
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defining the metal binding pathways of human metallothionein 1a: balancing zinc availability and cadmium seclusion
Gordon W.
Irvine
,
Tyler B. J.
Pinter
and
Martin J.
Stillman
*
Department of Chemistry, The University of Western Ontario, London, Ontario N6A 5B7, Canada. E-mail: martin.stillman@uwo.ca; Fax: +1-519-661-3022; Tel: +1-519-661-3821
First published on 13th November 2015
Abstract
Metallothioneins (MTs) are cysteine-rich, metal-binding proteins that are found throughout Nature. This ubiquity highlights their importance in essential metal regulation, heavy metal detoxification and cellular redox chemistry. Missing from the current description of MT function is the underlying mechanism by which MTs achieve their proposed biological functions. To date, there have been conflicting reports on the mechanism of metal binding and the structures of the metal binding intermediates formed during metalation of apoMTs. The form of the metal-bound intermediates dictates the metal sequestering and metal-donating properties of the protein. Through a detailed analysis of spectral data from electrospray ionization mass spectromeric and circular dichroism methods we report that Zn(II) and Cd(II) metalation of the human MT1a takes place through two distinct pathways. The first pathway involves formation of beaded structures with up to five metals bound terminally to the 20 cysteines of the protein via a noncooperative mechanism. The second pathway is dominated by the formation of the four-metal domain cluster structure M4SCYS11via a cooperative mechanism. We report that there are different pathway preferences for Zn(II) and Cd(II) metalation of apo-hMT1a. Cd(II) binding follows the beaded pathway above pH 7.1 but beginning below pH 7.1 the clustered (Cd4Scys11) pathway begins to dominate. In contrast, Zn(II) binding follows the terminal, “beaded”, pathway at all physiologically relevant pH (pH ≥ 5.2) only following the clustered pathway below pH 5.1. The results presented here allow us to reconcile the conflicting reports concerning the presence of different metalation intermediates of MTs. The conflict regarding cooperative versus noncooperative binding mechanisms is also reconciled with the experimental results described here. These two metal-specific pathways and the presence of radically different intermediate structures provide insight into the multi-functional nature of MT: binding Zn(II) terminally for donation to metalloenzymes and sequestering toxic Cd(II) in a cluster structure.
Introduction
Mammalian metallothioneins (MTs) are a family of small, cysteine rich metal-binding proteins that are involved in zinc and copper homeostasis,1–8 heavy metal detoxification9–16 and cellular redox chemistry.17–21 When saturated with seven divalent metals, mammalian MTs consist of an N-terminal β-domain and a C-terminal α-domain connected by a flexible linker region.22 The α-domain can accommodate up to 4 divalent metals and the β-domain 3 metals by forming MII4Scys11 and MII3Scys9 clusters, respectively. This domain description is only relevant in the fully metalated structure as there are no true binding domains in the partially-metalated and apo-structures under most conditions.23–27 The more biologically relevant structures of the apo- and partially metalated MTs are considered to be poorly defined.23,27,28 Currently, these apo- and partially metalated MT species are thought to adopt a fluxional, globular structure.23,29,30Metalation of the 20 Cys apo-peptide can take place in a number of different ways.24,31 As there is no pre-existing domain structure in the absence of bound metals, the thermodynamics and kinetics associated with metal binding control the structures adopted at each metalation step.27 For each species (i.e. M1βαMT, M2βαMT, etc.) numerous structures are possible and complex rearrangement of the bound metals can occur.32,33 The mechanism of divalent metal binding to MTs, specifically that of Zn(II) and Cd(II), has been a topic of great interest but the results have generated conflicting reports concerning the identity and description of the intermediate structures formed before the complete complement of seven metals has been bound.2,34–38 Because of the spectroscopic accessibility of Cd(II), much of what is currently known about Zn(II) and Cd(II) binding to MT has been based largely on the chemistry of Cd(II) or other spectroscopically active metals, such as Co(II).25,39 Indeed, 113Cd is often used to probe structural properties of Zn(II) binding sites in proteins other than MTs.40
When discussing metal binding mechanisms of MTs, the terms cooperative and noncooperative are often used. In a fully cooperative mechanism, only the apoMT and end-product should be detected at any point during the reaction. In a noncooperative mechanism, intermediates can be measured and become the most abundant species during the early to mid-stages of the metal titration. The kinetic data for As(III) binding to MT1a clearly follow this noncooperative, stochastic model.31
Metalation details from early studies of cadmium binding relied on 113Cd NMR26 or protein modification41 and reported the formation of cluster dominated products. With the development of new technologies, most notably electrospray ionization mass spectrometry (ESI-MS), the description of a purely cluster dominated mechanism became less convincing. Some groups have reported a cluster dominated mechanism with ESI-MS37 and reported that the Cd-thiolate cluster formation occurs first in the α-domain.30 However, reports by our group and others have shown that the binding pathway involves non-clustered intermediates.35,36,38,42 It should be noted that these studies used different solution conditions and often different isoforms of human MT, which possess different metalation properties.43 It has also been suggested that differing ESI-MS settings may cause the discrepancy in mechanistic details.30
In this paper, we report definitive evidence for two parallel, competing pathways that are dependent on metal identity, Zn(II) or Cd(II), and the pH. We have examined the pH dependence of the Zn(II) and Cd(II) metalation pathways of human MT1a using ESI-MS and circular dichroism spectroscopy at the important early stages of metalation where the largest number of conformations are possible. The presence of these two distinct metalation pathways was determined by monitoring the formation of the partially metalated intermediates during the Zn(II) and Cd(II) metalation reactions of the apoMT1a. We discuss how significant differences in the intermediates formed during metalation for each metal may explain how MT1a functions as a multi-purpose protein: regulating zinc levels, providing zinc to other apo-enzymes,44 and sequestering toxic heavy metals.45,46 From the data presented here it is clear that metalation can occur via one of two major pathways: a cooperative, α-cluster driven pathway or a noncooperative, terminally-bound, beaded pathway (Fig. 1).
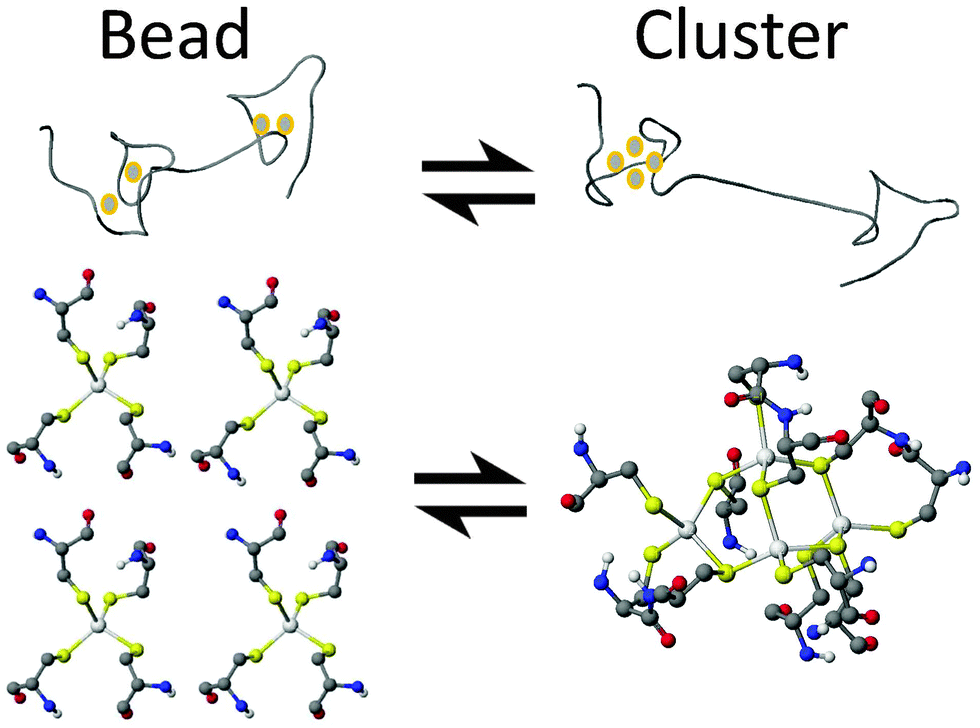 | ||
| Fig. 1 Cartoon ribbon representation of proposed MT1a metalation intermediates. The “beaded” intermediate and a ball-and-stick representation of the terminal coordination of the metals with up to 5 MSCYS4 “beads” (left; 4 beads shown). The cluster structure M4SCYS11 is shown in the α-domain of the ribbon structure and as a ball-and-stick model of the bridged and terminal Cys that make up the cluster (right). | ||
Methods
Protein preparation
Recombinant human metallothionein 1a (MGKAAAACSC ATGGSCTCTG SCKCKECKCN SCKKAAAACC SCCPMSCAKC AQGCVCKGAS EKCSCCK KAA AA) was expressed with an S-tag in BL21 E. coli cells which has been described in detail elsewhere.47 In brief, cells containing the plasmid for the full protein (βα-MT1a) were plated on to growth media containing kanamycin from a stock culture stored at −80 °C and grown for 16 hours at 37 °C. The grown cells were then inoculated into 4 × 1L broth cultures enriched with 50 μL of 1 M cadmium and incubated in a shaker for 4 hours until OD600 absorbance was 0.8. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was then added to induce expression of MT and 30 minutes later 150 μL of 1 M cadmium sulfate solution was added to the broth. The cells were collected 3.5 hours after induction, centrifuged and stored at −80 °C.The recombinant cells were lysed using a cell disruptor (Constant Systems, UK) shot at 20k psi. From there, the cell lysate was centrifuged for 1 h to pellet out cellular debris. The supernatant was filtered and loaded on to a GE healthcare SP ion exchange columns with a total volume of 10 mL. The columns were washed with pH 7.4 10 mM Tris(tris-hydroxymethyl-aminomethane) buffer for approximately 2 h to remove loosely bound proteins and other organic compounds. MT was eluted using an increasing gradient of 1 M NaCl + 10 mM Tris buffer at pH 7.4. The eluted MT was concentrated down to <20 mL and the S-tag cleaved using a Thrombin Clean-Cleave kit as per the manufacturers' instructions (Sigma-Aldrich). The mixture was then diluted, desalted and placed on another SP ion exchange column. The S-tag does not bind as strongly as MT and thus elutes at low salt concentrations. The protein and S-tag were separated in this fashion. The eluted MT was concentrated to approximately 120 μM and stored at −20 °C.
To prepare MT for the pH titration experiments, aliquots were first demetalated and desalted using centrifugal filter tubes with a 3 kDa membrane (Millipore) and a 10 mM pH 2.8 ammonium formate buffer. The low pH solutions contained 1 mM Dithiothreitol (DTT) to prevent oxidation of the free thiols in MT. The pH was raised by buffer exchange with argon saturated, pH 7.0 10 mM ammonium formate solutions that did not contain reductant. The protein solutions were checked for final concentration by remetalation of a small aliquot with cadmium using the metal-to-ligand charge transfer band at 250 nm (ε250 = 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L mol−1 cm−1). The solutions were also monitored for oxidation using UV-visible absorption spectroscopy to monitor absorption corresponding to 280 nm from oxidized disulfide. Once demetalated and desalted, the MT concentration was determined, all concentrations were between 40–90 μM to ensure good signal to noise ratios In the ESI-MS experiment. We note that this was a concern because the titration introduced salt into the solution which suppresses the MT signal at high concentrations.
000 L mol−1 cm−1). The solutions were also monitored for oxidation using UV-visible absorption spectroscopy to monitor absorption corresponding to 280 nm from oxidized disulfide. Once demetalated and desalted, the MT concentration was determined, all concentrations were between 40–90 μM to ensure good signal to noise ratios In the ESI-MS experiment. We note that this was a concern because the titration introduced salt into the solution which suppresses the MT signal at high concentrations.
In addition to demetalating MT in the presence of DTT, the solutions were vacuum degassed and bubbled with Argon to displace any dissolved oxygen. This was done for the 10 mM Cd2+ and Zn2+ and the 0.5% NH3 and 0.5% formic acid solutions as well to ensure no oxygen was introduced into the system during the titration. Great care was taken to reduce the possibility of oxidation pf the protein at neutral pH.
ESI-MS and circular dichroism pH titrations
Mass spectra were collected on a micrOTOF II electrospray-ionization time-of-flight mass spectrometer (Bruker Daltonics) in the positive ion mode. NaI was used as the mass calibrant. The scan conditions for the spectrometer were: end plate offset, −500 V; capillary, +4200 V; nebulizer, 2.0 bar; dry gas flow, 8.0 L min−1; dry temperature, 30 °C; capillary exit, 180 V; skimmer 1, 22.0 V; hexapole 1, 22.5 V; hexapole RF, 600 Vpp; skimmer 2, 22 V; lens 1 transfer, 88 μs; lens 1 pre-pulse storage, 23 μs. The mass range was 500.0–3000.0 m/z. Spectra were assembled and deconvoluted using the Bruker Compass data analysis software package. ESI-mass spectrometry was used to monitor all stages of the pH titration of Cd-MT and Zn-MT. First, approximately 2.5 molar equivalents of Cd(II) acetate and Zn(II) acetate were added to the MT solution at pH 5.5. This caused a drop in pH due to the displacement of H+ from the thiol groups. After the metal solution was added, the ESI-MS spectra were recorded and averaged over 2 minutes. Then aliquots of 0.5% NH3 were added to raise the pH and the spectra recorded. The pH was confirmed using a micro-pH probe (Accumet). This was repeated in steps until the salt peaks became more intense than the peaks corresponding to MT species. No change of the M2+/MT ratio was observed meaning no precipitation of Cd(OH)2 or Zn(OH)2 occurred.The pH titrations of the partially metalated Cd-MT and Zn-MT were performed at least 6 separate times with different protein preparations and starting at slightly different pH values in the region of 4.5–5.5. Thus separate data points were obtained for each pH reported increasing in increments of 0.1 pH units and serve as a statistical check. The separate preparations were run under the same solution and ESI-MS conditions. The error associated with these ESI-MS measurements is estimated to be about ±10%.
For the circular dichroism (CD) spectra, 2.5 molar equivalents of cadmium acetate were added to apo-MT in a pH 7.0 10 mM ammonium formate solution in the manner described above for the ESI-MS data. The CD spectra (Jasco J810) were measured over the range of 200–300 nm at various stages in the pH titration. Below 220 nm, the CD spectrum is skewed due to the absorbance of the ammonium formate buffer and is not shown. The significant Cd-dependent CD spectral bands lie in the 240–280 nm region. The CD spectra obtained were compared to spectra of apo-MT and Cd4-MT that were obtained previously by our group and have been extensively discussed in other works.48,49 We note that the Zn–S charge transfer band lies at 220–230 nm under the protein bands.
To determine the degree of cluster formation vs. terminally bound metal, the ratios of the abundances of the intermediates (Cd1–3βαMT) to the initial apo-MT and final product (Cd4βαMT) were determined and normalized for each ESI mass spectrum in the pH titration. The most clustered spectra (at lower pH) had only a minimal fraction of intermediates compared to the spectra recorded at more basic pH. The sigmoidal fit of the set of ratios was generated using a least squares method. The lack of saturation of the sigmoidal fit is likely due to the 100% terminal coordination binding mechanism occurring at the highest pH range of the titration and spectra were not measured above pH 8.2.
The simulated ESI-MS data were calculated using a set of 4 equilibrium constants that correspond to each of the bimolecular metalation reactions up to Cd4MT (Scheme 1). Each sequential bimolecular reaction was controlled by an individual Kf. The values of the log(Kf) of the formation constants all average to 14.4 which is within the range reported in the literature for Cd(II) binding to MT.50 It should be noted that the binding constants presented in this work and those referenced are conditional binding constants that amongst other parameters, are dependent on pH.
 | ||
| Scheme 1 The possible metalation pathways for the first four metals bound to human MT1a ending in either a clustered (cooperative mechanism) or beaded (terminally-bound, noncooperative) structure. | ||
Results
pH dependence of Cd(II) binding to apo-MT1a
Fig. 2 shows a series of deconvoluted ESI-MS spectra recorded for a single sample of apoMT1a metalated with 2.5 equivalents of Cd(II) at increasing pH (A–E). This metal loading of 2.5 molar equivalents (mol eq.) was chosen to challenge the formation of the Cd4 intermediate. This is the point at which the spectra corresponding to the cooperative and non-cooperative mechanisms appear in stark contrast to each other. Each species present in solution can be identified and the change in its relative abundance monitored. At more acidic pH (<6.0) the relative abundance of the intermediates Cd1–3MT1a with a distribution of 1 to 3 Cd(II) bound is very low, the dominant species is the Cd4-MT1a. With increasing pH, the appearance of Cd1–3MT1a can be seen with their relative intensities increasing as the pH is raised. At pH 7.9 the distribution of species present is narrower, now completely dominated by the terminally-bound Cd1–3-MT1a species, which we describe as the “beaded” CdSCYS4 structures. Our assignment of these species being terminally-bound is supported by the loss of the specific spectral envelope motif in the circular dichroism spectral data seen in Fig. 3. The change in metalation pathway is shown by the complete loss of both the initial apo- and Cd4-MT1a masses in the ESI-mass spectrum (Fig. 2A). These two species are replaced by higher intensities of Cd1–3MT1a species (Fig. 2E). Fig. 2B–D show a blending of these two pathways leading to a mixture of intermediate products.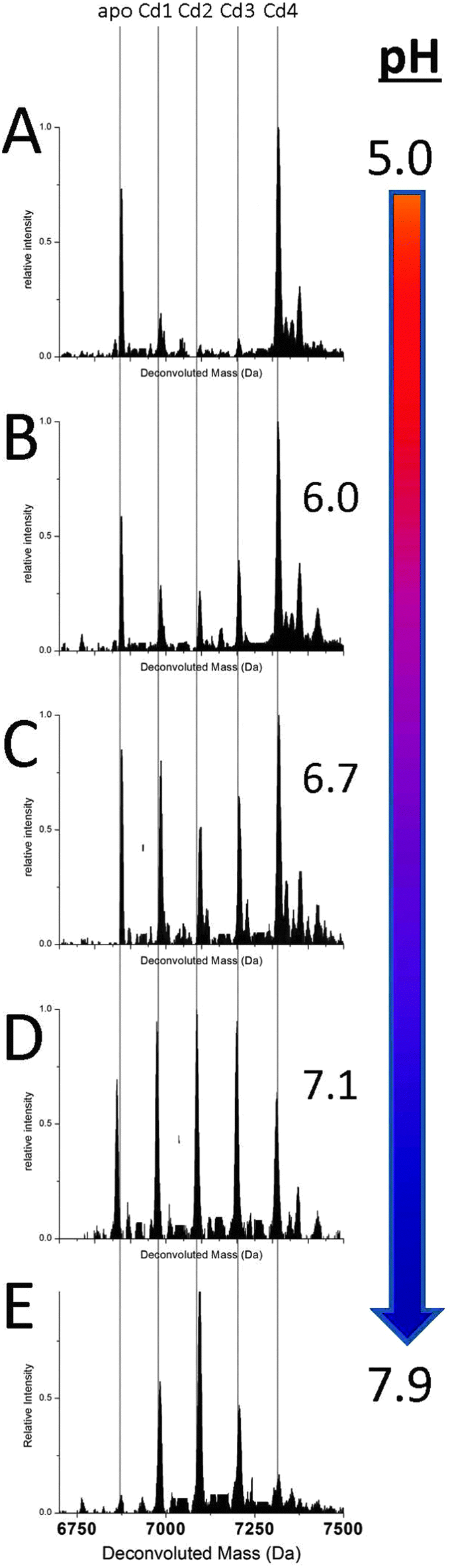 | ||
| Fig. 2 Representative ESI-MS data showing the relative abundances of CdxMT species (x = 0–4) as a function of pH. These are representative spectra from pH titrations after an aliquot of approx. 2.5 molar equivalents of Cd(II) was added to the apoMT solution. The species distribution at pH 5.0 (A) and 6.0 (B) represent a mostly cluster-dominated pathway for metal binding. The spectra at pH 6.7 (C) shows a blend of the pathways, and at pH 7.1 (D) and 7.9 (E) a terminal binding pathway dominates. Adduct peaks with a mass of +60 Da were present for each species and were removed for clarity. | ||
 | ||
| Fig. 3 Circular dichroism spectra of apoMT1a with 2.5 mol eq. of Cd(II) added as a function of increasing pH. The derivative envelope with a cross-over near 240 nm is characteristic of the Cd4-cluster whereas the maximum dichroic intensity near 242 nm is characteristic of partially-metalated species with terminal thiolate coordination or supermetallated species. | ||
The switch from cluster bound Cd(II) to terminally bound Cd(II) species was observed in the circular dichroism (CD) spectra, Fig. 3.54–57 When the Cd(II) are bound only as Cd4α-clusters the spectra show a characteristic derivative envelope with a crossover point near 240 nm and a maximum near 260 nm. The shape of this CD envelope has been assigned previously as arising from exciton coupling of Cd(II) from the ligand to metal charge transfer band.51–54 The spectrum shown here is a combination of contributions from apoMT1a and the clustered intermediate, Cd4MT1a. As the pH is raised, the terminally bound contribution increases and the envelope from the clustered species disappears. This neutral pH spectra matches that of previous work where 1–3 mol eq. Cd(II) had been added to mouse apoMT1.51 When the cluster structure is disrupted by either excess or insufficient Cd(II) the spectrum is blue shifted and that is what is observed in Fig. 3. The CD spectral data correlate closely with the species measured in the ESI-mass spectra. The Cd4 cluster, dominant at low pH in the ESI-MS data (Fig. 2) is associated with the derivative envelope in Fig. 3. The terminally bound Cd(II) at neutral pH exhibit a CD envelope centered on the absorption band of CdMT (240–250 nm). The CD data confirm that the ESI-MS data are reporting the solution species accurately.
The pH dependence of the Cd-metalation pathway is plotted in Fig. 4. The trend is similar to a standard pH titration with a sharp change just before pH 7.0. Between pH 5.0–6.4 the change from cluster to terminal pathway is gradual, then the terminally-bound fraction increases sharply between 6.5–7.2. The binding mechanism is mostly terminal above pH 7.2, the noncooperative metalation pathway.
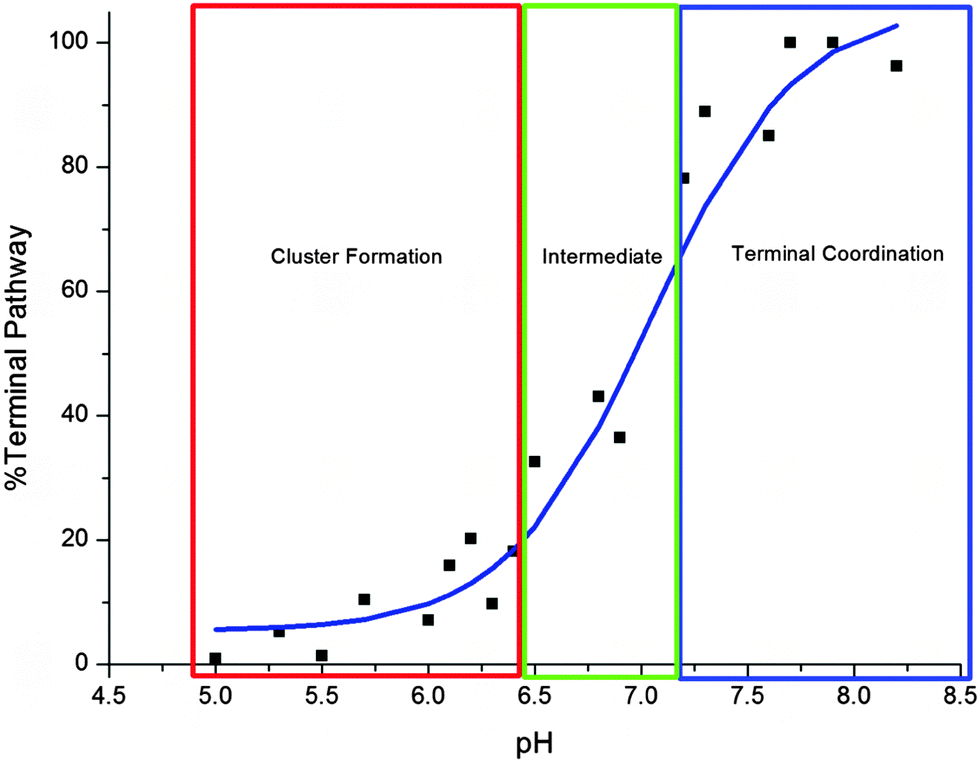 | ||
| Fig. 4 Change in cadmium binding pathway as a function of pH. The “%Terminal Pathway” was calculated from the ESI-MS data shown in Fig. 2. The cluster formation pathway dominates below pH 6.5 (red box), between pH 6.5–7.2 (green) the two pathways compete and the terminal pathway based on “beads” dominates above pH 7.2 (blue). | ||
pH dependence of Zn(II) binding to apoMT1a
MT1a-speciation during Zn(II) binding differs drastically from that of Cd(II) at all pH values, Fig. 5. This is a very significant result when Cd(II) is considered as a model for Zn(II). Clustering does not become dominant to the extent that it does with the Cd(II) metalation at any pH tested here. Only at very low pH, less than 5.0, does some cluster formation (Zn4SCYS11) become evident. Even at this low pH, the Zn(II) spectra are comparable to the blended pathway shown in Fig. 2B or C for Cd(II). Such a low pH (≤5.0) is not physiologically relevant except in low pH cellular compartments such as the lysosome.55,56 Therefore, we can conclude that the Zn(II) binding pathway is dominated by the formation of terminally-bound Zn(II) intermediates (Fig. 5B and C) over a physiologically relevant pH range, in sharp contrast to Cd(II) binding.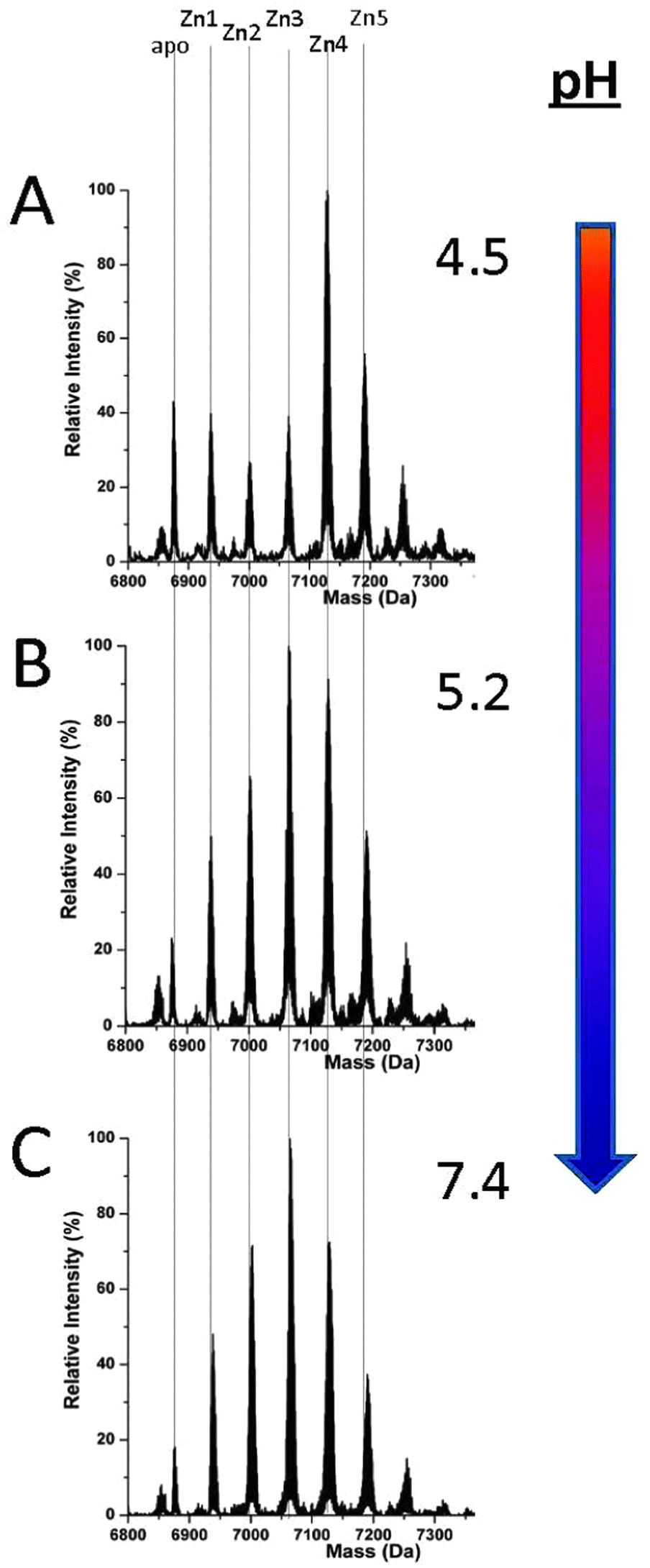 | ||
| Fig. 5 Representative ESI-MS spectra recorded during the pH titration of ZnxMT (x = 0–5) after approximately 2.5 equivalents of Zn(II) had been added. The spectra at pH 4.5 (A) shows a mixed binding mechanism whereas by pH 5.2 (B) it is largely distributive and terminally bound remaining unchanged to pH 7.4 (C). | ||
Discussion
The metalation mechanism of MT1a is critical to our understanding of its in vivo function and to the fundamental description of protein metal-thiolate chemistry. MT1a provides a unique example of a highly flexible metal-binding protein that can coordinate many metals in a number of conformations. This flexibility makes defining a specific binding mechanism difficult as there may be many possible mechanisms and conformations that can be adopted. Adding to this difficulty, metallothioneins lack spectroscopic features common in other proteins like aromatic amino acids or well-defined secondary structural elements.57 Thus, spectroscopically active metals like Cd(II) have been used as a model for Zn(II) binding which is more difficult to monitor. From the experiments presented here, we show that MT1a metalation can begin via two main pathways and that Zn(II) and Cd(II) show divergence in their pathway preference and pH sensitivity. The presence of a pH dependent equilibrium between structures of the partially metalated species has fundamental importance in the assessment of the role of MT1a in vivo.The MT1a-Cd(II) binding pathway is pH dependent
The data shown in Fig. 2 and 3 demonstrate the pH dependence of the Cd(II) binding pathway. The distributed pattern of the Cd1–3MT1a species at pH 7.9, and reported for more basic pH,38 shows a stochastic distribution of metals over 7 possible binding sites. Scheme 1 shows the equilibria for the binding of the first four metals to MT with different Ks to indicate that the values of these constants change depending on conditions and control pathway selection.The declining stoichiometric binding constants (K1 > K2 > K3 > K4), calculated to fit the data are shown in Fig. 6C. From the binding constants, simulated ESI-MS spectra were generated which closely match the experimental data. At pH 5.0, the data do not show a distributed pattern and we sought to replicate the experimental data by modifying the binding constants in the simulation. Fig. 6A shows that an increasing series of binding constants (K1 < K2 < K3 < K4) generates a close fit to the low pH data. In Fig. 6B the intermediate or mixed pathway is replicated by binding constants that are approximately equal. These simulations confirm the presence of two major, pH dependent metalation pathways: one leading to a clustered Cd4MT1a and the other a series of terminally-bound CdxMT1a (x = 1–5) species. The analysis shows that the pH controls pathway selection based on modification of the relative magnitude of the apparent binding constants associated with formation of bead or cluster structures. The important feature is the relative magnitude of the constants and the trend that they follow, not the absolute magnitude, as concentrations of each species were not measured.
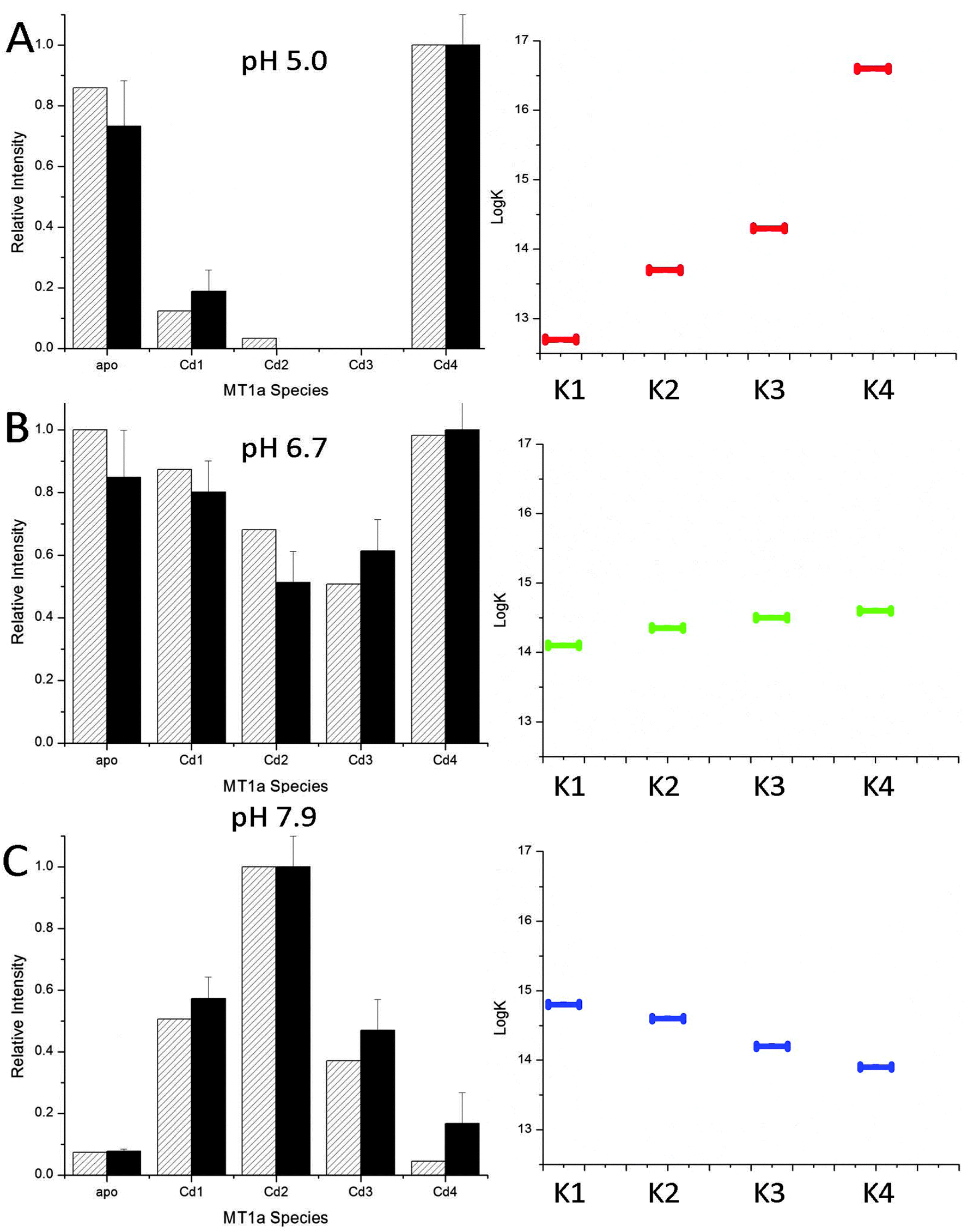 | ||
| Fig. 6 Simulated (hatched) and experimental (solid) ESI-MS data for binding of 2.5 mol equivalent of Cd(II) to apoMT1a at pH 5.0 (A), 6.7 (B) and 7.9 (C). The experimental ESI-MS data are shown by the solid black bars and the simulated data by the hatched bars (left). The simulated data were calculated based on the relative log(K) values for 4 consecutive bimolecular reactions assuming 2.5 mol eq. Cd(II) had been added. The log(K) values average to 14.4 in A–C, a value similar to that previously reported for the MT Cd-binding affinity.50 The trend in the relative log(K) values are shown on the right. K1 → K4 are the equilibrium constants for the addition of 1 to 4 Cd(II) to MT1a. It should be noted that these values are relatively correct but not absolute and decrease at lower pH. | ||
The MT1a-Zn(II) binding pathway always proceeds via terminally-bound, beaded species near physiological pH
The pH sensitivity over a physiologically relevant range, as shown for the Cd(II) data, was not observed for Zn(II). The ESI-MS spectra for Zn(II)-binding (Fig. 5) are essentially unchanged between pH 5.2 and 7.4. This means that the declining series of formation constants associated with the sequential Zn(II) metalation reactions are much less sensitive to pH and only begin to change below pH 5.2 which is not physiologically relevant. The pathway selection for the Zn(II) metalation is dominated by the noncooperative, terminally-bound beaded structure. Fig. 7, specific to Zn(II) binding, was generated in the same way as Fig. 6 and gives an accurate description of the changes in Kf that are involved in the pathway selection. Unlike for the Cd(II) data, the Kf values do not steadily increase for the lowest pH tested, but instead slightly increase then fall. This matches previous work that showed the last Zn(II) atoms are bound weakly by MT1a and are available for donation.44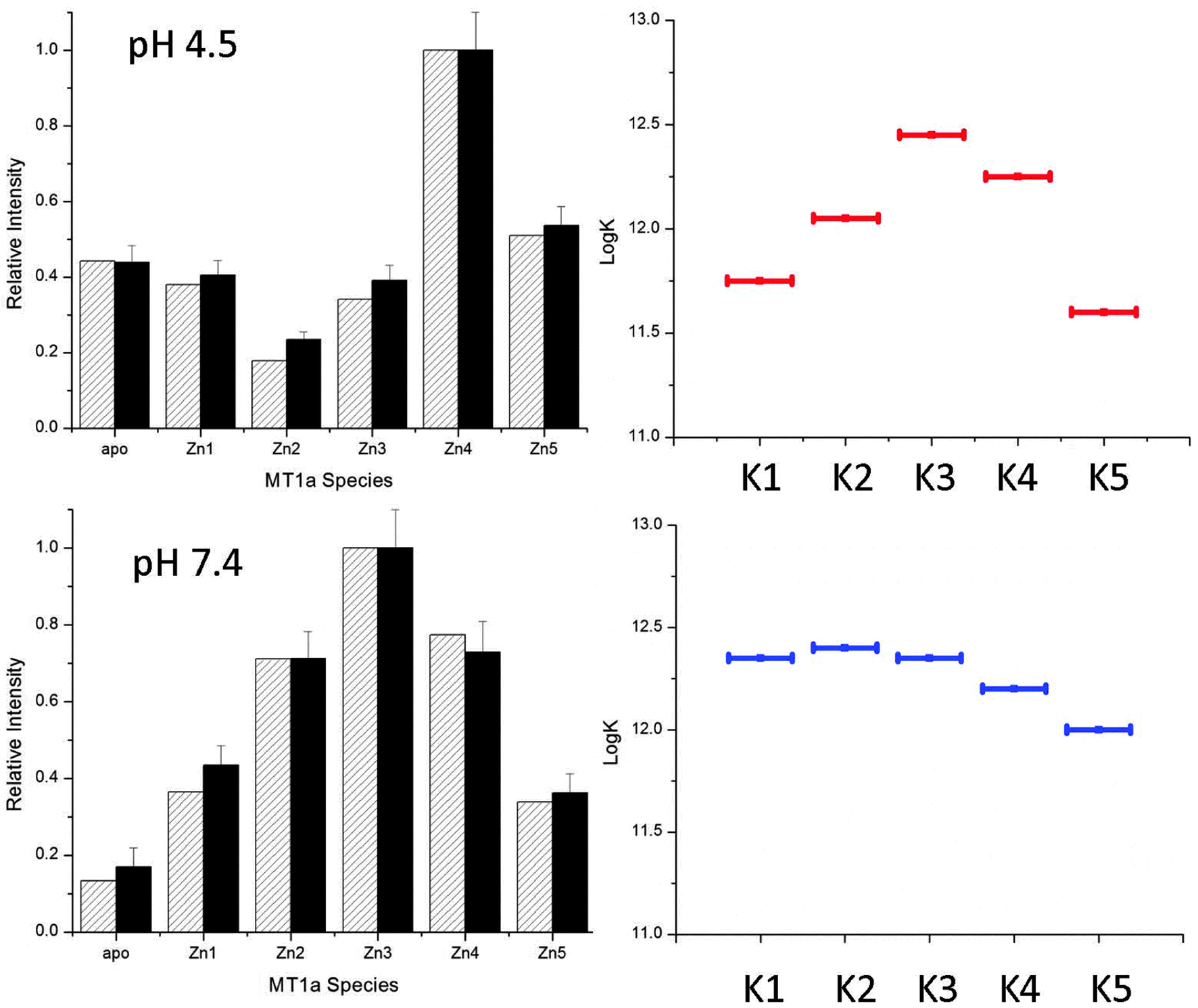 | ||
| Fig. 7 Simulated (hatched) and experimental (solid) ESI-MS data for MT metalation with 2.5 mol equivalents of Zn(II) at pH 4.5 (A) and 7.4 (B). The simulated data were calculated using the relative log(K) values for 5 consecutive bimolecular reactions with relative log(K) values similar to those reported in the literature.44 | ||
Zinc(II) and cadmium(II) show different metalation pathway preferences
Critical to the biological functions of MTs are the structures adopted when coordinating various metal ions. The proposed in vivo functions of MTs require this family of structurally homogenous proteins to bind essential and toxic metals for different purposes; namely to act as a metallochaperone for Zn(II) and Cu(II) while aiding in the detoxification of heavy metals like Hg(II) and Cd(II). On the surface, this appears to be a difficult task for a protein that lacks defined binding sites and formal secondary structure in the absence of bound metals. This is especially true for Zn(II) and Cd(II) since they both preferentially adopt tetrahedral coordination geometries and both form M4-Cys11 and M3-Cys9 clusters in the α and β domains of MTs. Critical to the use of Cd(II) as a model for Zn(II) is that the two metals follow the same metalation pathway, form the same intermediates and exhibit similar structural properties. However, our results show that the assumption that the binding pathway is the same for both Zn(II) and Cd(II) is flawed.Fig. 8 shows the two competing pathways that account for the metalation of apoMT1a to the fully metalated species. At slightly basic pH, Cd(II) passes through intermediates to form species similar to those shown in Fig. 8A and B before all Cys are involved in coordination and clustering must occur to accommodate further metal binding. At slightly acidic pH, Cd(II) metalation proceeds through the pathway shown by red arrows where the reaction is dominated by the formation of the Cd4-cluster structure. The metalation step leading to D may be part of a cooperative pathway that only features a clustered intermediate (Fig. 8C) and a fully metalated, two-domain end-product (Fig. 8E).
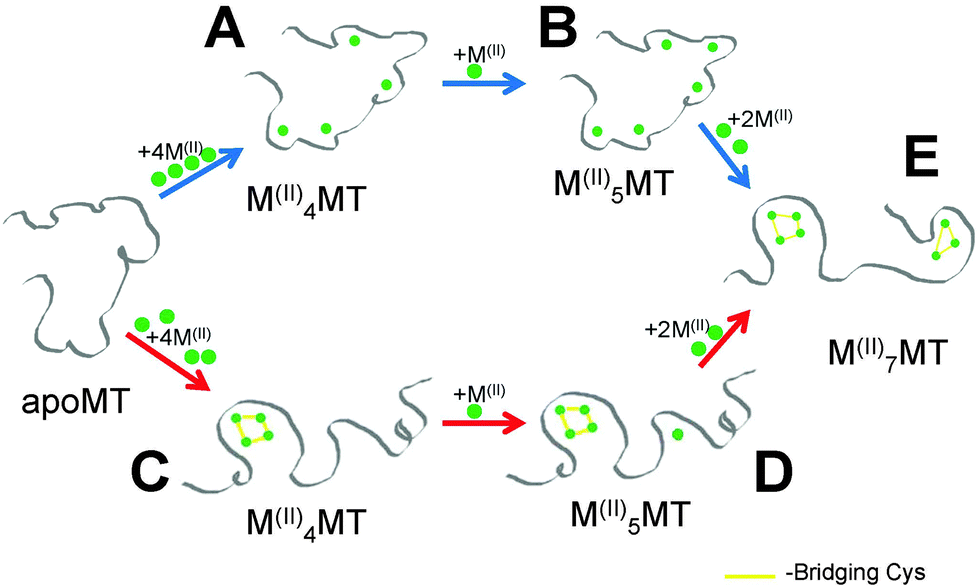 | ||
| Fig. 8 Representation of the two possible pathways for metalation with Zn(II) or Cd(II). The cooperative cluster driven pathway (red arrows) and the noncooperative beaded pathway (blue arrows). The terminally-bound, beaded structure (A) is able to coordinate an additional metal terminally, forming (B), which utilizes all twenty Cys residues, after which clusters are formed to accommodate the last two metals and form the stable two-cluster M7MT (E). The cooperatively formed cluster (C) must coordinate additional metals in the β-domain (D). | ||
The pathway selection for Zn(II) is strongly biased towards the beaded pathway shown by the blue arrows in Fig. 8. At very low pH, a mixture of the two pathways is seen with some clustering occurring but with a large fraction of Zn5MT1a being formed, compared to the low pH Cd(II) titration where no Cd5MT1a was observed. This suggests the presence of both clustered (Fig. 8C) and terminally-bound species (Fig. 8B) in equilibrium at low pH for Zn(II) metalation. The Zn4MT1a cluster does not have a stability advantage over terminally bound Zn1–5MT1a. This supports our previous model where we suggested that Zn(II) binding to apoMT1a results in a beaded structure with 5 terminally bound zinc.59
Fig. 9 summarizes the experimental data and compares the pH dependence of the Zn(II) and Cd(II) metalation pathways. Significantly, even at neutral pH, there is a considerable contribution from the clustered pathway of Cd(II). This suggests that, in vivo, MT1a can sequester Cd(II) into clusters with relatively higher binding constants, making it less available for transfer to zinc-dependent enzymes which mostly involve terminal coordination.58
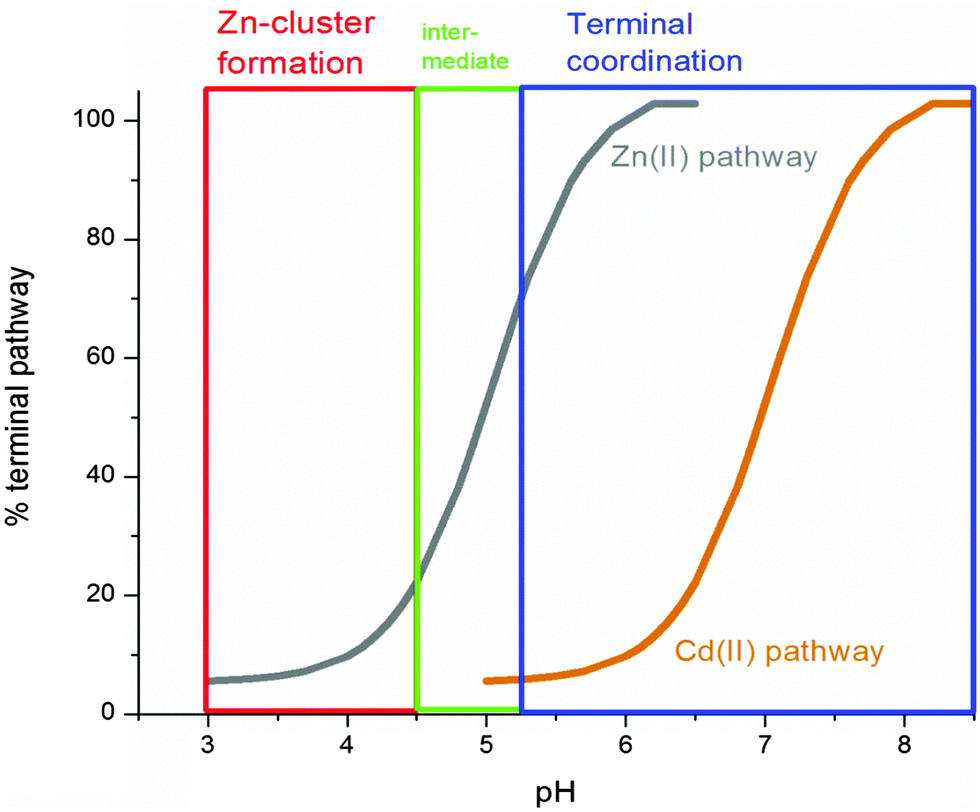 | ||
| Fig. 9 Change in Zn(II) metalation pathway as a function of pH (grey line) compared to Cd (orange line). Demetalation begins to occur at pH < 4.5 so the curve was extrapolated to pH 3 based on the Cd metalation data. At the lowest physiologically relevant pH around 5.5 the binding pathway is greater than 80% terminal (blue box) and only forms clusters below pH 4.5 (red box). | ||
Cadmium completely switches pathway preference between pH 6.5 and 7.2, where cluster formation is favored at lower pH and terminal coordination at higher pH. At lower pH, H+ effectively compete with incoming metals for the thiolates and the stability associated with the Cd4-thiolate cluster becomes the driving force behind cadmium binding. At higher pH, cadmium is bound terminally as the competition between metal and protons for thiolates is less intense.
In light of the results presented here, the use of exogenous metals as models for zinc and copper binding with respect to cluster formation need to be carried out with caution. Due to its spectroscopic properties, many studies on cluster formation in MT used Co(II) as a spectroscopic probe.25,39,60 Although Zn(II) and Cd(II) are both d10 metals, their Lewis acidities differ resulting in chemistries that change the mechanism by which they are bound to thiolates. It is likely that the differences between Zn(II) and Co(II) are even greater.
Possible origin of the proton sensitivity of the beads vs. cluster binding constants
The ESI-MS data show that the [Cd4(Cys)11]3− cluster dominates as an intermediate below pH 6.5 in the metalation of MT1a. DFT calculations of Ohanessian et al.61 on the proton affinities of thiolates in a wide range of zinc-thiolate compounds, have shown the terminal thiolates in [Zn(Cys)4]2− are significantly more basic than in the clustered [Zn4(Cys)11]3−. In our context, this suggests that the terminally-coordinated, beaded structures will be less stable under acidic conditions. We show the points at which the terminal structures become less stable in MT are approximately pH 5.0 for Zn(II) and 7.0 for Cd(II). The increased nephelauxetic effect or propensity for covalency of Cd(II) compared with Zn(II) supports the stability of the [Cd4(Cys)11α-clusters] over terminally bound Cd(II) below neutral pH and the different speciation observed here by ESI-MS between Zn(II) and Cd(II) for partially metalated MT1a. (Where the nephelauxetic effect refers to the increased propensity for covalency going down the Zn–Cd–Hg triad.)Understanding MT1a`s multitude of biological functions
The fact that Zn(II) does not cluster as readily as Cd(II) is relevant to its in vivo function. MT1a acts as a zinc-chaperone by delivering Zn(II) to newly synthesized metalloenzymes which requires a constant shuffling of metal ions.44,62 This function is facilitated by terminally bound Zn(II), increasing accessibility for donation compared to clustered Zn(II). On the other hand, the donation of Cd(II) often causes Zn-metalloenzymes to partially or completely lose function.63–65 By locking most of the Cd(II) in a cluster structure, donation may be discouraged. Only a small change in pH would induce clustering and many biological compartments have a pH well below 7.2 where clustering begins to take over as the preferred Cd-binding pathway.55,56,66 This is especially true in the medulla and proximal tubules of the kidneys which are known to be at low pH.67,68 Cd(II) has also been shown to accumulate in those areas69–71 and the clustered MT1a may aid in this accumulation. Wolff et al. have also demonstrated that Cd-MT is localized to lysosomes in proximal tubule cells, which is typically in the pH range that favors cluster formation.72 The clustered Cd-MT is known to be more resistant to proteolysis than apo- or partially metalated MT.73–75 This resistance to degradation, especially in low pH compartment to which MT1a is localized, may provide a mechanism by which cadmium persists in the kidneys for long periods of time, with a half-life of 10–30 years.76 Contrary to the concept of detoxification of heavy metals, this tendency to cluster and persist in the kidneys may cause more damage. It is for this reason that mammalian MTs may not be thought of as being “real” cadmium detoxifiers, but bind cadmium as a secondary, intrinsic property which exacerbates the toxicity in mammalian systems.In addition to specialized cells that are typically found at slight acidic pH, organelles responsible for protein maturation, such as the golgi, are also slightly acidic (pH 6.4–7.0).77 With ZnMTs being less prone to clustering under slightly acidic conditions, they would still be able to effectively donate zinc ions to maturing metalloproteins. The clustering of CdMTs at these pH ranges may provide a mechanism of protection by which the maturing metalloproteins are not improperly metalated with cadmium.
In addition to metal sequestration, MTs plays a role in cellular redox chemistry due to its abundance of thiols.78 For these thiols to be active they need to be exposed, so only apo- and partially metalated MT forms in the MT pool are redox active.79 The higher propensity to cluster in slightly acidic environments exposes more thiols which may affect cellular redox balance. This role as a redox active species is not necessarily separate from its main function as a zinc chaperone, and many reports have shown that these functions are intrinsically linked.21,80
Importance of pH control during experimental measurements
Fig. 4 and 7 highlight the potential complication when determining the metal speciation of MT, particularly when partially metalated, due to the pH effect on exact intermediate formation. For example, during Cd(II) metalation, a slight change in pH from 7.2 to 7.0 results in a large increase in the cluster pathway contribution. Experimentally, this small pH change could be caused by the displacement of cysteinyl protons during metalation so it is essential to monitor pH before and after each metal addition. It is also clear that this is much less of a problem during Zn(II) metalation because of the decreased pH sensitivity at physiological pH. MT also binds a number of exogenous heavy metals involved in chemotherapeutics81–83 and water contamination84 and the pH considerations raised in this paper likely apply to other MT-metal binding systems. Similar metalation experiments with human MT2a show a mixed mechanism at pH 7.430 and rabbit liver MT2a has been shown by NMR to cluster at pH 7.2, with the signal disappearing at higher pH.26 The pH dependence curve can likely be modestly shifted for different isoforms of MT. Indeed small differences in binding properties were found between human MT2 and MT3 isoforms, although the overall trend remained the same.85Conclusions
The considerable debate in the literature over the mechanisms behind MT metalation can be reconciled by the results presented in this paper. MT1a can bind Cd(II) and, to a lesser degree, Zn(II) via two distinct pathways that feature either a cooperative (clustered) or noncooperative (beaded) mechanism. Thus, the discrepancy between experimental results was likely due to small changes in pH conditions and the specific pH at which the cluster pathway dominates for each MT isoform.Of the two metalation pathways presented here, Zn(II) has a clear preference for the noncooperative, terminally-coordinated pathway under most pH conditions. The trend in Kf values associated with Zn(II) metalation of MT is largely unchanged over the range of physiologically relevant pH where most experiments are carried out. In contrast, Cd(II) showed a change in pathway preference between pH 6.8–7.2 where clustering became dominant at slightly acidic pH. This highlights the potential pitfalls in using Cd(II) as a model for Zn(II) binding.
The pH dependence of metal binding pathway and the structures adopted in those pathways provide insight into how MT functions as a metallochaperone (for Zn(II)) and sequesters toxic Cd(II). The cluster binds Cd(II) with a high affinity preventing the donation to lower affinity, terminal sites for Cd(II) coordination. This structure, resistant to degradation, may provide the mechanism by which Cd(II) persists in lower pH compartments in renal cells for many years. For Zn(II), these results strengthen our previously proposed model, where the terminally-coordinated beaded structure in dominant and the Zn5MT species is much more stable compared to the clustered structures. The last two Zn(II) are bound weakly and can easily be donated to other metalloenzymes.
Abbreviations
| MT | Mammalian metallothionein |
| MT1a | Human metallothionein 1a isoform |
| ESI-MS | Electrospray ionization mass spectrometry |
| CD | Circular dichroism |
| apoMT | Metal-free metallothionein |
Acknowledgements
We gratefully acknowledge financial support from NSERC of Canada through a CGS and PGS-D scholarships to G. W. I. and T. B. J. P., a Discovery Grant and a Research Tools and Instruments Grant, and the University of Western Ontario through an Academic Development Fund grant to M. J. S. We thank Ms. Brittany Lewis for preliminary studies.References
- V. Hodgkinson and M. J. Petris, J. Biol. Chem., 2012, 287, 13549 CrossRef CAS PubMed.
- D. E. Sutherland and M. J. Stillman, Metallomics, 2011, 3, 444 RSC.
- R. A. Festa and D. J. Thiele, Curr. Biol., 2011, 21, R877 CrossRef CAS PubMed.
- K. Balamurugan and W. Schaffner, Biochim. Biophys. Acta, Mol. Cell Res., 2006, 1763, 737 CrossRef CAS PubMed.
- T. Miyayama, Y. Ishizuka, T. Iijima, D. Hiraoka and Y. Ogra, Metallomics, 2011, 3, 693 RSC.
- E. Artells, Ò. Palacios, M. Capdevila and S. Atrian, FEBS J., 2014, 281, 1659 CrossRef CAS PubMed.
- F. Carmona, D. Mendoza, S. Kord, M. Asperti, P. Arosio, S. Atrian, M. Capdevila and J. M. Dominguez-Vera, Chem. – Eur. J., 2015, 21, 808 CrossRef CAS PubMed.
- C. A. Blindauer, Chem. Commun., 2015, 51, 4544 RSC.
- J. D. Park, Y. Liu and C. D. Klaassen, Toxicology, 2001, 163, 93 CrossRef CAS PubMed.
- A. M. Ronco, F. Garrido and M. N. Llanos, Toxicology, 2006, 223, 46 CrossRef CAS PubMed.
- E. Grill, E.-L. Winnacker and M. H. Zenk, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 439 CrossRef CAS.
- M. Vašák and D. W. Hasler, Curr. Opin. Chem. Biol., 2000, 4, 177 CrossRef.
- Y. Kondo, S.-M. Kuo, S. C. Watkins and J. S. Lazo, Cancer Res., 1995, 55, 474 CAS.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869 CrossRef CAS PubMed.
- R. J. Person, N. N. O. Ngalame, N. L. Makia, M. W. Bell, M. P. Waalkes and E. J. Tokar, Toxicol. Appl. Pharmacol., 2015, 36 CrossRef CAS PubMed.
- D. E. Sutherland, M. J. Willans and M. J. Stillman, J. Am. Chem. Soc., 2012, 134, 3290 CrossRef CAS PubMed.
- W. Maret, Exp. Gerontol., 2008, 43, 363 CrossRef CAS PubMed.
- S. G. Bell and B. L. Vallee, ChemBioChem, 2009, 10, 55 CrossRef CAS PubMed.
- H. Gonzalez-Iglesias, L. Alvarez, M. García, C. Petrash, A. Sanz-Medel and M. Coca-Prados, Metallomics, 2014, 6, 201 RSC.
- W. Qu and M. P. Waalkes, Toxicol. Appl. Pharmacol., 2015, 282, 267 CrossRef CAS PubMed.
- W. Maret, J. Nutr., 2000, 130, 1455S CAS.
- W. Braun, M. Vasak, A. Robbins, C. Stout, G. Wagner, J. Kägi and K. Wüthrich, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 10124 CrossRef CAS.
- G. W. Irvine, K. E. Duncan, M. Gullons and M. J. Stillman, Chem. – Eur. J., 2015, 21, 1269 CrossRef CAS PubMed.
- J. Ejnik, J. Robinson, J. Zhu, H. Försterling, C. F. Shaw and D. H. Petering, J. Inorg. Biochem., 2002, 88, 144 CrossRef CAS PubMed.
- I. Bertini, C. Luchinat, L. Messori and M. Vasak, J. Am. Chem. Soc., 1989, 111, 7296 CrossRef CAS.
- M. Good, R. Hollenstein, P. J. Sadler and M. Vasak, Biochemistry, 1988, 27, 7163 CrossRef CAS PubMed.
- S.-H. Chen, L. Chen and D. H. Russell, J. Am. Chem. Soc., 2014, 136, 9499 CrossRef CAS PubMed.
- G. W. Irvine and M. J. Stillman, Biochem. Biophys. Res. Commun., 2013, 441, 208 CrossRef CAS PubMed.
- K. E. Rigby and M. J. Stillman, Biochem. Biophys. Res. Commun., 2004, 325, 1271 CrossRef CAS PubMed.
- S.-H. Chen, W. K. Russell and D. H. Russell, Anal. Chem., 2013, 85, 3229 CrossRef CAS PubMed.
- T. T. Ngu, A. Easton and M. J. Stillman, J. Am. Chem. Soc., 2008, 130, 17016 CrossRef CAS PubMed.
- C. A. Blindauer, J. Inorg. Biochem., 2013, 121, 145 CrossRef CAS PubMed.
- C. Afonso, Y. Hathout and C. Fenselau, Int. J. Mass Spectrom., 2004, 231, 207 CrossRef CAS.
- M. Vaher, N. Romero-Isart, M. Vašák and P. Palumaa, J. Inorg. Biochem., 2001, 83, 1 CrossRef CAS PubMed.
- K. E. Duncan and M. J. Stillman, FEBS J., 2007, 274, 2253 CrossRef CAS PubMed.
- P. Palumaa, E. Eriste, O. Njunkova, L. Pokras, H. Jörnvall and R. Sillard, Biochemistry, 2002, 41, 6158 CrossRef CAS PubMed.
- P. M. Gehrig, C. You, R. Dallinger, C. Gruber, M. Brouwer, J. H. Kägi and P. E. Hunziker, Protein Sci., 2000, 9, 395 CrossRef CAS PubMed.
- D. E. Sutherland and M. J. Stillman, Biochem. Biophys. Res. Commun., 2008, 372, 840 CrossRef CAS PubMed.
- M. Vasak, J. Am. Chem. Soc., 1980, 102, 3953 CrossRef CAS.
- A. M. M. van Roon, J. C. Yang, D. Mathieu, W. Bermel, K. Nagai and D. Neuhaus, Angew. Chem., 2015, 127, 4943 CrossRef.
- W. R. Bernhard, M. Vasak and J. H. Kagi, Biochemistry, 1986, 25, 1975 CrossRef CAS PubMed.
- A. Krȩzel and W. Maret, J. Am. Chem. Soc., 2007, 129, 10911 CrossRef PubMed.
- E. Artells, Ò. Palacios, M. Capdevila and S. Atrian, Metallomics, 2013, 5, 1397 RSC.
- T. B. Pinter and M. J. Stillman, Biochemistry, 2014, 53, 6276 CrossRef CAS PubMed.
- H. Baba, K. Tsuneyama, M. Yazaki, K. Nagata, T. Minamisaka, T. Tsuda, K. Nomoto, S. Hayashi, S. Miwa and T. Nakajima, Mod. Pathol., 2013, 26, 1228 CrossRef CAS PubMed.
- J. Loebus, B. Leitenmaier, D. Meissner, B. Braha, G.-J. Krauss, D. Dobritzsch and E. Freisinger, J. Inorg. Biochem., 2013, 127, 253 CrossRef CAS PubMed.
- J. Chan, Z. Huang, I. Watt, P. Kille and M. J. Stillman, Can. J. Chem., 2007, 85, 898 CrossRef CAS.
- M. J. Stillman, Coord. Chem. Rev., 1995, 144, 461 CrossRef CAS.
- K. E. Duncan, C. W. Kirby and M. J. Stillman, FEBS J., 2008, 275, 2227 CrossRef CAS PubMed.
- D. W. Hasler, L. T. Jensen, O. Zerbe, D. R. Winge and M. Vašák, Biochemistry, 2000, 39, 14567 CrossRef CAS PubMed.
- N. Cols, N. Romero-Isart, M. Capdevila, B. Oliva, P. Gonzàlez-Duarte, R. Gonzàlez-Duarte and S. Atrian, J. Inorg. Biochem., 1997, 68, 157 CrossRef CAS PubMed.
- W. Lu and M. J. Stillman, J. Am. Chem. Soc., 1993, 115, 3291 CrossRef CAS.
- M. Stillman, W. Cai and A. Zelazowski, J. Biol. Chem., 1987, 262, 4538 CAS.
- J. Pande, C. Pande, D. Gilg, M. Vasak, R. Callender and J. Kägi, Biochemistry, 1986, 25, 5526 CrossRef CAS PubMed.
- N. Demaurex, Physiology, 2002, 17, 1 CAS.
- C. Settembre, A. Fraldi, D. L. Medina and A. Ballabio, Nat. Rev. Mol. Cell Biol., 2013, 14, 283 CrossRef CAS PubMed.
- S.-H. Hong, Q. Hao and W. Maret, Protein Eng., Des. Sel., 2005, 18, 255 CrossRef CAS PubMed.
- W. Maret, BioMetals, 2011, 24, 411 CrossRef CAS PubMed.
- K. L. Summers, D. E. Sutherland and M. J. Stillman, Biochemistry, 2013, 52, 2461 CrossRef CAS PubMed.
- G. Meloni, K. Zovo, J. Kazantseva, P. Palumaa and M. Vašák, J. Biol. Chem., 2006, 281, 14588 CrossRef CAS PubMed.
- G. Ohanessian, D. Picot and G. Frison, Int. J. Quantum Chem., 2011, 111, 1239 CrossRef CAS.
- Y. Hathout, D. Fabris and C. Fenselau, Int. J. Mass Spectrom., 2001, 204, 1 CrossRef CAS.
- D. R. Holland, A. C. Hausrath, D. Juers and B. W. Matthews, Protein Sci., 1995, 4, 1955 CrossRef CAS PubMed.
- S. Y. Lo, C. E. Säbel, M. I. Webb, C. J. Walsby and S. Siemann, J. Inorg. Biochem., 2014, 140, 12 CrossRef CAS PubMed.
- M. F. Dunn, H. Dietrich, A. K. MacGibbon, S. C. Koerber and M. Zeppezauer, Biochemistry, 1982, 21, 354 CrossRef CAS PubMed.
- R. Gagescu, N. Demaurex, R. G. Parton, W. Hunziker, L. A. Huber and J. Gruenberg, Mol. Biol. Cell, 2000, 11, 2775 CrossRef CAS PubMed.
- J. Kleinman, W. Brown, R. Ware and J. Schwartz, Am. J. Physiol.: Renal, Physiol., 1980, 239, F440 CAS.
- N. Raghunand, C. Howison, A. D. Sherry, S. Zhang and R. J. Gillies, Magn. Reson. Med., 2003, 49, 249 CrossRef CAS PubMed.
- C. Erfurt, E. Roussa and F. Thévenod, Am. J. Physiol.: Cell Physiol., 2003, 285, C1367 CrossRef CAS PubMed.
- G. Nordberg, R. Goyer and M. Nordberg, Arch. Pathol., 1975, 99, 192 CAS.
- C. Dorian, V. H. Gattone and C. D. Klaasen, Toxicol. Appl. Pharmacol., 1992, 114, 173 CrossRef CAS PubMed.
- N. A. Wolff, W.-K. Lee and F. Thévenod, Toxicol. Lett., 2011, 203, 210 CrossRef CAS PubMed.
- K. B. Nielson, C. Atkin and D. Winge, J. Biol. Chem., 1985, 260, 5342 CAS.
- K. B. Nielson and D. Winge, J. Biol. Chem., 1985, 260, 8698 CAS.
- D. Winge and K.-A. Miklossy, J. Biol. Chem., 1982, 257, 3471 CAS.
- L. Järup and A. Åkesson, Toxicol. Appl. Pharmacol., 2009, 238, 201 CrossRef PubMed.
- J. Llopis, J. M. McCaffery, A. Miyawaki, M. G. Farquhar and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6803 CrossRef CAS.
- B. Ruttkay-Nedecky, L. Nejdl, J. Gumulec, O. Zitka, M. Masarik, T. Eckschlager, M. Stiborova, V. Adam and R. Kizek, Int. J. Mol. Sci., 2013, 14, 6044 CrossRef CAS PubMed.
- D. H. Petering, J. Zhu, S. Krezoski, J. Meeusen, C. Kiekenbush, S. Krull, T. Specher and M. Dughish, Exp. Biol. Med., 2006, 231, 1528 CAS.
- W. Maret, J. Nutr., 2003, 133, 1460S CAS.
- A. Casini, A. Karotki, C. Gabbiani, F. Rugi, M. Vašák, L. Messori and P. J. Dyson, Metallomics, 2009, 1, 434 RSC.
- J. Lecina, Ò. Palacios, S. Atrian, M. Capdevila and J. Suades, JBIC, J. Biol. Inorg. Chem., 2014, 465 Search PubMed.
- L. Liu, B. E. Rogers, N. Aladyshkina, B. Cheng, S. J. Lokitz, D. T. Curiel and J. M. Mathis, Mol. Imaging, 2014, 13, 1 Search PubMed.
- A. P. Esser-Kahn, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 15820 CrossRef CAS PubMed.
- P. Palumaa, I. Tammiste, K. Kruusel, L. Kangur, H. Jörnvall and R. Sillard, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1747, 205 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |