
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lead and manganese levels in serum and erythrocytes in Alzheimer's disease and mild cognitive impairment: results from the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing†
Dominic J.
Hare‡
 *ab,
Noel G.
Faux‡
bc,
Blaine R.
Roberts
bd,
Irene
Volitakis
b,
Ralph N.
Martins
defg and
Ashley I.
Bush
*bd
*ab,
Noel G.
Faux‡
bc,
Blaine R.
Roberts
bd,
Irene
Volitakis
b,
Ralph N.
Martins
defg and
Ashley I.
Bush
*bd
aElemental Bio-imaging Facility, University of Technology Sydney, Broadway, New South Wales, Australia. E-mail: dominic.hare@uts.edu.au; Fax: +61 2 9514 1460; Tel: +61 3 9035 9459
bThe Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, Victoria, Australia. E-mail: ashley.bush@florey.edu.au; Fax: +61 3 9035 3107; Tel: +61 3 9035 6532
cIBM Research – Australia, Carlton, Victoria, Australia
dCooperative Research Centre for Mental Health, Carlton South, Victoria, Australia
eThe School of Medical Sciences, Edith Cowan University, Joondalup, Western Australia, Australia
fThe McCusker Alzheimer's Research Foundation, Hollywood Private Hospital, Nedlands, Western Australia, Australia
gThe Centre of Excellence for Alzheimer's Disease Research and Care, Edith Cowan University, Joondalup, Western Australia, Australia
First published on 4th March 2016
Abstract
We examined serum and erythrocyte lead and manganese levels in the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing (AIBL), which contains over 1000 registrants including over 200 cases of Alzheimer's disease (AD) and 100 mildly cognitively impaired (MCI) individuals. After correcting for confounding effects of age, collection site and sex, we found a significant decrease in serum manganese levels in AD subjects compared to healthy controls. Analysis of smaller subset of erythrocytes revealed no difference in either lead or manganese levels in AD. Although lead and manganese have neurotoxic effects and may be involved in AD pathology, our results showed that neither metal in serum nor erythrocytes are suitable biomarkers in our cohort. However, prospective studies might reveal whether the burden of either metal modifies disease outcomes.
Introduction
Lead is a potent and chronic toxin in the brain, where it exerts adverse effects through a range of mechanisms, including inducing mitochondrial dysfunction, impairing normal neurotransmitter activity, and substituting calcium and zinc ions to cause inappropriate neuronal responses.1 Although most attention is paid to the neurotoxicity of lead during development,2–4 this heavy metal is able to cross the fully-formed blood–brain barrier with relative ease,5 and thus retains significant potential for neurotoxicity in adults. Manganese is an essential element, though it can also exert neurotoxicity when present in excess. Much attention has been paid to manganism, a Parkinson's disease-like condition that is highly prevalent in cases of occupational exposure.6 However, the proposed mechanism of manganese neurotoxicity stems from its ability to induce heightened oxidative stress and mitochondrial dysfunction,7 which is not necessarily specific to the degenerating dopaminergic neurons common to Parkinson's disease and may be involved in other neurodegenerative disorders, including Alzheimer's disease (AD).Around 95% of the total body burden of lead is in bone,8 where it exchanges for calcium and is stored in hydroxyapatite. There is some conjecture regarding the influence of bone lead concentrations as it relates to circulating lead versus acute exogenous exposure,9,10 though it is certain that some contribution to circulating lead levels occurs through remobilisation of lead from mineralised storage. Bone lead has a half-life measured in decades, whilst blood lead has an average turnover of around 30 days.11 There are potentially many confounding factors that influence lead release from bone into circulation,12 and thus large cohorts of subjects are required to study potential relationships between disease and circulating lead levels. This is further compounded by the low levels at which endogenous lead is found within blood products. However, these issues aside, blood lead levels do appear to reflect the steady-state lead concentrations at a given time point,13 and with sufficiently sensitive analytical methods for measuring lead in biological fluids it can be used to assess potential relationships between metal levels and a clinical classification.
Circulating manganese is differentially distributed throughout the components of blood, with 66% found in erythrocytes and 4.4% in plasma.14 Although manganese is an essential cofactor in several enzymes in the brain,15 it is also a chronic neurotoxicant, and prolonged low-level environmental exposure causes significant degeneration throughout the basal ganglia.16 Manganese transport in the periphery is closely related to that of iron, another redox-active metal that has been widely implicated in AD pathology,17 which may present an avenue through which manganese can also impart neurotoxicity in the disease.
We analysed the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing (AIBL) cohort to examine possible relationships between cognitive decline and serum lead and manganese levels. Metals have long been associated with AD,18 and AIBL provides a well-characterised cohort of over 1000 individuals including over 200 cases of AD and 100 cases of mild cognitive impairment (MCI),19 which is an age-related decline in memory and cognition that has been described as ‘prodromal AD’.20 This significant cohort provides a unique opportunity to examine blood lead and manganese levels and how they relate to AD.
Experimental
Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing
Details of clinical review and diagnosis of MCI and AD in AIBL can be found in Ellis et al.19 AIBL has two collection sites in Australia: Melbourne, Victoria and Perth, Western Australia. Demographic details are shown in Table 1.| HC | MCI | AD | |
|---|---|---|---|
| Clinical classification (n) | 758 | 128 | 206 |
| Site of collection | |||
| Melbourne (n) | 422 | 80 | 145 |
| Perth (n) | 322 | 48 | 61 |
| Sex (n) | F = 236; M = 222 | F = 73; M = 55 | F = 127; M = 79 |
| Age (years ± SD) | 70.0 ± 7.0 | 75.7 ± 7.6 | 78.0 ± 8.6 |
Sample collection and preparation
Serum was extracted from whole blood drawn by venepuncture from overnight fasted participants. Filled Sarstedt s-monovette serum-gel 7.5 mL tubes (Sarstedt, Nümbrecht, Germany) were left standing upright for a minimum of 20 minutes at room temperature. Samples were centrifuged at 1800g for 15 minutes at 20 °C with braking on. The serum was then split into 250 μL aliquots, and stored in Nunc Cryobank polypropylene tubes (Nunc, Rochester, NY, USA) in liquid nitrogen vapour tanks until required for metal analysis. For erythrocytes, whole blood was collected from overnight fasted participants with a 27 gauge needle, into Sarstedt S-Monovette® Lithium-Heparin 7.5 mL tubes. The blood was spun at 3200g for 30 minutes at room temperature and the plasma was carefully removed. Erythrocytes were then washed 3 times in 0.9% (w/v) normal saline. Erythrocytes were distributed by tube inversion, and then centrifuged at 650g for 10 minutes at 20 °C. The supernatant was removed and the tubes centrifuged at 1500g for a further 10 minutes at 20 °C. Erythrocytes were then resuspended to a volume of 6 mL in phosphate buffered saline (PBS) (pH 7.4) and aliquoted into polypropylene (Nunc) tubes for storage in liquid nitrogen.Lead and manganese analysis
For lead and manganese analysis, we used inductively coupled plasma-mass spectrometry according to the methods previously described.21 After cryogenically-stored serum was retrieved on the day of analysis, it was briefly spun at 1800g, transferred to 1.5 mL polypropylene tubes (Techno Plas, St Marys, Australia) and then diluted 1 in 10 with 1% HNO3 (Merck Millipore, Bayswater, Australia). For a subset of lead and manganese levels in red blood cells (nHC = 40; nAD = 40), 50 μL of washed erythrocytes were digested in equivalent volumes of concentrated (65%) HNO3 and H2O2 (Merck Milipore) in 1.5 mL polypropylene tubes (Techno Plas) at 80 °C for 5 minutes, then diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 with 1% HNO3. Samples were analysed using an Agilent 7700x Series ICP-MS (Mulgrave, Australia). Lead-206 (206Pb) was the isotope chosen for analysis, as was the monoisotopic manganese-55 (55Mn). Analytical validity was confirmed using commercial standard serum (Seronorm L1 and L2) as previously described.21 All analysis was performed on baseline samples taken at the study's inception.
:20 with 1% HNO3. Samples were analysed using an Agilent 7700x Series ICP-MS (Mulgrave, Australia). Lead-206 (206Pb) was the isotope chosen for analysis, as was the monoisotopic manganese-55 (55Mn). Analytical validity was confirmed using commercial standard serum (Seronorm L1 and L2) as previously described.21 All analysis was performed on baseline samples taken at the study's inception.
Statistical analysis
All analysis was performed in R (Version 3.2.2),22 using the following packages: NADA,23 ggplot2,24 car25 and multcomp.26 As the serum lead levels contained 38% left censored data (data points below lower limit of detection, <0.063 μg L−1), the mean, medians and standard deviations of these data were estimated by maximum likelihood estimation, in a log-normal distribution (assumption confirmed by inspection of the probability plot, Fig. S1, ESI†). To assess the effects of age, sex, diagnosis and collection site, log likelihood tests were performed. All other data used was complete and above limits of detection. Serum manganese and erythrocyte lead and manganese were not normally distributed. Box–Cox analyses27 indicated the suitability of an inverse transformation for serum manganese, natural log transform for erythrocyte lead and square root transform for erythrocyte manganese. Analysis of covariance (ANCOVA – type II SS) was performed to assess the effect of age, sex, diagnosis, collection site, ApoE ε4 carrier presence, and for serum manganese batch effects. All models reported are the parsimonious models using the Akaike's Information Criterion (AIC).28 Cook's distance and leverage plots of residuals29 did not identify outliers or influential data points.Results
Analysis of the serum lead data revealed that the collection site produced a measurable effect; with Melbourne-based AIBL subjects having significantly higher serum lead levels than Perth-based counterparts. Age had a slight positive relationship with serum lead (Table 2). After correcting for these two variables, there was no apparent difference in serum lead levels according to clinical classification (Fig. 1; Table S1, ESI†). Examination of the serum manganese data (Table S2, ESI†) revealed effects according to diagnostic classification (p < 0.001), sex (p < 0.05), collection site (p < 0.001), and a batch effect (p < 0.001; Table S3, ESI†). Using simultaneous tests for a general linear hypothesis to correct for this variation, a significant decrease in serum manganese levels was observed between healthy controls and AD subjects, though not in MCI subjects (p < 0.001, Fig. 2; Table 3; Table S3, ESI†).| p-Value | |
|---|---|
| Clinical classification | 0.374 |
| Site of collection | <0.001 |
| Sex | 0.096 |
| Age | 0.012 |
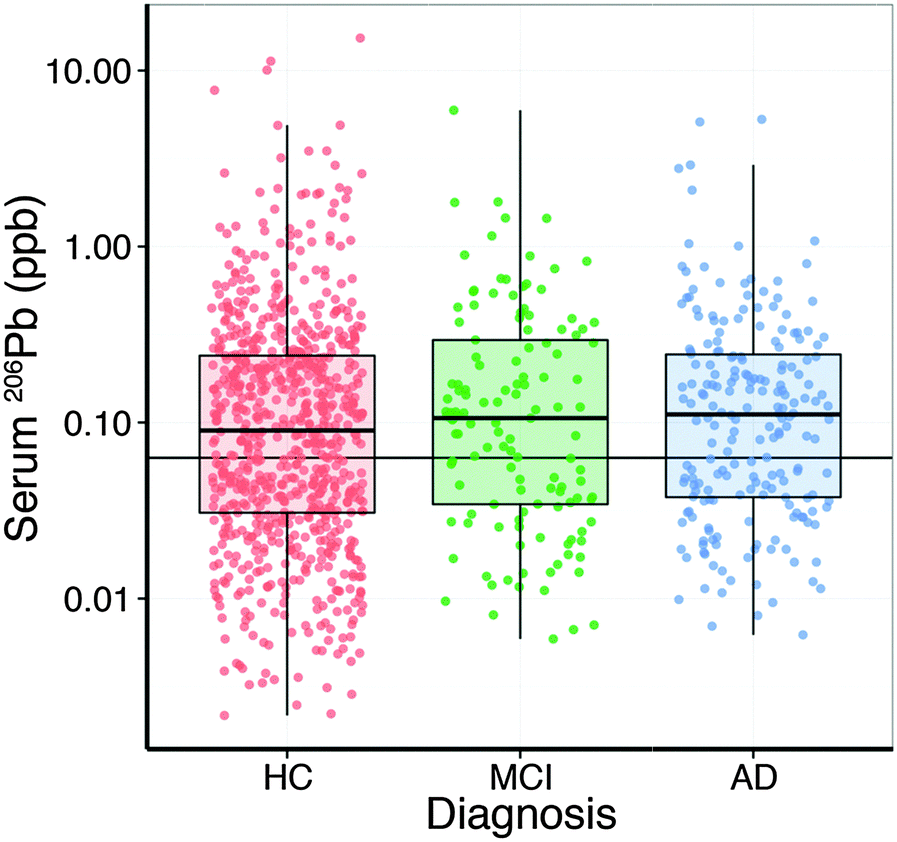 | ||
| Fig. 1 Boxplot of all analysed AIBL serum samples for serum lead concentration (n = 1093), including left censored values (beneath the thin black horizontal line; 0.063 μg L−1) based on maximum likelihood (ncent = 411; 37.6% of all measured values). No significant difference was identified between clinical classifications. Data reported as median ± SD. HC = healthy control (n = 758); MCI = mild cognitive impairment (n = 129); AD = Alzheimer's disease (n = 206). | ||
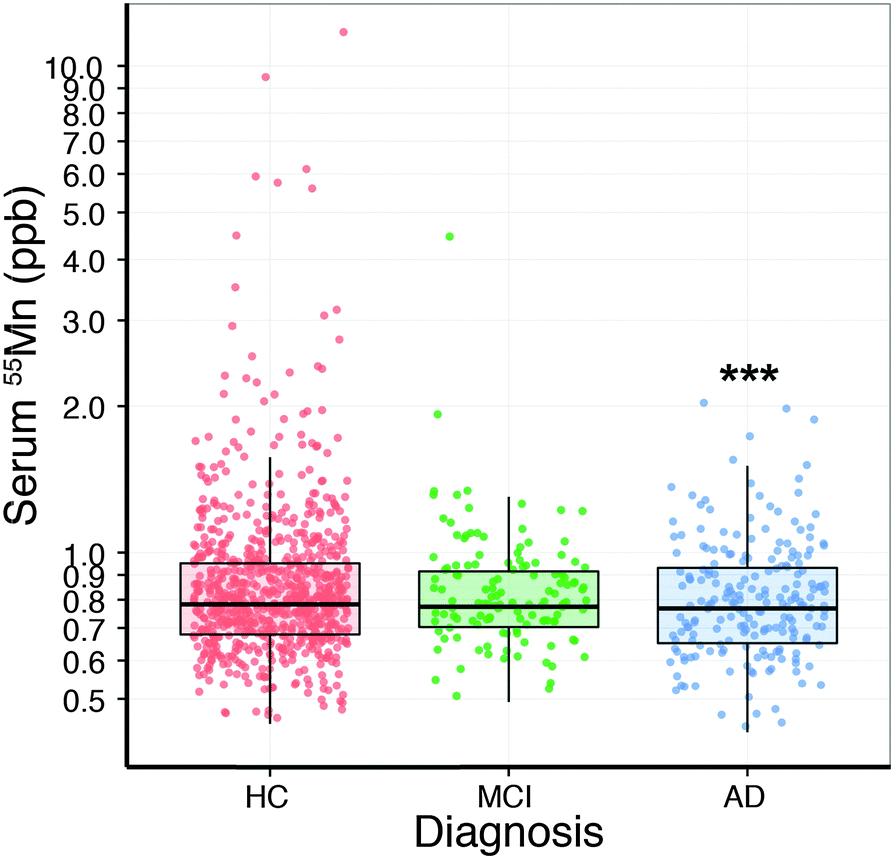 | ||
| Fig. 2 Boxplot of difference in serum manganese levels (mean ± SD; n = 1093) by clinical classification. The y-axis is presented as a log scale. Serum manganese was decreased (p < 0.001; ***) in AD compared to HC. HC = healthy control (n = 758); MCI = mild cognitive impairment (n = 129); AD = Alzheimer's disease (n = 206). | ||
| Comparison | Estimate | Standard error | t-Value | p-Value (>|t|) |
|---|---|---|---|---|
| MCI vs. HC | 0.0372 | 0.0324 | 1.147 | 0.4822 |
| AD vs. HC | 0.1005 | 0.0287 | 3.509 | <0.001 |
| AD vs. MCI | 0.0634 | 0.0366 | 0.1730 | 0.1921 |
In comparison to the serum levels, the subset of erythrocytes analysed (Table S4, ESI†) did not show a collection site difference or a relationship with age, but a slight sex difference for lead (p = 0.053). No difference between HC and AD was observed (Fig. 3a). Erythrocyte manganese did not show a difference across the diagnostic groups (Fig. 3b), nor age or site. However, males showed a small elevation in erythrocyte manganese (p = 0.053). Comparing serum and erythrocyte lead and manganese levels obtained from the same AIBL subjects revealed no measurable relationship between the sample types (ANOVA Type II sum of squares test).
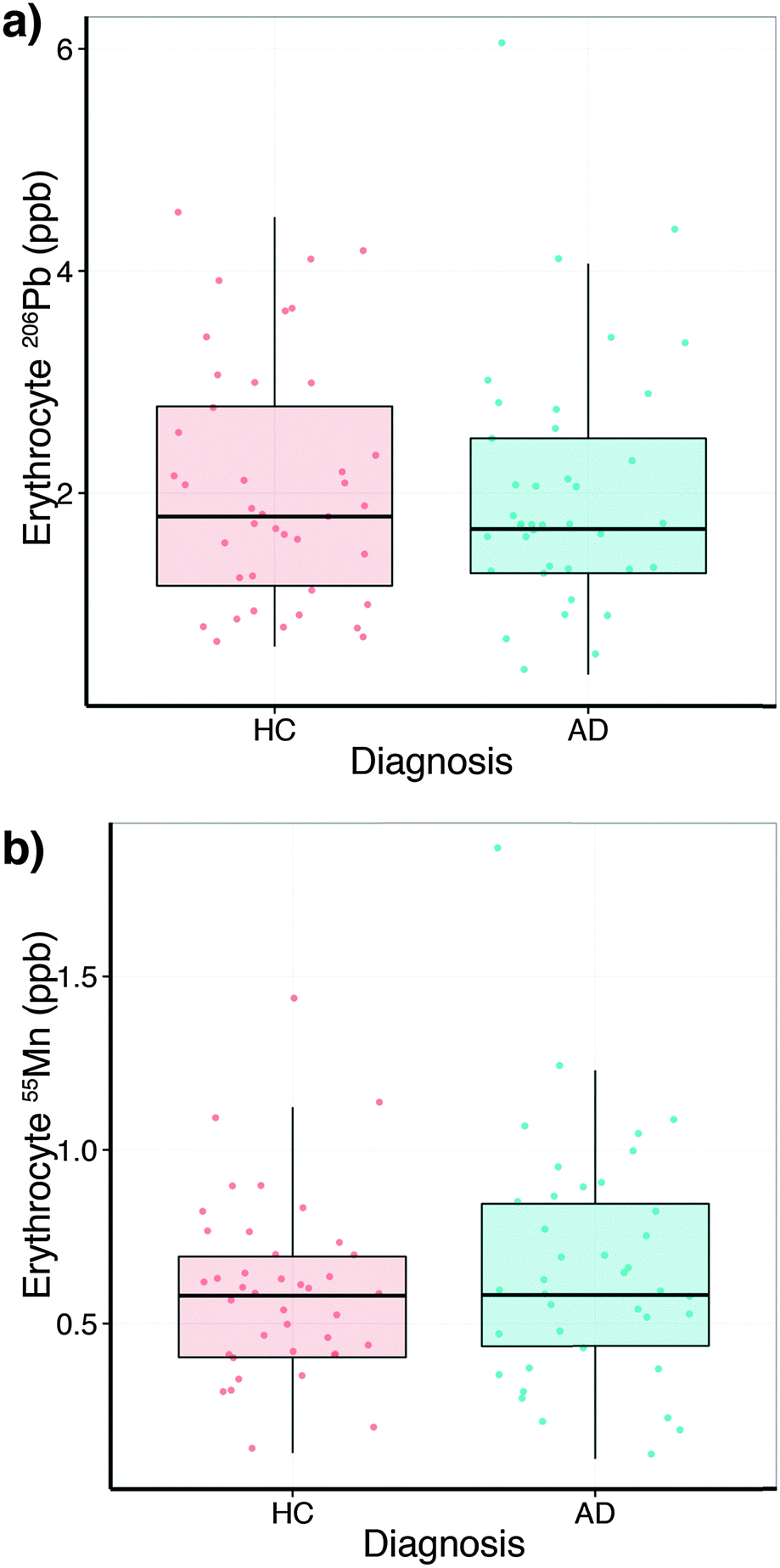 | ||
| Fig. 3 Boxplots of erythrocyte lead (a) and manganese (b) levels (mean ± SD) by clinical classification. No difference between healthy controls (HC; n = 40) and Alzheimer's disease (AD; n = 40) was observed. | ||
Discussion
To date, while there has been no causative relationship between AD and lead exposure found; studies in animals have yielded interesting results. Numerous transgenic animal models expressing a range of protein mutations associated with AD have shown that lead exposure can recapitulate AD disease phenotypes. In a particularly interesting study, monkeys exposed to lead during infancy showed marked neuropathology in the frontal cortex consistent with the characteristic β-amyloid deposition found in AD.30 Cumulative lead exposure, measured in bone, has been shown to have a negative effect on cognition in elderly males,31 and that accurate measures of historic lead exposure may be of use in a epidemiological setting for identifying a potential relationship between AD and heavy metal exposure.32 In the brain, substitution by lead of zinc ions in the zinc finger proteins DNA methyltransferase 1 (DNMT1) and presenilin 1 and 2 (PSEN1/2) has clear implications for AD;33 all three proteins are implicated in mishandling of amyloid precursor protein (APP), which leads to the formation of toxic β-amyloid oligomers and proteinaceous inclusions.Similarly, manganese has not been directly related to AD pathology, with no significant variation in metal levels identified in AD brain tissue.34 However, changes in expression of the manganese-dependent mitochondrial antioxidant superoxide dismutase-2 (SOD2) have been observed in circulating lymphocytes in AD.35 This potentially reflects a ‘double-edge sword’ paradigm regarding manganese in AD: in Tg19959 transgenic mice that carry two mutations to APP found in familial AD crossed with mice overexpressing SOD2, the increased activity of this enzyme appeared to reduce amyloid deposition, oxidative stress and improve memory impairment compared to the Tg19959 mutant alone.36 Mice with the SOD2 gene ablated do not survive past the first week of life, though treatment with antioxidants expands lifespan to reveal significant levels of tau hyperphosphorylation, which is characteristic of AD. Crossing this mouse with the Tg2576 APP mutation model also resulted in increased brain amyloid burden.37 Manganese, along with other biometals including zinc, copper, iron and chromium has also been shown to have an inverse correlation with human cerebrospinal fluid β-amyloid 1–42 levels.
Our data showed a small effect of decreased manganese in serum in AD patients compared to healthy controls, though confounding effects of age; collection site and sex were also observed for both lead and manganese. Though these could be statistically corrected, even in a cohort the size of AIBL a clear relationship between serum and erythrocyte lead and manganese was not obvious, and was not an indicator of disease status. Erythrocyte manganese levels have previously been correlated with increased signal intensity in T1-weighted magnetic resonance imaging (MRI) of globus pallidus manganese in exposed individuals.38 This region is a secondary site of cholinergic neurodegeneration in AD,39 though MRI imaging found evidence of increased pallidal manganese burden with the absence of clinical symptoms.38 Furthermore, high occupational exposure to manganese is not a feature of the AIBL cohort.
These results reflect that the partition of lead and manganese (as is the case with other metals involved in AD pathology, such as zinc21) is somewhat dichotomous, and are unlikely to have diagnostic potential when viewed in isolation. Studies identifying relationships between circulating metal levels and AD have also used more comprehensive data sets that encompass not only the metal itself, but also associated regulatory proteins, such as our reported anaemia of AD40 and possible association between decreased non-ceruloplasmin bound copper and MCI/AD.41 Circulating metal levels have previously been described as biomarkers of several health conditions, including total blood lead as an indicator of hypertension42 and plasma copper/zinc ratios as a marker of heavy metal toxicity in children with autism spectrum disorders.43 However, in this case, if neurotoxic metals like lead and manganese are involved in AD pathology within the brain, the relatively short temporal window provided by serum and erythrocyte metal levels does not appear to be reflective of the chronic nature of the disease.
Acknowledgements
We wish to thank the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing (http://www.aibl.csiro.au/), including all scientists, participants and their families. This work was supported by the Cooperative Research Centre for Mental Health and by Operational Infrastructure Support from the Victorian State Government.Notes and references
- L. H. Mason, J. P. Harp and D. Y. Han, BioMed Res. Int., 2014, 2014, 1–8 Search PubMed.
- P. Grandjean and P. J. Landrigan, Lancet, 2006, 368, 2167–2178 CrossRef CAS.
- P. Grandjean and P. J. Landrigan, Lancet Neurol., 2014, 13, 330–338 CrossRef CAS PubMed.
- G. Flora, D. Gupta and A. Tiwari, Interdiscip. Toxicol., 2012, 5, 47–58 CAS.
- W. Zheng, M. Aschner and J.-F. Ghersi-Egea, Toxicol. Appl. Pharmacol., 2003, 192, 1–11 CrossRef CAS PubMed.
- A. W. Dobson, K. M. Erikson and M. Aschner, Ann. N. Y. Acad. Sci., 2004, 1012, 115–128 CrossRef CAS PubMed.
- M. A. Verity, Neurotoxicology, 1999, 20, 489–497 CAS.
- P. S. Barry and D. B. Mossman, Br. J. Ind. Med., 1970, 27, 339–351 CAS.
- K. M. Cake, R. J. Bowins, C. Vaillancourt, C. L. Gordon, R. H. McNutt, R. Laporte, C. E. Webber and D. R. Chettle, Am. J. Ind. Med., 1996, 29, 440–445 CrossRef CAS PubMed.
- I. A. Bergdahl and S. Skerfving, Am. J. Ind. Med., 1997, 32, 317–318 CrossRef CAS PubMed.
- M. B. Rabinowitz, Environ. Health Perspect., 1991, 91, 33–37 CrossRef CAS PubMed.
- H. Hu, M. Rabinowitz and D. Smith, Environ. Health Perspect., 1998, 106, 1–8 CrossRef CAS PubMed.
- Lead in the Human Environment: A Report, National Research Council (U.S.) Committee on Lead in the Human Environment, 1980.
- D. B. Milne, R. L. Sims and N. V. Ralston, Clin. Chem., 1990, 36, 450–452 CAS.
- K. J. Horning, S. W. Caito, K. G. Tipps, A. B. Bowman and M. Aschner, Annu. Rev. Nutr., 2015, 35, 71–108 CrossRef CAS PubMed.
- B. A. Racette, M. Aschner, T. R. Guilarte, U. Dydak, S. R. Criswell and W. Zheng, Neurotoxicology, 2012, 33, 881–886 CrossRef CAS PubMed.
- R. A. Yokel, J. Alzheimer's Dis., 2006, 10, 223–253 Search PubMed.
- B. R. Roberts, T. M. Ryan, A. I. Bush, C. L. Masters and J. A. Duce, J. Neurochem., 2011, 120, 149–166 CrossRef PubMed.
- K. A. Ellis, A. I. Bush, D. Darby, D. De Fazio, J. Foster, P. Hudson, N. T. Lautenschlager, N. Lenzo, R. N. Martins, P. Maruff, C. Masters, A. Milner, K. Pike, C. Rowe, G. Savage, C. Szoeke, K. Taddei, V. Villemagne, M. Woodward, D. Ames and A. R. Group, Int. Psychogeriatr., 2009, 21, 672–687 CrossRef PubMed.
- B. Dubois and M. L. Albert, Lancet Neurol., 2004, 3, 246–248 CrossRef PubMed.
- A. Rembach, D. J. Hare, J. D. Doecke, S. C. Burnham, I. Volitakis, C. J. Fowler, R. A. Cherny, C. McLean, R. Grimm, R. Martins, D. Ames, C. L. Masters, A. I. Bush and B. R. Roberts, Metallomics, 2014, 6, 1216–1219 RSC.
- R. Development Core Team, R: A Language and Environment for Statistical Computing, The R Foundation for Statistical Computing, Vienna, Austria, 2011 Search PubMed.
- L. Lee, R package version 1.5-3, 2010 Search PubMed.
- H. Wickham, ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag, New York, USA, 2009 Search PubMed.
- J. Fox and H. S. Weisberg, An R Companion to Applied Regression, SAGE Publishing, Los Angeles, USA, 2nd edn, 2010 Search PubMed.
- T. Hothorn, F. Bretz and P. Westfall, Biom. J., 2008, 50, 346–363 CrossRef PubMed.
- G. E. P. Box and D. R. Cox, J. R. Statist. Soc. B, 1964, 26, 211–252 Search PubMed.
- H. Akaike, IEEE Trans. Autom. Control, 1974, 19, 716–723 CrossRef.
- R. D. Cook, J. Am. Stat. Assoc., 1979, 74, 169–174 CrossRef.
- J. Wu, M. R. Basha, B. Brock, D. P. Cox, F. Cardozo-Pelaez, C. A. McPherson, J. Harry, D. C. Rice, B. Maloney, D. Chen, D. K. Lahiri and N. H. Zawia, J. Neurosci., 2008, 28, 3–9 CrossRef CAS PubMed.
- M. G. Weisskopf, R. O. Wright, J. Schwartz, A. Spiro, D. Sparrow, A. Aro and H. Hu, Am. J. Epidemiol., 2004, 160, 1184–1193 CrossRef PubMed.
- K. M. Bakulski, L. S. Rozek, D. C. Dolinoy, H. L. Paulson and H. Hu, Curr. Alzheimer Res., 2012, 9, 563–573 CrossRef CAS PubMed.
- J. M. Ordemann and R. N. Austin, Metallomics, 2016 10.1039/C5MT00300H.
- W. R. Markesbery, W. D. Ehmann, T. I. Hossain and M. Alauddin, Neurotoxicology, 1984, 5, 49–57 CAS.
- M. E. De Leo, S. Borrello, M. Passantino, B. Palazzotti, A. Mordente, A. Daniele, V. Filippini, T. Galeotti and C. Masullo, Neurosci. Lett., 1998, 250, 173–176 CrossRef CAS PubMed.
- M. Dumont, E. Wille, C. Stack, N. Y. Calingasan, M. F. Beal and M. T. Lin, FASEB J., 2009, 23, 2459–2466 CrossRef CAS PubMed.
- S. Melov, P. A. Adlard, K. Morten, F. Johnson, T. R. Golden, D. Hinerfeld, B. Schilling, C. Mavros, C. L. Masters, I. Volitakis, Q.-X. Li, K. Laughton, A. Hubbard, R. A. Cherny, B. Gibson and A. I. Bush, PLoS One, 2007, 2, e536 Search PubMed.
- Y. Jiang, W. Zheng, L. Long, W. Zhao, X. Li, X. Mo, J. Lu, X. Fu, W. Li, S. Liu, Q. Long, J. Huang and E. Pira, Neurotoxicology, 2007, 28, 126–135 CrossRef CAS PubMed.
- S. Lehéricy, E. C. Hirsch, L. B. Hersh and Y. Agid, Neurosci. Lett., 1991, 123, 152–155 CrossRef.
- N. G. Faux, A. Rembach, J. Wiley, K. A. Ellis, D. Ames, C. J. Fowler, R. N. Martins, K. K. Pertile, R. L. Rumble, B. Trounson, C. L. Masters, A. R. Group and A. I. Bush, Mol. Psychiatry, 2014, 19, 1227–1234 CrossRef CAS PubMed.
- A. Rembach, J. D. Doecke, B. R. Roberts, A. D. Watt, N. G. Faux, I. Volitakis, K. K. Pertile, R. L. Rumble, B. O. Trounson, C. J. Fowler, W. Wilson, K. A. Ellis, R. N. Martins, C. C. Rowe, V. L. Villemagne, D. Ames, C. L. Masters and A. I. Bush, J. Alzheimer's Dis., 2013, 34, 171–182 CAS.
- S. Telišman, J. Jurasović, A. Pizent and P. Cvitković, Environ. Res., 2001, 87, 57–68 CrossRef PubMed.
- S. Faber, G. M. Zinn, J. C. Kern and H. M. S. Kingston, Biomarkers, 2009, 14, 171–180 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figure and tables. See DOI: 10.1039/c6mt00019c |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |