
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical phonons in methylammonium lead halide perovskites and implications for charge transport†
Michael
Sendner
ab,
Pabitra K.
Nayak
c,
David A.
Egger
d,
Sebastian
Beck
ab,
Christian
Müller
abe,
Bernd
Epding
ae,
Wolfgang
Kowalsky
abe,
Leeor
Kronik
d,
Henry J.
Snaith
c,
Annemarie
Pucci
abf and
Robert
Lovrinčić
*ae
aInnovationLab, Speyerer Str. 4, 69115 Heidelberg, Germany
bKirchhoff Institute for Physics, Heidelberg University, Im Neuenheimer Feld 227, 69120 Heidelberg, Germany
cClarendon Laboratory, Department of Physics, University of Oxford, Oxford OX1 3PU, UK
dDepartment of Materials and Interfaces, Weizmann Institute of Science, Rehovoth 76100, Israel
eInstitute for High Frequency Technology, TU Braunschweig, Schleinizstr. 22, 38106 Braunschweig, Germany. E-mail: r.lovrincic@tu-braunschweig.de
fCentre for Advanced Materials, Heidelberg University, Germany
First published on 7th October 2016
Abstract
Lead-halide perovskites are promising materials for opto-electronic applications. Recent reports indicated that their mechanical and electronic properties are strongly affected by the lattice vibrations. Herein we report far-infrared spectroscopy measurements of CH3NH3Pb(I/Br/Cl)3 thin films and single crystals at room temperature and a detailed quantitative analysis of the spectra. We find strong broadening and anharmonicity of the lattice vibrations for all three halide perovskites, which indicates dynamic disorder of the lead-halide cage at room temperature. We determine the frequencies of the transversal and longitudinal optical phonons, and use them to calculate, via appropriate models, the static dielectric constants, polaron masses, electron–phonon coupling constants, and upper limits for the phonon-scattering limited charge carrier mobilities. Within the limitations of the model used, we can place an upper limit of 200 cm2 V−1 s−1 for the room temperature charge carrier mobility in MAPbI3 single crystals. Our findings are important for the basic understanding of charge transport processes and mechanical properties in metal halide perovskites.
Conceptual insightsMetal halide perovskites are an outstanding material class for opto-electronics. The interplay of electronic and structural dynamics in these materials is currently under scrutiny. One of the open questions is the nature of electron–phonon interactions, which may bear important implications for charge-carrier generation and transport. We report here a detailed quantitative analysis of far-infrared spectra of the three methylammonium lead halide perovskites (CH3NH3Pb(I/Br/Cl)3). This allows us to deduce electron–phonon coupling constants and polaron masses, which are consistent with a large polaron picture for the free charges. Using the Feynman model in conjunction with our data, we set an upper limit for the optical phonon limited charge carrier mobilities at room temperature of ∼200 cm2 V−1 s−1 for CH3NH3PbI3. Our findings help to understand fundamental electrical transport mechanisms in metal halide perovskites. |
1 Introduction
Lead-halide perovskites, with the general chemical structure ABX3, have recently reemerged as promising materials for opto-electronic applications.1–3 The most prominent candidates to date for perovskite-based solar cells are the hybrid organic–inorganic perovskites ABX3,4 where A is the organic cation, B = Pb, and X is the halide. The most widely studied examples in this material class are the methylammonium (MA) lead halides5 (MAPb(I/Br/Cl)3). Their bandgaps range from approximately 1.6 eV (MAPbI3) to 3.0 eV (MAPbCl3).6 They exhibit structural phase transitions in an orthorhombic–tetragonal–cubic sequence, with MAPbI3 being tetragonal at room temperature, and MAPbBr3 and MAPbCl3 being both cubic.7 As these phase transitions can occur close to room temperature, structural fluctuations of the lead-halide cage have potentially strong impact on the opto-electronic properties.8Long charge carrier diffusion lengths exceeding 1 μm have been reported for MAPbI3 and MAPbBr3 single crystals and thin films.9–11 The Urbach energies in these materials are ∼15 meV, comparable to the values for typical inorganic crystalline semiconductor materials like GaAs,12 suggesting a low prevalence of bandgap states, which often lower the carrier mobilities in semiconductors. Despite the low number of band gap states and the often observed high structural order of the corner sharing lead halide octahedras, the charge carrier mobilities are modest, especially in view of the low effective masses.13,14 Moreover, the thermal conductivity of MAPbI3 has recently been calculated to be ultralow.15 All of these properties are intimately connected to the nature of the lattice vibrations in these materials. In particular, the modest charge carrier mobilities have been explained by either predominant electron scattering from acoustic16–19 or optical phonons20–22 or the formation of polarons.19,23–26 Furthermore, recently the influence of spin–orbit coupling on the temperature dependence of the mobility was shown theoretically.27 To better understand such fundamental material properties, detailed knowledge of the phonon spectra is mandatory. Such can in principle be obtained from Raman measurements, which for MAPbX3 are, however, experimentally challenging due to the ease of beam damage under laser irradiation. Far infrared (IR) spectroscopy circumvents the beam damage problem and reveals information complementary to Raman spectroscopy. Several far-IR studies of MAPbX3 have appeared over the last two years.20,28,29 Pérez-Osorio et al. measured the frequency of the transversal optical (TO) phonons and theoretically predicted a strong LO–TO splitting for the strongest vibration.29 Wright et al. estimated the longitudinal optical (LO) phonon frequencies indirectly via temperature dependent photoluminescence measurements (ωLO = 11.5 and 15.3 meV, 92 and 122 cm−1).22 However, a direct experimental determination of the LO phonon frequency is currently missing. In view of the fact that LO phonons can interact with charge carriers, it is important to understand charge transport close to room temperature.
Herein we report far-IR spectra of the three methylammonium lead halide perovskite (MAPb(I/Br/Cl)3) thin films and provide a detailed quantitative analysis of the lattice vibrations corroborated by reflectance measurements on single crystals. From these data, we determine the frequencies and damping coefficients of both LO and TO phonons and find strong broadening and anharmonicity of the lattice vibrations for all three halide perovskites. This indicates dynamic disorder of the lead-halide cage at room temperature, in agreement with recent low frequency, off-resonance Raman measurements of MAPbBr3.8 From our data we calculate the static dielectric constants of all three halide perovskites via the Cochran-Cowley relation.30 We use the derived dielectric constants and phonon frequencies to estimate electron–phonon coupling constants and polaron masses. To quantify the importance of optical vibrations for charge transport, we employ the Feynman polaron model at elevated temperatures in conjunction with exciton effective masses, which provides upper limits for the optical phonon-scattering limited charge carrier mobilities in methylammonium lead halide perovskites.
2 Results
We measured far IR spectra in the range of 30–300 cm−1, which contain information on the Pb–X lattice vibrations, in contrast to the mid IR range that is dominated by molecular vibrations of the MA cation.31–33Fig. 1 compares measurements of a MAPbI3 thin film under two angles of incidence with the measurement of the corresponding lead halide, PbI2. Whereas at normal incidence one can only determine the resonance frequency of the TO mode (ωTO), it is possible to additionally determine the position of the longitudinal optical vibration (ωLO) at non-normal incidence via the Berreman mode.34 For polar materials such as MAPbI3 the LO frequency is higher than the TO frequency, hence a clear LO–TO splitting occurs. The peak positions for MAPbI3 and PbI2, determined through detailed analysis of the data as discussed below, are indicated in Fig. 1 and differ by only ∼10 cm−1. This is reasonable given the similar bulk moduli B for the two materials,35 which are directly related to the LO frequency of the crystals (B ∝ ωLO2).36 The strong broadening and asymmetry of the main TO mode of MAPbI3 at 63 cm−1 (8 meV) is apparent when compared to the sharp and symmetric TO mode of PbI2. Since MAPbI3 is highly crystalline, as evidenced by typically very sharp XRD peaks at room temperature, the phonon broadening is unlikely to be explained by static disorder caused by the micro strain present in polycrystalline thin films, because the broadening is also present in single crystals as we show below. We rather attribute it to dynamic disorder of the perovskite structure,8,37–40 similar to certain oxide perovskites.41 Given the frequency range of our measurements and the similar peak positions of MAPbI3 and PbI2, this likely originates from disorder of the lead-halide framework that is coupled to fast reorientations of the methylammonium cation,31,42 and possibly accompanied by strongly anharmonic hopping of Pb and/or I.8,43 | ||
| Fig. 1 Relative transmittance of a 266 nm thick MAPbI3 (black) and a 130 nm thick PbI2 (red) thin film on a silicon substrate under normal incidence (solid), and under 70° angle of incidence (dashed) with unpolarized light. The positions of the strongest respective TO and LO modes are marked with dashed vertical lines. | ||
Fig. 2 shows the measured far IR spectra of all three methylammonium lead halides at two angles of incidence, together with optically modeled spectra. Given the strong LO–TO splitting and anharmonic phonon coupling apparent in our data, a harmonic potential of the vibrations cannot be assumed anymore. Therefore, we use the Gervais model43 to fit the spectra for the dielectric function, which extends the harmonic oscillator model to the case of strong oscillations and, in contrast to a Lorentz oscillator, allows for different damping coefficients for LO and TO phonons (γLO, γTO):43,44
 | (1) |
 | ||
| Fig. 2 Relative transmittance of 266 nm thick MAPbI3 (black), 287 nm thick MAPbBr3 (blue) and 226 nm thick MAPbCl3 (green) thin films on silicon substrates at normal incidence (solid) and at 70° angle of incidence (dashed) with unpolarized light. Model fits for both angles of incidence for every MAPbX3, are shown in red dashed lines, with the resulting dielectric function shown in Fig. 4. | ||
The model takes into account that TO modes appear as the poles of ε(ω), and LO modes as the zeros of ε(ω).43 The high frequency dielectric background ε∞ is taken from mid-infrared spectral modeling.32 Two oscillators were used in eqn (1) to model the spectra for each thin film, simultaneously for the two angles of incidence – one rather strong oscillator and a second weaker one on the low energy side of the spectrum. Although the fits show minor deviations from the measured data, the agreement is very good overall. Following previously published theoretical calculations,29 we assign the mode at 32 cm−1 in MAPbI3 mostly to a Pb–I–Pb rocking vibration, and the stronger mode at 63 cm−1 mostly to a Pb–I stretching vibration. A similar assignment of modes can be assumed for MAPbBr3 and MAPbCl3, given their chemical similarity to MAPbI3.
We performed additional far IR measurements on single crystals to corroborate the thin film results and exclude possible influence of grain boundaries. We note that due to their thickness and extinction coefficients these have to be measured in reflection geometry and that the spectra of the I and Br single crystals were scaled in intensity because the crystal facets were smaller than the IR beam diameter. Fig. 3 shows the measured reflection spectra of the single crystals together with simulated spectra that were calculated using the corresponding dielectric functions obtained from the thin film transmission measurements. All three halide perovskites display an increased reflectivity in the spectral range between the TO and LO modes. This so called Reststrahlen band is typical for polar materials with strong LO–TO splitting.44,45 Due to the increasing ωTO and ωLO values from I to Cl, also the Reststrahlen band is blueshifted, as can be seen in the single crystal measurements. Importantly, we observe very good agreement between the measurements and calculations for all three spectra, which underlines that the dielectric functions obtained from the thin film measurements are meaningful.
 | ||
| Fig. 3 Relative reflectance spectra of MAPbI3 (black), MAPbBr3 (red), and MAPbCl3 (green) single crystals at room temperature with unpolarized light under an 80° angle of incidence (gold mirror as reference). The corresponding solid lines show calculated reflectance spectra using the dielectric function, derived for the thin films shown in Fig. 4 (spectra with star were scaled in intensity). | ||
The derived dielectric functions are shown in Fig. 4. The increase of the phonon frequencies in the I–Br–Cl series can be well described by the decrease of the effective ionic masses, μX, of halides X involved in Pb–X oscillations (ωTO ∝ μ−0.5; μI−0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :μBr−0.5:μCl−0.5 = 1:1.16:1.61), which strongly corroborates our assignment of these modes to lead-halide stretch vibrations. Fig. 4 also shows the loss function, Im(−1/ε(ω)), the peaks of which mark the positions of frequencies corresponding to LO phonons.34
:μBr−0.5:μCl−0.5 = 1:1.16:1.61), which strongly corroborates our assignment of these modes to lead-halide stretch vibrations. Fig. 4 also shows the loss function, Im(−1/ε(ω)), the peaks of which mark the positions of frequencies corresponding to LO phonons.34
 | ||
| Fig. 4 Derived dielectric functions for MAPbI3 (black), MAPbBr3 (red) and MAPbCl3 (green). Left: Real part ε1 of the dielectric function. Right: Imaginary part ε2 together with the loss function Im(−1/ε(ω)) (dashed lines). Peaks in ε2 indicate positions of TO phonons, peaks in Im(−1/ε(ω)) positions of LO phonons. | ||
Table 1 summarizes the determined ωTO and ωLO frequencies for the different halide perovskites. Using the Cochran-Cowley relation,30
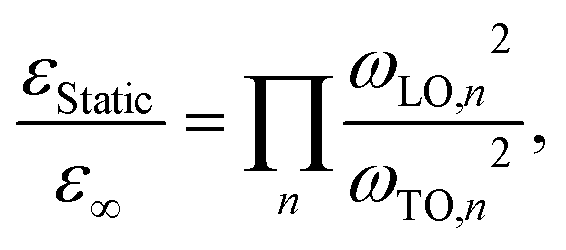 | (2) |
| ω TO [cm−1] | γ TO [cm−1] | ω LO [cm−1] | γ LO [cm−1] | ε ∞ | ε Static | |
|---|---|---|---|---|---|---|
| MAPbI3 | 32 | 9 | 40 | 11 | 5.0 | 33.5 |
| 63 | 20 | 133 | 30 | |||
| MAPbBr3 | 45 | 10 | 51 | 15 | 4.7 | 32.3 |
| 73 | 30 | 167 | 27 | |||
| MAPbCl3 | 66 | 15 | 70 | 16 | 4.0 | 29.8 |
| 89 | 52 | 225 | 34 |
We now examine the implications of our results for electron–phonon interactions and charge transport. Fröhlich described the movement of electrons in fields of polar lattice vibrations theoretically, introducing an interaction parameter49,50
 | (3) |
The coupling constant α quantifies the interaction between a charge carrier with an effective mass m*, and an optical phonon with frequency ωLO (in [cm−1] units). The ionic screening parameter, 1/ε* = 1/ε∞ − 1/εStatic, is used to describe an effective dielectric background. h is Planck's constant, c is the speed of light, and Ry the Rydberg energy. With multiple LO phonon branches, as is the case for the MAPbX3 studied here (see Table 1), an effective ωLO can be used, that is an average of the actual frequencies weighted by their spectral weight.51 We note that this is also reasonable given the possibly strong anharmonicity of modes we find in our data, which may result in more complex scattering processes rather than interaction of carriers with discrete optical modes.
Within Fröhlich's polaron theory, as extended by Feynman, the effective mass of the polaron mp can be calculated as52
 | (4) |
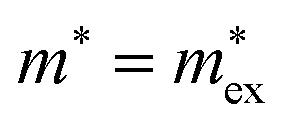 , with
, with  being the reduced exciton mass obtained from high quality magneto-optical measurements of MAPbI3 and MAPbBr3.53 For MAPbCl3, we performed density functional theory (DFT) based band-structure calculations using the VASP code,54 the PBE exchange–correlation functional55 and accounting for spin–orbit coupling (see Methods for details). We note that the reduced exciton effective mass obtained in this way is in good agreement with recent GW calculations,56 but individual electron and hole masses deviate more significantly. Our calculated polaron masses, as well as the coupling constants α, the ionic screening parameters 1/ε*, and the corresponding polaron radii
being the reduced exciton mass obtained from high quality magneto-optical measurements of MAPbI3 and MAPbBr3.53 For MAPbCl3, we performed density functional theory (DFT) based band-structure calculations using the VASP code,54 the PBE exchange–correlation functional55 and accounting for spin–orbit coupling (see Methods for details). We note that the reduced exciton effective mass obtained in this way is in good agreement with recent GW calculations,56 but individual electron and hole masses deviate more significantly. Our calculated polaron masses, as well as the coupling constants α, the ionic screening parameters 1/ε*, and the corresponding polaron radii | (5) |
| Effective massa | Ionic screening | Coupling constant | Polaron mass | Polaron radius | Mobility μ | |
|---|---|---|---|---|---|---|
|
|
1/ε* | α |
|
l p [Å] | [cm2 V−1 s−1] | |
| a In units of the free electron mass m0. | ||||||
| MAPbI3 | 0.10453 | 0.17 | 1.72 | 1.36 | 51 | 197 |
| MAPbBr3 | 0.11753 | 0.18 | 1.69 | 1.35 | 43 | 158 |
| MAPbCl3 | 0.20 | 0.22 | 2.17 | 1.48 | 27 | 58 |
| GaAs57 | 0.067 | 0.016 | 0.068 | 1.01 | 40 | 7000 |

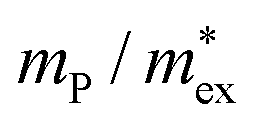
Importantly, these parameters can be used to estimate an upper limit for charge carrier mobilities μ in hybrid perovskites under the assumption that carriers are interacting only with optical phonons. Previously, Kadanoff's equation58 for weakly coupled polarons in the low temperature regime was used to estimate the charge carrier mobility in MAPbI3.20 In view of the rather low Debye temperatures of MAPbX359 and that for solar cell devices we are especially interested in μ at room temperature, we have to use the general result of Feynman et al.,52,60 obtained in the absence of the low temperature approximation:49
 | (6) |
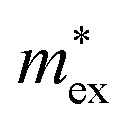 , which is related to the higher ionicity of the Pb–Cl bonds indicated by the higher LO–TO splitting.
, which is related to the higher ionicity of the Pb–Cl bonds indicated by the higher LO–TO splitting.
3 Discussion
The observed strong broadening and anharmonicity of the lattice vibrations imply a large dynamic disorder in methylammonium lead halides at room temperature. The fast reorientation of the methylammonium cation in the PbI3 host framework certainly contributes to this dynamic disorder.31,42,61,62 Moreover, it was recently reported that dynamic disorder is intrinsic to the general lead-halide perovskite structure even in the absence of organic cations.8 This effect may well contribute to the broadening of the vibrations observed here. We note that the addition of a central peak to the dielectric function that would indicate strongly anharmonic hopping41 of Pb or I, while consistent with our data, improves the quality of our fits only to a limited extent (see ESI†). Our obtained TO positions for MAPbI3 are very similar to those previously published by La-o-vorakiat et al.20 However, our oscillator strengths are roughly twice as large, possibly due to only partial surface coverage of the films used by La-o-vorakiat et al. (see ESI† for detailed comparison). We also find good qualitative agreement with a recently published dielectric function of MAPbI3 obtained from DFT and MD calculations.56 Our LO frequencies for MAPbI3 and MAPbBr3 are somewhat higher but still close to those indirectly estimated from temperature dependent photoluminescence measurements by Wright et al.,22 confirming their analysis.The determined polaron coupling constants for MAPbX3 of α ∼ 2 are at the higher end of the range typical for weak coupling (large polarons). Nevertheless, the polaron radii are consistent with large polarons, as they are far above the lattice constants of the materials and, in fact, show that polarons are spread over several unit cells. Consequently, the polaron masses are less than 50% higher than the reduced excitonic mass determined from magneto optical studies.53 Similar values are found by phonon calculations of MAPbI3 leading to a coupling constant of 2.4 in the large polaron regime.28,39
A comparison of our mobility estimations to literature reports is complicated by the wide spread of published values. For example, for MAPbI3 measured mobilities around 50 cm2 V−1 s−1 for thin films7,63 and 2.5 or 165 cm2 V−1 s−1 for single crystals9,64 have been reported. Overall, our estimates are at the higher end of reported values, which is what one would expect as our numbers are upper limits in the complete absence of defect scattering, acoustic phonon scattering, and the possible impact of local polaronic distortions.24 Our findings nevertheless imply that room temperature mobilities well below 100 cm2 V−1 s−1 obtained from MAPbI3 single crystal measurements may be strongly influenced by surface or interface effects. However, room temperature mobility values which are estimated to be significantly above 200 cm2 V−1 s−1 require careful scrutiny.65
We included the polaron parameters of GaAs57 in Table 2 for comparison. We can attribute the large difference in mobility between GaAs and the perovskites to two material properties: (i) the stronger ionic screening (indicative of a higher ionicity), and (ii) the lower LO phonon energy in the perovskites. The lead halide perovskites are a rare example for semiconductors with a Debye temperature of the LO phonon frequency, ΩD = hcωLO/kB, below room temperature, leading to a strong occupation of phonon states at 300 K which limits the mobility.
Finally, we briefly discuss the temperature dependence of the carrier mobility. For MAPbI3, a comparison is complicated because the cubic-to-tetragonal phase transition temperature is close to room temperature. For MAPbBr3, the transition occurs around 240 K and so it is generally an easier task. In the ESI† we demonstrate numerically that between 350 K to 250 K the mobility of MAPbBr3, including only the scattering due to optical phonons within the simple model used here, changes as T−0.62. Indeed, in contrast to this finding it has been discussed that in the cubic phase the overall temperature dependence of the measured Hall mobility is best described as T−1.4,19 the fingerprint of scattering due to acoustic phonons.13,16–19 However, several theoretical reports showed that mobilities limited by acoustic phonon scattering should be extremely high,66,67 which also conflicts with the experimental data. Therefore, we are currently facing the contradiction that, as shown here, the large polaron model provides the incorrect temperature dependence but the correct magnitude of the mobility, and models including scattering only due to acoustic phonons follow the correct temperature dependence but strongly overestimate the mobility itself. This may indicate that one or several assumptions underlying these textbook models of charge-carrier scattering are actually not valid for the case of lead-halide perovskites. Indeed, as we have emphasized throughout this paper, lead-halide perovskites show strongly anharmonic vibrations and dynamic disorder.8,37–40 Therefore, the canonical picture of charge carriers being scattered by harmonic vibrations of an otherwise rigid ionic lattice may not be valid for lead-halide perovskites. This situation calls for charge-carrier scattering models that do not start from the harmonic approximation and can take the intriguing structural dynamics of lead-halide perovskites fully into account. It also clearly highlights the need for more experimental data to better understand the transport of carriers in lead-halide perovskites.
4 Summary
In summary, we measured and analyzed quantitatively the lattice vibrations of the three methylammonium lead halide perovskites at room temperature. The spectra point to strong anharmonicity and dynamic disorder in these materials at room temperature, similar to certain oxide perovskites.41 Our detailed analysis of the obtained far IR spectra allows us to directly determine the LO–TO splitting, and deduce the strength of electron–phonon coupling in MAPbX3. Our estimated upper limit for the room temperature mobility in MAPbI3 of ∼200 cm2 V−1 s−1 indicates that there is still considerable room for further improvement of thin film quality, where mobilities of up to 50 cm2 V−1 s−1 have been achieved.7,63 However, we also show that despite only moderate polaron masses, the low LO phonon frequency and high ionicity of the lead halide perovskites fundamentally limit the charge carrier mobilities in these materials.5 Experimental methods
Thin film preparation
Silicon substrates were cut and cleaned by sonication in acetone and isopropyl alcohol for 10 minutes. Afterwards they were dried in a N2 stream. For perovskite film preparation we chose a vapor assisted evaporation process. Therefore we used as received PbCl2, PbBr2 (Alfa Aeser) and PbI2 (Sigma Aldrich) for evaporation. We used a high vacuum chamber with a pressure of 1.5 × 10−5 mbar and evaporated 176 nm thick layers of every one of those three materials at a rate of 8 nm min−1. After evaporation the as evaporated films were transferred into a nitrogen filled glovebox for the vapor assisted process. Accordingly we put the lead halide samples in a petri dish and surrounded it with the corresponding methylammonium halide. We used as received MACl (VWR), MABr, and MAI (DYESOL). For perovskite formation we heated the petri dish for 2 hours at 165 °C, 160 °C and 155 °C respectively. Thus we achieved perovskite films with a layer thickness of approximately 300 nm.Single crystal preparation
Single crystals of MAPbCl3, MAPbBr3, and MAPbI3 were prepared and characterized using the method described in the literature.68 In Fig. S5 (ESI†) we show the optical images of the crystals that were used in this study.IR spectroscopy
The as-fabricated perovskite thin films were transported in nitrogen atmosphere to a Bruker Vertex 80v Fourier transform IR (FTIR) spectrometer and measured under vacuum conditions (∼2 mbar). Spectra were acquired using a liquid Helium cooled Si-Bolometer with unpolarized light and a resolution of 4 cm−1. All spectra were referenced to the bare Silicon substrate with natural oxide at the respective angle of incidence. Spectra of the single crystals were measured in reflectance under an angle of incidence of 80° using unpolarized light and a gold mirror as reference. Scaling of the single crystal spectra was executed due to an improper matching of the focal plane of reference and single crystal.Optical modeling
Modeling of the measured thin-film spectra was performed using the commercially available software package SCOUT.69 The used optical model was made of a 1 mm thick Si substrate (described elsewhere32) and the MAPbX3 film on top. The film thicknesses of the perovskite layer was determined by mid-infrared FTIR measurement of the same samples and fitting their thin film spectrum with the published optical model.32 This thickness and the dielectric background ε∞ was used for the FIR optical model. The dielectric function of the perovskite thin films furthermore consisted of Gervais oscillators. The resonance frequencies of the transverse and longitudinal optical mode (ωTO, ωLO) and their damping (γTO, γLO) were fitted simultaneously for 0° and 70° angle of incidence.Computational methods
DFT calculations were performed using a cubic unit-cell of MAPbCl3 and a lattice constant of 5.71 Å as previously calculated, see ref. 70 for details. The plane-wave kinetic energy cutoff was set to 400 eV and an 8 × 8 × 8 Γ-centered k-point grid was used for self-consistently calculating the charge-density. The band-structure calculation was performed non-selfconsistently using an equally-spaced k-grid of 100 points (Δk ∼ 0.01 Å−1); the numerical convergence with respect to Δk was verified. To estimate the reduced exciton effective mass, we have fitted the equation, , to the onsets of the valence- and conduction band around R in the direction of Γ in the Brillouin zone, as the fundamental gap occurs at the R-point. The obtained reduced exciton mass is in good agreement with recent GW calculations for MAPbCl3,56 although individual electron and hole masses show somewhat stronger deviations. This may imply that our estimate for the reduced exaction mass benefits from error cancellation. To test the reliability of our PBE + SOC results, we have also performed HSE + SOC calculations71,72 on carefully reduced k-grids, from which we obtained a virtually identical reduced exciton mass, similar to what has been reported for MAPbI3 previously.73
, to the onsets of the valence- and conduction band around R in the direction of Γ in the Brillouin zone, as the fundamental gap occurs at the R-point. The obtained reduced exciton mass is in good agreement with recent GW calculations for MAPbCl3,56 although individual electron and hole masses show somewhat stronger deviations. This may imply that our estimate for the reduced exaction mass benefits from error cancellation. To test the reliability of our PBE + SOC results, we have also performed HSE + SOC calculations71,72 on carefully reduced k-grids, from which we obtained a virtually identical reduced exciton mass, similar to what has been reported for MAPbI3 previously.73
Acknowledgements
We acknowledge the German Federal Ministry of Education and Research for financial support within the InterPhase project (FKZ 13N13656, 13N13657). The work in Oxford was funded by EPSRC, UK. M. S. and C. M. acknowledge financial support by the Heidelberg Graduate School of Fundamental Physics. P. K. N. is supported by Marie-Curie actions individual fellowships (grant agreement number 653184). D. A. E. and L. K. were supported by the Austrian Science Fund (FWF): J3608-N20 and by a research grant from Dana and Yossie Hollander, in the framework of the WIS Alternative sustainable Energy Initiative.References
- W. Zhang, G. E. Eperon and H. J. Snaith, Nature Energy, 2016, 1, 16048 CrossRef
.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed
-
D. B. Mitzi, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc., 1999, pp. 1–121 Search PubMed
- D. Weber, Zeitschrift für Naturforschung B, 1978, 33, 1443 Search PubMed
- Y. Liu, Z. Yang, D. Cui, X. Ren, J. Sun, X. Liu, J. Zhang, Q. Wei, H. Fan, F. Yu, X. Zhang, C. Zhao and S. F. Liu, Adv. Mater., 2015, 27, 5176–5183 CrossRef CAS PubMed
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed
- O. Yaffe, Y. Guo, T. Hull, C. C. Stoumpos, L. Z. Tan, D. A. Egger, F. Zheng, G. Szpak, O. E. Semonin, A. N. Beecher, T. F. Heinz, L. Kronik, A. M. Rappe, M. G. Kanatzidis, J. S. Owen, M. A. Pimenta and L. E. Brus, 2016, arXiv:1604.08107 [cond-mat].
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Science, 2015, 347, 519–522 CrossRef CAS PubMed
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed
- Y. Chen, H. T. Yi, X. Wu, R. Haroldson, Y. N. Gartstein, Y. I. Rodionov, K. S. Tikhonov, A. Zakhidov, X. Y. Zhu and V. Podzorov, Nat. Commun., 2016, 7, 12253 CrossRef CAS PubMed
- S. De Wolf, J. Holovsky, S.-J. Moon, P. Löper, B. Niesen, M. Ledinsky, F.-J. Haug, J.-H. Yum and C. Ballif, J. Phys. Chem. Lett., 2014, 5, 1035–1039 CrossRef CAS PubMed
- T. M. Brenner, D. A. Egger, A. M. Rappe, L. Kronik, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2015, 6, 4754–4757 CrossRef CAS PubMed
- T. M. Brenner, D. A. Egger, L. Kronik, G. Hodes and D. Cahen, Nature Reviews Materials, 2016, 1, 15007 CrossRef
- M. Wang and S. Lin, Adv. Funct. Mater., 2016, 26, 5297–5306 CrossRef CAS
- R. L. Milot, G. E. Eperon, H. J. Snaith, M. B. Johnston and L. M. Herz, Adv. Funct. Mater., 2015, 25, 6218–6227 CrossRef CAS
- H. Oga, A. Saeki, Y. Ogomi, S. Hayase and S. Seki, J. Am. Chem. Soc., 2014, 136, 13818–13825 CrossRef CAS PubMed
- T. J. Savenije, C. S. Ponseca, L. Kunneman, M. Abdellah, K. Zheng, Y. Tian, Q. Zhu, S. E. Canton, I. G. Scheblykin, T. Pullerits, A. Yartsev and V. Sundström, J. Phys. Chem. Lett., 2014, 5, 2189–2194 CrossRef CAS PubMed
- H. T. Yi, X. Wu, X. Zhu and V. Podzorov, Adv. Mater., 2016, 28, 6509–6514 CrossRef CAS PubMed
- C. La-o-vorakiat, H. Xia, J. Kadro, T. Salim, D. Zhao, T. Ahmed, Y. M. Lam, J.-X. Zhu, R. A. Marcus, M.-E. Michel-Beyerle and E. E. M. Chia, J. Phys. Chem. Lett., 2016, 7, 1–6 CrossRef CAS PubMed
- E. Menéndez-Proupin, C. L. Beltrán Ríos and P. Wahnón, Phys. Status Solidi RRL, 2015, 9, 559–563 CrossRef
- A. D. Wright, C. Verdi, R. L. Milot, G. E. Eperon, M. A. Pérez-Osorio, H. J. Snaith, F. Giustino, M. B. Johnston and L. M. Herz, Nat. Commun., 2016, 7, 11755 Search PubMed
- X.-Y. Zhu and V. Podzorov, J. Phys. Chem. Lett., 2015, 6, 4758–4761 CrossRef CAS PubMed
- A. J. Neukirch, W. Nie, J.-C. Blancon, K. Appavoo, H. Tsai, M. Y. Sfeir, C. Katan, L. Pedesseau, J. Even, J. J. Crochet, G. Gupta, A. D. Mohite and S. Tretiak, Nano Lett., 2016, 16, 3809–3816 CrossRef CAS PubMed
- W. Nie, J.-C. Blancon, A. J. Neukirch, K. Appavoo, H. Tsai, M. Chhowalla, M. A. Alam, M. Y. Sfeir, C. Katan, J. Even, S. Tretiak, J. J. Crochet, G. Gupta and A. D. Mohite, Nat. Commun., 2016, 7, 11574 CrossRef CAS PubMed
- A. M. Soufiani, F. Huang, P. Reece, R. Sheng, A. Ho-Baillie and M. A. Green, Appl. Phys. Lett., 2015, 107, 231902 CrossRef
- Z.-G. Yu, J. Phys. Chem. Lett., 2016, 7, 3078–3083 CrossRef CAS PubMed
- F. Brivio, J. M. Frost, J. M. Skelton, A. J. Jackson, O. J. Weber, M. T. Weller, A. R. Goñi, A. M. A. Leguy, P. R. F. Barnes and A. Walsh, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 144308 CrossRef
- M. A. Pérez-Osorio, R. L. Milot, M. R. Filip, J. B. Patel, L. M. Herz, M. B. Johnston and F. Giustino, J. Phys. Chem. C, 2015, 119, 25703–25718 Search PubMed
- W. Cochran and R. A. Cowley, J. Phys. Chem. Solids, 1962, 23, 447–450 CrossRef CAS
- A. A. Bakulin, O. Selig, H. J. Bakker, Y. L. Rezus, C. Müller, T. Glaser, R. Lovrinčić, Z. Sun, Z. Chen, A. Walsh, J. M. Frost and T. L. C. Jansen, J. Phys. Chem. Lett., 2015, 6, 3663–3669 CrossRef CAS PubMed
- T. Glaser, C. Müller, M. Sendner, C. Krekeler, O. E. Semonin, T. D. Hull, O. Yaffe, J. S. Owen, W. Kowalsky, A. Pucci and R. Lovrinčić, J. Phys. Chem. Lett., 2015, 6, 2913–2918 CrossRef CAS PubMed
- C. Müller, T. Glaser, M. Plogmeyer, M. Sendner, S. Döring, A. A. Bakulin, C. Brzuska, R. Scheer, M. S. Pshenichnikov, W. Kowalsky, A. Pucci and R. Lovrinčić, Chem. Mater., 2015, 27, 7835–7841 CrossRef
- D. W. Berreman, Phys. Rev., 1963, 130, 2193–2198 CrossRef CAS
- Y. Rakita, S. R. Cohen, N. K. Kedem, G. Hodes and D. Cahen, MRS Commun., 2015, 5, 623–629 CrossRef CAS
- F. Aguado and V. G. Baonza, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 024111 CrossRef
- A. Poglitsch and D. Weber, J. Chem. Phys., 1987, 87, 6373–6378 CrossRef CAS
- M. T. Weller, O. J. Weber, P. F. Henry, A. M. D. Pumpo and T. C. Hansen, Chem. Commun., 2015, 51, 4180–4183 RSC
- J. M. Frost and A. Walsh, Acc. Chem. Res., 2016, 49, 528–535 CrossRef CAS PubMed
- D. A. Egger, A. M. Rappe and L. Kronik, Acc. Chem. Res., 2016, 49, 573–581 CrossRef CAS PubMed
- T. Ostapchuk, J. Petzelt, V. Zelezny, S. Kamba, V. Bovtun, V. Porokhonskyy, A. Pashkin, P. Kuzel, M. D. Glinchuk, I. P. Bykov, B. Gorshunov and M. Dressel, J. Phys.: Condens. Matter, 2001, 13, 2677 CrossRef CAS
- A. M. A. Leguy, J. M. Frost, A. P. McMahon, V. G. Sakai, W. Kochelmann, C. Law, X. Li, F. Foglia, A. Walsh, B. C. O'Regan, J. Nelson, J. T. Cabral and P. R. F. Barnes, Nat. Commun., 2015, 6, 7124 CrossRef PubMed
- F. Gervais and B. Piriou, J. Phys. C: Solid State Phys., 1974, 7, 2374 CrossRef CAS
- D. W. Berreman and F. C. Unterwald, Phys. Rev., 1968, 174, 791–799 CrossRef CAS
- H. Rubens and E. F. Nichols, Ann. Phys., 1897, 296, 418–462 CrossRef
- A. Poglitsch and D. Weber, J. Chem. Phys., 1987, 87, 6373–6378 CrossRef CAS
- N. Onoda-Yamamuro, T. Matsuo and H. Suga, J. Phys. Chem. Solids, 1990, 51, 1383–1395 CrossRef CAS
- T.-Y. Yang, G. Gregori, N. Pellet, M. Grätzel and J. Maier, Angew. Chem., 2015, 127, 8016–8021 CrossRef
- I. Biaggio, R. W. Hellwarth and J. P. Partanen, Phys. Rev. Lett., 1997, 78, 891–894 CrossRef CAS
- H. Fröhlich, Adv. Phys., 1954, 3, 325–361 CrossRef
- R. W. Hellwarth and I. Biaggio, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 299–307 CrossRef CAS
- R. P. Feynman, Phys. Rev., 1955, 97, 660–665 CrossRef CAS
- K. Galkowski, A. Mitioglu, A. Miyata, P. Plochocka, O. Portugall, G. E. Eperon, J. T.-W. Wang, T. Stergiopoulos, S. D. Stranks, H. J. Snaith and R. J. Nicholas, Energy Environ. Sci., 2016, 9, 962–970 CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- M. Bokdam, T. Sander, A. Stroppa, S. Picozzi, D. D. Sarma, C. Franchini and G. Kresse, Sci. Rep., 2016, 6, 28618 CrossRef PubMed
- S. Adachi, J. Appl. Phys., 1985, 58, R1–R29 CrossRef CAS
- L. P. Kadanoff, Phys. Rev., 1963, 130, 1364–1369 CrossRef
- J. Feng, APL Mater., 2014, 2, 081801 CrossRef
- R. P. Feynman, R. W. Hellwarth, C. K. Iddings and P. M. Platzman, Phys. Rev., 1962, 127, 1004–1017 CrossRef
- J.-H. Lee, N. C. Bristowe, P. D. Bristowe and A. K. Cheetham, Chem. Commun., 2015, 51, 6434–6437 RSC
- I. P. Swainson, C. Stock, S. F. Parker, L. Van Eijck, M. Russina and J. W. Taylor, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 100303 CrossRef
- C. Wehrenfennig, M. Liu, H. J. Snaith, M. B. Johnston and L. M. Herz, Energy Environ. Sci., 2014, 7, 2269–2275 CAS
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed
- D. A. Valverde-Chávez, C. S. Ponseca, C. C. Stoumpos, A. Yartsev, M. G. Kanatzidis, V. Sundström and D. G. Cooke, Energy Environ. Sci., 2015, 8, 3700–3707 Search PubMed
- Y. He and G. Galli, Chem. Mater., 2014, 26, 5394–5400 CrossRef CAS
- Y. Wang, Y. Zhang, P. Zhang and W. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 11516–11520 RSC
- P. K. Nayak, D. T. Moore, B. Wenger, S. Nayak, A. A. Haghighirad, A. Fineberg, N. K. Noel, O. G. Reid, G. Rumbles, P. Kukura, K. A. Vincent and H. J. Snaith, Nat. Communs., 2016 DOI:10.1038/ncomms13303
-
W. Theiss, Scout, 2015, http://www.mtheiss.com/ Search PubMed
- D. A. Egger and L. Kronik, J. Phys. Chem. Lett., 2014, 5, 2728–2733 CrossRef CAS PubMed
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef
- E. Menéndez-Proupin, P. Palacios, P. Wahnón and J. C. Conesa, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 045207 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mh00275g |
| This journal is © The Royal Society of Chemistry 2016 |