
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A time-course analysis of changes in cerebral metal levels following a controlled cortical impact†
Stuart D.
Portbury
a,
Dominic J.
Hare
 ab,
Charlotte
Sgambelloni
a,
David I.
Finkelstein
a and
Paul A.
Adlard
*a
ab,
Charlotte
Sgambelloni
a,
David I.
Finkelstein
a and
Paul A.
Adlard
*a
aThe Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Kenneth Myer Building, 30 Royal Parade, Parkville, Victoria 3052, Australia. E-mail: paul.adlard@florey.edu.au; Fax: +61 3 90353103; Tel: +61 3 90356775
bElemental Bio-imaging Facility, University of Technology Sydney, Thomas Street, Broadway, New South Wales 2007, Australia
First published on 11th December 2015
Abstract
Traumatic brain injury (TBI) is complicated by a sudden and dramatic change in brain metal levels, including iron (Fe), copper (Cu) and zinc (Zn). Specific ‘metallo-pathological’ features of TBI include increased non-heme bound Fe and the liberation of free Zn ions, both of which may contribute to the pathogenesis of TBI. To further characterise the metal dyshomeostasis that occurs following brain trauma, we performed a quantitative time-course survey of spatial Fe, Cu and Zn distribution in mice receiving a controlled cortical impact TBI. Images of brain metal levels produced using laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) in the upper quadrant of the ipsilateral hemisphere were compared to the corresponding contralateral hemisphere, together with regional areas radiating toward the center of the brain from the site of lesion. Significant regional and time point specific elevations in Fe, Zn and Cu were detected immediately and up to 28 days after TBI. The magnitude and timeframe of many of these changes suggest that TBI results in a pronounced and sustained alteration in normal metal levels within the brain. Such alterations are likely to play a role in both the short- and long-term consequences of head trauma and suggest that pharmacological modulation to normalize these metal levels may be efficacious in improving functional outcome.
Introduction
Traumatic Brain Injury (TBI) is a significant international health concern with diverse etiology and epidemiological patterns.1 It is the leading cause of mortality and disability in high-income countries, and the global incidence of TBI is constantly rising.2 Moreover, according to statistics compiled by the Brain Trauma Foundation of America, TBI is the leading cause of death and disability in children and adults from ages 1 to 44. In the United States alone, it is estimated that 1.7 million TBI's occur annually, and in 2006, 5.3 million people were currently living with long-term or permanent disabilities as a result of brain injury.3 Common causes of TBI include motor vehicle accidents, occupational hazards, physical violence and falls,3 and complications following TBI contribute to a significant public health burden and may be antecedent to alternate forms of mortality, particularly in the elderly.Similarly, contact sport engagement (e.g. American and Association football, ice hockey and combat sports) can also culminate in long-term cognitive disabilities.4–6 This is often in the form of chronic traumatic encephalopathy (CTE) and its subsequent behavioral and neuropathological effects,7 resulting from mild or repetitive TBI experienced during the normal conduct of the sport. Additionally, military blast-induced TBI in the theaters of war in Iraq and Afghanistan have been dubbed as the ‘signature injury’ of these conflicts (in a similar, yet better-characterized way to Gulf War Syndrome from the 1991 campaign), resulting in long-term cognitive deficits in affected individuals.8 Since 2003 over 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U.S. service members deployed to the Middle East and Central Asia have been officially diagnosed with TBI,9 accounting for approximately two thirds of the documented head injuries sustained in these conflicts.10
000 U.S. service members deployed to the Middle East and Central Asia have been officially diagnosed with TBI,9 accounting for approximately two thirds of the documented head injuries sustained in these conflicts.10
It is clear that cognitive impairment is one of the most predominant symptoms that occurs post-TBI, and this can occur even after a mild TBI, which is now a recognized source of morbidity and which has been demonstratively linked to long-term cognitive complaints.11 There are a breadth of other consequences of head injury, including somatic symptoms (fatigue, headaches, dizziness) and affective symptoms (irritability, impairments in executive function, depression, anxiety, emotional lability; all which may contribute to suicide). Moreover, TBI and mild TBI have been shown to precede and potentially initiate the onset of several neurodegenerative disorders such as Alzheimer's disease (AD),12 amyotrophic lateral sclerosis (ALS)13 and Parkinson's disease (PD).14
To date, and despite significant effort, there is no therapeutics available to prevent or alleviate cognitive dysfunction following TBI. One of the avenues that is being explored, however, is metal ion homeostasis.
Metals play a fundamental role in biological processes within the central nervous system,15 and the biological transition metals iron (Fe), copper (Cu) and zinc (Zn) have been proposed to play major roles in the pathogenesis of AD, PD and ALS.16,17 Subsequently, their modulation has been proposed as a potential pathway for therapeutic intervention in these and other neurodegenerative disorders.18 The literature further suggests that Zn homeostasis may be an important factor in the pathobiology of TBI, with accumulations of Zn shown at the site of brain injury in rodents19 with a specific loss of Zn from pre-synaptic boutons and a concomitant appearance of Zn in injured neurons20—the latter which may arise from a number of sources (such as from the pre-synaptic neuron or post-synaptic Zn-sequestering proteins). These fluctuations in Zn are hypothesised to be neurotoxic, potentially via excitotoxicity, and have been suggested to contribute to the neurodegeneration and cell death that follows multiple forms of brain injury.21 Recent reports have also suggested a potential role for Fe in brain injury (human TBI and stroke), where there is a reported acute elevation in Fe levels22,23 that is independent of the heme-bound Fe associated with the blood leakage within the site of injury. The cause of this is not understood, but it catalyzes the production of toxic reactive oxygen species and is a major compounding factor in oxidative stress, inflammation, and excitotoxicity; thus, similar to Zn, it is likely to contribute to cell loss. Finally, Cu, which is considered essential to proper myelination of axons, may be deficient following brain trauma, potentially playing a role in the development of the hallmark pathology of diffuse axonal injury (DAI) that is a feature of TBI.24
Considering the evidence supporting a link between the pathobiology of TBI and metal homeostasis,25 together with the potential for metal ions to be a tractable therapeutic target, in this study we utilised laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) imaging to further characterize the spatio-temporal modulation of metal ion homeostasis in response to a controlled cortical impact model of TBI in mice.
Experimental
Animal ethics
All procedures were carried out in accordance with protocols approved by the Howard Florey Animal Ethics Committee and were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes as described by the National Health and Medical Research Council of Australia.Controlled cortical impact model
Controlled cortical impact (CCI) was carried out using a Hatteras PCI 3000 precision cortical impactor (Hatteras Instruments, Cary, NC). 3 month-old male C57Bl6 mice were anesthetized via intraperitoneal injection of 100 mg kg−1 ketamine and 10 mg kg−1 xylazine. Anesthesia was monitored via respiration and pedal retraction reflexes. Upon adequate anesthesia the surgical site was clipped and shaved and cleansed with 70% ethanol. A 10 mm mid-line incision was made over the skull, and the skin and fascia were reflected to make a 4 mm craniotomy on the central aspect of the right parietal bone using a motorized drill. Excised bone was placed in sterile PBS until resituating post-injury. The animal was then positioned in a stereotaxic frame. The 3 mm tip of the impactor was lowered to the surface of the exposed dura, signified by an audible contact alarm, and the injury subsequently delivered (3 m s−1 velocity with a 1.5 mm penetration depth). The excised bone was then replaced and glued in position with super glue, the skin similarly glued together and the animal then placed in a 37 °C heated cage until completely ambulatory.Animals (n = 5 per group) were euthanized at 24 hours, 72 hours, 7 day, 14 day and 28 day-post surgeries. Animals were anesthetized with Nembutal, before a 50 ml pericardial perfusion with ice-cold 4% paraformaldehyde27 in PBS. Brains were rapidly dissected and further incubated in PFA (4% in PBS at 4 °C), followed by immersion in a cryoprotective solution for 7 days (30% sucrose in PBS, 4 °C). Sections were subsequently cryostat sectioned at 30 μm intervals at −20 °C and mounted on standard microscope slides. Sections were stored at −80 °C prior to analysis, when they were air-dried at room temperature. This specific fixation method and subsequent cryosectioning procedure has been previously described and demonstrated as a suitable method for biological metal analysis in murine neurological tissue.28 For Perls staining, tissue was blocked for endogenous peroxidase with 0.3% H2O2 for 10 minutes, washed with distilled H2O for five minutes, before a 2 hour 37 °C incubation in 7% potassium ferrocyanide in 3% HCl solution. Reaction was enhanced using DAB (DAKO DAB + Substrate Chromogen System #K3468) until brown reaction occurred. Tissue was counterstained with 5 dips of Harris haematoxylin followed by a 30 second immersion in Scotts tap water. Stained tissue was rinsed in running tap water before dehydration and mounting for further microscopic analysis.
Laser ablation-inductively coupled plasma-mass spectrometry imaging
Analysis was performed using a New Wave Research UP213 laser ablation system with a two-volume large format cell (ablation area 25 × 25 cm). This system was hyphenated to an Agilent Technologies 7500ce ICP-MS fitted with ‘cs’ lenses for enhanced sensitivity. Quantitative data was obtained by representative ablation of matrix-matched tissue standards produced according to the protocol previously reported.29The experiment was performed in a single batch. Horizontal lines of ablation were drawn across both ipsilateral and contralateral (CLS) upper quadrants of the selected section. A beam diameter of 30 μm was used; traversing the section at a speed of 90 μm s−1, laser fluence of 0.3 J cm−2 and repetition rate of 20 Hz. The IC-PMS was configured to collect 3 data points per second, thus the resultant images had pixel dimensions equivalent to 30 μm2 (total pixel area = 900 μm2).29
Image production and statistical analysis
Images were produced by reducing multiple ablation lines into ASCII data files for importing into ENVI 6.0 (Exelis Visual Information Solutions), in which regions of interest were extracted and statistically analysed. Each ROI represents a quadrant that radiates outwards from the impact site towards the center of the brain (Fig. 1). These regions were selected to allow the differentiation of discrete functional brain areas surrounding the injury site that may be differentially affected by the TBI. ROI1 has a diameter of 2152 ± 465 μm, ROI2 is 973.9 ± 323.8 μm (minus the diameter of ROI1) and ROI3 is 1680 ± 330 μm (minus ROIs 1 and 2). The entire hemisphere, which is the whole quadrant scanned, is 4706 ± 330 μm. The numbers apply equally to both the ipsilateral and contralateral side, so ideally they are equivalent.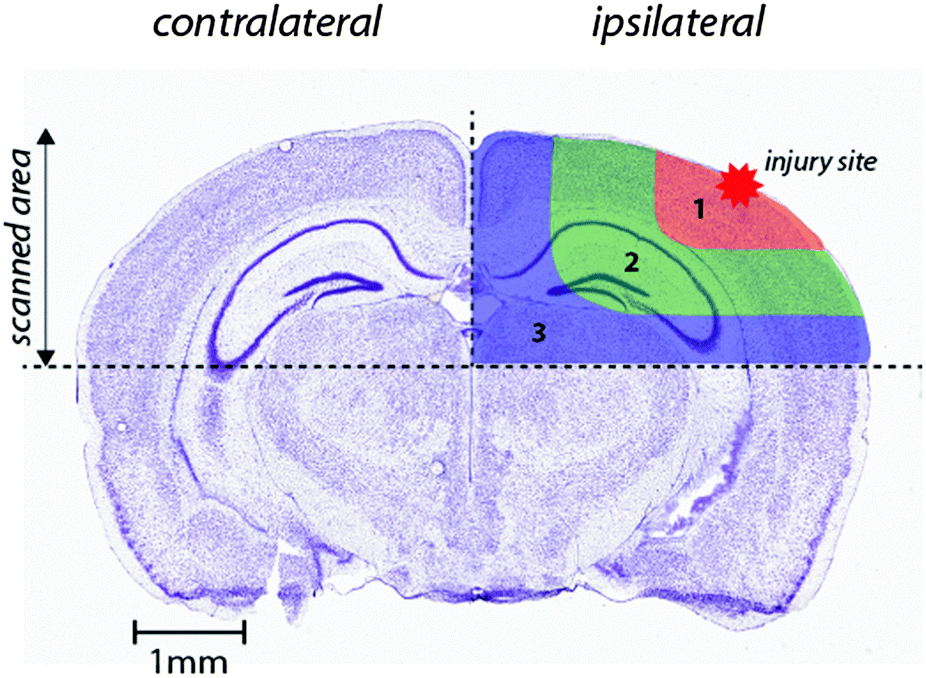 | ||
| Fig. 1 Schematic demonstrating regions of interest selection for assessment of changes in metal levels radial to the site of impact. Selected ROIs were applied to each measured metal and extracted for statistical comparisons. Whole-hemisphere metal concentrations and equivalent ROIs on the contralateral side were also extracted. Nissl stained reference section (bregma −2.255 mm) used from the Allen Brain Atlas.26 | ||
Statistical analysis was carried out in Prism (Graph-Pad). Analysis was carried out using either a two-tailed t-test with the level of significance set at p = 0.05 or a two-way ANOVA as appropriate. Where appropriate, a standard Q-test was used to identify outliers, which were then excluded from statistical analysis.
Results
Time course LA-ICPMS images of the upper left and right hemispheres are shown in Fig. 2. Full quantitative data is presented in the ESI.† Iron concentration was significantly increased within the immediate vicinity of the impact site (ROI1 p < 0.001; Fig. 3a) as well as in the two additional ROIs extending radially from the lesion (ROI2 p < 0.01; ROI3 p < 0.05; Fig. 3b and c). Furthermore, the entire hemisphere comparison between ipsilateral and contralateral revealed a concomitant significant increase in Fe (Fig. 3d), though it was obvious that the most significant variation occurred at the location of the induced injury. ROI1 revealed the largest increase in Fe, followed by ROI2, and then ROI3. Fe concentration increased in all ROIs and at every time point yielding the largest increase at 28 days post injury (Fig. 3).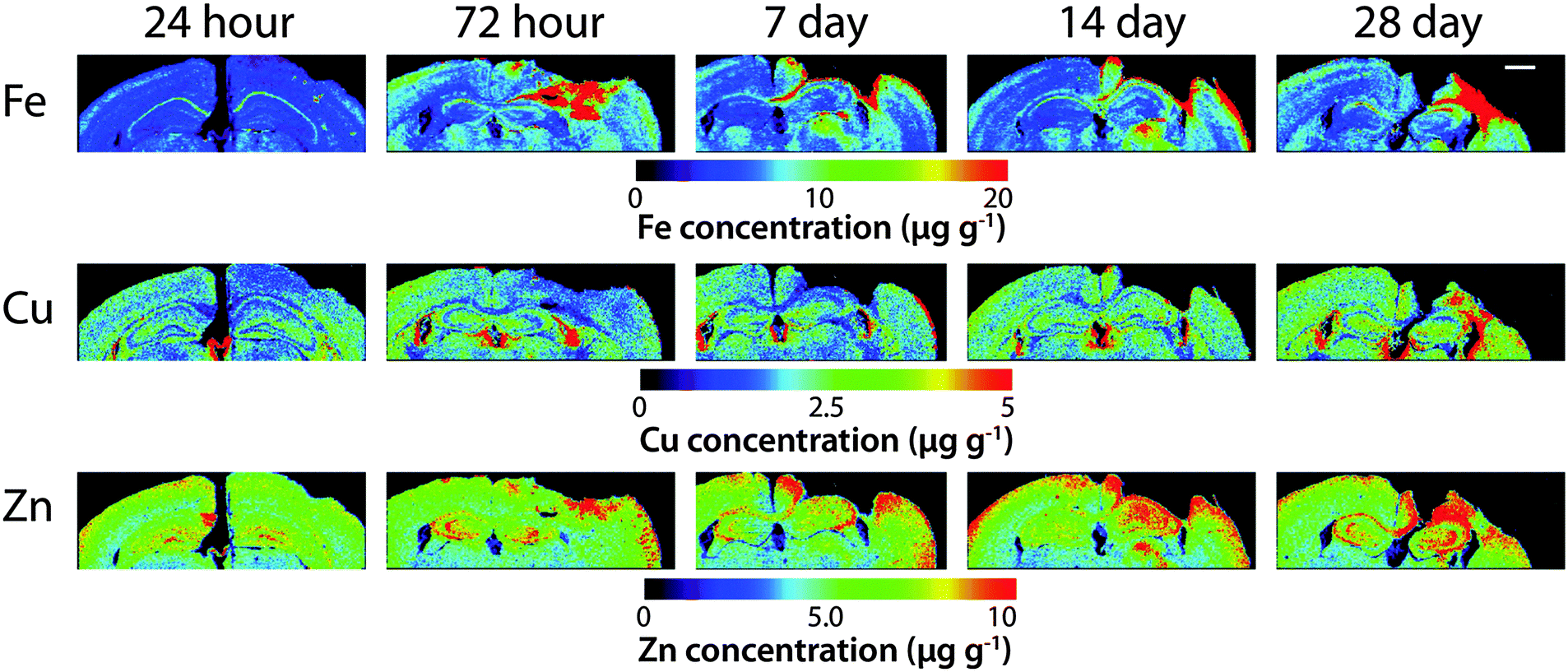 | ||
| Fig. 2 Representative LA-ICP-MS images of the upper two quadrants of both hemispheres from lesioned (right side) taken at approximately bregma −2.7 mm from 24 hours to 28 days post-lesion. Images were scanned at a spatial resolution of 30 μm. Scale bar = 1 mm. | ||
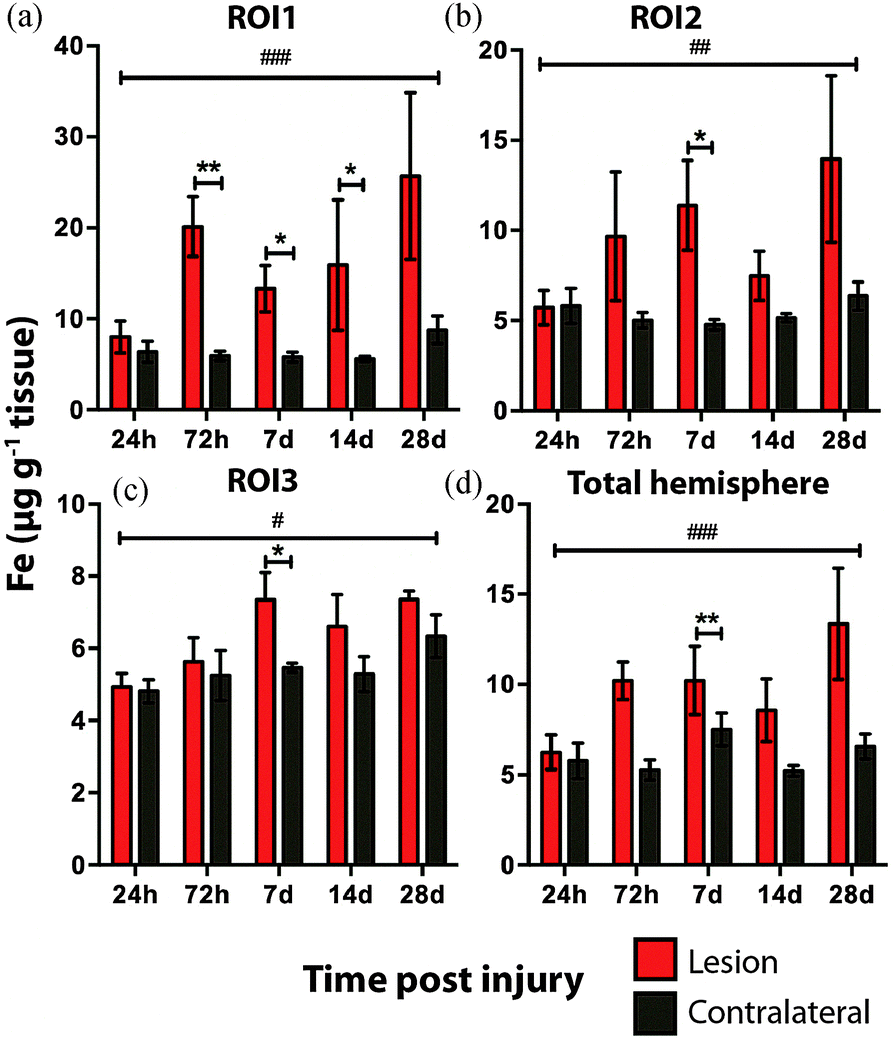 | ||
| Fig. 3 Iron concentrations across the time course following controlled cortical impact TBI. In ROI 1 there was a significant difference in Fe levels between the ipsilateral and contralateral sides across the time course (ANOVA, p = 0.0003). ROI1 intra-day comparisons also revealed significant differences between ipsilateral and contralateral sides at 72 h, 7 day and 14 day time points. There was also a significant trend to increasing Fe across the time course in ROI2 (ANOVA, p = 0.0018), ROI3 (ANOVA, p = 0.0161), and total hemisphere (ANOVA, p = 0.0002). Intra-day comparison revealed significant increase at day 7 in each region and over the total hemisphere. *p < 0.05, **p < 0.01, *p < 0.05. | ||
Analysis of the spatial distribution and concentration of Zn revealed no significant changes between the ipsilateral and contralateral regions of interest (Fig. 4a–c), nor was there a significant difference between the combined upper halves of the hemispheres (Fig. 4d). ROI1 showed a mild but insignificant increase in Zn from 24 hours to 28 days post TBI, as did ROI2 across the same time period. ROI3 similarly showed a mild but insignificant increase in Zn to 28 days, though this commenced at day 7 rather than within 24 hours of injury. Intra-day analysis revealed a significant increase in Zn between the ipsilateral and contralateral sides at day 7 in ROI1 and ROI2. Collectively, analysis of the entire hemisphere showed a mild but insignificant increase in Zn overall from day 7 to 28. Preceding the day 7 time point there was a mild yet insignificant decrease in Zn overall in the entire hemisphere (Fig. 4d).
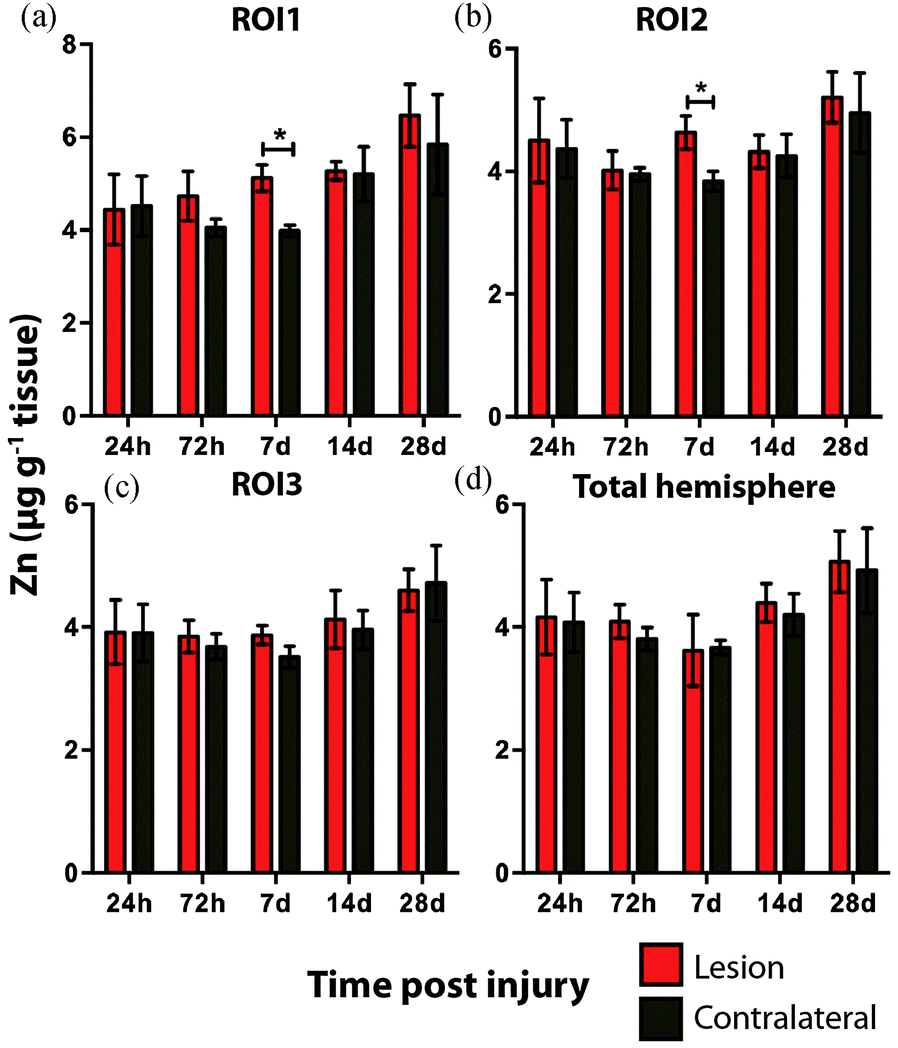 | ||
| Fig. 4 Zinc concentrations across the time course following controlled cortical impact TBI. There was no significant difference in Zn levels between the ipsilateral and contralateral sides in ROI1, ROI2, ROI3, and total hemisphere (2-way ANOVA). Intra-day analysis revealed a significant increase in Zn between the ipsilateral and contralateral sides at day 7 in ROI 1 and ROI 2. *p < 0.05. | ||
There was no overall change in Cu concentration in ROIs 1 (Fig. 5a), 2 (Fig. 5b), or 3 (Fig. 5c), nor was there any significant alteration in Cu levels when comparing the entire hemispheres (Fig. 5d). In each ROI Cu concentration was remarkably stable at 24 h, 72 h and 7 day time points. Thereafter Cu concentration trended to an increase when comparing ipsilateral to contralateral ROIs. Comparing the ipsilateral only ROIs, however, revealed a significant increase between time points 24 h and 28 days, in both ROI1 and ROI2, though not in ROI3. As expected a similar comparison of contralateral time points only revealed no significant differences in any region. Due to the low abundance of cobalt (Co) in the brain, our previous results in humans demonstrating an increase in response to TBI by solution nebulisation ICP-MS was not observed using this LA-ICP-MS method.30
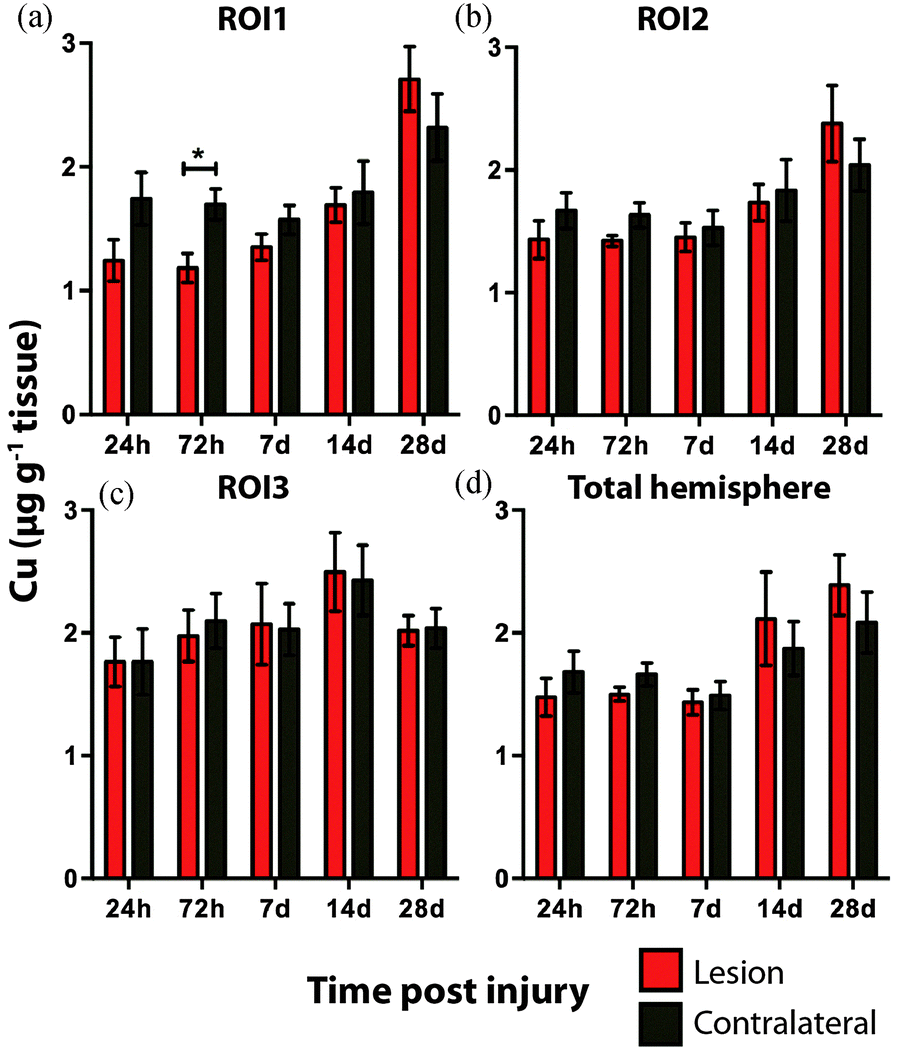 | ||
| Fig. 5 Copper concentrations across the time course following controlled cortical impact TBI. There was no significant difference in Cu levels between the ipsilateral and contralateral sides in ROI1, ROI2, ROI3, and total hemisphere (2-way ANOVA). Intra-day analysis revealed no significant increase in Cu between the ipsilateral and contralateral sides at any time point in any region. *p < 0.05. | ||
Discussion
The literature demonstratively indicates that Zn, Fe, and Cu may have pathological roles in TBI outcomes.23,24,31 Most studies, however, have only examined metal homeostasis in the acute response to trauma. Therefore, to further define how metals are regulated following TBI, we have utilised LA-ICPMS to quantitatively assess the spatial distribution of metals in the brain at up to 28 days post-injury. The metals Zn, Fe and Cu were selected for analysis because of their reported role not just in TBI, but also in other neurodegenerative disorders that exhibit similar clinico-pathological features as TBI.25Zinc is an integral part of normal brain function,32 however, when the brain is compromised through disease or injury, there is a resulting Zn dyshomeostasis that has been hypothesized to play an integral role in the pathogenesis of TBI.31 Zinc has been demonstrated to be both neuroprotective and neurotoxic in TBI models.33–36 Nevertheless there is sound evidence that the synaptic release of Zn from pre-synaptic boutons can cause injury to postsynaptic neurons under excitotoxic conditions.20,37
Hence, the literature indicates that Zn may have a critical yet somewhat temporally ill-defined role in the neuronal death that characterizes TBI. It is therefore curious that we did not observe any significant alterations in Zn following TBI. A plausible explanation for this could be that total Zn levels do not change post TBI, but rather the cellular translocation of Zn may be critical, as noted in other studies.20,37 A finer delineation of regions of interest at the sub-μm level, utilizing synchrotron XFM analysis,38 or micro-particle induced X-ray emission (μPIXE) analysis,39 may therefore be required to reveal alterations in Zn concentrations. There was the suggestion, however, of a slight decrease in Zn at the first three time points, only to then slightly yet non significantly increase for the final two time points. As Zn is postulated to play a role in depression and mood disorders,40 a common yet non-immediate outcome after TBI and mild TBI, it would be of interest to determine if Zn continues to increase beyond the 28 day time point. It is therefore possible that the interaction of Zn in TBI is unlikely to be defined by a simple linear relationship immediately post trauma, thus requiring further investigation.
In TBI, Fe elevation has been observed in the brain after injury23 with a concomitant increase in the Fe handling protein ferritin.22 In excess, Fe can induce neurotoxicity by its ability to promote the formation of reactive oxygen species (ROS) leading to oxidative stress and cell death. Moreover, Fe has been shown to associate with aggregation prone proteins that activate cell death pathways in neurodegenerative diseases such as AD, PD, and ALS.41
It is interesting to note that within the first 24 hours after TBI the Fe concentration appears to be unchanged in any ROI or within the entire injured hemisphere, even in the presence of haem associated Fe caused by blood infiltration at the injury site, indicating that a mechanism may exist initially to compensate for extra Fe accumulation. This time point observation correlates with a recent publication by Ayton et al.42 where cortical Fe measured by ICP-MS showed no difference between ipsilateral and contralateral hemispheres at 24 hours post injury, after a significant Fe elevation between hemispheres at 6 hours. Recent investigations in both animal models and humans have consistently implicated abnormal cellular Fe elevation in the pathogenesis of the disorder, in both TBI,43,44 and mild TBI.23,45,46 Moreover, several of these studies, using in vivo T2 MRI imaging technologies, clearly indicate a non-haem associated mechanism of cellular Fe accumulation, at sub-thalamic regions greatly distal to the cortical site of injury. T2 MRI has been shown to effectively differentiate Fe-protein co-ordination environments in vivo.47 To determine the likelihood of intracellular Fe accumulation in this study, a Perls Prussian blue stain for non-haem Fe was employed on 28 day post-TBI tissue samples from the same animals that underwent LA-ICP-MS analysis. The Perls stain clearly and prominently demonstrated intracellular Fe accumulation penetrating well into the depth of ROI3 (Fig. 6), correlating with the LA-ICP-MS data presented here, and is supportive of previous studies cited herein indicating abnormal cellular Fe elevation greatly distal to the cortical injury site.
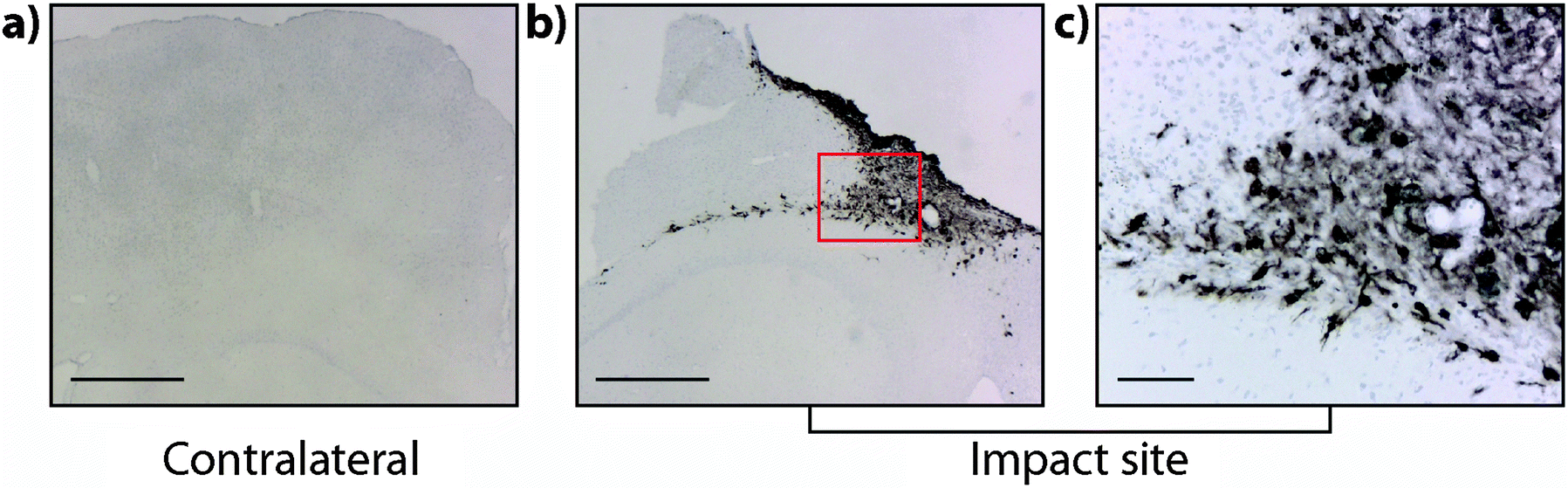 | ||
| Fig. 6 Perls Prussian blue stained 28 day TBI contralateral and ipsilateral hemispheres. (a) Perls Prussian blue stained contralateral hemisphere (40×; scale bar = 500 μm) demonstrating absence of intracellular Fe staining. (b) Perl's stained ipsilateral hemisphere (40× scale bar = 500 μm) showing pronounced Fe staining deep into cortex and hippocampus. (c) Higher magnification of highlighted area in box (b) clearly reveals prominent intracellular Perls staining (100×; scale bar = 100 μm). | ||
The current study is the first to our knowledge that demonstrates that there is a sustained elevation in Fe levels around the injury site that persists well beyond 24 hours. Such a chronic dyshomeostasis in Fe may lead to abnormal neuronal pathologies, as Fe accumulation has been shown to contribute to neuronal free radical damage48,49 and is widely acknowledged as a modulator of the degenerative cascade in TBI.50–52 Iron may, therefore, be a tractable therapeutic target in TBI. Indeed there are multiple selective Fe chelators currently approved for use in Fe metabolism disorders.53,54 Interestingly, a 1996 study by Long et al.,55 demonstrated that administration of the Fe chelator deferoxamine improved spatial memory performance following experimental brain injury in rats, indicating the potential of Fe chelation or modulation as a therapeutic avenue to be investigated. Moreover, and unexpectedly based on this analysis, a larger and unrecognized therapeutic window of up to 28 days may be acceptable for Fe chelation treatment to obtund chronic Fe accumulation. However, caution should be applied in the use of Fe chelation treatments due to potential off-target effects of Fe depletion. Alternative methods of modulating Fe regulatory function should therefore be investigated as an alternative avenue to restore post-TBI Fe homeostasis over chelation therapies.
Copper is required for essential metabolic processes including mitochondrial respiration, neurotransmitter biosynthesis and as a cofactor for antioxidant enzymes.56 Copper is a particularly potent pro-oxidant and can form hydrogen peroxide and subsequent hydroxyl radicals at a high rate, significantly greater than that of Fe.57,58 Conversely, a reduction in brain Cu has been shown to exacerbate tau hyperphosphorylation in a transgenic mouse model of AD.59 Hyperphosphorylated tau and the formation of NFTs are an integral pathological feature of all TBI types; having been shown in the neocortex of boxers,4 other contact sports athletes60 and are also evident in blast exposed military veterans.61 Concomitantly, and potentially in combination with Cu, Fe and Zn are also both intricately involved in the signaling pathways that regulate tau and the development of tau pathology.62–64
Additionally, DAI, the pathological hallmark of TBI, is the result of axonal shearing during acceleration and rotational forces derived from head impact. Myelin along axons is required for efficient neuronal conduction. TBI studies have shown the shearing of myelin in DAI is responsible for a reduction in the speed of axonal transmission along axons resulting in compromised neuronal function.65,66 As myelination is a Cu dependent process67 it stands to reason that adequate physiological levels of Cu would be a requirement for effective re-myelination after TBI. Our results indicate that Cu concentrations were not significantly altered out to 7 days post injury. Thereafter, however, Cu began to mildly elevate in the ipsilateral cortex. That this elevation was significant in the ipsilateral cortex adjacent to the impact zone (ROI 1 and 2) at 28 days may be indicative of a potential physiological mechanism related to demyelination or remyelination.
Importantly, the observation that an elevated Cu concentration is prominent and may continue beyond 28 days, should give pause for concern in relation to the potential chronic Cu toxicity as it relates to oxidative stress and subsequent neuronal damage. The altered Cu concentration is therefore of interest and should be further assessed at later time points with further experimentation.
Conclusions
Our results clearly indicate that metal dyshomeostasis continues over an extended period of time following brain injury. These data provide a more detailed spatial and temporal profile of metal changes in the brain following TBI and may consequently provide greater insight for both therapeutic development and the potential need for different metal-based therapeutic strategies at different stages post-injury. The data also indicate that further studies extending beyond 28 days are required.Acknowledgements
P. A. A. is supported by an Australian Research Council Future Fellowship (FT120100030). D. J. H. is supported by an Australian Research Council Linkage Project with Agilent Technologies (LP140100095). In addition, the Florey Institute of Neuroscience and Mental Health acknowledge the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant. The Elemental Bio-imaging Facility receives material support from Agilent Technologies.Notes and references
- B. Roozenbeek, A. I. Maas and D. K. Menon, Nat. Rev. Neurol., 2013, 9, 231–236 CrossRef PubMed.
- A. I. Maas, N. Stocchetti and R. Bullock, Lancet Neurol., 2008, 7, 728–741 CrossRef PubMed.
- M. Faul, L. Xu, M. M. Wald, V. Coronado and A. M. Dellinger, Inj. Prev., 2010, 16, A268 CrossRef.
- J. A. Corsellis, C. J. Bruton and D. Freeman-Browne, Psychol. Med., 1973, 3, 270–303 CrossRef CAS PubMed.
- B. Omalu, J. Bailes, R. L. Hamilton, M. I. Kamboh, J. Hammers, M. Case and R. Fitzsimmons, Neurosurgery, 2011, 69, 173–183 CrossRef PubMed ; discussion 183.
- B. I. Omalu, S. T. DeKosky, R. L. Minster, M. I. Kamboh, R. L. Hamilton and C. H. Wecht, Neurosurgery, 2005, 57, 128–134 CrossRef PubMed ; discussion 128–134.
- P. H. Montenigro, D. T. Corp, T. D. Stein, R. C. Cantu and R. A. Stern, Annu. Rev. Clin. Psychol., 2015, 11, 309–330 CrossRef PubMed.
- H. S. Levin and C. S. Robertson, J. Neurotrauma, 2013, 30, 610–617 CrossRef PubMed.
- M. W. Weiner, K. E. Friedl, A. Pacifico, J. C. Chapman, M. S. Jaffee, D. M. Little, G. T. Manley, A. McKee, R. C. Petersen, R. K. Pitman, K. Yaffe, H. Zetterberg, R. Obana, L. J. Bain and M. C. Carrillo, Alzheimer's Dementia, 2013, 9, 445–451 CrossRef PubMed.
- G. Miller, Science, 2012, 336, 790–791 CrossRef CAS PubMed.
- T. Skandsen, T. G. Finnanger, S. Andersson, S. Lydersen, J. F. Brunner and A. Vik, Arch. Phys. Med. Rehabil., 2010, 91, 1904–1913 CrossRef PubMed.
- C. Van Den Heuvel, E. Thornton and R. Vink, Prog. Brain Res., 2007, 161, 303–316 CAS.
- H. Chen, M. Richard, D. P. Sandler, D. M. Umbach and F. Kamel, Am. J. Epidemiol., 2007, 166, 810–816 CrossRef PubMed.
- S. A. Factor and W. J. Weiner, Mov. Disord., 1991, 6, 225–229 CrossRef CAS PubMed.
- S. Bolognin, L. Messori and P. Zatta, NeuroMol. Med., 2009, 11, 223–238 CrossRef CAS PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- S. Rivera-Mancia, I. Perez-Neri, C. Rios, L. Tristan-Lopez, L. Rivera-Espinosa and S. Montes, Chem.-Biol. Interact., 2010, 186, 184–199 CrossRef CAS PubMed.
- K. J. Barnham and A. I. Bush, Chem. Soc. Rev., 2014, 43, 6727–6749 RSC.
- S. W. Suh, J. W. Chen, M. Motamedi, B. Bell, K. Listiak, N. F. Pons, G. Danscher and C. J. Frederickson, Brain Res., 2000, 852, 268–273 CrossRef CAS PubMed.
- C. J. Frederickson, M. D. Hernandez and J. F. McGinty, Brain Res., 1989, 480, 317–321 CrossRef CAS PubMed.
- C. J. Frederickson, W. Maret and M. P. Cuajungco, Neuroscientist, 2004, 10, 18–25 CrossRef CAS PubMed.
- H. D. Liu, W. Li, Z. R. Chen, M. L. Zhou, Z. Zhuang, D. D. Zhang, L. Zhu and C. H. Hang, Neurol. Sci., 2013, 34, 1173–1180 CrossRef PubMed.
- E. Raz, J. H. Jensen, Y. Ge, J. S. Babb, L. Miles, J. Reaume, R. I. Grossman and M. Inglese, AJNR Am. J. Neuroradiol., 2011, 32, 1851–1856 CrossRef CAS PubMed.
- L. M. Klevay, Med. Hypotheses, 2013, 81, 995–998 CrossRef CAS PubMed.
- S. D. Portbury and P. A. Adlard, J. Alzheimer's Dis., 2015, 46, 297–311 CrossRef CAS PubMed.
- E. S. Lein, M. J. Hawrylycz, N. Ao, M. Ayres, A. Bensinger, A. Bernard, A. F. Boe, M. S. Boguski, K. S. Brockway, E. J. Byrnes, L. Chen, L. Chen, T. M. Chen, M. C. Chin, J. Chong, B. E. Crook, A. Czaplinska, C. N. Dang, S. Datta, N. R. Dee, A. L. Desaki, T. Desta, E. Diep, T. A. Dolbeare, M. J. Donelan, H. W. Dong, J. G. Dougherty, B. J. Duncan, A. J. Ebbert, G. Eichele, L. K. Estin, C. Faber, B. A. Facer, R. Fields, S. R. Fischer, T. P. Fliss, C. Frensley, S. N. Gates, K. J. Glattfelder, K. R. Halverson, M. R. Hart, J. G. Hohmann, M. P. Howell, D. P. Jeung, R. A. Johnson, P. T. Karr, R. Kawal, J. M. Kidney, R. H. Knapik, C. L. Kuan, J. H. Lake, A. R. Laramee, K. D. Larsen, C. Lau, T. A. Lemon, A. J. Liang, Y. Liu, L. T. Luong, J. Michaels, J. J. Morgan, R. J. Morgan, M. T. Mortrud, N. F. Mosqueda, L. L. Ng, R. Ng, G. J. Orta, C. C. Overly, T. H. Pak, S. E. Parry, S. D. Pathak, O. C. Pearson, R. B. Puchalski, Z. L. Riley, H. R. Rockett, S. A. Rowland, J. J. Royall, M. J. Ruiz, N. R. Sarno, K. Schaffnit, N. V. Shapovalova, T. Sivisay, C. R. Slaughterbeck, S. C. Smith, K. A. Smith, B. I. Smith, A. J. Sodt, N. N. Stewart, K. R. Stumpf, S. M. Sunkin, M. Sutram, A. Tam, C. D. Teemer, C. Thaller, C. L. Thompson, L. R. Varnam, A. Visel, R. M. Whitlock, P. E. Wohnoutka, C. K. Wolkey, V. Y. Wong, M. Wood, M. B. Yaylaoglu, R. C. Young, B. L. Youngstrom, X. F. Yuan, B. Zhang, T. A. Zwingman and A. R. Jones, Nature, 2007, 445, 168–176 CrossRef CAS PubMed.
- E. S. O’Meara, W. A. Kukull, L. Sheppard, J. D. Bowen, W. C. McCormick, L. Teri, M. Pfanschmidt, J. D. Thompson, G. D. Schellenberg and E. B. Larson, Am. J. Epidemiol., 1997, 146, 373–384 CrossRef.
- D. J. Hare, J. L. George, L. Bray, I. Volitakis, A. Vais, T. M. Ryan, R. A. Cherny, A. I. Bush, C. L. Masters, P. A. Adlard, P. A. Doble and D. I. Finkelstein, J. Anal. At. Spectrom., 2014, 29, 1726 RSC.
- J. Lear, D. J. Hare, F. Fryer, P. A. Adlard, D. I. Finkelstein and P. A. Doble, Anal. Chem., 2012, 84, 6707–6714 CrossRef CAS PubMed.
- B. R. Roberts, D. J. Hare, C. A. McLean, A. Conquest, M. Lind, Q.-X. Li, A. I. Bush, C. L. Masters, M.-C. Morganti-Kossmann and T. Frugier, Metallomics, 2015, 7, 124–128 RSC.
- D. R. Morris and C. W. Levenson, Curr. Opin. Clin. Nutr. Metab. Care, 2013, 16, 708–711 CrossRef CAS PubMed.
- B. Szewczyk, Front. Aging Neurosci., 2013, 5, 33 Search PubMed.
- E. C. Cope, D. R. Morris and C. W. Levenson, Nutr. Rev., 2012, 70, 410–413 CrossRef PubMed.
- E. C. Cope, D. R. Morris, A. G. Scrimgeour and C. W. Levenson, Neurorehabil. Neural Repair, 2012, 26, 907–913 CrossRef PubMed.
- E. C. Cope, D. R. Morris, A. G. Scrimgeour, J. W. VanLandingham and C. W. Levenson, Physiol. Behav., 2011, 104, 942–947 CrossRef CAS PubMed.
- C. J. McClain, D. L. Twyman, L. G. Ott, R. P. Rapp, P. A. Tibbs, J. A. Norton, E. J. Kasarskis, R. J. Dempsey and B. Young, J. Neurosurg., 1986, 64, 224–230 CrossRef CAS PubMed.
- N. Tonder, F. F. Johansen, C. J. Frederickson, J. Zimmer and N. H. Diemer, Neurosci. Lett., 1990, 109, 247–252 CrossRef CAS PubMed.
- P. A. Adlard, J. Parncutt, V. Lal, S. James, D. Hare, P. Doble, D. I. Finkelstein and A. I. Bush, Neurobiol. Dis., 2015, 81, 196–202 CrossRef CAS PubMed.
- D. J. Hare, E. J. New, M. D. de Jonge and G. McColl, Chem. Soc. Rev., 2015, 44, 5941–5958 RSC.
- E. C. Cope and C. W. Levenson, Curr. Opin. Clin. Nutr. Metab. Care, 2010, 13, 685–689 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- S. Ayton, M. Zhang, B. R. Roberts, L. Q. Lam, M. Lind, C. McLean, A. I. Bush, T. Frugier, P. J. Crack and J. A. Duce, Free Radical Biol. Med., 2014, 69, 331–337 CrossRef CAS PubMed.
- H. Bayir, P. M. Kochanek and V. E. Kagan, Dev. Neurosci., 2006, 28, 420–431 CrossRef CAS PubMed.
- G. Onyszchuk, S. M. LeVine, W. M. Brooks and N. E. Berman, Neurosci. Lett., 2009, 452, 204–208 CrossRef CAS PubMed.
- C. Bouras, P. Giannakopoulos, P. F. Good, A. Hsu, P. R. Hof and D. P. Perl, Eur. Neurol., 1997, 38, 53–58 CrossRef CAS PubMed.
- Q. G. Zhang, M. D. Laird, D. Han, K. Nguyen, E. Scott, Y. Dong, K. M. Dhandapani and D. W. Brann, PLoS One, 2012, 7, e34504 CAS.
- E. P. Raven, P. H. Lu, T. A. Tishler, P. Heydari and G. Bartzokis, J. Alzheimer's Dis., 2013, 37, 127–136 CAS.
- J. Caliaperumal, Y. Ma and F. Colbourne, Exp. Neurol., 2012, 237, 363–369 CrossRef CAS PubMed.
- T. Carbonell and R. Rama, Curr. Med. Chem., 2007, 14, 857–874 CrossRef CAS PubMed.
- M. A. Ansari, K. N. Roberts and S. W. Scheff, J. Neurotrauma, 2008, 25, 513–526 CrossRef PubMed.
- M. A. Ansari, K. N. Roberts and S. W. Scheff, Free Radical Biol. Med., 2008, 45, 443–452 CrossRef CAS PubMed.
- K. Zhang, B. Johnson, M. Gay, S. G. Horovitz, M. Hallett, W. Sebastianelli and S. Slobounov, J. Neurotrauma, 2012, 29, 756–765 CrossRef PubMed.
- D. Hare, S. Ayton, A. Bush and P. Lei, Front. Aging Neurosci., 2013, 5, 34 Search PubMed.
- O. Weinreb, S. Mandel, M. B. Youdim and T. Amit, Free Radical Biol. Med., 2013, 62, 52–64 CrossRef CAS PubMed.
- D. A. Long, K. Ghosh, A. N. Moore, C. E. Dixon and P. K. Dash, Brain Res., 1996, 717, 109–117 CrossRef CAS PubMed.
- M. M. Pena, J. Lee and D. J. Thiele, J. Nutr., 1999, 129, 1251–1260 CAS.
- X. Huang, M. P. Cuajungco, C. S. Atwood, M. A. Hartshorn, J. D. Tyndall, G. R. Hanson, K. C. Stokes, M. Leopold, G. Multhaup, L. E. Goldstein, R. C. Scarpa, A. J. Saunders, J. Lim, R. D. Moir, C. Glabe, E. F. Bowden, C. L. Masters, D. P. Fairlie, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1999, 274, 37111–37116 CrossRef CAS PubMed.
- C. Opazo, X. Huang, R. A. Cherny, R. D. Moir, A. E. Roher, A. R. White, R. Cappai, C. L. Masters, R. E. Tanzi, N. C. Inestrosa and A. I. Bush, J. Biol. Chem., 2002, 277, 40302–40308 CrossRef CAS PubMed.
- K. Voss, C. Harris, M. Ralle, M. Duffy, C. Murchison and J. F. Quinn, Transl. Neurodegener., 2014, 3, 24 CrossRef PubMed.
- A. C. McKee, R. C. Cantu, C. J. Nowinski, E. T. Hedley-Whyte, B. E. Gavett, A. E. Budson, V. E. Santini, H. S. Lee, C. A. Kubilus and R. A. Stern, J. Neuropathol. Exp. Neurol., 2009, 68, 709–735 CrossRef PubMed.
- L. E. Goldstein, A. M. Fisher, C. A. Tagge, X. L. Zhang, L. Velisek, J. A. Sullivan, C. Upreti, J. M. Kracht, M. Ericsson, M. W. Wojnarowicz, C. J. Goletiani, G. M. Maglakelidze, N. Casey, J. A. Moncaster, O. Minaeva, R. D. Moir, C. J. Nowinski, R. A. Stern, R. C. Cantu, J. Geiling, J. K. Blusztajn, B. L. Wolozin, T. Ikezu, T. D. Stein, A. E. Budson, N. W. Kowall, D. Chargin, A. Sharon, S. Saman, G. F. Hall, W. C. Moss, R. O. Cleveland, R. E. Tanzi, P. K. Stanton and A. C. McKee, Sci. Transl. Med., 2012, 4, 134ra160 Search PubMed.
- J. T. Egana, C. Zambrano, M. T. Nunez, C. Gonzalez-Billault and R. B. Maccioni, BioMetals, 2003, 16, 215–223 CrossRef CAS PubMed.
- Y. Huang, Z. Wu, Y. Cao, M. Lang, B. Lu and B. Zhou, Cell Rep., 2014, 8, 831–842 CrossRef CAS PubMed.
- X. Y. Sun, Y. P. Wei, Y. Xiong, X. C. Wang, A. J. Xie, X. L. Wang, Y. Yang, Q. Wang, Y. M. Lu, R. Liu and J. Z. Wang, J. Biol. Chem., 2012, 287, 11174–11182 CrossRef CAS PubMed.
- J. A. Creed, A. M. DiLeonardi, D. P. Fox, A. R. Tessler and R. Raghupathi, J. Neurotrauma, 2011, 28, 547–563 CrossRef PubMed.
- J. Flygt, A. Djupsjo, F. Lenne and N. Marklund, Eur. J. Neurosci., 2013, 38, 2153–2165 CrossRef CAS PubMed.
- A. W. Zimmerman, J. M. Matthieu, R. H. Quarles, R. O. Brady and J. M. Hsu, Arch. Neurol., 1976, 33, 111–119 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary tables. See DOI: 10.1039/c5mt00234f |
| This journal is © The Royal Society of Chemistry 2016 |