
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combinatorial phenotypic screen uncovers unrecognized family of extended thiourea inhibitors with copper-dependent anti-staphylococcal activity
Alex G.
Dalecki
a,
Aruni P.
Malalasekera
b,
Kaitlyn
Schaaf
a,
Olaf
Kutsch
a,
Stefan H.
Bossmann
b and
Frank
Wolschendorf
*a
aDepartment of Medicine, University of Alabama at Birmingham, 845 19th Street S, Birmingham, AL 35294, USA. E-mail: fwolsche@uab.edu; Tel: +1-205-975-2760
bDepartment of Chemistry, Kansas State University, Manhattan, KS, USA
First published on 26th February 2016
Abstract
The continuous rise of multi-drug resistant pathogenic bacteria has become a significant challenge for the health care system. In particular, novel drugs to treat infections of methicillin-resistant Staphylococcus aureus strains (MRSA) are needed, but traditional drug discovery campaigns have largely failed to deliver clinically suitable antibiotics. More than simply new drugs, new drug discovery approaches are needed to combat bacterial resistance. The recently described phenomenon of copper-dependent inhibitors has galvanized research exploring the use of metal-coordinating molecules to harness copper's natural antibacterial properties for therapeutic purposes. Here, we describe the results of the first concerted screening effort to identify copper-dependent inhibitors of Staphylococcus aureus. A standard library of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds was assayed for anti-staphylococcal activity, with hits defined as those compounds with a strict copper-dependent inhibitory activity. A total of 53 copper-dependent hit molecules were uncovered, similar to the copper independent hit rate of a traditionally executed campaign conducted in parallel on the same library. Most prominent was a hit family with an extended thiourea core structure, termed the NNSN motif. This motif resulted in copper-dependent and copper-specific S. aureus inhibition, while simultaneously being well tolerated by eukaryotic cells. Importantly, we could demonstrate that copper binding by the NNSN motif is highly unusual and likely responsible for the promising biological qualities of these compounds. A subsequent chemoinformatic meta-analysis of the ChEMBL chemical database confirmed the NNSNs as an unrecognized staphylococcal inhibitor, despite the family's presence in many chemical screening libraries. Thus, our copper-biased screen has proven able to discover inhibitors within previously screened libraries, offering a mechanism to reinvigorate exhausted molecular collections.
000 compounds was assayed for anti-staphylococcal activity, with hits defined as those compounds with a strict copper-dependent inhibitory activity. A total of 53 copper-dependent hit molecules were uncovered, similar to the copper independent hit rate of a traditionally executed campaign conducted in parallel on the same library. Most prominent was a hit family with an extended thiourea core structure, termed the NNSN motif. This motif resulted in copper-dependent and copper-specific S. aureus inhibition, while simultaneously being well tolerated by eukaryotic cells. Importantly, we could demonstrate that copper binding by the NNSN motif is highly unusual and likely responsible for the promising biological qualities of these compounds. A subsequent chemoinformatic meta-analysis of the ChEMBL chemical database confirmed the NNSNs as an unrecognized staphylococcal inhibitor, despite the family's presence in many chemical screening libraries. Thus, our copper-biased screen has proven able to discover inhibitors within previously screened libraries, offering a mechanism to reinvigorate exhausted molecular collections.
Introduction
The advent of high throughput screening (HTS) technologies over thirty years ago revolutionized drug discovery efforts. Having apparently exhausted the readily identifiable repertoire of natural antibacterials derived from soil bacteria, HTS strategies began a renaissance in drug discovery, promising an effectively unlimited supply of novel compounds to combat the emerging threat of antibiotic resistance.1 However, despite ever-expanding compound libraries and highly efficient screening methodologies, new classes of synthetic antibiotics have yet to materialize. The increasing concerns of returning to a pre-antibiotic era have proven severe enough to warrant attention and action by top governmental agencies.2,3Of the sixteen antibiotic classes used clinically, all but two were derived from environmental sources, and there is growing interest in returning to natural inspirations.1,4–6 Among these inspirations lies metal-mediated innate immunity, by which the innate immune system directly modulates environmental levels of metals such as manganese, iron, zinc, and copper at the site of infection.7,8 Through limitation, in the case of iron, zinc, and manganese or oversaturation, in the case of copper, the intrinsic properties of these ions are utilized to form a crucial line of defense against pathogens. Growing bodies of evidence point toward copper's essentiality in particular. In many systems, including Mycobacterium tuberculosis, Pseudomonas aeruginosa, Listeria monocytogenes, and Streptococcus pneumoniae, bacterial copper resistance is linked to virulence;9 conversely, attenuation of the “copper burst” within macrophages weakens the phagocytic response, promoting bacterial survival.10,11 All sequenced bacteria possess at least a rudimentary level of copper resistance machinery,12 varying from simple expression of an efflux pump,13 to a complex network including pumps, sequestration proteins, oxidases, chaperones, and transcriptional regulators.14 As free copper levels are buffered below one ion per cell in both prokaryotes15 and eukaryotes,16 that even obligate intracellular bacteria retain resistance machinery further underscores copper's profound role as an environmental and immunological insult.12
Recent advances in our understanding of copper's role in immunity have paralleled the rise of reports detailing the phenomenon of copper-dependent antibiotics. These compounds are highly inhibitory in the presence of copper, yet impotent in its absence. Example inhibitors have been described for Gram negatives,17 Gram positives,18 and mycobacteria,19 as well as eukaryotic targets including pathogenic fungi20 and cancer cells.21–23 The broad range of potential targetable pathogens, coupled with the wide array of novel mechanisms of action, has generated interest in exploiting copper's antibiotic properties.24,25 However, until now, discoveries have been largely serendipitous, or based upon established metal-binding motifs; though well suited for chemical probes, these scaffolds are often poorly adaptable to therapeutic uses. Exploring the full potential of copper-mediated therapeutics requires new motifs and a directed discovery effort to identify proof-of-principle compounds.
Here, we demonstrate for the first time the power of a copper-focused HTS screening campaign. This approach is uniquely able to uncover new interactions between copper ions and compounds in existing chemical libraries, and their subsequent synergistic inhibitory activities. A copper-biased combinatorial screen against Staphylococcus aureus revealed nearly twice as many hits as a traditional, copper-blind campaign. A reoccurring extended thiourea motif, dubbed NNSN, featured novel copper-binding properties, and was revealed by UV/Vis and NMR analysis to participate in a new interaction between the ligand and copper ion. This motif was highly inhibitory against S. aureus in a copper-dependent and copper-specific manner, yet was well tolerated in cell culture. Finally, despite their presence in many screening libraries, a chemoinformatic meta-analysis demonstrated NNSNs as previously unrecognized antistaphylococcal agents, confirming the ability of copper-biased screens to discover new compounds hiding in existing chemical space.
Methods
Bacterial strains, antibiotics and compounds
S. aureus clinical isolate SA3 (resistant to ampicillin, clindamycin, erythromycin, penicillin, and tetracycline) was characterized by and obtained in a de-identified manner from UAB Laboratory Medicine. Bacteria were routinely grown in Mueller Hinton (MH) medium (Oxoid Ltd, Basingstoke, Hampshire, England) overnight at 37 °C before inoculating plates according to assay conditions unless otherwise stated. All experiments were performed in 96-well plates using MH medium or RPMI1640 medium supplemented with trace metals. The trace metal mix was prepared as a 1000-fold stock solution containing 3 mM EDTA, 50 mM MgCl2, 0.7 mM CaCl2, 80 μM NaMoO4, 168 μM CoCl2, 0.55 mM MnCl2, 0.7 mM ZnSO4, 2 mM FeSO4. All screened compounds were randomly taken from our 43000 in-house compound library (Chembridge) or Chembridge's Hit2Lead online library (http://www.hit2lead.com). Copper sulfate and all other commercial compounds were purchased from Sigma-Aldrich. Copper treated assays were supplemented with 50 μM CuSO4 unless otherwise indicated. Other metals were used at 100 μM or as indicated.
High throughput screening assay
The HTS assay was conducted as published previously.18 Briefly, all compounds were screened at 10 μM in duplicate plates run in parallel, one containing only medium (trace-copper conditions) and one containing medium supplemented with 50 μM CuSO4. S. aureus was added to each well to achieve a final optical density (OD600) of ∼0.001 to 0.004 (1:1000 dilution of an overnight culture) in a total volume of 160 μl. All steps were performed using the Precision Power automated microplate pipetting system (BioTek). Plates were sealed with parafilm (Millipore) to minimize evaporation and incubated on a Heidolph Titramax 1000 plate shaker at 450 rpm at 37 °C for 8 hours. Optical density, as a quantitative surrogate marker of bacterial growth, was determined using a Synergy HT plate reader (BioTek). Background correction was performed against wells containing only medium and compounds that decreased the growth of S. aureus were identified and further analyzed.
HTS data analysis
An in-house algorithm was utilized for analysis. The software package used MySQL and PHP as a server backend, and a web accessible HTML interface as the front end. Hits were defined based upon number of standard deviations (1 SD = 20%) from the overall mean, which averaged 94% across all copper-replete samples after blank correction and normalization to positive controls. Independent hits were compounds with less than 34% growth (3 SDs from the mean) in both standard and copper supplemented conditions. Copper dependent hits had under 34% growth in the copper replete plate, with at least 40 percentage points (2 SD) more growth in copper trace plates (e.g., 20% growth in Cu-replete and >60% growth in Cu-trace plates). Inverse hits were the opposite, with under 34% growth in trace plates and at least 40 percentage points more growth in replete plates. Substructure analysis was completed using the Open Babel 2.3.2 chemistry toolbox, as installed on a Linux server.26 The SMARTS search string for non-cyclic thioureas was “[N;!R]C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)[N;!R],” while the NNSN substructure search was extended to “[#7][#6][N;!R]C(S)[N;!R]” to include both cyclic and non-cyclic carbons and nitrogens.
S)[N;!R],” while the NNSN substructure search was extended to “[#7][#6][N;!R]C(S)[N;!R]” to include both cyclic and non-cyclic carbons and nitrogens.
Structure activity relationship (SAR) studies (SAR-by-catalogue)
Active molecules were grouped based on the occurrence of structural motifs identified during visual and computational inspection of their chemical structures. Representative compounds and additional derivatives of each group were ordered from http://www.hit2lead.com. Activity was confirmed in dose response curves, of which we also determined the MIC (for details see below). Additional compounds featuring respective key motifs were identified in the Chembridge Hit2Lead online library using the search feature provided on the website (http://www.hit2lead.com).Determination of the minimal inhibitory concentration (MIC)
Compounds were reconstituted in sterile DMSO (Sigma-Aldrich) typically at a concentration of 10 mM, then aliquotted, and stored at −80 °C. In some instances, a decline in potency of the compounds was noted over a period of 6 months and after repeatedly freezing and thawing reconstituted compounds. Compounds were diluted in 96-well in 2-fold increments, typically covering a concentration range from ∼0.07 to 10 μM. Control wells containing media only (sterility control) and media with cells in the absence of compound with or without 50 μM copper were included as well. Assay conditions and incubation procedure were similar to that of the pilot screen. Dose–response curves were analyzed by determining the OD600 using a plate reader (Synergy HT, BioTek). Data was analyzed as the average value of 3 wells with identical conditions and normalized to the proper growth controls. MIC was defined as the concentration at which growth was reduced by at least 85%. Additional transition metals were assayed in Roswell Park Memorial Institute 1640 medium without phenol red (RPMI; Life Technologies) and supplemented with a 1:1000 dilution of a copper-free trace metal mix (RPMI1640 + TM) to promote growth.27
Eukaryotic toxicity assessment
THP-1 cells were grown in standard RPMI plus 10% FBS. Cells were plated in RPMI at 100000 cells per well in a total volume of 200 μL in a 96 well plate, and challenged with compound in the presence or absence of 15 μM Cu for 24 hours. This copper concentration was well tolerated by THP-1 cells in our system and lies within the physiological range of copper levels in blood (10–25 μM).28 Viability was read using a Guava flow cytometer, through gating on live populations as assessed through forward and side scatter measurements.
Characterization of metal complexation and structure modeling
The binding constants of CuBr in Trizma-HCl (pH = 7)/methanol (90/10 v/v) to compounds were determined according to the method of Benesi and Hildebrand.29 The binding of Cu(II) and Cu(I) enhances the UV-absorption band of APT-6i at λmax = 235 nm. The differences in UV-absorption at λmax = 235 nm were used to calculate the binding constants KB. UV/Vis spectra of 8.3, 17, 41, 83, 170 and 410 nM of APT-6i were recorded in the absence and presence of 10 μM CuBr (Sigma-Aldrich, ACS grade) at 283 K in Trizma-HCl (pH = 7.0)/methanol (1:1 v/v) using a Varian Cary 500 UV/Vis-NIR spectrophotometer and 4.0 mL quartz cuvettes. The measurements were performed under argon to avoid Cu(I) oxidation to Cu(II).
NMR titrations
1H-NMR spectra were recorded using a Bruker Avance III, 600 MHz NMR-spectrometer at 298 K. Compound APT-6i was dissolved in deuterated acetone (6.25 mM), total volume: 400 μl. Cu(I) was added from a stock solution of 1.5 × 10−4 M CuBr in deuterated acetone. 5 mL of CH3OD was added as internal standard (peak not shown).ChEMBL database meta-analysis
The ChEMBLdb relational database, version 20, was downloaded as the provided virtual machine (MyChEMBL).30,31 All SQL searches were conducted from a Linux command line interface.Results
Copper-biased HTS environment uncovers numerous copper-dependent hits
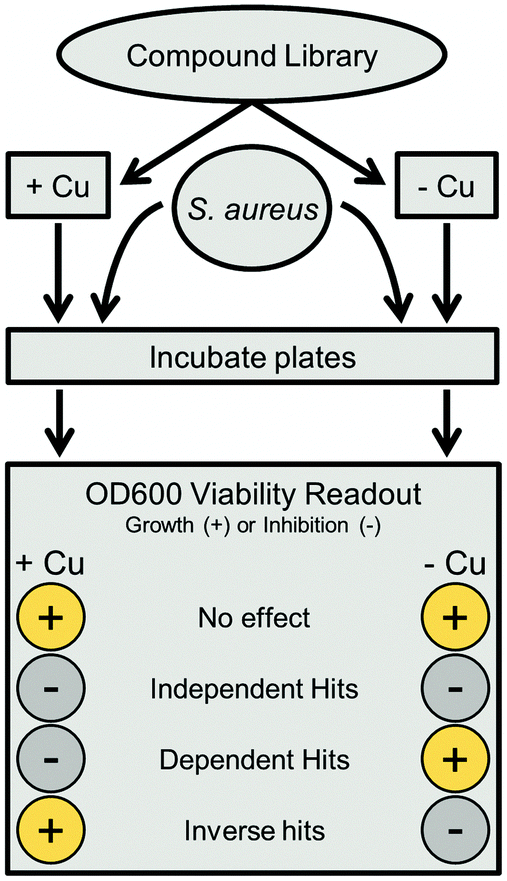 | ||
| Fig. 1 Parallel combinatorial scheme. Compounds from a master library are assayed twice in parallel, with one plate containing added copper sulfate, and the other containing only base medium. After adding S. aureus, both plates are incubated and viability is determined via OD600. Hits were classified by comparing growth values in both plates; the specific criteria used are detailed in the Methods. | ||
Screening our 10000 compound test library against a clinically isolated drug-resistant S. aureus strain (SA3) identified 129 total hits, or 1.29% of the overall number of compounds. Comparison of the hits from both the trace-copper screen (Fig. 2A) and copper-supplemented screen (Fig. 2B) revealed 70 compounds (54%) that were similarly inhibitory under both screening conditions (copper independent hits) and 6 compounds (5%) that lost their antibacterial properties in the presence of copper (copper inverse hits). Importantly, 53 molecules (41%) were found only in the presence of copper, demonstrating that copper-activated antibacterial activities occur rather frequently. Of the copper-dependent hits, over half (28) met our criteria for a primary hit, i.e., more than 90% inhibition. These copper-dependent hits represent hitherto unrecognized inhibitors, given that conventional screening campaigns for antibiotics do not contain sufficient and physiological quantities of copper ions.
 | ||
| Fig. 2 Combinatorial screening results. (A) Growth values of all 10000 compounds in standard screening medium as normalized to plate controls. The red lines represent three standard deviations above and below the mean growth value as a cutoff for hits. (B) Growth values of all compounds in medium with copper added, as normalized to plate controls. Red, green, and blue circles are Primary (growth ≤ 10%, and greater than 2 SD below the –Cu plate), Secondary (10% > growth ≤ 20%, and greater than 2 SD below the –Cu plate), and Tertiary (20% > growth ≤ 34%, and greater than 2 SD below the –Cu plate) dependent hits; grey boxes are independent hits; and orange diamonds are inverse hits. Some outliers may have fallen below the lower cut-off line but were classified as inactive because they did not meet hit criteria. | ||
logP) ≤ 5, hydrogen bond donors ≤ 5, and hydrogen bond acceptors ≤ 10. The criteria were later extended to include topological polar surface area (tPSA) ≤ 140 Å2 and rotational bonds (RB) ≤ 10. To visualize our results in aggregate, we compared Ro5 values of dependent hits, independent hits, inverse hits, and the screened library as a whole. Molecular weight only varied slightly between groups, and all were statistically indistinguishable from the library as a whole (Fig. 3A). Rotational bonds and clogP values were slightly significantly different when comparing copper-dependent hits to the screened library (p = 0.0122 and 0.0116, respectively), though all three categories clustered toward the top of the clogP and bottom of the RB ranges (Fig. 3B and C). Topological polar surface area (tPSA) differed greatly between dependent hits and all other categories (p < 0.0001), with the dependent hit median tPSA 38% lower than the library average (Fig. 3D). The most striking comparison, however, came from numbers of hydrogen bond donors and acceptors. The screened library had a relatively even distribution of both acceptors and donors (Fig. 3E), with a median of 4 acceptors and 1 donor; while the median of copper-dependent hits clustered tightly at only 1 acceptor and 2 donors (Fig. 3G). These structural differences may enable assembly or construction of custom libraries targeted towards enhanced copper-binding molecules.
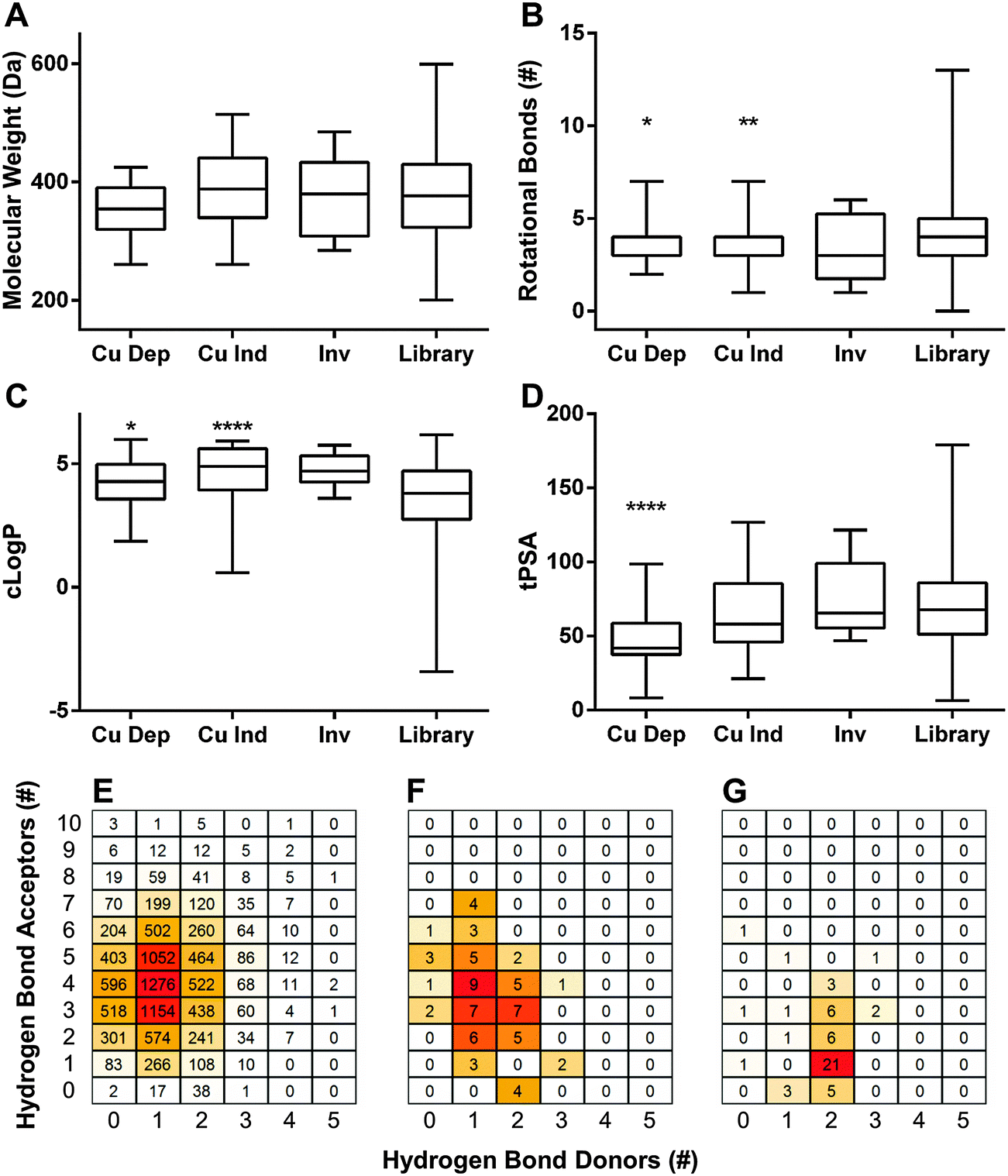 | ||
| Fig. 3 Lipinski Rule of Five characteristics. Aggregate (A) molecular weight, (B) rotational bonds, (C) calculated logP (clogP), and (D) topological polar surface area (tPSA) properties of copper-dependent hits (Cu Dep), copper-independent hits (Cu Ind), and inverse hits (Inv). All groups were compared to the screened library as a whole (Library) using a one way ANOVA and Dunnett's multiple comparisons test. * p < 0.05, ** p < 0.01, **** p < 0.0001. (E) Heatmap comparisons of the hydrogen bond acceptors and donors within the entire screened library, (F) copper-independent hits, and (G) copper-dependent hits. | ||
Extended thiourea structure comprised a well-established hit cluster
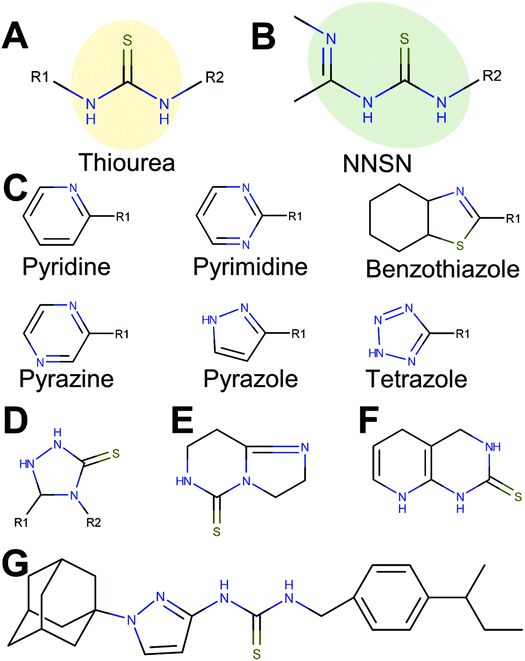 | ||
| Fig. 4 A novel copper-binding NNSN motif and side groups. 47 copper-dependent hits contained a thiourea (A), with 12 featuring an extended NNSN motif (B) that consists of the thiourea base group paired with one of 6 possible heterocyclic nitrogen-containing ring structures (C). Compounds with a rigid and inflexible NNSN motif (D, E and F) conferred no activity. (G) Full structure of APT-6i. | ||
Interestingly, 12 of the 45 thioureas featured an extended thiourea structure, which we dubbed NNSN-motif (Fig. 4B). No NNSN-motif was found among the copper-independent hits. All 12 copper-dependent NNSN molecules featured a linear thiourea structure, complemented by a heterocyclic ring system (pyrazolyl, tetrazolyl, thiazolyl, pyridinyl, pyrimidinyl or pyrazinyl) to form the full NNSN motif (Fig. 4C). Molecular flexibility appeared to be essential for activity since rigid NNSN motifs with a cyclic thiourea structure as in triazolethiones (Fig. 4D), imidazo-pyrimidine-thiones (Fig. 4E) or pyrido-pyrimidine-thiones (Fig. 4F) were inactive. A substructure search within the structures of all 10000 randomly picked molecules that were included in our screen identified 30 total non-cyclic NNSN motifs; thus, the screen discovered 40% of all possible NNSN molecules.
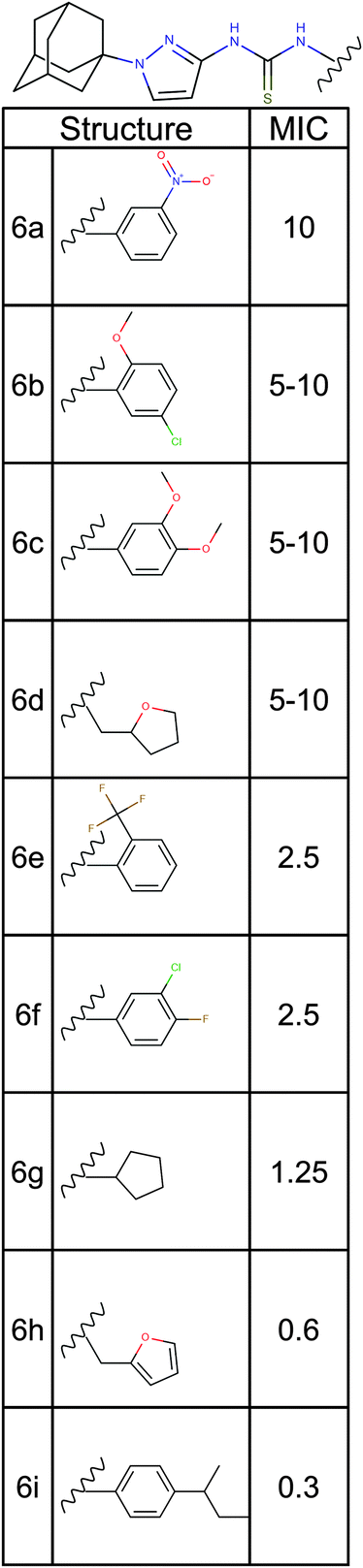 | ||
| Fig. 5 Structure activity relationship analysis. Nine analogs of a promising hit molecule were purchased from the supplier (ChemBridge) and examined for antimicrobial activity. | ||
Among the analogs tested, APT-6i (Fig. 4G) exhibited excellent copper-dependent inhibition of S. aureus (Fig. 6A), with a minimum inhibitory concentration of 0.3 μM. Additionally, APT-6i was relatively benign toward THP-1 cells, a human monocyte cell line (Fig. 6A). Activity was also entirely copper-specific, with no observed inhibition when growth media was supplemented with other transition metals such as Mn, Fe, Co and Zn (Fig. 6B). However, the presence of additional metals did not preclude activity, as full inhibition was restored upon co-incubation of Zn and Cu with APT-6i (Fig. 6B).
 | ||
| Fig. 6 APT-6i exhibits stark copper dependency and specificity. (A) Inhibitory effects of APT-6i against S. aureus (black squares) and THP-1 cells, a human monocyte line (open circles). APT-6i is active only in the presence of copper (orange line). (B) Activity of 10 μM APT-6i against S. aureus grown in RPMI to better resolve metal dependencies. Cu is included at 50 μM, and Fe, Mn, Co, and Zn are included at 100 μM. Inhibition is strictly copper-specific, and occurs in the presence of other ions, such as a Cu/Zn coincubation. All values are normalized to the respective 0 μM compound control (either with or without copper), and expressed as percentages of survival. | ||
Pyrazolyl-thioureas are functionalized through unique copper-coordination chemistry
To further examine the interaction between APT-6i and copper, we analyzed the metal–ligand complex using UV-Vis spectroscopy and 1H-NMR. UV-Vis titrations are a straightforward method to visualize metal–ligand complex formation, and revealed a main absorption peak at λ = 235 nm, with intensity strongly dependent on the APT-6i concentration (Fig. 7A). This peak represents an energy shift in electron orbitals, indicative of complex formation and subsequent π bonding between the copper ion and ligand. Further analysis through the Benesi–Hildebrand method29 produced linear plots, confirming a 1:1 stoichiometry within the ligand–metal complex (Fig. 7B). The binding constant of CuBr and APT-6i in Trizma/methanol was calculated to KB = 476700 ± 1200 (L mol−1).
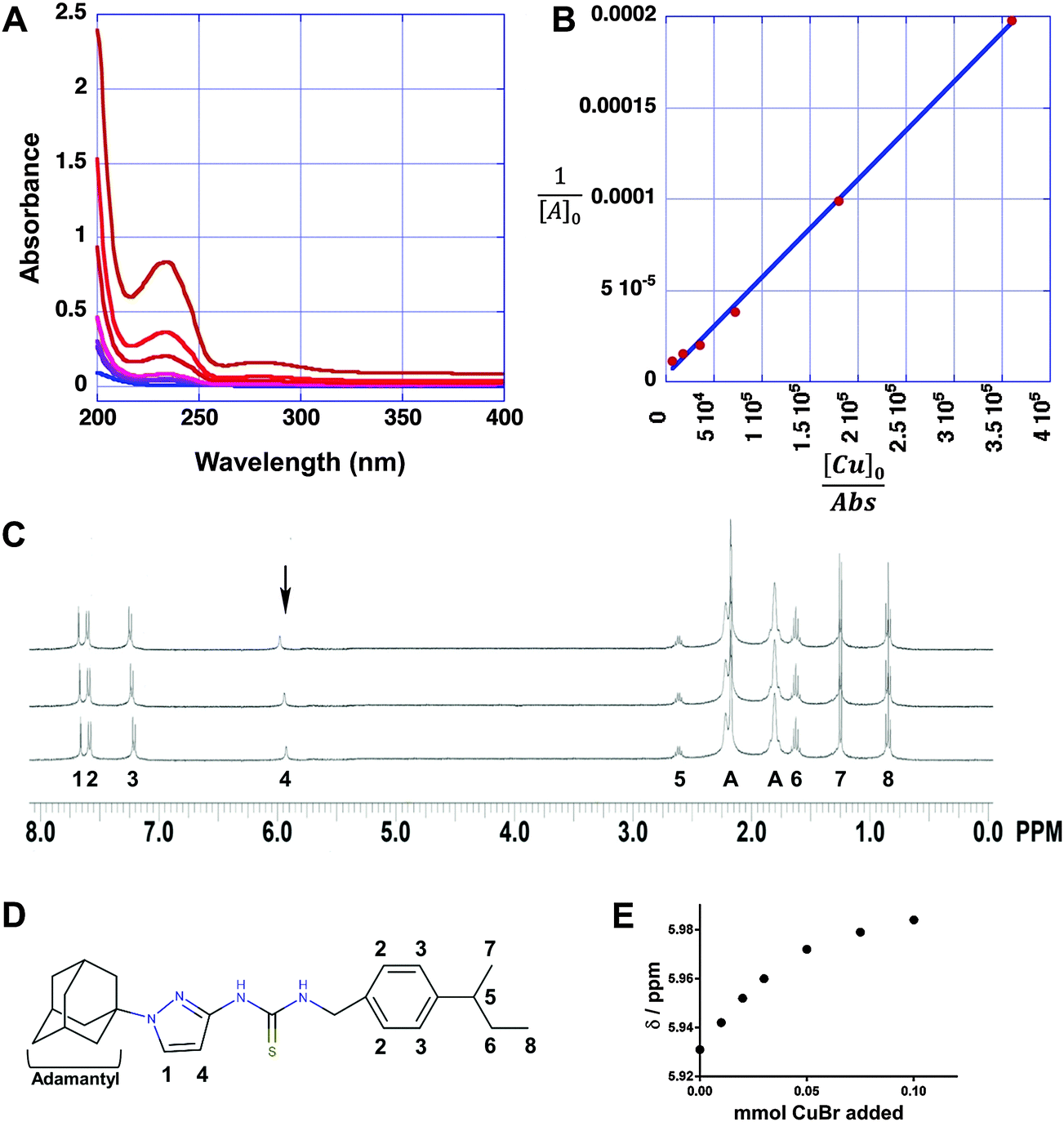 | ||
| Fig. 7 APT-6i forms a unique copper complex. (A) UV/Vis absorption spectra of compound APT-6i in the presence of 3.3 μM Cu(I)Br in Trizma-HCl (pH = 7)/methanol (90/10 v/v). Concentrations of APT-6i were 8.3 nM, 17 nM, 41 nM, 83 nM, 170 nM and 410 nM. (B) Benesi Hildebrand plots for determining the binding constant and molar absorption coefficients of APT-6i with CuBr. (C) Overlay of the 1H-NMR spectra (Bruker Avance III, 600 MHz, 298 K) of compound APT-6i without CuBr (bottom), with 50 μM CuBr (middle), and with 100 μM CuBr (top) in deuterated acetone. Peak assignments correspond to panel D with A = adamantyl. The arrow indicates a copper responsive peak shift. (D) Structure of APT-6i showing relevant peak assignments and match with panel C. (E) Shift of highlighted peak 4 with increasing concentrations of CuBr from panel C, indicating an interaction with copper ions at these sites. | ||
As we were unable to obtain crystal structures of the Cu(I) and APT-6i complex, we relied on 1H-NMR-titration with CuBr in deuterated acetone to discern the structure formed in solution (Fig. 7C). Intriguingly, the 1H-NMR titration confirmed that the observed 1:1 complexation geometry is clearly different from canonical Cu(I)-thiourea complexes, which usually feature at least two thiourea ligands per metal cation. It is especially noteworthy that both thioamide groups, but not the thiocarbonyl group, take part in the observed complexation. Following the shift of peak position 4 from δ = 5.931 to 5.984 ppm with increasing CuBr concentration (Fig. 7E), it is apparent that one of the aromatic carbons of the pyrazole unit is in close proximity to Cu(I) in solution, assuming that a CH3OD molecule (added as internal standard) is also coordinated to Cu(I) in order to obtain a slightly distorted tetrahedral geometry. The solution structure of the APT-6i-Cu(I) complex is the basis for the molecular modeling (Fig. 8A–C). From our modeling, it becomes clear that significant conformational changes are required to accommodate the complexation of a central Cu(I) cation (Fig. 8B and C).
 | ||
| Fig. 8 The APT-6i and copper complex has unique coordination chemistry. The complex's geometry was determined using UV-Vis and 1H-NMR, shown in Fig. 7. (A) The (minor) resonance structure of APT-6i is able to form a complex with Cu(I). (B) 3D representation of uncomplexed APT-6i using the CHARMM force field, showing a relatively linear structure. Non-polar hydrogens are removed for clarity. (C) 3D model of the APT-6i/Cu(I)/CH3OD complex. Coordination twists the molecule from a linear structure to a bent configuration. Copper is represented as the orange sphere, with a D1-methanol added to the coordination complex. Yellow sphere represents sulfur, blue are nitrogen atoms, grey are carbon atoms and white are polar hydrogens. | ||
More importantly to drug discovery efforts, this novel complexation could potentially reopen the door to a wide array of otherwise unattractive compounds. Although thioureas readily complexes metal ions, and are thus logical candidates for discovery by the paradigm detailed here, they are often regarded as undesirable due to common hepatotoxicity and thyroid peroxidase inhibition.37 Sulfur's lack of participation in the complex suggests the possibility of bioisoteric substitution of other functional groups or atoms, negating toxicity while retaining complexation ability and antibacterial activity. Previous reports offer precedent, though results are expectedly mixed: some substitutions, such as cyanoguanidines, retained activity or increased therapeutic indices,38 while others lost biological activity.39 A concerted SAR effort would likely produce new metal-complexing non-thioureas.
Meta-analysis reveals NNSNs as previously unrecognized antistaphylococcal agents
Having identified the NNSN motif as a promising and novel copper-dependent antibacterial structure, we examined whether this motif had previously been recognized for its therapeutic potential. We conducted a meta-analysis of published activities using the ChEMBL database, a publically available relational database containing over 13 million activity records of 1.4 million compounds, taken from multiple sources including 2700 PubChem BioAssays and tens of thousands of primary literature reports.30 The database facilitates tracking individual compounds through a large number of systems, such as biochemical assays, whole cell screens, and in vivo data, against prokaryotic and eukaryotic targets. Although ChEMBL does not generally include inactive hits, it is a rich repository of bioactive compounds in a variety of contexts.Querying the NNSN motif against the database returned 608 hits with recorded activity (Fig. 9). Narrowing the search to only whole-cell activities against S. aureus returned only 57 hits. Most of these activities, however, were pulled from batch synthesis efforts reported in the literature, rather than from a directed screening campaign. Of the entire set, 39 had reported MICs below 500 μM, and only 5 had MICs below 50 μM; of these, only a single compound had a MIC below 10 μM.40 Given that HTS are limited to testing only one concentration, often at 10 μM, it is likely none of these compounds would have been found individually in a screening campaign, and, hence, would not have been identified as a hit series.
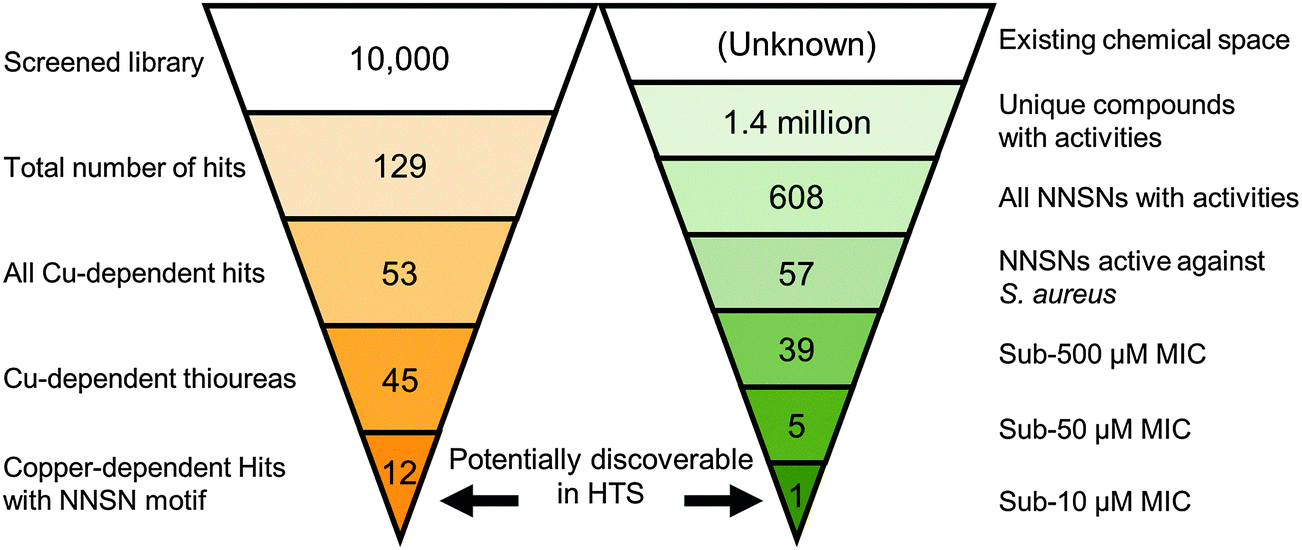 | ||
| Fig. 9 Chemoinformatic search of the ChEMBL database. The ChEMBL chemoinformatic database was queried with the NNSN motif for similar molecules. Though the limited screen described here revealed 12 NNSNs with significant antibacterial effects, only a single NNSN with activity against S. aureus below 10 μM was found within the database. | ||
Discussion
In this study, we report the results of the first high throughput screening (HTS) campaign for the discovery of copper-dependent antibacterial inhibitors. This strategy not only resulted in a much greater number of promising hit molecules than traditional screening methods, but also specifically revealed a previously unknown hit series, whose anti-staphylococcal activity escaped previous detection despite being present in many screening libraries.Though metal complexation screens could hypothetically proceed using any of the first row transition metals (Mn, Fe, Co, Ni, Cu, and Zn), copper offers the most attractive and most viable vehicle for synergistic inhibition. Copper's antimicrobial properties are multifaceted, but ultimately stem from its relative proclivity toward stable ligand complexation when compared to other transition metal ions. This phenomenon is described by the Irving–Williams series, where complex stabilities are their lowest with manganese, increase in periodic fashion until maximum stability at copper, and finally decline at zinc.41 Functionally, this gives copper a higher affinity for sulfur, oxygen, and nitrogen than any other physiologically relevant transition metal.42 This affinity, combined with copper's high redox potential, is often exploited in metallo-proteins such as superoxide dismutases or cytochrome c oxidases, and as such is key to the normal function of both prokaryotic and eukaryotic cells. However, copper ions in excess readily displace or replace crucial metalloenzyme cofactors, attack vulnerable iron–sulfur clusters, and directly damage accessible amino acid residues.15 Cumulatively, these effects heavily interfere with cellular function; as a result, all sequenced bacteria possess at least some level of copper resistance.12
As an antimicrobial, copper assaults numerous sites within the cell. Many bacteria have had specific targets identified (e.g., dehydratases in E. coli,43 aerobic nucleotide synthesis in Streptococcus pneumoniae,44 and heme biosynthesis in Neisseria gonorrhoeae45), but outside of general mechanisms such as attacking iron–sulfur clusters in proteins, specific points of failure vary widely from organism to organism. The detected source of toxicity is simply the “weakest link,” or first component to fail. It is very likely some copper-dependent inhibitors feature a similar variety of mechanisms, especially if acting through general copper overload (as in the case of 8-hydroxyquinoline20). More targeted compounds may be capable of additional mechanisms as well. GTSM, for example, acts upon the electron transport chain of N. gonorrhoeae, with its spectrum of activity hypothesized to be explained through a particular bacterium's reliance on aerobic respiration.17,25 Yet, GTSM is also a potent copper-dependent inhibitor of S. pneumoniae (ref. 25; unpublished observations), a facultative anaerobe lacking an electron transport chain. Thus, additional modes of action must be at play, indicating a multi-faceted mechanism of activity. Though the “magic bullet” paradigm of one drug-one target has dominated since its proposal by Paul Ehrlich a century ago,46 copper-dependent inhibitors may offer an alternative: since multiple individual targets could be vulnerable, development of resistance would be much more difficult than against traditional, single target antibiotics.
The allure of copper's anti-bacterial properties has not gone unnoticed.17–20,25,47–49 Unfortunately, free copper ions have little therapeutic value due to their erratic reactivity.50–53 To pharmacologically control the activity of copper ions and direct them to a specific target, numerous chemosynthetic efforts have synthesized copper complexes with greatly enhanced antibacterial activities.47,54–58 However, many of these rely on metal binding motifs that had analytical and technical purposes, rather than medicinal applications. Subsequent synthesis efforts often prioritize advancing the frontiers of organo-metallo-chemistry (e.g. mixed ligand complexes), leaving biological considerations as a secondary focus. Consequently, structure activity relationship studies, such as exemplified by the clinically used metalloantibiotic bacitracin,59 are extremely rare. While powerful in its own right, the agnosticism of this chemosynthetic strategy toward biological considerations impedes its applicability to HTS discovery. Though we have begun exploring how to harness the potential of copper in chelate based metalloantibiotics, we still need HTS solutions to probe the existing chemical space for novel metal-related activities to facilitate discovery and development of innovative metal-oriented therapeutics.
Conclusion
In summation, our work details the first concerted antibiotic HTS discovery effort to harness the activity of an unconventional antimicrobial, copper. These unique inhibitors have, until now, largely gone unnoticed within conventional screening libraries, offering a way to repurpose and reprobe existing chemical collections. Though therapeutically infeasible on its own, copper's potential can be readily exploited through combination with small organic molecules, offering a promising new approach in the battle for novel antibacterials.Acknowledgements
The authors would like to thank Saran Kupul for excellent technical assistance and lab management. Some research of this study was supported by the University of Alabama at Birmingham's (UAB) Department of Medicine, the Center for AIDS Research (UAB CFAR) and its Cytometry Core/Joint UAB Flow Cytometry Core which are funded by NIH/NIAID P30 AI027767 and by NIH 5P30 AR048311, and in part by NIH grant RO1 AI104952 to F. W. In addition, S. H. B. thanks the National Science Foundation for supporting this study (DMR-1242765 and CBET-1337438), as well as Kansas State University, Department of Chemistry. The funding agencies had no role in study design, data collection and interpretation, or the decision to submit the work for publication.Notes and references
- K. Lewis, Nat. Rev. Drug Discovery, 2013, 12, 371–387 CrossRef CAS PubMed.
- T. W. House, http://www.whitehouse.gov/the-press-office/2014/09/18/executive-order-combating-antibiotic-resistant-bacteria, 2014.
- CDC, http://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf, 2013.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089–1093 CrossRef CAS PubMed.
- J. Clardy, M. A. Fischbach and C. T. Walsh, Nat. Biotechnol., 2006, 24, 1541–1550 CrossRef CAS PubMed.
- A. L. Demain, Nat. Biotechnol., 2002, 20, 331 CrossRef PubMed.
- M. I. Hood and E. P. Skaar, Nat. Rev. Microbiol., 2012, 10, 525–537 CrossRef CAS PubMed.
- Y. Fu, F. M. Chang and D. P. Giedroc, Acc. Chem. Res., 2014, 47, 3605–3613 CrossRef CAS PubMed.
- V. Hodgkinson and M. J. Petris, J. Biol. Chem., 2012, 287, 13549–13555 CrossRef CAS PubMed.
- C. White, T. Kambe, Y. G. Fulcher, S. W. Sachdev, A. I. Bush, K. Fritsche, J. Lee, T. P. Quinn and M. J. Petris, J. Cell Sci., 2009, 122, 1315–1321 CrossRef CAS PubMed.
- M. D. Johnson, T. E. Kehl-Fie, R. Klein, J. Kelly, C. Burnham, B. Mann and J. W. Rosch, Infect. Immun., 2015, 83, 1684–1694 CrossRef CAS PubMed.
- M. Solioz, H. K. Abicht, M. Mermod and S. Mancini, JBIC, J. Biol. Inorg. Chem., 2010, 15, 3–14 CrossRef CAS PubMed.
- K. Y. Djoko, J. A. Franiek, J. L. Edwards, M. L. Falsetta, S. P. Kidd, A. J. Potter, N. H. Chen, M. A. Apicella, M. P. Jennings and A. G. McEwan, Infect. Immun., 2012, 80, 1065–1071 CrossRef CAS PubMed.
- X. Shi and K. H. Darwin, Metallomics, 2015, 7, 929–934 RSC.
- A. W. Foster, D. Osman and N. J. Robinson, J. Biol. Chem., 2014, 289, 28095–28103 CrossRef CAS PubMed.
- T. D. Rae, P. J. Schmidt, R. A. Pufahl, V. C. Culotta and T. V. O'Halloran, Science, 1999, 284, 805–808 CrossRef CAS PubMed.
- K. Y. Djoko, B. M. Paterson, P. S. Donnelly and A. G. McEwan, Metallomics, 2014, 6, 854–863 RSC.
- M. Haeili, C. Moore, C. J. C. Davis, J. B. Cochran, S. Shah, T. B. Shrestha, Y. Zhang, S. H. Bossmann, W. H. Benjamin, O. Kutsch and F. Wolschendorf, Antimicrob. Agents Chemother., 2014, 58, 3727–3736 CrossRef PubMed.
- A. G. Dalecki, M. Haeili, S. Shah, A. Speer, M. Niederweis, O. Kutsch and F. Wolschendorf, Antimicrob. Agents Chemother., 2015, 59, 4835–4844 CrossRef CAS PubMed.
- R. A. Festa, M. E. Helsel, K. J. Franz and D. J. Thiele, Chem. Biol., 2014, 21, 977–987 CrossRef CAS PubMed.
- S. M. Hadi, M. F. Ullah, A. S. Azmi, A. Ahmad, U. Shamim, H. Zubair and H. Y. Khan, Pharm. Res., 2010, 27, 979–988 CrossRef CAS PubMed.
- S. M. Schmitt, M. Frezza and Q. P. Dou, Front. Biosci., 2012, 4, 375–391 CrossRef.
- M. A. Cater, H. B. Pearson, K. Wolyniec, P. Klaver, M. Bilandzic, B. M. Paterson, A. I. Bush, P. O. Humbert, S. La Fontaine, P. S. Donnelly and Y. Haupt, ACS Chem. Biol., 2013, 8, 1621–1631 CrossRef CAS PubMed.
- O. Neyrolles, F. Wolschendorf, A. Mitra and M. Niederweis, Immunol. Rev., 2015, 264, 249–263 CrossRef CAS PubMed.
- K. Y. Djoko, M. M. Goytia, P. S. Donnelly, M. A. Schembri, W. M. Shafer and A. G. McEwan, Antimicrob. Agents Chemother., 2015, 59, 6444–6453 CrossRef CAS PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- A. Speer, T. B. Shrestha, S. H. Bossmann, R. J. Basaraba, G. J. Harber, S. M. Michalek, M. Niederweis, O. Kutsch and F. Wolschendorf, Antimicrob. Agents Chemother., 2013, 57, 1089–1091 CrossRef CAS PubMed.
- T. Lech and J. K. Sadlik, Biol. Trace Elem. Res., 2007, 118, 16–20 CrossRef CAS PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- A. P. Bento, A. Gaulton, A. Hersey, L. J. Bellis, J. Chambers, M. Davies, F. A. Krüger, Y. Light, L. Mak, S. McGlinchey, M. Nowotka, G. Papadatos, R. Santos and J. P. Overington, Nucleic Acids Res., 2014, 42, D1083–1090 CrossRef CAS PubMed.
- M. Davies, M. Nowotka, G. Papadatos, F. Atkinson, G. van Westen, N. Dedman, R. Ochoa and J. Overington, Challenges, 2014, 5, 334 CrossRef.
- J. P. Lisher and D. P. Giedroc, Front. Cell. Infect. Microbiol., 2013, 3, 91 Search PubMed.
- O. M. Goudouri, E. Kontonasaki, U. Lohbauer and A. R. Boccaccini, Acta Biomater., 2014, 10, 3795–3810 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- T. H. Keller, A. Pichota and Z. Yin, Curr. Opin. Chem. Biol., 2006, 10, 357–361 CrossRef CAS PubMed.
- J. Liu, D. Obando, V. Liao, T. Lifa and R. Codd, Eur. J. Med. Chem., 2011, 46, 1949–1963 CrossRef CAS PubMed.
- G. F. Smith, in Progress in Medicinal Chemistry, ed. G. Lawton and D. R. Witty, Elsevier, 2011, vol. 50, pp. 1–47 Search PubMed.
- H. J. Petersen, C. K. Nielsen and E. Arrigoni-Martelli, J. Med. Chem., 1978, 21, 773–781 CrossRef CAS PubMed.
- M. Högberg, P. Engelhardt, L. Vrang and H. Zhang, Bioorg. Med. Chem. Lett., 2000, 10, 265–268 CrossRef.
- G. S. Hassan, S. M. El-Messery, F. A. M. Al-Omary, S. T. Al-Rashood, M. I. Shabayek, Y. S. Abulfadl, E.-S. E. Habib, S. M. El-Hallouty, W. Fayad, K. M. Mohamed, B. S. El-Menshawi and H. I. El-Subbagh, Eur. J. Med. Chem., 2013, 66, 135–145 CrossRef CAS PubMed.
- H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192–3210, 10.1039/JR9530003192.
- D. Nies, in Molecular Microbiology of Heavy Metals, ed. D. Nies and S. Silver, Springer Berlin Heidelberg, 2007, ch. 75, vol. 6, pp. 117–142 Search PubMed.
- L. Macomber and J. A. Imlay, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8344–8349 CrossRef CAS PubMed.
- M. D. Johnson, T. E. Kehl-Fie and J. W. Rosch, Metallomics, 2015, 7, 786–794 RSC.
- K. Y. Djoko and A. G. McEwan, ACS Chem. Biol., 2013, 8, 2217–2223 CrossRef CAS PubMed.
- K. Strebhardt and A. Ullrich, Nat. Rev. Cancer, 2008, 8, 473–480 CrossRef CAS PubMed.
- K. Y. Djoko, M. M. Goytia, P. S. Donnelly, M. A. Schembri, W. M. Shafer and A. G. McEwan, Antimicrob. Agents Chemother., 2015, 59, 6444–6453 CrossRef CAS PubMed.
- G. Grass, C. Rensing and M. Solioz, Appl. Environ. Microbiol., 2011, 77, 1541–1547 CrossRef CAS PubMed.
- X. Liu, X. Li, Z. Zhang, Y. Dong, P. Liu and C. Zhang, Biol. Trace Elem. Res., 2013, 154, 150–155 CrossRef CAS PubMed.
- G. J. Cooper, Drugs, 2011, 71, 1281–1320 CrossRef CAS PubMed.
- L. Macomber, C. Rensing and J. A. Imlay, J. Bacteriol., 2007, 189, 1616–1626 CrossRef CAS PubMed.
- C. Rensing and G. Grass, FEMS Microbiol. Rev., 2003, 27, 197–213 CrossRef CAS PubMed.
- P. Szymanski, T. Fraczek, M. Markowicz and E. Mikiciuk-Olasik, BioMetals, 2012, 25, 1089–1112 CrossRef CAS PubMed.
- M. L. Beeton, J. R. Aldrich-Wright and A. Bolhuis, J. Inorg. Biochem., 2014, 140, 167–172 CrossRef CAS PubMed.
- B. Bottari, R. Maccari, F. Monforte, R. Ottana, E. Rotondo and M. G. Vigorita, Bioorg. Med. Chem. Lett., 2000, 10, 657–660 CrossRef CAS PubMed.
- Z. H. Chohan, M. Arif, Z. Shafiq, M. Yaqub and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2006, 21, 95–103 CrossRef CAS PubMed.
- Z. H. Chohan, H. Pervez, A. Rauf, A. Scozzafava and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2002, 17, 117–122 CrossRef CAS PubMed.
- H. Gershon, R. Parmegianir and W. J. Nickerson, Appl. Microbiol., 1962, 10, 556–560 CAS.
- L. J. Ming and J. D. Epperson, J. Inorg. Biochem., 2002, 91, 46–58 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |