
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrinciples and implementations of electrolysis systems for water splitting
Chengxiang
Xiang
*a,
Kimberly M.
Papadantonakis
ab and
Nathan S.
Lewis
*abcd
aJoint Center for Artificial Photosynthesis, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: nslewis@caltech.edu; cxx@caltech.edu
bDivision of Chemistry and Chemical Engineering, California Institute of Technology, 210 Noyes Laboratory, 127-72, Pasadena, CA 91125, USA
cBeckman Institute and Molecular Materials Research Center, California Institute of Technology, Pasadena, CA 91125, USA
dKavli Nanoscience Institute, California Institute of Technology, Pasadena, CA 91125, USA
First published on 12th February 2016
Abstract
Efforts to develop renewable sources of carbon-neutral fuels have brought a renewed focus to research and development of sunlight-driven water-splitting systems. Electrolysis of water to produce H2 and O2 gases is the foundation of such systems, is conceptually and practically simple, and has been practiced both in the laboratory and industrially for many decades. In this Focus article, we present the fundamentals of water splitting and describe practices which distinguish commercial water-electrolysis systems from simple laboratory-scale demonstrations.
Introduction
Electrolysis of water produces H2 and O2 gases, is conceptually and practically simple, and requires only two metal electrodes, salt water, and a power supply. When a simple U-tube and two inverted burettes are incorporated into the cell set-up, electrolysis provides a straightforward method to produce H2 at 1 atm of pressure as well as to demonstrate the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H:O stoichiometry of water. In such systems, electrons are consumed at a metal cathode, where water molecules or hydrogen ions are converted to H2 gas (reduction half-reaction), and are liberated at a metal anode, where water molecules or oxygen-containing anions are converted to O2 gas (oxidation half-reaction). Thermodynamically, application of at least 1.23 V is required to electrolyze water under standard conditions.1 The kinetic barriers associated with making and breaking chemical bonds during each half-reaction manifest as overpotentials at each electrode, increasing the voltage required to pass a given current density through each electrode/solution interface relative to the thermodynamic voltage required to split water. An additional voltage is required to overcome the ohmic resistance of the aqueous electrolyte. Moreover, concentration overpotentials result from depletion of the reactant concentration relative to the product concentration near each electrode during operation. Hence the total overpotential of the system is the sum of the kinetic overpotential for the hydrogen-evolution reaction (HER), the kinetic overpotential for the oxygen-evolution reaction (OER), the concentration overpotential, and the overpotential required to overcome ohmic resistance (Fig. 1a). Simple electrolysis systems with overpotentials (∼0.7–1.0 V) and resistive losses (0.5 V) that are large relative to those of state-of-the-art systems can exhibit 40–50% efficiency for the conversion of input electrical energy into chemical energy in the form of H2(g) and O2(g).2
:1 H:O stoichiometry of water. In such systems, electrons are consumed at a metal cathode, where water molecules or hydrogen ions are converted to H2 gas (reduction half-reaction), and are liberated at a metal anode, where water molecules or oxygen-containing anions are converted to O2 gas (oxidation half-reaction). Thermodynamically, application of at least 1.23 V is required to electrolyze water under standard conditions.1 The kinetic barriers associated with making and breaking chemical bonds during each half-reaction manifest as overpotentials at each electrode, increasing the voltage required to pass a given current density through each electrode/solution interface relative to the thermodynamic voltage required to split water. An additional voltage is required to overcome the ohmic resistance of the aqueous electrolyte. Moreover, concentration overpotentials result from depletion of the reactant concentration relative to the product concentration near each electrode during operation. Hence the total overpotential of the system is the sum of the kinetic overpotential for the hydrogen-evolution reaction (HER), the kinetic overpotential for the oxygen-evolution reaction (OER), the concentration overpotential, and the overpotential required to overcome ohmic resistance (Fig. 1a). Simple electrolysis systems with overpotentials (∼0.7–1.0 V) and resistive losses (0.5 V) that are large relative to those of state-of-the-art systems can exhibit 40–50% efficiency for the conversion of input electrical energy into chemical energy in the form of H2(g) and O2(g).2
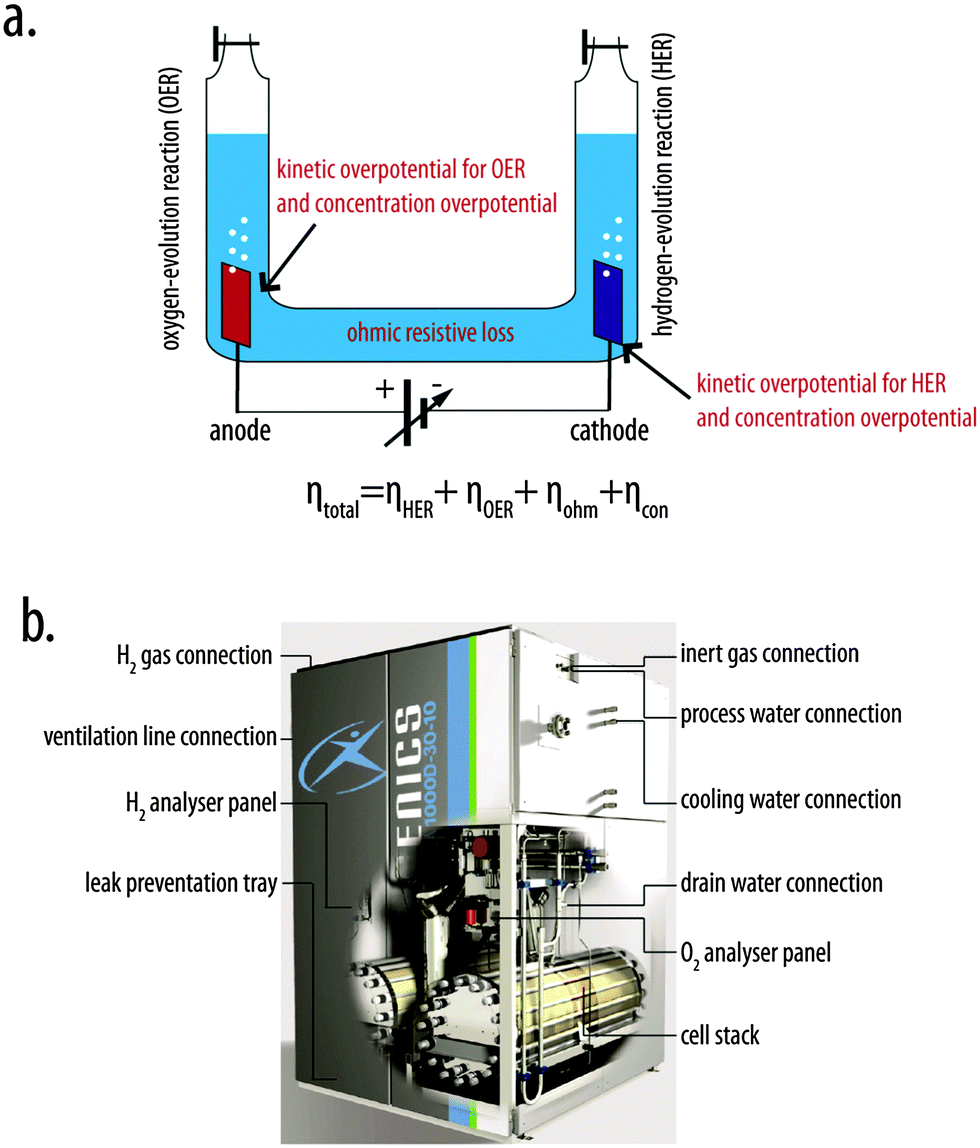 | ||
| Fig. 1 (a) A schematic illustration of a simple U-tube configuration for water electrolysis. Voltage penalties (overpotentials) increase the total voltage required to operate the system at a given rate relative to the thermodynamically required potential, and include the kinetic overpotentials associated with the hydrogen-evolution and oxygen-evolution reactions (ηHER and ηOER, respectively), the concentration overpotential (ηcon), and ohmic resistance loss (ηohm). The total operating voltage of the system is given by the sum of the thermodynamic voltage for the water-splitting reaction and the total overpotential, ηtotal, for the system. (b) A schematic illustration of a commercially available (Hydrogenics) alkaline water electrolyzer with auxiliary components.7 | ||
Commercial electrolyzers differ significantly from simple demonstration systems, and typically incorporate flow fields, precisely engineered electrodes, nanostructured electrocatalysts, membranes, strongly acidic or strongly alkaline electrolytes, multiple electrolysis cells arranged in stacks, water-purification systems, gas-conditioning units, heat-management units, controls for flows of input-fluid and output-gas streams, and power-management electronics.3–6Fig. 1b shows a schematic illustration of a commercially available alkaline water electrolyzer, HySTAT™, from Hydrogenics.7 Commercial water-splitting systems must be simultaneously safe, efficient, robust, cost-effective, and capable of producing H2 under pressure.8–11 These operational constraints dictate the types of electrodes, membranes, electrocatalysts, and electrolytes that are useful in commercial electrolyzers.
Efficiency
Essentially all of the operating costs of commercial electrolyzers are associated with the cost of electricity; hence increases in efficiency directly lower the operating costs.8,9 Efficiency can be increased relative to demonstration systems by employing optimized electrocatalysts, heat, and strongly acidic or strongly alkaline electrolytes.Electrocatalysts
Electrocatalysts are key components of commercial electrolyzers, and increase the efficiency of the system by reducing the kinetic overpotentials for the HER and OER. The rates of electron transfer between most metal electrodes and a species in solution are slow for complex, multi-step electrochemical reactions, including reactions as simple as the two-electron reduction of two protons to form a single molecule of H2.12,13 The overpotential partially overcomes the activation barrier for such processes, with the exact partitioning of the potential between the liquid and the kinetic barrier depending on the details of the system. Assuming an Arrhenius-type relationship for the interfacial charge-transfer kinetics yields an exponential relationship between the overpotential and the rate, or current density, j, of the reaction of interest: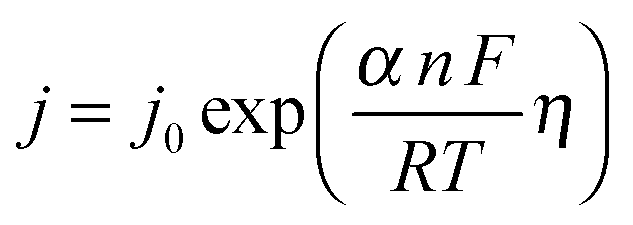 | (1) |
Kinetic overpotentials for the water-splitting half-reactions generally are lower for electrocatalysts in contact with strongly acidic or strongly alkaline electrolytes than for electrocatalysts in contact with neutral electrolytes. Reduction of a positively charged proton is easier than reduction of a neutral species, water, so electrocatalysts for the HER exhibit much lower overpotentials in acid than at neutral pH.12,13 Conversely, oxidation of a negatively charged hydroxide ion is easier than oxidation of a neutral species, water, so electrocatalysts for the OER exhibit much lower overpotentials in alkaline media than at neutral pH.12,13
In commercial electrolyzers, the water-oxidation and water-reduction electrocatalysts are deposited either directly onto the anode and cathode, respectively, or onto opposite sides of a gas-impermeable membrane separator, forming a membrane-electrode assembly (MEA). Electrical contact to the MEA is made using metal plates that contain channels to supply the input-water feedstock and that separately collect the H2(g) at the cathode and the O2(g) at the anode.8 The electrocatalysts should not be soluble in the electrolyte and should not be able to debond or desorb from the electrode, because such processes will lead to loss of the electrocatalyst from the MEA (or the electrode) when the input-water feed is continually provided to the electrolyzer. Commercial electrolyzers also contain systems for purifying the input-water feed, to minimize the concentration of trace metals that can plate onto the cathode and thereby poison its catalytic properties. For commercial electrolyzers, the area-related balance-of-systems costs far exceed the catalyst or MEA costs. Highly active catalysts drive higher current densities, reducing the electrode area required to produce a fixed amount of H2(g) per unit time, and thus reducing the area-related balance-of-systems costs.
Temperature
The gaseous products of water splitting are favored entropically relative to the reactants, so the thermodynamically required voltage to split water decreases as the temperature is increased. For example, the thermodynamic voltage required to perform the water-splitting reaction is reduced from 1.23 V at 298 K to 0.91 V at 1300 K.1 The kinetic overpotentials for water splitting also decrease (exponentially, per eqn (1)) with increased temperature. Thus, the efficiency of an electrolyzer improves substantially as the temperature increases. Commercial electrolyzers generally operate at temperatures above room temperature, and increased temperatures can be readily obtained by utilizing the heat produced by the ohmic and overpotential-derived voltage losses of the operating electrolysis unit.3,12,13,15Electrolytes
Increased temperatures also generally increase the conductivity of electrolytes. Strongly acidic or strongly alkaline electrolytes are compatible with optimally efficient water splitting because such electrolytes have high ionic conductivity, produce low ohmic resistance losses, and allow for the necessary transfer of protons or hydroxide ions between the compartments of the electrolyzer. Addition of a buffer species and/or salt to a near-neutral pH solution decreases the ohmic resistance losses for neutral electrolytes, but introduces other voltage penalties. The charged buffer or salt species in solution will be transported between the electrodes when current is flowing in the cell. Energy is consumed in driving this electrodialysis of the solution, resulting in reduced system efficiency while also producing a concentration gradient of the buffer or salt species. Gradual development of a concentration gradient in the electrolyte introduces additional voltage losses that increase over time, decreasing efficiency, and potentially leading to the eventual cessation of cell operation.Safety
Water-splitting systems that do not incorporate a robust method for separating the gaseous products of electrolysis are not “intrinsically safe”, a criterion which requires that a flammable, potentially explosive mixture of H2 and O2 is not formed at any point in space or time in the reactor. In the absence of a robust separator, convection or diffusion will transport product molecules from one side of the cell to the other, potentially resulting in an unsafe mixture of gases and decreasing the system efficiency due to product recombination or back-reactions (e.g., reduction of O2 at the cathode). Robust separation of the gaseous products of water-splitting can be accomplished by placing a porous separator or gas-impermeable membrane between the electrodes.Porous separators
Porous separators, such those made from asbestos, glass fibers, porous carbon, etc., prevent physical crossover of gas bubbles from one side of the cell to another, but do not prevent fluid flow. Two molecules of H2 are produced for each molecule of O2 produced during water splitting, so a pressure differential will develop between the two sides of the cell and will push liquid from the catholyte to the anolyte, eventually resulting in a catastrophic failure of the electrolyzer system. Therefore, water-splitting systems that use porous separators require active pressure control.16 Porous separators will allow some product crossover, but can provide intrinsically safe operation, provided that the rate of crossover of the dissolved gases is small compared to the rate of gas evolution at the electrodes.Gas-impermeable membranes
Gas-impermeable ion-exchange membranes, such as Nafion,17–19 can suppress product crossover in addition to blocking the transport of gas bubbles from one side of the cell to the other. Non-porous separators must be highly ionically conductive so as to add minimal ohmic resistance to the cell, and should be capable of holding back pressure, allowing for intrinsically safe operation in the absence of active pressure management. Because hydrogen evolution consumes protons and oxygen evolution liberates protons, water-splitting cells require a means for transporting protons (or hydroxide ions) from one side of the cell to the other. Membranes with fixed negatively charged sites, such as Nafion, can facilitate the selective transport of positively charged species such as protons, and are used in electrolyzers that operate in locally acidic conditions. Conversely, membranes that have fixed positively charged sites, such as a SELEMION membrane, can support the selective transport of negatively charged species such as hydroxide ions, and are thus appropriate for use in electrolyzers that operate in locally alkaline conditions. Note that the electrolyzer need not contain appreciable liquid solutions of either strong acid or strong base, because at steady state, pure water, either as a liquid or as a vapor, can be fed to the electrolyzer to produce H2(g) and O2(g) as a consequence of the electrolysis process.20Examples of commercial systems
Two types of low-temperature electrolyzers, alkaline water electrolyzers and polymer-electrolyte-membrane (PEM) electrolyzers, are the primary existing technologies for electrolytic water splitting (Fig. 2a and b). A third type of electrolyzer, the solid-oxide electrolyzer is available for high-temperature water splitting.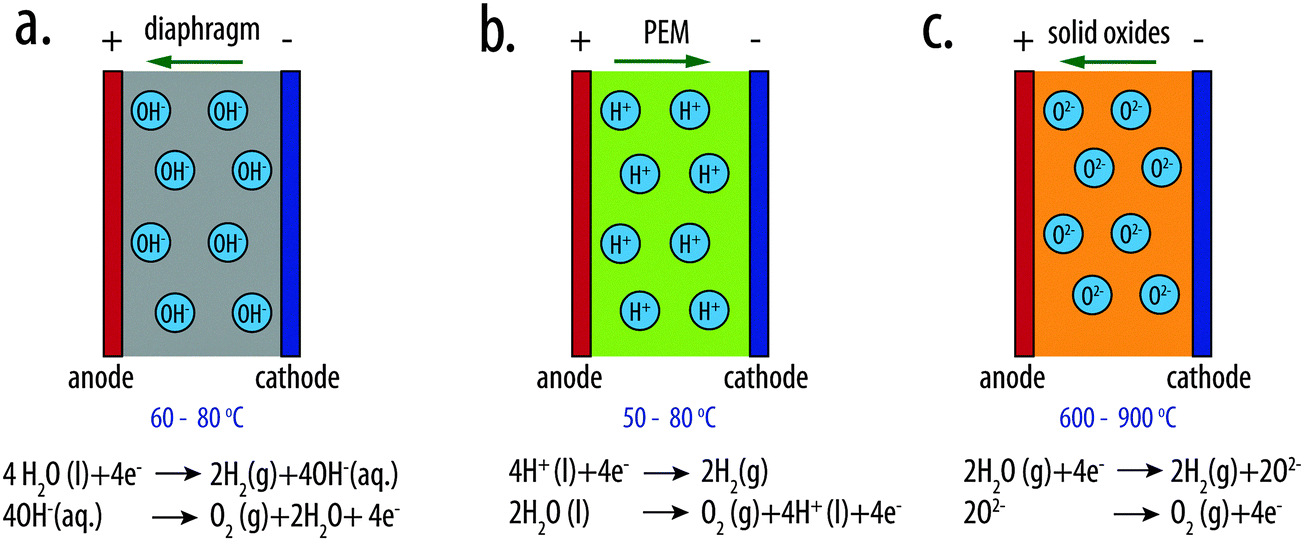 | ||
| Fig. 2 Schematic illustrations of (a) an alkaline water electrolyzer, (b) a polymer-electrolyte-membrane (PEM) electrolyzer and (c) a high-temperature solid-oxide electrolyzer. | ||
Alkaline water electrolyzers
The alkaline water electrolyzer is a mature technology that has been commercialized and used for decades for large-scale hydrogen production. The electrolysis cell consists of two electrodes separated by a gas-tight diaphragm that serves as the porous separator. A highly concentrated aqueous solution of KOH (25–30 wt%) is used as the electrolyte. The diaphragm generally consists of composites based on ceramics or microporous materials, such as microporous polymer membranes, glass-reinforced polyphenylene sulfide (PSS) compounds, or metal oxide composites.16 Non-platinum-group metals are generally used as the electrocatalysts for the HER and OER. High-surface-area, porous Ni or Ni-based metal alloys, such as RANEY® Ni or NiMo, are often used to minimize the catalytic overpotential for HER. For example, RANEY®-type NiMo electrodes exhibit an HER overpotential as low as 67 mV at 250 mA cm−2 of current density in 1 M KOH(aq).21 For the anode materials, oxides with a spinel structure, such as NiCo2O422 or Li-doped Co3O4,23 or oxides with a perovskite structure, such as SrFeO324 or La1−xSrxCoO3,25–27 or mixed metal oxides, such as (Ni, Fe)Ox,28–31 have been extensively studied and used as OER electrocatalysts in alkaline media. The alkaline water electrolyzer typically operates at ∼60–80 °C with a corresponding thermodynamic voltage for water splitting of 1.20–1.18 V. The terminal cell voltage of an alkaline water electrolyzer is 1.8–2.4 V at the typical operational current density of 0.2 to 0.4 A cm−2.32,33Polymer-electrolyte-membrane electrolyzers
In PEM-based electrolyzers, a solid polymer electrolyte (SPE) supports transport of protons between the anode and cathode. SPEs typically are made from perfluorosulfonic acid polymers, such as Nafion®, Felmion®, Fumapem® or Aciplex®. These polymers possess high oxidative stability, high mechanical strength and durability, and exhibit high conductivity for proton transport. The low permeability of the polymeric membranes precludes the formation of flammable gas mixtures and even allows for safe operation at the low current densities that are present during cycling operations. At membrane thicknesses of 100 μm to 200 μm, the proton conductivity is >0.1 S cm−2, which minimizes the resistive loss and allows operation at higher operating current densities than are possible for cost-effective alkaline electrolyzers. Platinum black, with a loading of 0.2–1 mg cm−2, and mixed Ir and Ru oxides with a typical loading of 1–2 mg cm−2, are generally used as the HER and OER electrocatalysts, respectively in PEM electrolyzers.15 PEM electrolyzers operate within the same temperature range (50–80 °C) as alkaline water electrolyzers. The terminal cell voltage of PEM electrolyzers ranges between 1.8 V to 2.2 V under the typical operational current density of 0.6 to 2 A cm−2.15 A state-of-the-art PEM electrolyzer exhibits a terminal cell voltage of ∼1.70 V (total overpotential of ∼520 mV) at an operational current density of 1.0 A cm−2 at 80 °C.3,15Solid-oxide electrolyzers
A third type of electrolyzer, the solid-oxide electrolyzer, can be used to effect high-temperature water splitting (Fig. 2c). Solid-oxide electrolyzers operate at temperatures of 600–900 °C. The thermodynamically required potential for water splitting using such systems is thus significantly lower (<1.0 V) than for other water-splitting systems. In this system, instead of a liquid electrolyte, steam is fed to the cathode chamber. The O2− anions generated at the cathode are transported through the solid electrolyte to the anode, where O2 is formed. The solid-oxide electrolyte generally consists of mixed yttrium zirconium oxides (YSZ). The cathode material is usually nickel and YSZ, whereas the anode material is generally a perovskite such as LaMnO3, LaFeO3, etc.34–37 High temperatures are needed to lower the resistivity of the electrolyte to O2− ions, but enable the use of non-noble-metal electrocatalysts. The solid-oxide electrolyte is a particularly attractive, cost-effective approach to water electrolysis when a high-temperature heat source is available.Summary
In summary, while the operational principles for water-electrolysis systems are straightforward, commercially available electrolyzers incorporate various auxiliary components and sophisticated design considerations, based on well-established principles of electrochemical engineering, to ensure stable, safe, cost-effective, scalable operation. These electrolyzer units exclusively use strongly acidic or alkaline electrolyte media (liquid electrolytes, polymer electrolytes or solid electrolytes) to minimize the resistive losses, concentration-overpotential losses and kinetic overpotentials for water splitting, and to ensure a unity transference number of protons and/or hydroxide ions thereby neutralizing any pH gradients that would otherwise result as a consequence of the electrolysis process. Separators with pressure-control systems, or membranes that hold back pressure, are necessary to ensure low crossover rates between the product gases at elevated operational temperatures and to support the pressure differentials characteristic of commercial electrolyzers. State-of-the-art electrolyzers operate at 70–80% electricity-to-hydrogen efficiency, and produce high-purity (>99.9%) hydrogen at ∼10 bar pressure while providing intrinsically safe operation at all times.Acknowledgements
This material is based upon work performed by the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award Number DE-SC0004993.References
- CRC Handbook of Chemistry and Physics, 96th edn, 2015 Search PubMed.
- K. Harrison, R. Remick, A. Hoskin and G. Martin, Hydrogen Production: Fundamentals and Case Study Summaries, Report NREL/PR-560-48269, National Renewable Energy Laboratory, The 18th World Hydrogen Energy Conference, Essen, Germany, 2010.
- A. Ursua, L. M. Gandia and P. Sanchis, Proc. IEEE, 2012, 100, 410–426 CrossRef CAS.
- T. H. Muster, A. Trinchi, T. A. Markley, D. Lau, P. Martin, A. Bradbury, A. Bendavid and S. Dligatch, Electrochim. Acta, 2011, 56, 9679–9699 CrossRef CAS.
- E. Reddington, A. Sapienza, B. Gurau, R. Viswanathan, S. Sarangapani, E. S. Smotkin and T. E. Mallouk, Science, 1998, 280, 1735–1737 CrossRef CAS PubMed.
- H. Wendt and G. Imarisio, J. Appl. Electrochem., 1988, 18, 1–14 CrossRef CAS.
- Hystat™ 10, http://www.hydrogenics.com/hydrogen-products-solutions/industrial-hydrogen-generators-by-electrolysis/indoor-installation/hystat-trade-10, accessed January 17, 2016.
- K. E. Ayers, E. B. Anderson, C. B. Capuano, B. D. Carter, L. T. Dalton, G. Hanlon, J. Manco and M. Niedzwiecki, ECS Trans., 2010, 33, 3–15 CAS.
- C. Ainscough, D. Peterson and K. Randolph, 2014 Electrolytic Hydrogen Production Workshop Summary Report, U. S. Department of Energy, Washington, D. C., 2014.
- C. Ainscough, D. Peterson and E. Miller, Hydrogen Production Cost from PEM Electrolysis, DOE Hydrogen and Fuel Cells Program Record, United States Department of Energy, Washington, D. C., 2014 Search PubMed.
- W. G. Colella, B. D. James and J. M. Moton, Hydrogen Pathways Analysis for Polymer Electrolyte Membrane (PEM) Electrolysis, Strategic Analysis, Inc., U. S. Department of Energy, Washington, D. C., 2014 Search PubMed.
- B. V. Tilak, P. W. T. Lu, J. E. Colman and S. Srinivasan, in Comprehensive Treatise of Electrochemistry, ed. J. Bockris, Springer, 1981, ch. 1, vol. 2, pp. 1–104 Search PubMed.
- J. O. M. Bockris and A. K. N. Reddy, Modern Electrochemistry, Perseus Publishing, Cambridge, MA, 1970 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn, 2000 Search PubMed.
- M. Carmo, D. L. Fritz, J. Merge and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- R. Renaud and R. L. Leroy, Int. J. Hydrogen Energy, 1982, 7, 155–166 CrossRef CAS.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4585 CrossRef CAS PubMed.
- T. A. Zawodzinski, C. Derouin, S. Radzinski, R. J. Sherman, V. T. Smith, T. E. Springer and S. Gottesfeld, J. Electrochem. Soc., 1993, 140, 1041–1047 CrossRef CAS.
- Y. Sone, P. Ekdunge and D. Simonsson, J. Electrochem. Soc., 1996, 143, 1254–1259 CrossRef CAS.
- S. D. Greenway, E. B. Fox and A. A. Ekechukwu, Int. J. Hydrogen Energy, 2009, 34, 6603–6608 CrossRef CAS.
- L. Birry and A. Lasia, J. Appl. Electrochem., 2004, 34, 735–749 CrossRef CAS.
- P. Rasiyah and A. C. C. Tseung, J. Electrochem. Soc., 1983, 130, 2384–2386 CrossRef CAS.
- P. Rasiyah and A. C. C. Tseung, J. Electrochem. Soc., 1983, 130, 365–368 CrossRef CAS.
- Y. Matsumoto, J. Kurimoto and E. Sato, J. Electroanal. Chem., 1979, 102, 77–83 CrossRef CAS.
- H. Michishita, Y. Misumi, D. Haruta, T. Masaki, N. Yamamoto, H. Matsumoto and T. Ishihara, J. Electrochem. Soc., 2008, 155, B969–B971 CrossRef CAS.
- Y. Matsumoto and E. Sato, Electrochim. Acta, 1979, 24, 421–423 CrossRef CAS.
- R. N. Singh and B. Lal, Int. J. Hydrogen Energy, 2002, 27, 45–55 CrossRef CAS.
- E. L. Miller and R. E. Rocheleau, J. Electrochem. Soc., 1997, 144, 1995–2003 CrossRef CAS.
- M. D. Merrill and R. C. Dougherty, J. Phys. Chem. C, 2008, 112, 3655–3666 CAS.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377–384 CrossRef CAS.
- X. H. Li, F. C. Walsh and D. Pletcher, Phys. Chem. Chem. Phys., 2011, 13, 1162–1167 RSC.
- K. Zeng and D. K. Zhang, Prog. Energy Combust. Sci., 2011, 37, 631 CrossRef.
- I. Abe, Abstr. Pap. Am. Chem. Soc., 1979, 254 Search PubMed.
- H. S. Hong, U. S. Chae, S. T. Choo and K. S. Lee, J. Power Sources, 2005, 149, 84–89 CrossRef CAS.
- F. L. Chen and M. L. Liu, J. Eur. Ceram. Soc., 2001, 21, 127–134 CrossRef CAS.
- J. R. Kong, Y. Zhang, C. S. Deng and J. M. Xu, J. Power Sources, 2009, 186, 485–489 CrossRef CAS.
- C. H. Yang, A. Coffin and F. L. Chen, Int. J. Hydrogen Energy, 2010, 35, 3221–3226 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |