following chromatographic method: isocratic wash (2% B, 0–2 min), linear gradient from 2–100% B (2–12 min), followed by an isocratic wash (100% B, 12–14 min) and column re-equilibration (2% B, 14–15 min). MS acquisition parameters were as follows: dry gas flow 8 L min−1, nebuliser gas pressure 0.8 bar, dry gas 240 °C, capillary voltage 4500 V, offset 500 V, collision RF 650 Vpp. Processing and analysis of MS experimental data was carried out using Compass DataAnalysis version 4.1 (Bruker Daltonik, Bremen, Germany). Neutral mass spectra were generated using the ESI Compass version 1.3 Maximum Entropy deconvolution algorithm over a mass range of 7000–16000 Da. Overlapping peak envelopes were deconvoluted manually. 3-point Gaussian smoothing was applied to mass spectra of samples containing BSH and GSH. Exact masses are reported from peak centroids representing the isotope average neutral mass. Predicted masses are given as the isotope average of the neutral protein or protein complex, in which Cu(I)-binding is expected to be charge compensated.
Results
Detection of apo- and Cu(I)-bound forms of B. subtilis CopZ by MS
Mass spectra of both apo- and Cu-bound CopZ, before and after deconvolution, recorded under denaturing and non-denaturing (native) conditions are shown in Fig. 1. Before deconvolution, the mass spectra show an envelope of multiply charged molecular ions with m/z values corresponding to [M + x(Cu) + (n − x)H]n+/n, where M is the molecular mass of the protein and x the number of coordinated Cu+ ions, which offsets the number of protons required to achieve the observed charge state (n+). Under denaturing conditions, apo-CopZ and 1 Cu/CopZ samples both gave rise to a charge envelope containing six peaks with charges +5, +6, +7, +8, +9 (Fig. 1A and B). Deconvolution of the apo-CopZ and 1 Cu/CopZ spectra resulted in peaks with mass 7338.1 Da and 7337.0 Da, respectively, in good agreement with the predicted mass of CopZ (7338.1 Da).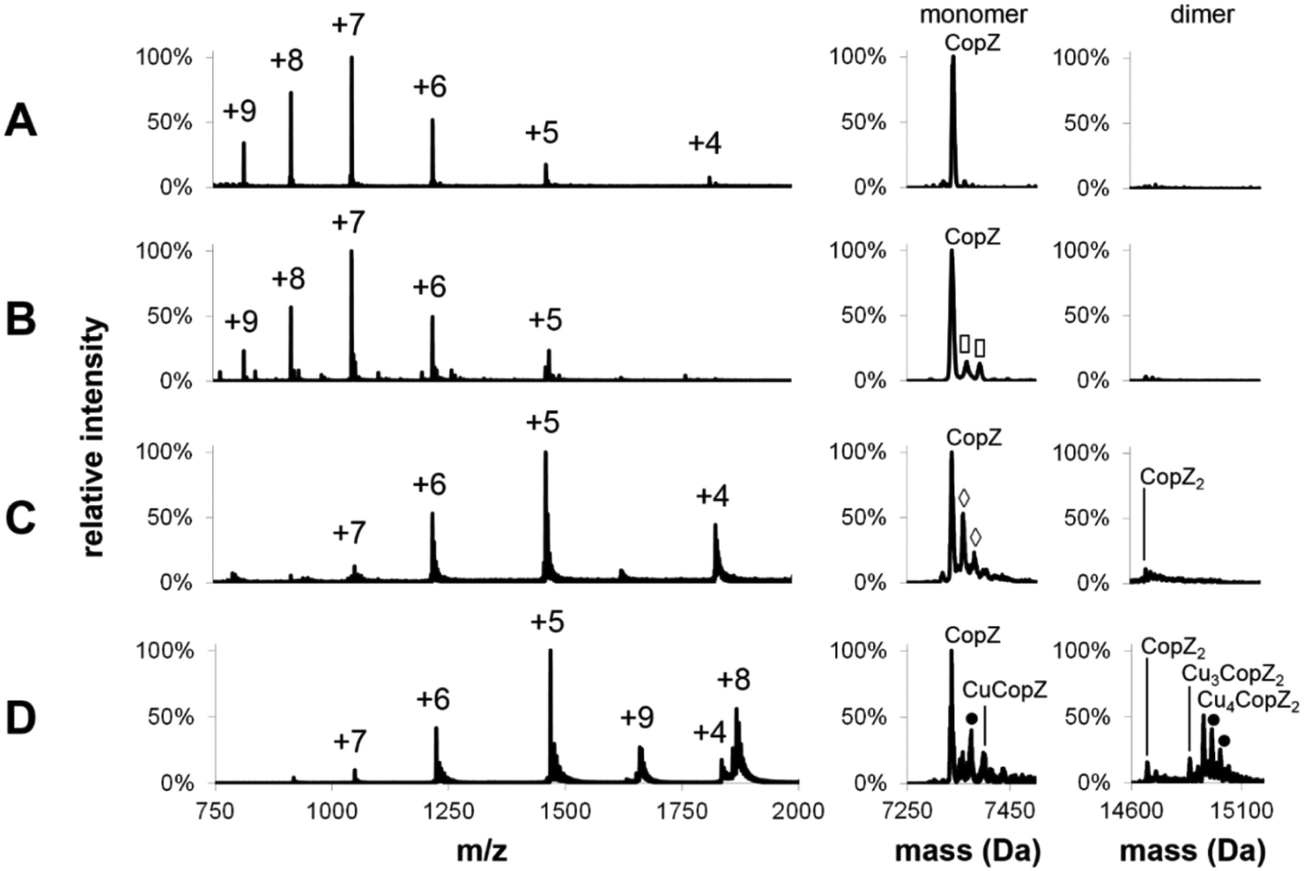 | ||
Fig. 1 ESI-MS charge state analysis of CopZ. Mass spectra acquired for (A) apo-CopZ and (B) 1.0 Cu/CopZ by LC-MS. (C and D) apo-CopZ and 1.0 Cu/CopZ by MS of CopZ under native conditions. Mass spectra prior to and after deconvolution are shown on the left (with the charge state of each peak labelled) and right, respectively. The monomer and dimer regions of the corresponding mass spectra are shown on the right. Protein species are labelled and symbols indicate adducts as follows: (◇) = Na adduct, (![[rectangle open, vertical]](https://www.rsc.org/images/entities/char_25af.gif) ) = acetonitrile adduct, (●) = K adduct. LC-MS experiments carried out in acetonitrile/H2O, 0.1% formic acid; MS experiments under native conditions carried out in 20 mM ammonium acetate, pH 7.4. ) = acetonitrile adduct, (●) = K adduct. LC-MS experiments carried out in acetonitrile/H2O, 0.1% formic acid; MS experiments under native conditions carried out in 20 mM ammonium acetate, pH 7.4. | ||
Under native conditions, apo-CopZ (Fig. 1C) gave rise to a charge envelope containing peaks of charge +4, +5 and +6 which, compared to the denatured protein spectra, displays fewer major peaks, with the peak envelope shifted toward increased m/z values. These changes reflect differences under native conditions, where the protein is folded. Fewer charges are a result of the decreased surface area of folded protein, and fewer major peaks result from a more tightly folded structure.35 Deconvolution of the apo-CopZ peaks gave a molecular mass of 7336.5 Da, 1.6 Da lower than the predicted neutral mass of CopZ (7338.1 Da). The spectrum of Cu(I)-bound CopZ (containing 1.0 Cu/protein), Fig. 1D, contained an additional peak envelope with charges +8 and +9, which indicates the presence of a distinct species. Deconvolution of the spectrum gave molecular masses of 7335.9 Da (2.2 Da lower than the predicted mass of CopZ) and 14926.6 Da, which agrees well with the predicted mass of Cu4(CopZ)2 (14926.4 Da). Also, upon binding of Cu(I), no change in charge state distribution was observed (Fig. 1C and D), indicating that CopZ is folded before and after copper binding occurs. The mass spectra of CopZ under native conditions (Fig. 1C and D) contain sodium adduct peaks of low intensity which, compared to the mass spectra generated under denaturing conditions (Fig. 1A and B), illustrates one of the issues arising when directly infusing protein samples in aqueous buffer. Nevertheless, speciation of the protein can be clearly determined.
Cooperative formation of Cu4(CopZ)2 detected by MS
To investigate the Cu(I)-dependent speciation of CopZ and, in particular, the formation of dimeric CopZ, MS experiments with CopZ under native conditions were performed at 0.5, 1.0, 1.5 and 2.0 Cu(I) per protein. Deconvoluted mass spectra (monomer and dimer regions) and bar graphs representing the distribution of CopZ species present are shown in Fig. 2; mass spectra before deconvolution are shown in Fig. S1 (ESI†). The addition of 0.5 equivalents of Cu(I) per CopZ (Fig. 2A) generated a mixture of metalloforms, with the mass spectra displaying the typical monomer peak envelope, consisting of three major peaks with charges +4, +5 and +6, and also a small amount of dimer peak envelope with charges +8 and +9. Deconvolution revealed the predominant species to have a mass of 7401.1 Da, close to the predicted neutral mass of CuCopZ (7400.6 Da). Also present were peaks due to apo-CopZ, and trace amounts of CopZ dimer with 1–4 Cu(I) ions bound.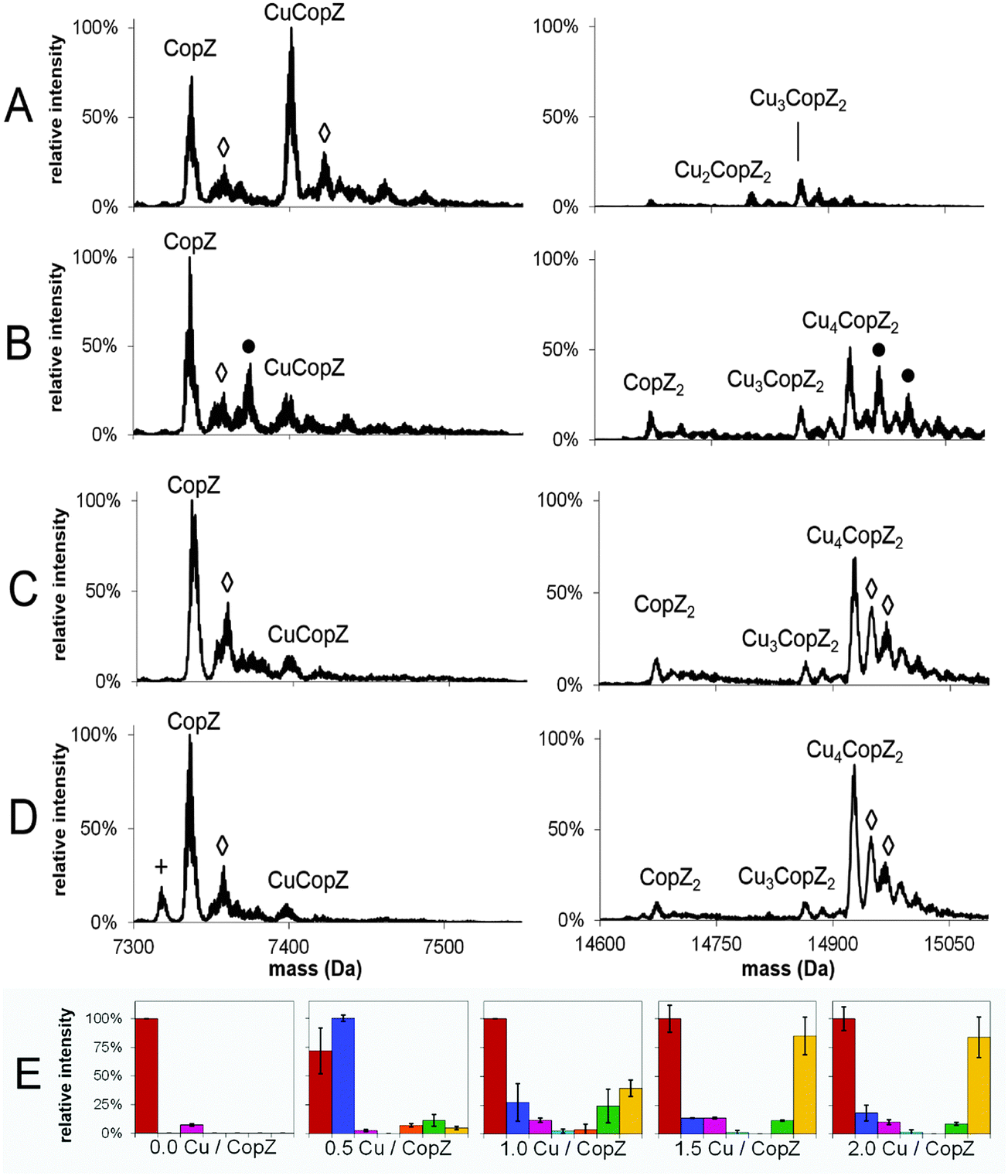 | ||
| Fig. 2 MS distribution of Cu(I)–CopZ species with increasing Cu(I). ESI-MS mass spectra of CopZ in 20 mM ammonium acetate, pH 7.4, reconstituted with (A) 0.5 Cu/CopZ, (B) 1.0 Cu/CopZ, (C) 1.5 Cu/CopZ and (D) 2.0 Cu/CopZ. (E) Bar graphs representing relative intensities of CopZ reconstituted with 0, 0.5, 1.0, 1.5 and 2.0 Cu/CopZ, as indicated, detected by MS. In (A–D) protein adduct species are labelled with symbols as follows: (◇) = Na adduct; (+) = loss of H2O; (●) = K adduct. In (E), protein species are: apo-CopZ (red); CuCopZ (blue); CopZ2 (magenta); Cu(CopZ)2 (cyan); Cu2(CopZ)2 (orange); Cu3(CopZ)2 (green); Cu4(CopZ)2 (yellow). Error bars represent standard deviation for at least three independent measurements. | ||
The addition of 1.0 equivalents of Cu(I) per CopZ (Fig. 2B and E) also generated a mixture of metalloforms, with a substantial monomer charge envelope present but also a significant increase in peaks due to the dimer form. Upon deconvolution, the mass spectra contained a peak due to apo-CopZ (of highest intensity), with the predominant copper-bound species being Cu4(CopZ)2. Also present was a peak at a mass of 14864.2 Da, in agreement with the predicted mass of Cu3(CopZ)2 (14863.7 Da), and small peaks corresponding to Cu(CopZ)2 and Cu2(CopZ)2.
The progression toward increased dimer and decreased monomer peak intensities continued in the sample containing 1.5 equivalents of Cu(I) per CopZ (Fig. 2C and E). Deconvolution revealed predominant species at masses of 7336.2 Da and 14927.1 Da, corresponding to apo-CopZ and Cu4(CopZ)2, respectively. Minor amounts of CuCopZ, Cu(CopZ)2 and Cu3(CopZ)2 were also observed. The sample containing 2.0 equivalents of Cu(I) per CopZ (Fig. 2D) displayed roughly equal proportions of monomer and dimer peak envelopes. Deconvolution revealed the predominant species to be apo-CopZ (7336.0 Da) and the major copper-bound species to be Cu4(CopZ)2, at 14927.2 Da with a small amount of Cu3(CopZ)2 and CuCopZ also present.
These data illustrate that, with increased copper loading, the relative amount of dimerised, metallated CopZ species increased. Above 1.0 Cu/CopZ, the relative amount of monomer species was significantly reduced, while the subsequent predominance of the tetra-copper species indicates cooperative binding of Cu(I) to the CopZ dimer, resulting in relatively low amounts of the Cu2(CopZ)2 and Cu3(CopZ)2 species.
Bacillithiol inhibits higher order Cu(I)-forms of dimeric CopZ
Previous spectroscopic studies of the effects of the LMWTs DTT, cysteine and GSH revealed that, though initial Cu(I)-binding behaviour is not perturbed, the formation of higher order Cu(I)-bound forms of dimeric CopZ is inhibited.10,29 These studies pre-dated the discovery of BSH as the major LMWT in the B. subtilis cytoplasm23 and so it was of interest to investigate the effects of BSH on Cu(I)-binding to CopZ.The first aim here was to determine the affinity of BSH for Cu(I), so that results with CopZ could be readily interpreted. Affinities associated with Cu(I)-binding to thiolate ligands can be obtained through competition experiments with a high affinity Cu(I) ligand, such as BCS. Previously, we have used this Cu(I) chelator to determine the binding affinities of Cu(I) for both CopZ and the soluble domains of its cognate membrane protein, CopAab.33,36 The spectra recorded during titration of BSH into the solution Cu(BCS)2 illustrated a steady decrease in A483nm as shown in Fig. 3, indicating the transfer of copper from BCS to BSH. By analogy with other LMWTs, it is expected that BSH binds Cu(I) in a 2:1 ratio to form Cu(BSH)2. Data were analysed according to eqn (1) and (2), giving an apparent formation constant at pH 7.5 of β2(BSH) = ∼4 × 1017 M−2 (Table 1).
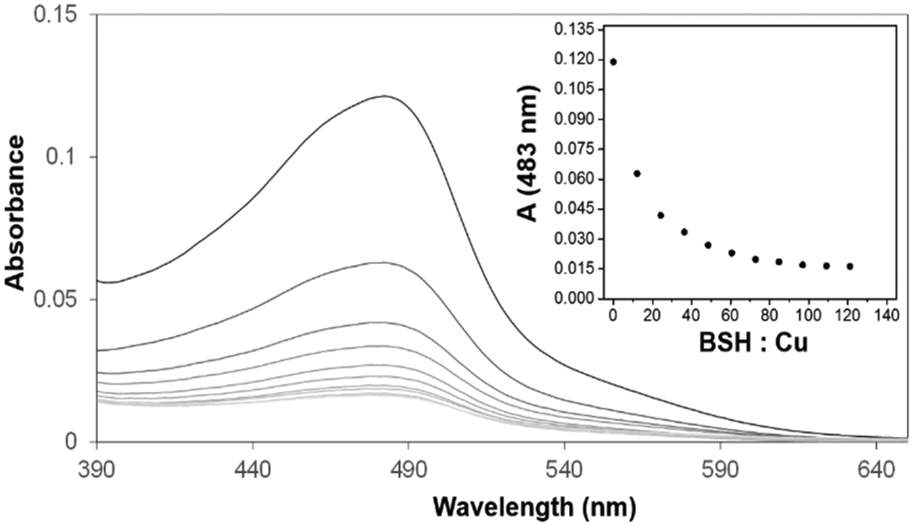 | ||
| Fig. 3 Affinity of BSH for Cu(I) determined through competition with BCS. Absorbance spectra of solution containing 8.95 μM Cu(BCS)2, 2 μM dithionite, following sequential additions of BSH. Inset is a plot of A483nm (measuring concentration of Cu(BCS)2) against the stoichiometric ratio of BSH:Cu(I). Similar data (not shown) were obtained with ascorbate in place of dithionite. | ||
| [BSH]t (μM) | [Cu]t (μM) | BSH/Cu(I) | [BCS]t (μM) | [Cu(BCS)2]3− (μM) | [Cu(BSH)2] (μM) | β 2 BSH (×1017) | Average β2 BSH (×1017) |
|---|---|---|---|---|---|---|---|
| 240 | 9.88 | 24 | 23.7 | 3.14 | 6.74 | 8.03 | 4.1 ± 1.5 |
| 359 | 9.86 | 36 | 23.7 | 2.52 | 7.34 | 5.40 | |
| 477 | 9.84 | 48 | 23.6 | 2.02 | 7.82 | 4.38 | |
| 595 | 9.82 | 61 | 23.6 | 1.73 | 8.09 | 3.57 | |
| 713 | 9.80 | 73 | 23.5 | 1.49 | 8.31 | 3.07 | |
| 830 | 9.78 | 85 | 23.5 | 1.40 | 8.38 | 2.44 | |
| 947 | 9.76 | 97 | 23.4 | 1.28 | 8.49 | 2.12 |
Methods previously used to study the effects of LMWTs on Cu(I)-binding to CopZ were employed for BSH. UV-visible spectra arising from additions of Cu(I) to CopZ samples containing a 20-fold excess BSH in the range of 0–2 Cu(I)/protein are shown in Fig. S2A, while Fig. S2B (ESI†) shows a plot of ΔA265nm against the increasing Cu(I)/protein ratio. Addition of Cu(I) ions led to a gradual increase in absorbance between 260–360 nm, and absorbance changes at 265 nm increased essentially linearly with respect to Cu(I). Cu(I)-binding in the absence of LMWTs has been well-characterised, consisting of a series of distinct binding phases with break points at ∼0.5, 1.0 and 1.5 Cu(I) per protein.29 Clearly, here, multiphasic Cu(I) binding to CopZ was not observed in the presence of excess BSH. However, because Cu(I) bound to LMWT also gives rise to absorbance in this spectral range, absorbance does not discriminate between protein-associated and non-protein-associated copper. CD spectroscopy, which provides more discriminating information about Cu(I)-binding to the protein was therefore used.
CD spectra of CopZ in the presence of 20-fold excess BSH resulting from anaerobic additions of Cu(I) in the range of 0–2 Cu(I)/protein are shown in Fig. 4A and B shows plots of ΔΔε values at 265 nm, 290 nm and 335 nm as a function of Cu(I) added. The initial Cu-free spectrum of CopZ with BSH is consistent with the previously reported spectrum of apo-CopZ with only a very shallow signal around 276 nm, due to its two tyrosine residues. Thus, CD intensity changes are attributed to Cu(I) ions bound at chiral centres within the protein. Initially, additions of Cu(I) gave rise to signal intensity changes at (+)265 nm and (−)295 nm, due to ligand–metal charge transfer bands. A plot of intensity versus Cu(I) shows binding in the range from 0–0.7 Cu/CopZ, after which there are only minor changes in intensity at (−)265 nm and (+)295 nm, in each case levelling off at ∼2 Cu/CopZ. A minor band at (−)335 nm, was observed to form above 0.7 Cu/CopZ. Intensity in this region was previously attributed to spin forbidden 3d–4s metal cluster-centred transitions arising due to Cu(I)–Cu(I) interactions in multinuclear Cu(I) complexes.37,38 This behaviour is broadly similar to that previously observed for Cu(I)-binding to CopZ in the presence of the same excess of GSH and cysteine.10 In the absence of LMWTs, initial binding results in the formation of the Cu(CopZ)2 species up to 0.5 Cu(I) per CopZ, to which further Cu(I) ions can bind. The presence of an excess of LMWT leads to a change in behaviour, with the first Cu(I)-binding phase continuing until ∼0.7 Cu/CopZ. This likely reflects a competition between formation of Cu(CopZ)2 and a Cu–CopZ–LMWT heterocomplex (as observed by NMR for CopZ in the presence of DTT where the third sulfur ligand to the Cu(I) was from the exogenous ligand).20
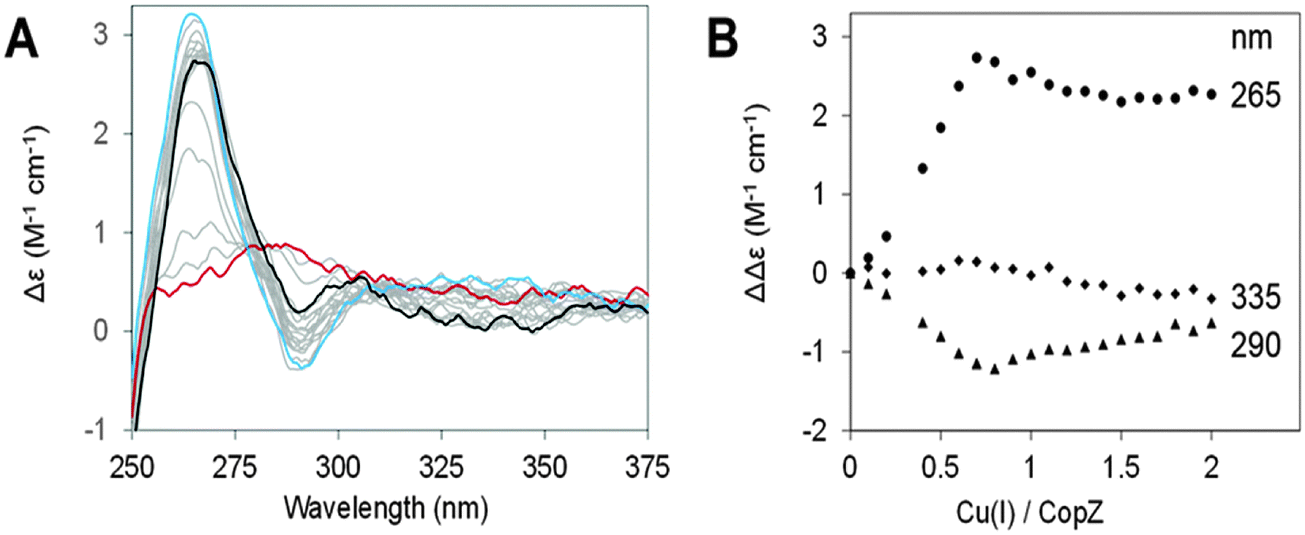 | ||
| Fig. 4 Cu(I) titration of CopZ in presence of BSH. Changes resulting from additions of Cu(I) to 35 μM CopZ in 100 mM MOPS, 100 mM NaCl, pH 7.5, and 20-fold excess BSH were monitored by circular dichroism. Spectra are shown in (A), starting with apo-CopZ (red line) and finishing at 2.0 Cu/CopZ (black line). A major inflection point was observed at 0.7 Cu/CopZ (blue line). Spectra due to intervening ratios are shown in grey. (B) Shows CD changes plotted against Cu(I) per CopZ at 265 nm (circles), 290 nm (triangles) and 335 nm (squares). | ||
MS affords the opportunity to gain high resolution insight into the effects of LMWTs on Cu(I)-binding to CopZ under native conditions. The mass spectra of CopZ containing 1 Cu/protein and progressively increasing concentrations of the three LMWTs DTT, GSH and BSH are shown in Fig. 5 and Fig. S3–S5 (ESI†), and bar graphs presenting the relative intensities of the various CopZ species present in each sample are shown in Fig. 6.
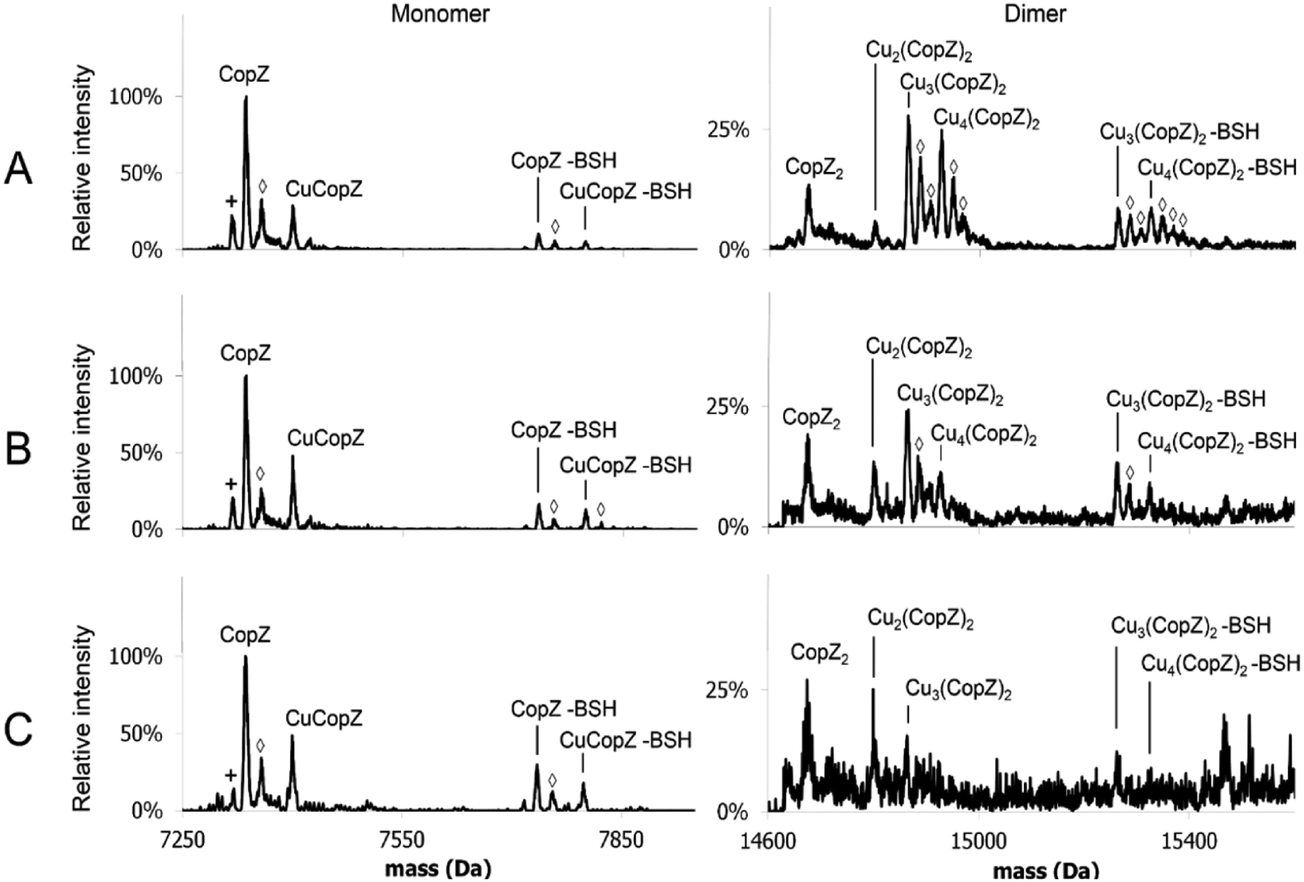 | ||
| Fig. 5 MS of 1 Cu/CopZ with increasing BSH. ESI-MS of CopZ in 20 mM ammonium acetate, pH 7.4, reconstituted with 1 Cu(I)/protein, in the presence of (A) 5 BSH/CopZ, (B) 10 BSH/CopZ and (C) 25 BSH/CopZ. Monomer and dimer regions of the deconvoluted mass spectra are shown (as indicated), with protein species labelled. Symbols indicate the following: (◇) = Na adduct, (+) = loss of H2O. | ||
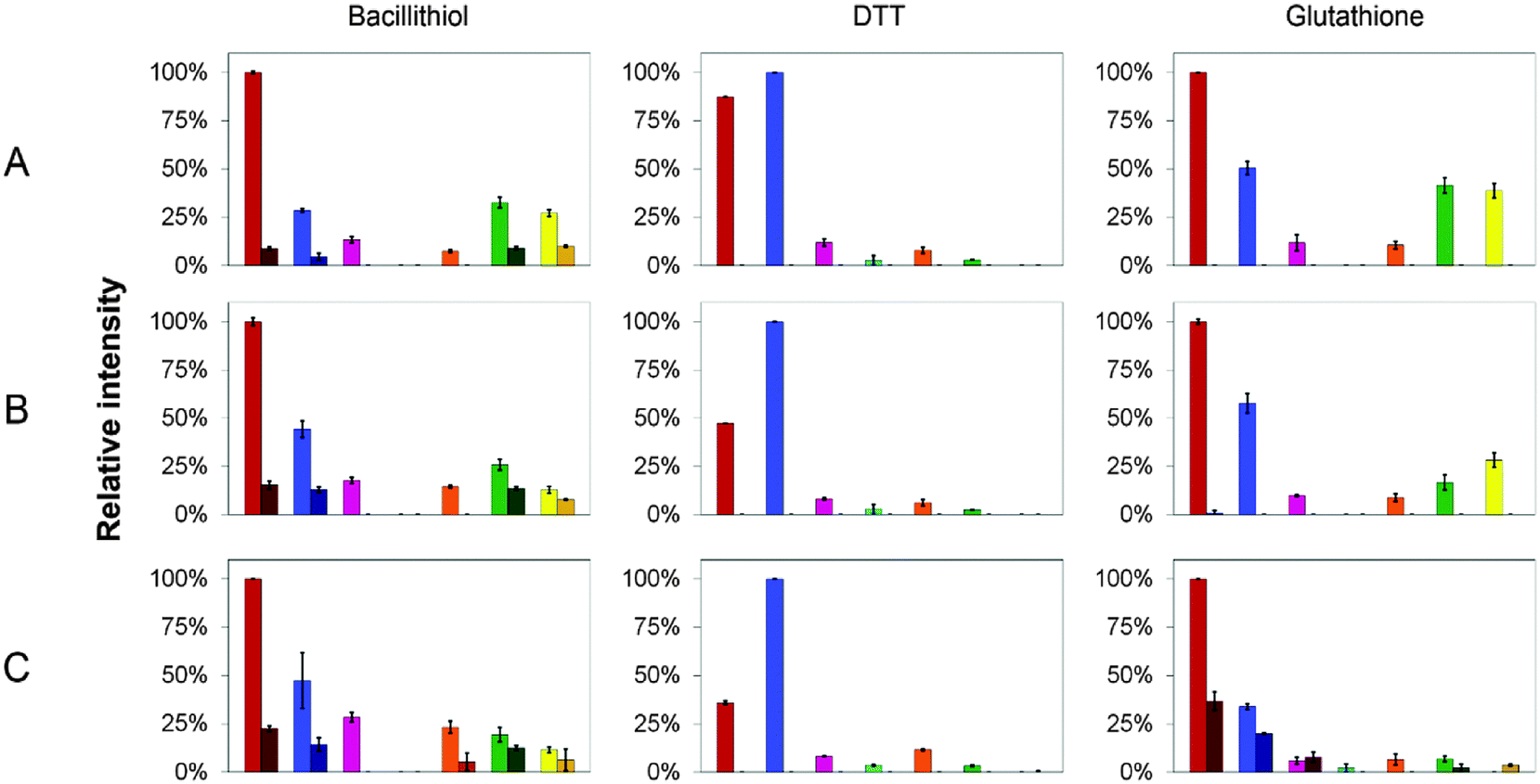 | ||
| Fig. 6 CopZ speciation in the presence of DTT, GSH and BSH. Relative proportions of CopZ species observed in ESI-MS experiments using CopZ reconstituted with 1 Cu(I)/protein in the presence of (A) 5 thiol/CopZ, (B) 10 thiol/CopZ and (C) 25 thiol/CopZ. The bar graphs represent the relative proportion of CopZ species observed in each sample, normalised to the greatest total ionisation intensity. Protein species are: apo-CopZ (red); CuCopZ (blue); CopZ2 (magenta); Cu(CopZ)2 (cyan); Cu2(CopZ)2 (orange); Cu3(CopZ)2 (green); Cu4(CopZ)2 (yellow). The darker portion represents the proportion of species containing a thiol adduct. Error bars represent standard deviation for at least three independent measurements. | ||
In the DTT experiment (Fig. 6 and Fig. S3, ESI†) mass spectra after deconvolution showed that at 5 DTT/protein (Fig. S3B, ESI†) the peaks corresponding to dimer CopZ species Cu3(CopZ)2 (14864.7 Da) and Cu4(CopZ)2 (14925.0 Da) were significantly decreased in intensity compared to those in the absence of the thiol. There was a corresponding increase in the CuCopZ peak (7400.4 Da), which is the most prominent copper-bound species, with apo-CopZ (7336.6 Da) next most abundant (89% relative intensity). At 10 DTT/protein and 25 DTT/protein (Fig. S3C and D, ESI†), the intensity of dimeric CopZ species were further reduced, and the most prominent peak was that of CuCopZ (7400.8 Da). Interestingly, the relative intensity of the apo-CopZ monomer (7337.0 Da) decreased to 55% and 37% at 10 and 25 DTT/protein, respectively. Such behaviour would not be expected if DTT was simply outcompeting CopZ for Cu(I), suggesting instead that the presence of DTT has a stabilising effect on the CuCopZ species. This could be through formation of a hetero-complex (e.g. DTT–Cu–CopZ)20 that dissociates to Cu–CopZ in the MS, or through an indirect effect, for example, a reduced ionisation efficiency of apo-CopZ.
In the case of both GSH (Fig. S4, ESI†) and BSH (Fig. 5 and Fig. S5, ESI†), behaviour was similar in that dimeric forms of CopZ, Cu3(CopZ)2 (14864.6 Da) and Cu4(CopZ)2 (14926.7 Da), decreased with increasing thiol, but the effect was not as marked as for DTT, with significant amounts of dimer remaining at 10 and even 25 thiol/CopZ, particularly in the case of BSH (Fig. S2D and S5D, ESI†).
Glutathionylation, bacillithiolation and CopZ/thiol hetero-complex formation
Experiments conducted in the presence of GSH and BSH resulted in the observation of thiol adducts of CopZ, whereas in the presence of DTT no such adducts were observed. For apo-CopZ species, these adducts correspond to glutathionylation (+306.3 Da) or bacillithiolation (+397.3 Da), whereby a mixed disulfide forms between a Cys residue of CopZ and the LMWT. For Cu(I)-bound forms of CopZ, the adducts are unlikely to be mixed disulfides, as the Cys side chains are involved in coordination of the metal. Thus, these are most likely hetero-complexes of CuCopZ and BSH in which the LMWT is coordinated to Cu(I). The extent to which these species were observed was different for GSH and BSH, with significantly more observed with the physiologically relevant BSH. Furthermore, the extent of complex formation for each CopZ species increased with the concentration of thiol, see Fig. 6 and Table 2.| Protein speciesa | Proportion of BSH adduct in native MS experiment | ||
|---|---|---|---|
| 5 BSH/CopZ | 10 BSH/CopZ | 25 BSH/CopZ | |
|
a CopZ was reconstituted with 1 Cu(I)/protein, in the presence of three different ratios of BSH:CopZ.
b Values represent the proportion of each species' BSH adduct relative to the total observed intensity of that particular species (see Fig. 5).
|
|||
| CopZ–BSH | 8% ± 1%b | 13% ± 1% | 18% ± 1% |
| CuCopZ–BSH | 14% ± 3% | 23% ± 1% | 24% ± 7% |
| Cu3(CopZ)2–BSH | 22% ± 2% | 35% ± 1% | 39% ± 3% |
| Cu4(CopZ)2–BSH | 27% ± 1% | 38% ± 4% | 35% ± 21% |
Discussion
Cu(I)-binding to B. subtilis CopZ has been shown previously to be complex. Initial binding of Cu(I) results in dimerisation of CopZ to form a Cu(CopZ)2 species, which has the capacity to bind three further Cu(I) ions at the monomer–monomer interface.29 While spectroscopic studies have revealed the different phases of Cu(I)-binding, and X-ray crystallography has provided a high resolution structure of the Cu4(CopZ)2 species,21 none of these techniques provided detailed information about the nature of the range of species present under different conditions of copper loading. Here we have shown that MS can provide detailed, high resolution information about the distribution of CopZ and its metallo-forms that exist at different stoichiometric ratios of Cu(I) in solution. Importantly, all of the species detected by MS have been either shown or predicted to exist, based on spectroscopic and structural data.21,29 While it cannot be assumed that all species ionise with the same efficiency, and so distribution of species within a single spectrum may not mirror the solution situation, it is the case that changes in MS intensities as the sample composition changes generally reflect changes occurring in solution.No Cu(I)-bound CopZ species were observed when a Cu(I)-loaded CopZ sample was subjected to the denaturing conditions of LC-MS. Here, both the increased number of charge states and greater average charge state reflect the unfolding of the protein in acidified solvent, disrupting the Cu-binding site. Under native conditions, apo-CopZ was, or close to, the most abundant species, even in copper-loaded samples. For a loading of 0.5 Cu/CopZ, this can be understood because solution studies previously showed that CuCopZ recruits a second CopZ molecule to form the dimeric species Cu(CopZ)2.29 However, the association constant for the binding of the second CopZ is not high (estimated to be ∼105 M−1)33 and so it is not surprising that the Cu(CopZ)2 species was observed only at low intensity in the mass spectrum, with apo-CopZ and CuCopZ the major species, resulting from dissociation of the Cu(CopZ)2 species. At higher levels of Cu(I), the appearance of apo-CopZ may in part be a consequence of the cooperative formation of the Cu4(CopZ)2 species. However, even at a loading of 2 Cu/CopZ, which should saturate the formation of the Cu4(CopZ)2 species, significant amounts of apo-CopZ were observed. Although unexpected because electrostatic interactions are normally strengthened in the gas phase, the data appear to suggest that the Cu(I)–CopZ interaction does not efficiently survive the ionisation process. While masses observed for apo-CopZ under denaturing conditions were as predicted, those observed under native conditions were consistently lower than the predicted mass of the neutral protein by 1–2 Da. The most obvious explanation for this is that a significant proportion of the protein undergoes oxidation in the MS experiment to an intramolecular disulfide-bonded form, as observed for Cys-containing peptides.39
Though it has been demonstrated that apo-CopZ exhibits reduced thermal and chemical stability,40 no change in ESI-MS charge state distribution was observed between apo-CopZ and Cu-loaded CopZ, which indicates that no major protein conformational change occurs upon addition of copper. The reduced charge state distribution compared to denatured CopZ further indicates that both apo-CopZ and Cu(I)-bound CopZ remain folded upon ionisation. Addition of higher amounts of Cu led to the emergence of a dimer peak envelope with charges +8, +9, resulting in higher m/z values than the monomer or denatured protein peak envelopes. This change in charge state behaviour is consistent with the well characterised relationship between protein association state and charge state.41
While at low Cu(I) loadings (0.5 Cu/CopZ) the major Cu(I)-bound species detected in the mass spectrum was CuCopZ, at higher levels of Cu(I) (≥1 Cu/CopZ), several dimeric forms of CopZ were readily detected. This is consistent with previous solution studies which showed that binding of ≥1 Cu(I) at the CopZ monomer–monomer interface stabilised the complex significantly.29 Cu2(CopZ)2, Cu3(CopZ)2 and Cu4(CopZ)2 species were all detected, with Cu3(CopZ)2 marginally the most abundant of these dimeric species at 1 Cu/CopZ. At Cu(I)-loadings above 1 per CopZ, the major Cu(I)-bound form of CopZ was Cu4(CopZ)2, at the expense of all the other dimeric forms. This demonstrates a cooperativity of Cu(I)-binding to the dimeric form of CopZ, indicating that the arrangement of four Cu(I) ions in a cluster at the monomer interface is a particularly thermodynamically stable form. This is consistent with previous structural studies, in which the structure of Cu4(CopZ)2 was solved, but from crystallisation solutions containing a 1:1 ratio of Cu:CopZ (i.e., the Cu2(CopZ)2 species was the intended structural target of that study).21E. hirae CopZ was previously studied by MS,42 with a similar narrow charge state distribution observed for both apo- and Cu(I)-bound protein. In that case, however, although dimeric CopZ was detected in solution it was not observed by MS (and so neither were higher Cu(I)-bound forms).
Previous spectroscopic and bioanalytical studies of the effects of LMWTs showed that DTT, GSH and Cys interfere with Cu(I)-binding to CopZ, inhibiting the formation of higher order Cu(I)-bound forms of the CopZ dimer.10,29 Here, we investigated the effects on Cu(I)-binding of BSH, the recently discovered physiological LMWT in B. subtilis.23 The CD data revealed a similar effect to that observed with GSH and Cys, with initial binding similar to that in the absence of thiol, but at high Cu(I) levels exhibiting a significant departure from thiol-free behaviour. The MS data revealed much more detail about the effects of thiols. Because the Cu(CopZ)2 species was not detected at significant abundance in the spectrum, the effect of DTT (and other thiols) on the formation of this species cannot be discerned. However, DTT severely inhibited the formation of higher order Cu(I)-bound forms of CopZ. It has been demonstrated that DTT is capable of serving as an additional ligand for the CuCopZ monomer species,20 and perhaps this is reflected by the increased proportion of CuCopZ with increasing DTT levels. However, DTT adducts of CuCopZ were not observed, and so we cannot rule out that this effect is indirect.
The effects of GSH and BSH were similar but less severe, with a marked decrease in the abundance of dimer observed only at the highest ratio of thiols. The data are consistent with previous investigations which indicated the presence of thiols (particularly GSH and Cys) does not appreciably disrupt the initial binding of Cu(I) to CopZ, but does inhibit the formation of higher order Cu(I)-bound CopZ species.10,29 Here, however, particularly in the presence of BSH, dimeric CopZ species persisted even at the highest ratios of thiol to protein tested.
At higher ratios of thiol and Cu(I) to protein, LMWTs increasingly compete for Cu(I). Because of the high Cu(I)-binding affinities involved, competition effects would most likely occur through the formation of transient hetero-complexes in which Cu(I) is at least temporarily coordinated by both CopZ and a LMWT. For GSH, and particularly BSH, it was found that such hetero-complexes are stable and can be readily detected. Interestingly, in addition to hetero-complexes formed between BSH and CuCopZ, BSH complexes of all the dimeric species were readily observed, suggesting that Cu(I) bound in clusters at the CopZ monomer–monomer interface can accommodate BSH coordination as well. The affinity of BSH for Cu(I), determined here as β2 = ∼4 × 1017 M−2, is significantly higher than of GSH, which was unable to compete for Cu(I) with BCS even when in large excess,43 consistent with a potential role in buffering Cu(I) levels in the cell. This affinity, however, is several orders of magnitude lower than that of CopZ33 and so it is unlikely that BSH alone buffers Cu(I). Consistent with this, B. subtilis cells lacking BSH were recently found to be unaffected in their overall sensitivity to Cu(I) stress compared to wild-type cells under equivalent conditions, but the expression of copZA was found to be elevated.28 These data suggest that BSH is not an essential component of the copper trafficking pathways of B. subtilis, but are consistent with BSH playing a role in copper-buffering. The observation of BSH complexes of Cu(I)-bound CopZ species indicates that BSH could function together with CopZ (and possibly CopA) via the formation of hetero-complexes to buffer Cu(I) levels in the cytoplasm of B. subtilis. Interestingly, BSH was recently found to be a major buffer of the cytoplasmic zinc pool in B. subtilis28 in which a dedicated Zn(II) chaperone has not been identified.
We note that apo-CopZ was also observed as an S-bacillithiolated form. Bacillithiolation of proteins in B. subtilis is known to occur as a reversible response to prevent over-oxidation of functionally important Cys residues that may occur under oxidative stress conditions. Bacillithiolation has been observed to protect proteins such as the redox-sensor OhrR44 and the methionine synthase MetE.26 Here, as for oxidation of apo-CopZ to a disulfide form, the oxidative driving force may be ionisation in the MS experiment.39 The fact that these species are observed, even at relatively low ratios of BSH to protein, suggests that this may be a physiologically important process for the protection of CopZ.
Conclusion
MS studies of CopZ revealed a range of Cu(I)-bound forms of CopZ that is entirely consistent with previous studies, and enabled changes in the distribution of CopZ species to be followed as Cu(I) concentration increased. This not only validated MS as a technique that provides information relevant to copper–protein interactions in solution, but also demonstrated that it is able to distinguish between species that cannot be resolved by spectroscopy, and which cannot be observed simultaneously by structural methods. Emerging from this is an unprecedented high resolution overview of Cu(I)-binding by CopZ, showing that Cu4(CopZ)2 forms cooperatively at levels of Cu(I) > 1/CopZ. MS was also able to provide a clear picture of the effects of the Bacillus LMWT BSH on CopZ, revealing the partial inhibition of formation of Cu4(CopZ)2 as well as other multinuclear Cu(I) forms of CopZ, along with bacillithiolation of CopZ and the formation of CopZ–Cu(I)–BSH hetero-complexes. The data are consistent with interactions between CopZ and BSH in the cytoplasm of B. subtilis, and a possible joint role in copper buffering in the B. subtilis cytoplasm.Abbreviations
| BSH | Bacillithiol |
| BCS | Bathocuproine disulfonate |
| DTT | Dithiothreitol |
| ESI | Electrospray ionisation |
| GSH | Glutathione |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazine-ethanesulfonic acid |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| LMWT | Low molecular weight thiol |
| MS | Mass spectrometry |
| m/z | Mass to charge ratio |
| TOF | Time of flight |
Acknowledgements
We thank UEA for funding the purchase of the mass spectrometer in our lab and for the award of a PhD studentship to KK. We thank Dr Myles Cheesman for access to the CD spectrometer.References
- S. Banerjee and S. Mazumdar, Int. J. Anal. Chem., 2012, 2012, 282574 CrossRef PubMed.
- J. T. Hopper and C. V. Robinson, Angew. Chem., Int. Ed., 2014, 53, 14002–14015 CrossRef CAS PubMed.
- N. G. Housden, J. T. Hopper, N. Lukoyanova, D. Rodriguez-Larrea, J. A. Wojdyla, A. Klein, R. Kaminska, H. Bayley, H. R. Saibil, C. V. Robinson and C. Kleanthous, Science, 2013, 340, 1570–1574 CrossRef CAS PubMed.
- D. E. Sutherland, K. L. Summers and M. J. Stillman, Biochem. Biophys. Res. Commun., 2012, 426, 601–607 CrossRef CAS PubMed.
- C. A. Blindauer, N. C. Polfer, S. E. Keiper, M. D. Harrison, N. J. Robinson, P. R. Langridge-Smith and P. J. Sadler, J. Am. Chem. Soc., 2003, 125, 3226–3227 CrossRef CAS PubMed.
- P. Palumaa, L. Kangur, A. Voronova and R. Sillard, Biochem. J., 2004, 382, 307–314 CrossRef CAS PubMed.
- F. D. Kondrat, G. R. Kowald, C. A. Scarff, J. H. Scrivens and C. A. Blindauer, Chem. Commun., 2013, 49, 813–815 RSC.
- W. Kaim and J. Rall, Angew. Chem., Int. Ed., 1996, 35, 43–60 CrossRef CAS.
- B. E. Kim, T. Nevitt and D. J. Thiele, Nat. Chem. Biol., 2008, 4, 176–185 CrossRef CAS PubMed.
- C. Singleton and N. E. Le Brun, BioMetals, 2007, 20, 275–289 CrossRef CAS PubMed.
- C. Andreini, L. Banci, I. Bertini and A. Rosato, J. Proteome Res., 2008, 7, 209–216 CrossRef CAS PubMed.
- T. V. O'Halloran and V. C. Culotta, J. Biol. Chem., 2000, 275, 25057–25060 CrossRef PubMed.
- L. Banci, I. Bertini, F. Cantini, I. C. Felli, L. Gonnelli, N. Hadjiliadis, R. Pierattelli, A. Rosato and P. Voulgaris, Nat. Chem. Biol., 2006, 2, 367–368 CrossRef CAS PubMed.
- J. M. Arguello, D. Raimunda and T. Padilla-Benavides, Front. Cell. Infect. Microbiol., 2013, 3, 73 Search PubMed.
- M. Luczkowski, B. A. Zeider, A. V. Hinz, M. Stachura, S. Chakraborty, L. Hemmingsen, D. L. Huffman and V. L. Pecoraro, Chemistry, 2013, 19, 9042–9049 CrossRef CAS PubMed.
- I. Morin, S. Gudin, E. Mintz and M. Cuillel, FEBS J., 2009, 276, 4483–4495 CrossRef CAS PubMed.
- Z. Ma, D. M. Cowart, R. A. Scott and D. P. Giedroc, Biochemistry, 2009, 48, 3325–3334 CrossRef CAS PubMed.
- A. N. Barry, U. Shinde and S. Lutsenko, JBIC, J. Biol. Inorg. Chem., 2010, 15, 47–59 CrossRef CAS PubMed.
- R. Wimmer, T. Herrmann, M. Solioz and K. Wuthrich, J. Biol. Chem., 1999, 274, 22597–22603 CrossRef CAS PubMed.
- L. Banci, I. Bertini, R. Del Conte, J. Markey and F. J. Ruiz-Duenas, Biochemistry, 2001, 40, 15660–15668 CrossRef CAS PubMed.
- S. Hearnshaw, C. West, C. Singleton, L. Zhou, M. A. Kihlken, R. W. Strange, N. E. Le Brun and A. M. Hemmings, Biochemistry, 2009, 48, 9324–9326 CrossRef CAS PubMed.
- D. S. Radford, M. A. Kihlken, G. P. Borrelly, C. R. Harwood, N. E. Le Brun and J. S. Cavet, FEMS Microbiol. Lett., 2003, 220, 105–112 CrossRef CAS PubMed.
- G. L. Newton, M. Rawat, J. J. La Clair, V. K. Jothivasan, T. Budiarto, C. J. Hamilton, A. Claiborne, J. D. Helmann and R. C. Fahey, Nat. Chem. Biol., 2009, 5, 625–627 CrossRef CAS PubMed.
- H. Antelmann and J. D. Helmann, Antioxid. Redox Signaling, 2011, 14, 1049–1063 CrossRef CAS PubMed.
- I. Dalle-Donne, R. Rossi, G. Colombo, D. Giustarini and A. Milzani, Trends Biochem. Sci., 2009, 34, 85–96 CrossRef CAS PubMed.
- B. K. Chi, K. Gronau, U. Mader, B. Hessling, D. Becher and H. Antelmann, Mol. Cell. Proteomics, 2011, 10, M111.009506 Search PubMed.
- A. Gaballa, B. K. Chi, A. A. Roberts, D. Becher, C. J. Hamilton, H. Antelmann and J. D. Helmann, Antioxid. Redox Signaling, 2014, 21, 357–367 CrossRef CAS PubMed.
- Z. Ma, P. Chandrangsu, T. C. Helmann, A. Romsang, A. Gaballa and J. D. Helmann, Mol. Microbiol., 2014, 94, 756–770 CrossRef CAS PubMed.
- M. A. Kihlken, A. P. Leech and N. E. Le Brun, Biochem. J., 2002, 368, 729–739 CrossRef CAS PubMed.
- C. Singleton, L. Banci, S. Ciofi-Baffoni, L. Tenori, M. A. Kihlken, R. Boetzel and N. E. Le Brun, Biochem. J., 2008, 411, 571–579 CrossRef CAS PubMed.
- S. V. Sharma, V. K. Jothivasan, G. L. Newton, H. Upton, J. I. Wakabayashi, M. G. Kane, A. A. Roberts, M. Rawat, J. J. La Clair and C. J. Hamilton, Angew. Chem., Int. Ed., 2011, 50, 7101–7104 CrossRef CAS PubMed.
- Z. Xiao, F. Loughlin, G. N. George, G. J. Howlett and A. G. Wedd, J. Am. Chem. Soc., 2004, 126, 3081–3090 CrossRef CAS PubMed.
- L. Zhou, C. Singleton and N. E. Le Brun, Biochem. J., 2008, 413, 459–465 CrossRef CAS PubMed.
- S. V. Sharma, M. Arbach, A. A. Roberts, C. J. Macdonald, M. Groom and C. J. Hamilton, ChemBioChem, 2013, 14, 2160–2168 CrossRef CAS PubMed.
- A. Dobo and I. A. Kaltashov, Anal. Chem., 2001, 73, 4763–4773 CrossRef CAS PubMed.
- C. Singleton and N. E. Le Brun, Dalton Trans., 2009, 688–696 RSC.
- D. W. Hasler, P. Faller and M. Vasak, Biochemistry, 1998, 37, 14966–14973 CrossRef CAS PubMed.
- D. L. Pountney, I. Schauwecker, J. Zarn and M. Vasak, Biochemistry, 1994, 33, 9699–9705 CrossRef CAS PubMed.
- M. Prudent and H. H. Girault, Metallomics, 2009, 1, 157–165 RSC.
- F. Hussain and P. Wittung-Stafshede, Biochim. Biophys. Acta, 2007, 1774, 1316–1322 CrossRef CAS PubMed.
- N. Felitsyn, M. Peschke and P. Kebarle, Int. J. Mass Spectrom., 2002, 219, 39–62 CrossRef CAS.
- A. Urvoas, B. Amekraz, C. Moulin, L. Le Clainche, R. Stocklin and M. Moutiez, Rapid Commun. Mass Spectrom., 2003, 17, 1889–1896 CrossRef CAS PubMed.
- Z. Xiao, J. Brose, S. Schimo, S. M. Ackland, S. La Fontaine and A. G. Wedd, J. Biol. Chem., 2011, 286, 11047–11055 CrossRef CAS PubMed.
- J. W. Lee, S. Soonsanga and J. D. Helmann, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8743–8748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mt00036c |
| This journal is © The Royal Society of Chemistry 2016 |