
The living dead – common misconceptions about reversible deactivation radical polymerization
Martina H.
Stenzel
*a and
Christopher
Barner-Kowollik
*bcd
aCentre for Advanced Macromolecular Design (CAMD), School of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: m.stenzel@unsw.edu.au
bSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
cPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu
dInstitut für Biologische Grenzflächen, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 4th August 2016
Abstract
‘Living/controlled’ radical polymerization, preferably termed ‘reversible deactivation radical polymerization’ (RDRP), is now a well-established technique to generate functional and complex polymer architectures for the design of next generation soft matter materials. We have noticed, however, that there exists some confusion in the literature on how the living character of an RDRP process is adequately established. The purpose of the current article is therefore to provide a tutorial on the basic principles governing RDRP processes.
Reversible-deactivation radical polymerization (RDRP), frequently known by the term ‘living’ – or sometimes ‘living/controlled’ – radical polymerization,1 has led to a paradigm shift in the synthesis of macromolecular structures and soft matter materials. The development of techniques such as atom transfer radical polymerization (ATRP),2,3 nitroxide mediated polymerization (NMP),4,5 reversible addition fragmentation chain transfer (RAFT) polymerization6–8 and other related processes9 initially led to a wealth of studies investigating their underpinning mechanism and reaction kinetics.10,11 Concomitantly, the synthetic ease and versatility of these processes soon opened access routes to complex macromolecular materials.12,13 It was therefore only a matter of time before the literature was filled with reports describing various polymer architectures based on new monomers that were not necessarily commercially available. Furthermore, industrial processes have been developed, implementing RDRP processes on a larger scale, using homogeneous (bulk and solution) and heterogeneous (emulsion, mini-emulsion) systems.14,15 As a result – 20 years on – reversible-deactivation radical polymerization is an established technique that is employed on a daily basis by researchers from a wide array of fields for advanced materials synthesis. Arguably, RDRP has influenced all soft matter materials fields ranging from biomedical applications,16,17 analytical devices,18 optoelectronic applications19 to nanocomposites20 and many more.21 Thus, the significance of RDRP in material design does not only lie in the possibility of generating novel complex polymer architectures. Advanced macromolecular designs using a large variety of monomers that could not be polymerized in a controlled manner otherwise are readily accessible, offering access routes to novel materials. For example, polymers with bioactive groups can now be directly obtained on a large scale for medical applications. New block copolymers are accessible that can be used to generate devices with nano-sized patterns, ready to be used as membranes or for photonic application. The high end group fidelity of these polymers combined with almost any conceivable polymer structure can be used to modify surfaces of e.g. implants or help creating better interfaces in polymer nanocomposites. More importantly, RDRP allows for high throughput polymer synthesis and thus the rapid design of a polymer library for optimized material properties.
The fundamental principle of reversible-deactivation radical polymerization has its conceptual roots in living anionic polymerization, pioneered by Szwarc,22 and is based on the key idea that polymerizations – in particular the molecular weight of the resulting polymer – can be controlled by elimination (or in the case of RDRP the limitation) of transfer and termination reactions. Combined with rapid initiation compared to the rate of propagation, the resulting macromolecules feature a low dispersity, while the molecular weight can be selected by adjusting the ratio of monomer and initiator concentration and the monomer conversion according to
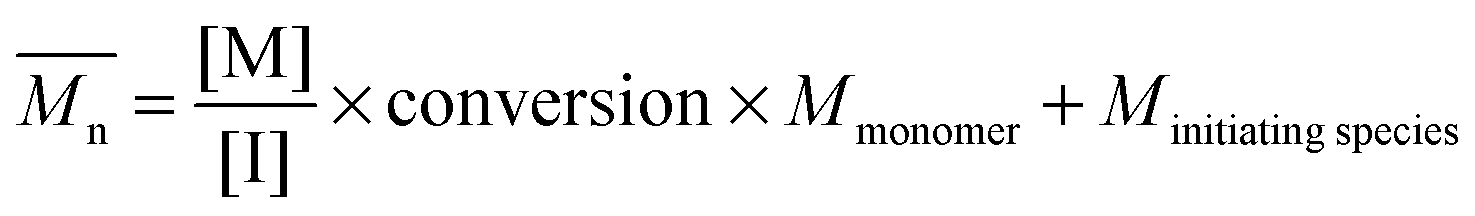 | (1) |
Firstly, it is paramount to realize that reversible-deactivation radical polymerizations are in fact radical reactions and Szwarc's principle – the absence of transfer and termination reactions – can never hold entirely true for a radical polymerization. Thus, one has to keep in mind that the controlling process, may it be ATRP, RAFT, NMP or others, is only superimposed onto a radical process where chain transfer reactions to monomer, polymer, solvent as well as a termination processes can take place at any time, albeit at a reduced rate at least in NMP and ATRP.
Before we embark on a discussion of the most common misconceptions that beset the interpretation of experimental polymerization data, it is important to briefly review the key characteristics of a process with living characteristics, which are derived from the original Szwarc definition. The below first three points are essential for a process with living characteristics, the fourth point is often observed yet not essential, while the points 5 to 7 are specific for the type of controlling process that is operational.
1. The molecular weight is determined by the monomer conversion and the ratio of the concentration of monomer to controlling species.
2. The lifetime of a propagating chain is increased from seconds (conventional radical polymerization) to several hours.
3. Rapid consumption of initiating species (ATRP, NMP) or chain transfer agent (RAFT).
4. Low dispersity (Đ).
5. Constant concentration of growing chains throughout the polymerization (however, this point is not unique to RDRP processes alone and is also associated with conventional free radical polymerizations).
6. The rate of termination and chain transfer reactions are strongly reduced (ATRP and NMP), while they are outcompeted by reversible chain transfer in RAFT.
7. Radicals are predominantly in a dormant state in the case of ATRP and NMP, while they are constantly reshuffled in the RAFT process, yet the overall radical concentration is not reduced.
While some of these characteristics can only be identified in detailed mechanistic studies, others (1, 4, 5) can be readily accessed by measuring molecular weights, usually by size exclusion chromatography, or by determining the monomer to polymer conversion.
How robust are RDRP processes?
In contrast to anionic polymerization, which requires stringent conditions in term of monomer and solvent purity, RDRP polymerizations are indeed more robust. The polymerization processes are orthogonal to many functional groups and function under variable conditions, yet there are limits to this freedom. RDRP initiators (as well as ligands used in the polymerization process) or chain transfer agents can be susceptible to chemical alteration by the reaction environment.23 Although many functional groups are orthogonal towards initiators, ligands and controlling agents, there are still a range of side reactions that can occur such as the hydrolysis or aminolysis of thioesters under alkaline conditions (e.g. by amines)24,25 or the replacement of ligands in ATRP by other functional groups or the elimination of halides within dormant species.26 Damages by peroxides, which are commonly present in solvents such as THF, dioxane or diethyl ether, cannot be neglected as they lead to the oxidation of RAFT controlling agents,27 a process that is often indicated by a colour change.28 In addition, light can activate or cleave the often labile bonds of the controlling agent29 resulting in changes in the rate of polymerization or can lead to a loss of molecular weight and end group control. Further, there are limitations with regard to the applicable temperature range, which in the case of RAFT and ATRP, should typically not exceed 120 °C.30In conclusion, although RDRPs are more robust than anionic polymerization, potential interference by impurities, functional groups and external fields need to be considered.
What can I learn from a conversion vs. time evolution or a first-order kinetic plot in an RDRP process?
After a polymerization has been carried out, the conversion-time profile (and the associated rate) has to be assessed. Typical phenomena that are observed are an initial delay of the reaction (inhibition) or an overall slower polymerization compared to a conventional free radical polymerization (retardation). Nota bene: ATRP and NMP both feature inherent retardation by virtue of their mechanism, while the RAFT process is in principle retardation free as the radical concentration is not lowered.31 Thus, in a RAFT process both phenomena (inhibition and retardation) can contain important mechanistic information.31 A range of techniques are available to determine the monomer consumption ranging from simple gravimetry (by precipitation or monomer evaporation), FT-NIR, NMR, dilatometry and many more. All techniques are suitable as long as their limitations are carefully considered. Gravimetry is only useful if polymer and monomer can be readily separated. On-line FT-NIR offers the unique possibility of recording many data points per unit of time, yet a traceable NIR band must be available, free of overlap with other absorptions. 1H-NMR is probably the most versatile technique to determine the monomer conversion as long as the polymer is soluble in the solvent. However, the main pitfall of this technique can be a loss of the quantitative integration to concentration relation due to inadequately selected relaxation delays. This cannot only be an issue when attempting to calculate the monomer conversion, but also the composition of a copolymer (see below).When interpreting conversion vs. time evolutions or – alternatively the associated pseudo first-order plot – great care is advised. In NMP and ATRP processes the rate of polymerization is much slower than in a comparable conventional free radical process and a linear first-order plot simply implies that a constant radical concentration is present. In a RAFT process, the rate of polymerization is essentially not reduced compared to the associated conventional process, as illustrated in Fig. 1. Here, a linear first-order plot is also only indicative of a constant radical concentration.
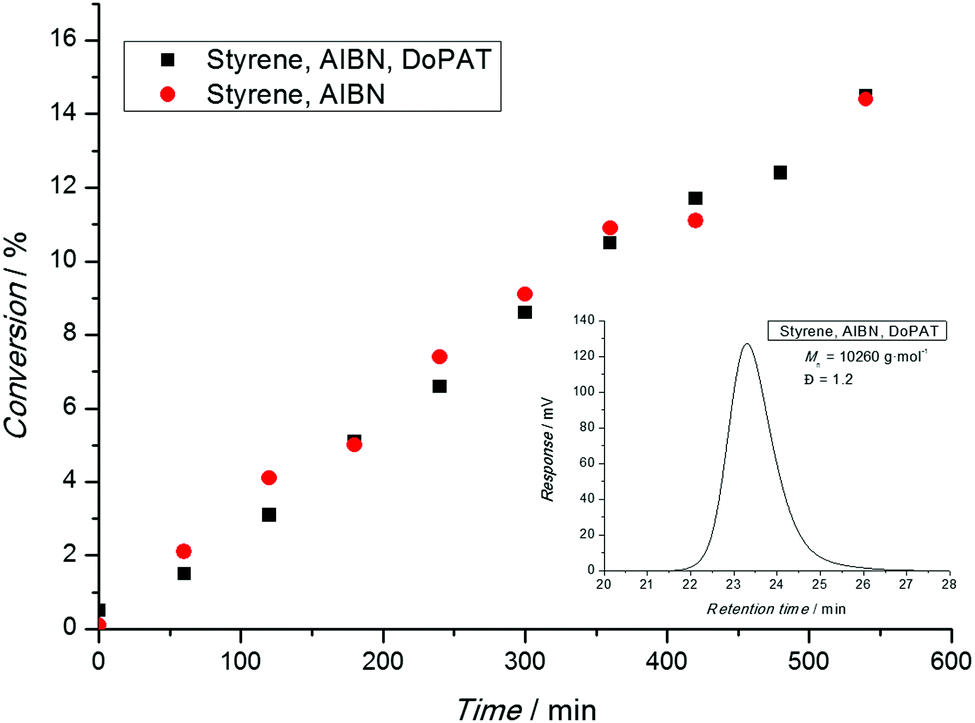 | ||
| Fig. 1 Conversion vs. time evolution of a trithiocarbonate (1 × 10−2 mol L−1) mediated radical polymerization of styrene (black symbols) and the associated conventional radical polymerization process (red symbols). Both polymerizations employ an initiator concentration (azobisisobutyronitrile (AIBN)) of 5 × 10−3 mol L−1 and were carried out at 60 °C. The final molecular distribution resulting from the RAFT process after 550 min is shown in the inset. | ||
In conclusion, a linear first order-plot can never be an indicator for living characteristics of an RDRP process. Claims such as ‘the linear first order plot underpins the living character of the polymerization’ or similar are simply incorrect and must be avoided.
How can I confirm the livingness of my polymerization?
As a linear first-order kinetic plot is therefore not a measure for livingness, one indicator for livingness is a linear relationship between molecular weight and conversion following the theoretical molecular weight evolution (eqn (1)). Thus, it is essential to carry out a range of polymerizations to different time points and therefore monomer conversions. Drawing conclusions from a single experiment (i.e. one point in time) is fraught with danger as the molecular weight might be close to the theoretical value by coincidence. Depicted in Fig. 2 is the molecular weight development of polystyrene during a RAFT polymerization experiment conducted in two solvents. The reaction in 1,4-dioxane did not proceed in good agreement with the theoretical value calculated usingprobably due to the oxidation of some RAFT agents by peroxides, lowering the RAFT concentration and inducing hybrid behaviour,32 that can be present in solvents such as 1,4-dioxanes and THF. However, at close to 30% monomer conversion the measured molecular weight is close to the theoretical value giving the (false) impression that the process is proceeding according to the expected mechanism.

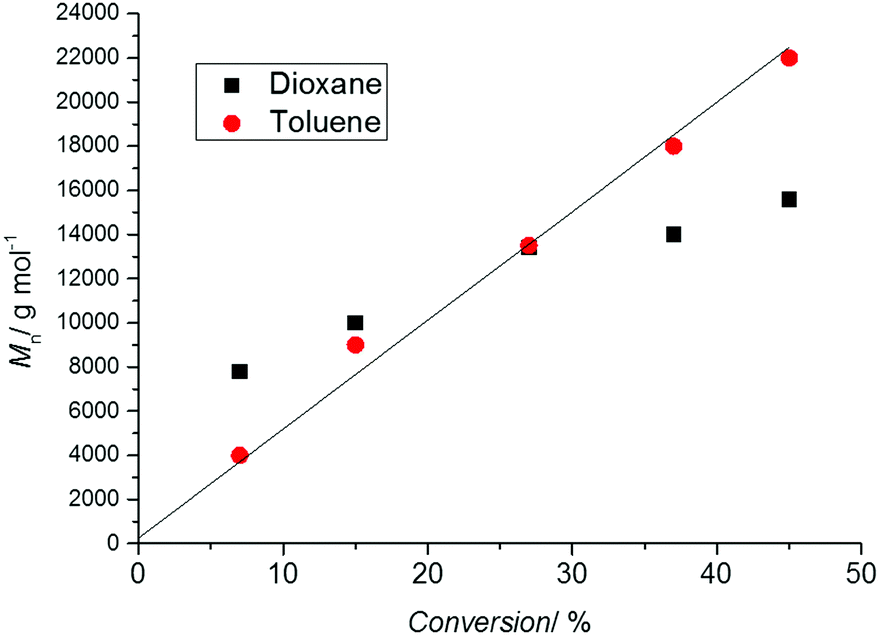 | ||
| Fig. 2 Molecular weight evolution (measured using SEC with polystyrene standards) vs. conversion during the polymerization of styrene (60 °C) in the presence of the RAFT agent 3-benzylsulfanylthiocarbonylsulfanyl-propionic acid in 1,4-dioxane (black symbols) as well as toluene (red symbols), [STY]0 = 2.9 mol L−1, [RAFT]0 = 6.1 × 10−3 mol L−1, [AIBN]0 = 2.0 × 10−3 mol L−1. The solid line represents the theoretical molecular weight evolution calculated using eqn (1). | ||
It must also be noted that the linear relationship between molecular weight and conversion indicated by eqn (1) only holds in ideal situations and especially in the RAFT process an improper choice of the RAFT agent can lead to a more complex behaviour.32
An important indication of the living character of a polymerization is a high end group fidelity that allows for further chain extensions. Direct characterization of the polymer structure including end group analysis is feasible via mass spectrometry,33 although this technique is not suitable for all polymers, in particular high molecular weight polymers. For details on block copolymer formation, refer to the last point in this article “How can I Judge if my Block Copolymer Synthesis is Successful?”
In conclusion, a linear evolution of the absolute number average molecular weight with conversion in conjunction with a clearly shifting full molecular weight distribution without bimodalities or tailing is a reliable indication for the living character of a radical polymerization.
Can I use NMR to determine the molecular weight?
Although SEC analysis is the most common technique to determine molecular weights, the method is not without problems. SEC requires suitable standards for calibration or detectors such as light scattering systems to access absolute molecular weights, with the latter often not being routinely available. Furthermore, the polymer may not be soluble in the mobile phase of the available SEC system. It is then tempting to employ the 1H NMR resonances associated with the polymer end group as a means to determine the molecular weight. In particular, 1H-NMR spectroscopy emerged as a popular tool to retrieve quantitative information about a polymer, including its absolute composition. As noted, for molecular weight determination, the integral value of resonances associated with the end groups is compared to suitable resonances associated with the repeating units, with this ratio subsequently being used to determine the number of repeating units in the polymer. However, caution is essential when employing the above strategy: while 1H-NMR analysis reveals the amount of end group moieties relative to a number of repeating units, it cannot be employed to assess if each polymer chain is end-capped with the functionality in question. Only a preceding diffusion ordered NMR spectrum (DOSY) can provide – under specific circumstances – information on the connectivities within a polymer, yet DOSY on polymer systems requires substantial NMR experience.34–36 These circumstances include end groups that have unambiguously assignable resonances that do not coincide with resonances associated with the lateral polymer chain. An identical diffusion coefficient derived from the end groups and the main chain proton resonances is an indication of direct connectivity between the two if at the same time (see below) the SEC trace is monomodal. As discussed above, RDRP is not strictly living and a detailed analysis of the final product will reveal a fraction of polymer generated by bimolecular termination or chain transfer events to solvent, monomer, polymer and other species. While this fraction can be small (close to 5 mol%), it can constitute a significant amount when the polymerization conditions are not ideal, for example when the RAFT agent or alkoxyamine is poorly selected or the radical flux is too high.5,9,32 The amount of ‘dead’ polymer will contribute to the signal intensity of the polymer repeating units, falsifying the molecular weight determination. To illustrate the problematic nature of 1H-NMR derived molecular weights in detail, we consider the case of poly(n-butyl acrylate) (pBA) prepared by RAFT polymerization. The 1H NMR spectrum of the pBA suggests a ratio of the BA units and RAFT agent functionality of 31.5 (Fig. 3b). Based on this ratio, the molecular weight can be inferred, which is in good agreement with the SEC derived molecular weight (using the MHKS parameters of pBA), i.e. 4300 g mol−1 (1H-NMR) vs. 4400 g mol−1 (SEC) (Fig. 3a). Next, we add conventionally generated pBA to a RAFT prepared pBA, to mimic the presence of ‘dead’ polymer. The polymer with Mn = 2100 g mol−1 (Đ = 1.18) is now mixed with pBA of high molecular weight and broad distribution (Mn = 151![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Đ = 2.55) at a ratio to effect the ratio between BA repeating units and RAFT agent of 31.5, resulting in an NMR spectrum that is virtually identical on first sight. However, the corresponding SEC analysis reveals a bimodal distribution highlighting that end group analysis alone, without supporting evidence from SEC analysis, may lead to incorrect conclusions (Fig. 3).
000 g mol−1, Đ = 2.55) at a ratio to effect the ratio between BA repeating units and RAFT agent of 31.5, resulting in an NMR spectrum that is virtually identical on first sight. However, the corresponding SEC analysis reveals a bimodal distribution highlighting that end group analysis alone, without supporting evidence from SEC analysis, may lead to incorrect conclusions (Fig. 3).
 | ||
| Fig. 3 (a and b) 1H-NMR and SEC analysis of a well-controlled RAFT polymerization of BA, where end group analysis using NMR leads to the same molecular weight as the measured value; (c and d) 1H-NMR and SEC analysis of an experiment mimicking side reactions, where the NMR analysis does not provide any hint with regard to the bimodal distribution. | ||
In conclusion, 1H-NMR analysis of polymers prepared by RDRP processes with the aim of determining the molecular weight is fraught with danger. It should only be employed in conjunction with supporting SEC distributions and – if possible – with DOSY NMR data.
Do broad molecular weight distributions imply that my RDRP was not successful?
When carrying out an RDRP process, one is often faced with a certain degree of uncertainty with regard to the dispersity of the resulting molecular weight distribution and what constitutes an acceptable dispersity. First, one has to note that the SEC recorded dispersity is strongly dependent – just as the molecular weight evolution – on the employed calibration. Only when absolute molecular weight detection or direct calibration has been employed, can the dispersity be regarded as reliable. Secondly, a broad distribution with a dispersity of e.g. 1.4 does not necessarily imply that the end group fidelity is not high. This is especially important for the RAFT process and can be readily understood when considering a non-degenerative transfer polymerization, where a dispersity of 2 is reached yet the end group fidelity is near perfect. Confirming end group fidelity is a complex exercise – yet must be carried out – typically consisting of the determination of the absolute molecular weight distribution, NMR analysis including DOSY as well as the recording of mass spectrometric data ideally from two ionization protocols (ESI and MALDI).In conclusion, while a (true) dispersity of below 1.1 or below is certainly associated with a high end group fidelity in RDRP, the reverse is not necessarily true, as a higher dispersity polymer can still have near perfect end group fidelity. Statements such as “The observed relatively high dispersity suggests poor end group control” can be incorrect.
How can I judge if my block copolymer synthesis is successful?
One of the main advantages of RDRP is the easy access to complex architectures such as block copolymers and star polymers.12,13 In particular, block copolymers are now routinely prepared using RDRP. Similar to homopolymerizations, one needs to consider all the potential reactions that can take place during the chain extension, which is the growth of the second block onto an existing endfunctional polymer. Prerequisite is therefore an extremely high end group fidelity of the first block and the careful purification prior to the chain extension to avoid the loss of reactive groups due to light, peroxides, heat and alkaline or acidic conditions. Addition of a second block is then subject to the same – above noted – indicators for living behaviour. Many block copolymers presented in literature have disparate polarities since these materials are often of interest in field of self-assembled materials. Once again, we urge caution when interpreting 1H-NMR data of block copolymers. Different solvents can lead to changes in polymer mobility, thus affecting the relaxation time as displayed in Fig. 4, where the ratio between both blocks can vary with the choice of solvent, leading to false interpretations with regard to the polymer composition and monomer consumption during the chain extension. Careful optimization of relaxation times is therefore paramount. If in doubt, the reader can always complement the results using other characterization techniques such as FT-IR, DSC and TGA.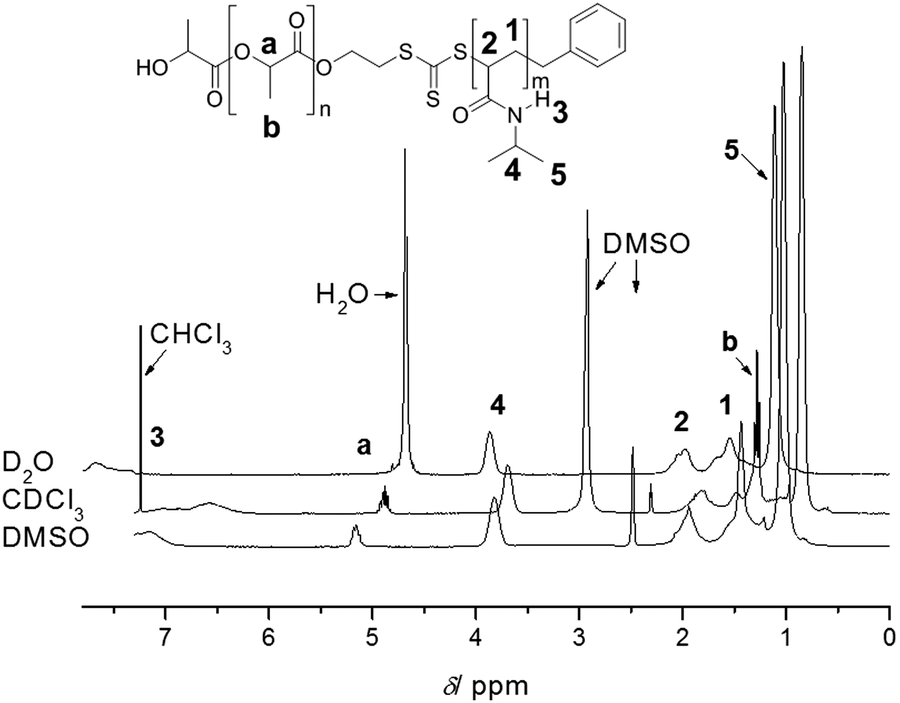 | ||
| Fig. 4 1H-NMR analysis of PLA-b-PNIPAAm in various solvents highlighting the importance of solvent choice to prevent suppression of signals employed for composition analysis. Reprinted with permission from ref. 37. | ||
Successful chain extension is usually confirmed by the shift of molecular weight and the literature abounds with examples where SEC appears to suggest a successful chain extension, yet only a small fraction of block copolymer has been formed. We again urge severe caution: the refractive index signal of an SEC analysis and the subsequent data processing provides a weight distribution, which is a quadratically (!) folded transformation of the number distribution. Thus, small molecular weight materials, i.e. the first prepared block, will be strongly suppressed in its signal by the presence of any higher molecular weight material. Thus, the SEC trace suggests that all low molecular weight material of the block copolymer has been chain extended, when in reality a very large percentage of the initial materials remains. Depending on the molecular weight range in question, the effect can be so substantial that no block copolymer has been formed, although the SEC only shows one monomodal trace and no material at the molecular weight of the initial polymer.
We have not covered here any aspects relating to more complex architectures, dispersed media and to polymer brushes grafted from surfaces, as these lie outside the scope of the current tutorial. The reader is referred to the relevant review articles.12,13,38,39
In conclusion, the assessment of block copolymer formation must – in the strictest terms – only proceed via the inspection of the number distribution of the block copolymers. 1H-NMR spectrometry can provide useful additional support for the polymer composition, provided the solvent is carefully selected, yet it cannot confirm block copolymer formation by itself.
Conclusions
The development of RDRP methods was and is critical for the design of advanced polymeric materials. RDRP does allow the design of new polymer structures, but more importantly, if the polymerization is optimized displaying living characteristics, the resulting polymers are well-defined in terms of molecular weight distribution and fraction of functional groups in complex architectures. The precise knowledge of the polymer structure can be crucial for understanding and tailoring material properties as the structure of each polymer holds the key to the macroscopic properties. For example, self-assembly processes of amphiphilic block copolymers in the solid state as well as in solution reply on equal length block copolymer segments when highly regular structures are targeted. Further, the folding of single polymer chains into defined morphologies via specific orthogonal folding points within the chain can only yield constant nanoparticle morphologies if the precursor polymers are of very narrow dispersity and the folding points are placed at identical points within the chain, including on the chain ends. Similarly, the efficient grafting of functional macromolecules to surfaces can only function if the to-be grafted chains all carry the identical and correct functionality. Despite the robustness of these processes, we advise significant caution when analysing these results and making statements what classifies as ‘living characteristics’. A careful assessment is especially important in the context of soft matter materials science, where the provision of e.g. well-defined copolymers or homopolymers with near perfect end group fidelity is often essential. Advanced high performance materials can only be prepared on the basis of well-understood polymeric precursors.Acknowledgements
C. B.-K. and M. H. S. are grateful to Kilian Wüst (KIT and UNSW) as well as Vincent Schüler and Katharina Elies (KIT) for carrying out selected experiments. C. B.-K. is grateful for support from the Queensland University of Technology (QUT), the Australian Research Council (ARC) as well as the Karlsruhe Institute of Technology (KIT) in the context of the STN Helmholtz and BIFTM programs. M. H. S. thanks the Australian Research Council (ARC) and the University of New South Wales (UNSW) for support. Both C. B.-K. and M. H. S. acknowledge the Sonderforschungsbereich 1176 funded by the German Research Council for support.Notes and references
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483–491 CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- C. Barner-Kowollik, M. Buback, B. Charleux, M. L. Coote, M. Drache, T. Fukuda, A. Goto, B. Klumperman, A. B. Lowe, J. B. McLeary, G. Moad, M. J. Monteiro, R. D. Sanderson, M. P. Tonge and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5809–5831 CrossRef CAS.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- X. Li, E. Mastan, W.-J. Wang, B.-G. Li and S. Zhu, React. Chem. Eng., 2016, 1, 23–59 CAS.
- M. Destarac, Macromol. React. Eng., 2010, 4, 165–179 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Q. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- H. S. Wang, M. Song and T. J. Hang, ACS Appl. Mater. Interfaces, 2016, 8, 2881–2898 CAS.
- G. Moad, M. Chen, M. Haussler, A. Postma, E. Rizzardo and S. H. Thang, Polym. Chem., 2011, 2, 492–519 RSC.
- S. J. Eichhorn, Soft Matter, 2011, 7, 303–315 RSC.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- R. Plummer, Y.-K. Goh, A. K. Whittaker and M. J. Monteiro, Macromolecules, 2005, 38, 5352–5355 CrossRef CAS.
- J. Xu, J. He, D. Fan, X. Wang and Y. Yang, Macromolecules, 2006, 39, 8616–8624 CrossRef CAS.
- D. B. Thomas, A. J. Convertine, R. D. Hester, A. B. Lowe and C. L. McCormick, Macromolecules, 2004, 37, 1735–1741 CrossRef CAS.
- N. V. Tsarevsky, W. A. Braunecker and K. Matyjaszewski, J. Organomet. Chem., 2007, 692, 3212–3222 CrossRef CAS.
- T. Gruendling, R. Pickford, M. Guilhaus and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7447–7461 CrossRef CAS.
- M. Dietrich, M. Glassner, T. Gruendling, C. Schmid, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010, 1, 634–644 RSC.
- T. G. Ribelli, D. Konkolewicz, S. Bernhard and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 13303–13312 CrossRef CAS PubMed.
- Y. Zhou, J. He, C. Li, L. Hong and Y. Yang, Macromolecules, 2011, 44, 8446–8457 CrossRef CAS.
- P. Vana, T. P. Davis and C. Barner-Kowollik, Macromol. Theory Simul., 2002, 11, 823–835 CrossRef CAS.
- C. Barner-Kowollik, J. F. Quinn, T. L. U. Nguyen, J. P. A. Heuts and T. P. Davis, Macromolecules, 2001, 34, 7849–7857 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Polymer, 2004, 45, 7791–7805 CrossRef CAS.
- T. S. Fischer, D. Schulze-Sunninghausen, B. Luy, O. Altintas and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2016 DOI:10.1002/anie.201602894.
- J. M. Zayed, F. Biedermann, U. Rauwald and O. A. Scherman, Polym. Chem., 2010, 1, 1434–1436 RSC.
- W. Li, H. Chung, C. Daeffler, J. A. Johnson and R. H. Grubbs, Macromolecules, 2012, 45, 9595–9603 CrossRef CAS PubMed.
- M. Hales, C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Langmuir, 2004, 20, 10809–10817 CrossRef CAS PubMed.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed.
- M. H. Stenzel, Macromol. Rapid Commun., 2009, 30, 1603–1624 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |