
Understanding the molecular basis for the controlled design of ruthenium nanoparticles in microporous aluminophosphates†
Matthew E.
Potter
ab,
Jamie M.
Purkis
a,
Michal
Perdjon
cd,
Peter P.
Wells
cd and
Robert
Raja
 *a
*a
aSchool of Chemistry, University of Southampton, Southampton, Hants SO17 1BJ, UK. E-mail: R.Raja@soton.ac.uk
bDepartment of Chemical and Biochemical Engineering, Georgia Institute of Technology, Atlanta, GA 30318, USA
cThe UK Catalysis Hub, Research Complex at Harwell, Harwell, Oxon OX11 0FA, UK
dDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
First published on 16th September 2016
Abstract
Controlling the structural properties of nanoparticle catalysts within a microporous framework is a major challenge. Using in situ X-ray absorption fine structure (XAFS) spectroscopy we detail the influence of activation parameters on the nature of ruthenium particles that are located within the confines of a nanoporous aluminophosphate (RuAlPO-5) architecture. These in situ studies confirm that controlled annealing conditions can tailor the formation of specific ruthenium species, which alter the catalytic performance towards the oxidation of cyclohexane to KA oil (a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cyclohexanone and cyclohexanol), the precursor for Nylon-6 and Nylon-6,6.
:1 mixture of cyclohexanone and cyclohexanol), the precursor for Nylon-6 and Nylon-6,6.
Design, System, ApplicationHeterogeneous nanoporous catalysts are widely used in the chemical industry for the production of bulk and fine chemicals. By introducing a few atom-percent of dopant metals or nanoparticles into the framework, it is possible to engineer these systems for more demanding C–H activation reactions. In order to optimise such a process, the nature of the active metal site in the catalyst must be designed and engineered at a molecular level. In this work, we demonstrate how modifying the post-synthesis annealing treatments of a ruthenium-doped microporous aluminophosphate can be used to design a range of the desired active sites. These were probed using in situ X-ray absorption spectroscopy to establish structure–property correlations at the molecular level, facilitating a greater understanding of the local structural environment in porous heterogeneous solids. In situ X-ray absorption spectroscopy studies, complemented with transmission electron microscopy to rationalise the precise location and coordination geometry of ruthenium heteroatoms within the nanoporous frameworks, has proved invaluable in tracking the transition of isolated framework precursor species to metal nanoparticles. The validity of this design strategy was confirmed by correlating the catalytic efficiency of these materials for the direct oxidation of cyclohexane to a mixture of cyclohexanol and cyclohexanone (known as KA-oil), which is an important precursor for the industrial production of Nylon-6 and Nylon 6,6. |
Introduction
Given the chemical industry's dependence on crude oil, the activation of inert hydrocarbons is of fundamental interest.1–4 Once oxidized these molecules gain significant value as precursors in the fine-chemical and polymer industries. Heterogenized ruthenium species have been used for a variety of sustainable oxidation processes,5 as the wide-range of available oxidation states makes ruthenium highly versatile for performing transformations of activated functional groups, e.g. alcohols to ketones and amines to nitriles.6–8 Many approaches have been used to heterogenize ruthenium onto porous supports, thereby combining the selectivity control of a nanoporous material (commonly oxidic or carbon-based) with the catalytic potential of the metallic species.9–11 A variety of metal precursors and post-synthesis thermal treatments have been used, which have proved effective in enhancing the catalytic activity of Ru.12–17In order to optimize catalytic performance many synthesis procedures have been developed with a view to creating uniform ruthenium sites. Ionic liquids are increasingly employed to generate nanoparticles (NPs) with a narrow size distribution, as the ionic liquid hinders particle agglomeration.18–20 Similarly polymers such as poly(vinylpyrrolidone) are used as capping agents to restrict the size of the NPs formed via a micellar method,21 before they are bound to a surface. The size and ensuing stability of ruthenium NPs has also been modified by inorganic additives, whereby a secondary metal can coat ruthenium NPs, maintaining metallic state and hindering subsequent formation of oxidic species.22 A range of ruthenium-containing complexes have also been developed, aiming to preserve the integrity and isolated (<10 atoms) nature of the precursor. Arguably, the most common of these are multimetallic nanoclusters, which form defined nanoparticles with distinct stoichiometry, concordant with the original cluster.23,24
However, these strategies are commonly hindered by complex and sensitive precursors, such as multimetallic metal clusters, or precisely tailored organometallic species.23,24 The use of isomorphously substituted metal atoms into a microporous framework (as outlined in Scheme S1,† where small quantities of dopant transition metals can be used to replace Al(III) and P(V) T-atom tetrahedra) provides an alternative design approach, which is not hampered by the drawbacks of the methods described above. This approach requires precise synthetic protocols, in conjunction with specific combinations of zeotype frameworks and metals.25–29 Such strategies have been effective for transition-metal-substituted zeotype materials with a range of redox species.25–29 These methods are complicated by the disparity in size of ruthenium atoms, and the framework substituents in appropriate support materials that are available; such as silicates, zeolites and aluminophosphates (AlPOs). We have already reported on the simultaneous bimetallic substitution of ruthenium and tin into an AlPO and the enhanced catalytic potential it demonstrated towards the epoxidation of olefins.30 Although these materials were active there was a variation in the nature of the Ru sites produced, which complicated elucidating accurate structure–function relationships.
This study extends the design rationale of isomorphously substituting metal atoms into a microporous frameworks, to prepare a series of Ru-AlPO-5 catalysts. The use of in situ X-ray absorption fine structure (XAFS), allows the thermal activation of Ru-AlPO materials to be followed and offer valuable insights in tuning the catalytic potential for the more demanding C–H activation of cyclohexane to KA oil (precursor for Nylon-6 and Nylon-6,6). XAFS is well-suited to this particular study, as the element-specific nature of the technique can steer focus solely on ruthenium species, whilst ignoring contributions from the rest of the inert AlPO framework. The study demonstrates how the nature of the ruthenium active centres, which are generated during the activation procedure, influences the catalytic performance towards the oxidation of cyclohexane.
Experimental procedures and methods
The hydrothermal synthesis of RuAlPO-5 (ref. 30) was performed with aluminium hydroxide hydrate, phosphoric acid, ruthenium(III)chloride hydrate and N,N-methyldicyclohexylamine, which were combined to yield a gel ratio of: 1.0Al:1.05P:0.84MDCHA:25H2O:0.06Ru. The homogeneous gel was heated under autogeneous pressure for 2 hours at 175 °C, washed with de-ionized water and dried overnight at 60 °C. Samples were then annealed under a flow of air or nitrogen and held at the specified temperature for 2 hours. The as-synthesized sample is henceforth referred to as RuAlPO-5-AS, and annealed samples are labelled RuAlPO-5-(O/I)-X, where X is the annealing temperature of the sample, under oxidative (O) or inert (I) annealing conditions.
Ru K-edge XAFS studies were carried out on the B18 beamline at the Diamond Light Source, Didcot, UK. Measurements were performed using a QEXAFS set-up with a fast-scanning Si (311) double crystal monochromator. All ex situ samples were diluted with cellulose and pressed into pellets to optimize the effective edge-step of the XAFS data and measured in transmission mode using ion-chamber detectors. All transmission XAFS spectra were acquired concurrently with a Ru foil placed between It and Iref. The time resolution of the spectra reported herein was 1 min per spectrum (kmax = 16), on average three scans were acquired to improve the signal to noise level of the data.
In situ activation studies were performed in transmission mode with a quartz capillary (OD 6 mm, wall thickness 250 μm), micro-reactor. The micro-reactor is equipped with a gas supply system, integrated heating system and a Cirrus 100 quadrupole mass spectrometer to monitor the outlet gas composition. Activation studies used a ramp rate of 6 °C min−1 up to 400 °C. XAFS data were recorded throughout the experiment, at a typical rate of 1 min per spectrum. On reaching the maximum temperature, the sample was held isothermally for 10 minutes to ensure no further change, after which it was cooled to room temperature. XAFS data processing and EXAFS analysis were performed using IFEFFIT (ref. 31) with the Horae package32 (Athena and Artemis). The amplitude reduction factor, S02, was derived from EXAFS data analysis of known reference compound (i.e. RuO2).
Powder X-ray diffraction patterns were obtained using a Siemens D5000 diffractometer using Cu Kα1 radiation, whereby λ = 1.54056 Å. BET surface area measurements were performed using a Micromeritics Gemini 2375 surface area analyser and prepared using flow gas preparation. ICP-MS (induced coupled plasma mass spectrometry) analysis was performed using a Perkin-Elmer Optimum 3000 DV. Annealed samples prepared and fully digested in 10 mL of deionized water and 10 mL of ACS Plus Certified H2SO4. Solutions of standard concentrations were used for calibration. Transmission electron microscopy images were obtained with a Jeol 2100-JEM fitted with Oxford Instruments X-MAXN 80-T EDX analyser. The powdered sample was dispersed in methanol and deposited upon 300 mesh copper grids covered with the holey carbon film.
Oxidation catalysis was performed in a glass-reactor, to which 13 mmol of cyclohexane, 13 mmol of tert-butyl-hydrogen-peroxide (TBHP, 70 wt% in H2O) were mixed with 50 mg of RuAlPO-5 and 5 ml of acetone (solvent). The system was then stirred and heated to 70 °C for 6 hours. The samples were analysed using a Perkin Elmer 3400CX gas chromatogram with flame ionization detector (FID). Samples were analysed using a HP1 cross linked methylsiloxane (30 m × 0.32 mm × 1 μm film thickness) column. Products were identified against authenticated standards and quantified by calibration to obtain response factors (RF) against the known internal standard (diglyme). Peroxide efficiency (PE) was defined as:
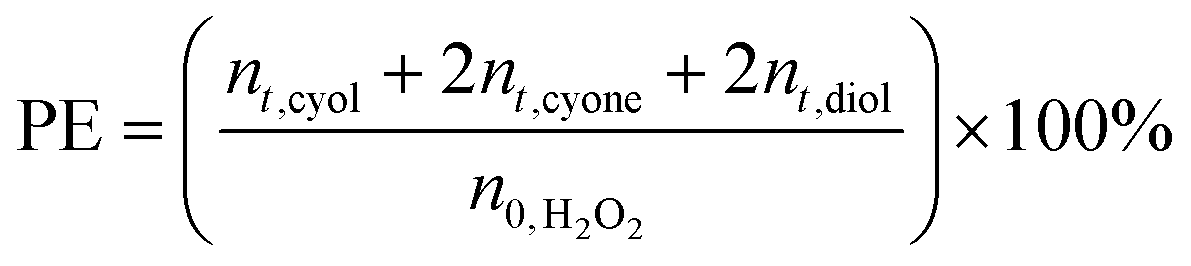
Results and discussions
Textural and crystalline properties
The structural integrity of the annealed species (RuAlPO-5-O-400) was confirmed using an array of physico-chemical techniques and contrasted with undoped AlPO-5 to study the effect of introducing ruthenium (which replaces framework Al(III) sites via isomorphous substitution, as outlined in Scheme S1†) into the framework. The ICP-MS derived loadings (Table S1†) of the undoped AlPO-5 and RuAlPO-5-O-400 species are in good agreement. A small change in the molar Al/P ratio is observed; however this value still falls within the expected range for an AlPO material. Given that AlPOs are composed of strictly alternating AlO4 and PO4 primary building units, the ratio should ideally be close to 1. Deviation from the theoretical value could be the due to extra-framework aluminium sites, or the isomorphous substitution of one specific T-site via type I or type II substitution (Scheme S1†).30The specific surface area is similar for the doped and undoped systems, both lying within the range expected for the one-dimensional AFI framework. This suggests minimal, if any, pore occlusion has occurred upon introducing the ruthenium into the system (Table S1†), and that metal clustering is not occurring around the pore mouths of the 1D channel. The powder X-ray diffraction and resulting unit cell parameters are also in good agreement with the undoped system, suggesting little framework distortion has occurred (Table S1 and Fig. S1†). The radius of cationic Ru is significantly larger than other cation sites within the AlPO framework and will locally distort the framework (and unit cell). However, the low Ru loading employed in this study has a negligible effect on the macroscale properties of the porous architecture.30
Designing the metal site
XAFS data of RuAlPO-5-AS was compared ex situ with a selection of well-known standards to probe the initial state (Fig. 1). The XANES (X-ray absorption near edge spectroscopy) data shows that RuAlPO-5-AS most closely resembles RuO2, with similar features, characteristic of oxidic Ru species.33 RuAlPO-5-AS shows a pre-edge feature at 22119 eV, along with RuO2 and RuCl3·xH2O, which is shifted to a higher energy than the metallic Ru foil (22117 eV). The energy value at half the normalized intensity for RuAlPO-5-AS (22127 eV) lies between that of the Ru(III)Cl3·xH2O species (22125 eV) and the oxidic Ru(IV)O2 species (22128 eV). Closer examination of the main-edge peak shows at higher energy values (>22120 eV) the RuAlPO-5-AS resembles the RuO2 species, however there is a deviation below 22120 eV, suggesting some small contribution from lower oxidation states, such as Ru(III) or Ru(0). More specifically, the XANES 1st derivative spectra show that RuAlPO-5-AS more closely resembles the Ru(IV)O2 species (Fig. S2†). Inspection of the XANES beyond the absorption edge shows that RuO2 has the characteristic two maxima, associated with anhydrous rutile RuO2. The two maxima are significantly less pronounced in the RuAlPO-5-AS species, showing that the oxidic environment has become less ordered. Commonly this is seen in hydrated RuO2 samples, as the presence of water introduces new features that deviate from the crystalline structure.34 These observations confirm that ruthenium species in RuAlPO-5-AS primarily possess characteristics of Ru(IV)O2, with the possibility of some contribution from lower oxidation states.35,36 This is not uncommon in AlPO chemistry, as the catalytic potential of substituted first row transition metals (Co, Mn, etc.) derives from their ability to exist in multiple oxidation states.37–39 Possessing a mixture of M(III/IV) states suggests that ruthenium may have undergone both type I and type II substitution to some degree, into the AlPO framework (Scheme S1†). The χ data again shows the similarity between RuO2 and RuAlPO-5-AS (Fig. 2A), although the differences at higher k (less structural features for RuAlPO-5-AS) suggest a less ordered system. The Fourier transform data (Fig. 2B) confirms this, as the dehydrated RuO2 species shows Ru–Ru features at 3.2 Å, indicative of long range order, which are common in both hydrated and dehydrated RuO2 species.34 These are not present in RuAlPO-5-AS.
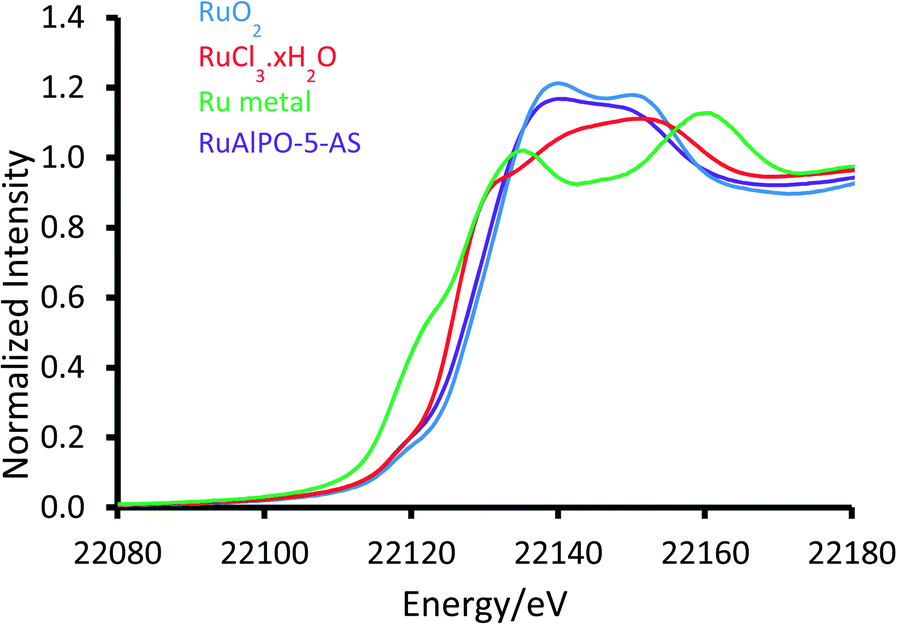 | ||
| Fig. 1 Normalized XANES spectra of RuAlPO-5-AS with Ru(IV)O2, Ru(III)Cl3·xH2O and metallic Ru foil. | ||
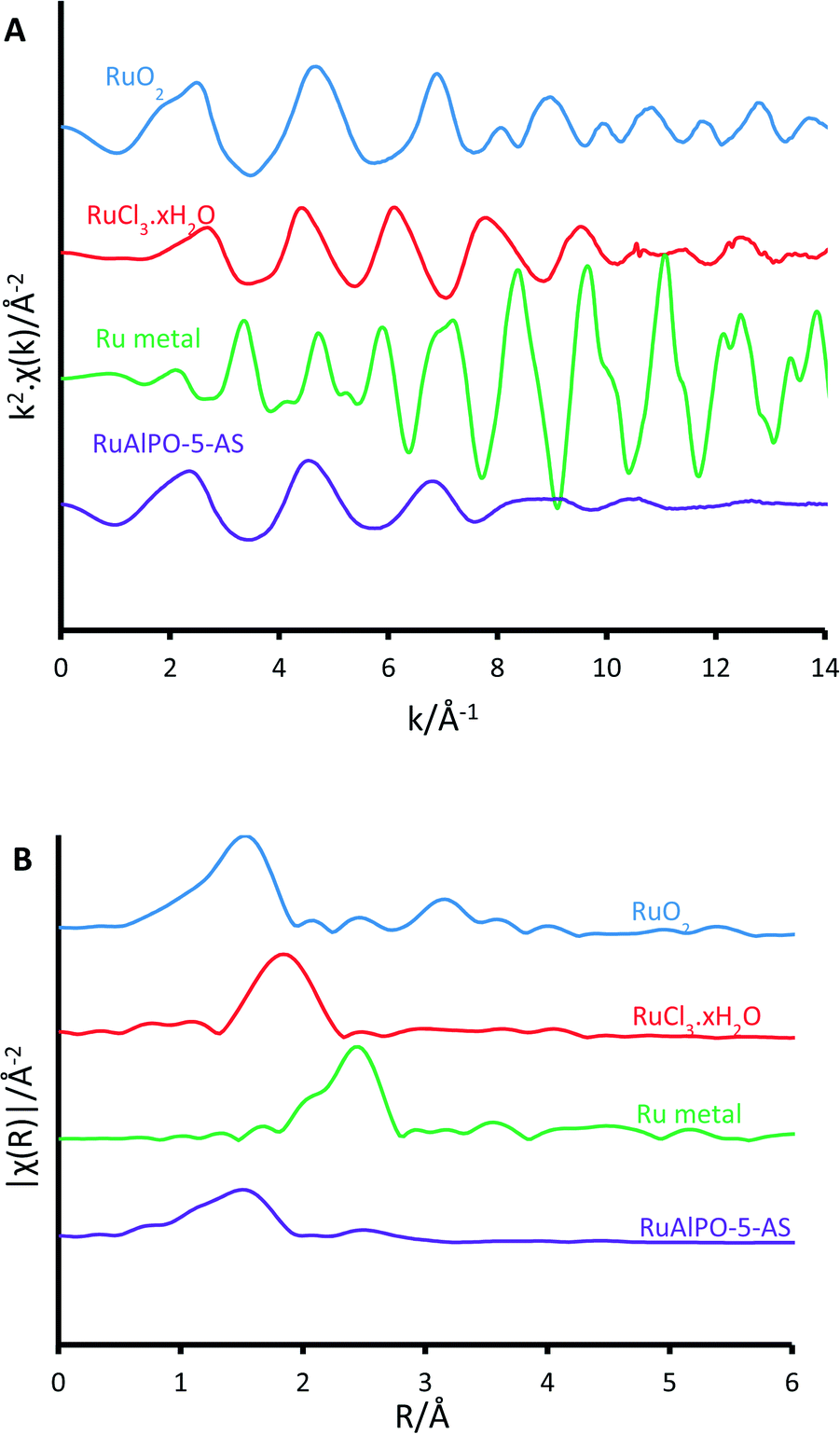 | ||
| Fig. 2 Non-phase corrected EXAFS data, showing A) k2·χ(k) data and B) magnitude of the k2 weighted Fourier transform for the EXAFS data, contrasting the environment of bulk RuO2 (blue), the RuCl3·xH2O precursor (red), bulk ruthenium metal (green) and RuAlPO-5-AS (purple). | ||
The non-phase corrected Fourier transform (Fig. 2B) of both RuO2 and RuAlPO-5-AS have a strong signal at 1.6 Å, assigned to a Ru–O scattering path, confirming the oxidic nature of RuAlPO-5-AS. The width of this signal suggests that multiple Ru–O scattering paths are present in RuAlPO-5-AS, shown by the need for two Ru–O paths in the EXAFS fit (Table 1 and Fig. 3). This behaviour is typical of substituted tetravalent AlPO dopants, as the net charge imbalance protonates an oxygen adjacent to the ruthenium, leading to a larger M–O distance. Similarly adsorbed water can also extend Ru–O distances in zeotype species.8 Differences between RuO2 and RuAlPO-5-AS occur at higher R values, where the Ru–Ru feature at 3.2 Å is absent in RuAlPO-5-AS. Instead only a single signal at 2.5 Å is present. This feature could not be simulated using Ru–Ru distances, however, a good level of fit was achieved with a Ru–Al distance (Table 1 and Fig. 3). This value is slightly larger than previous literature values for other doped AlPOs, though these exclusively correspond to smaller dopants.9 This distance is in good agreement with a M–O–Al feature, as expected in substituted AlPO materials, having undergone type II substitution. The lack of Ru–Ru features confirms that isolated sites have formed (in combination with enhanced TONs – see later) and are either anchored onto, or isomorphously substituted into, the AlPO-5 framework.
| Abs Sc | N | R/Å | 2σ2/Å2 | E f/eV | R factor |
|---|---|---|---|---|---|
| a Fitting parameters: S02 = 0.85, as deduced by Ru foil standard; fit range: 3 < k < 10, 1.15 < R < 3.85; number of independent points = 11.8. b Fitting parameters: 1.15 < R < k < 8.5, number of independent points = 8.4. | |||||
| Ru–O1a | 2.1(4) | 1.89(1) | 0.003(4) | 0.3(16) | 0.015 |
| Ru–O2a | 3.1(5) | 2.03(1) | 0.001(3) | ||
| Ru–Alb | 1.5(5) | 3.17(1) | 0.006(6) | ||
 | ||
| Fig. 3 Magnitude and imaginary component of the k2 weighted Fourier transform EXAFS data for RuAlPO-5-AS showing the influence of different fitted paths. | ||
Transferring the system on-line and annealing in situ with air as an oxidative environment showed no significant differences in the data up to 179 °C (RuAlPO-5-O-179). At these temperatures all adsorbed water would have been desorbed from the ruthenium atoms. The spectrum retains the feature between 2.5–3.0 Å, confirming this is due to framework aluminium and not water (Fig. S3†). Further there is still no feature attributed to the Ru–Ru signal at 3.1 Å, which would appear on dehydrating RuO2·xH2O, thus confirming the difference between the RuAlPO-5-AS species and RuO2·xH2O.35
Above 179 °C a rapid change in environment occurs (Fig. 4). A proportion of the ruthenium becomes reduced to isolated ruthenium metal (Fig. 4), as shown by the appearance of a peak at 2.4 Å in Fig. 2B, indicative of a metallic Ru–Ru scattering path. The observed reduction of the ruthenium species coincides with the removal of the amine structure-directing agent (SDA, Fig. S4†).29,30 It is known that framework species are stabilized through interactions with the nitrogen of the amine-based SDA, and removing the SDA has been shown to extrude strained dopants from framework positions to extra-framework species in other metal-substituted AlPO species.31 Further, the decomposition of the SDA under oxidative conditions was shown to liberate H2 gas, accounting for the reduction of ruthenium, even under oxidative conditions (Fig. S4†).
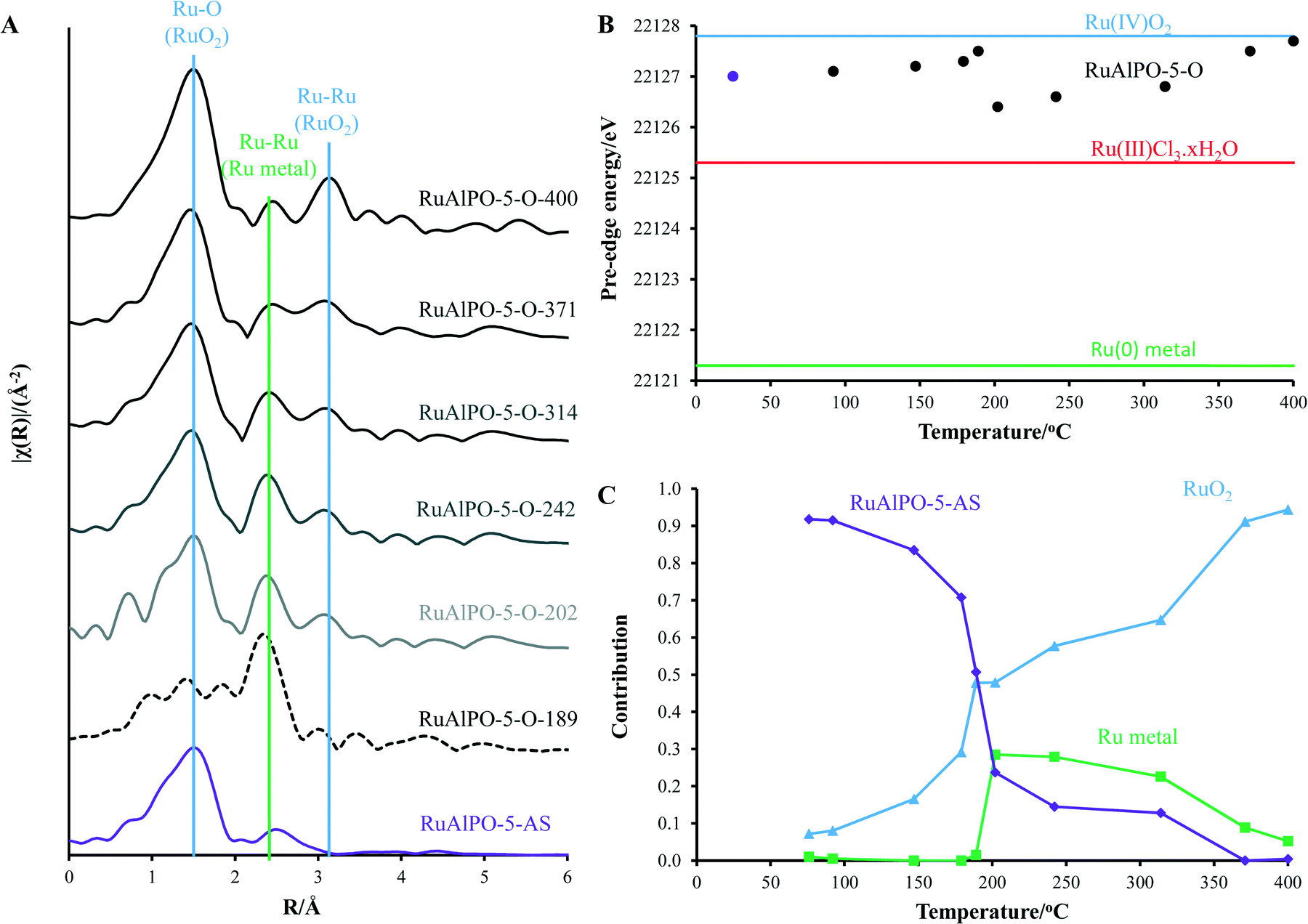 | ||
| Fig. 4 The influence of increasing temperature on A) magnitude of the k2 weighted Fourier transform, B) pre-edge position (assigned as normalized absorption equal to 0.5) and C) linear combination coefficients for RuAlPO-5 annealed under oxidative conditions at different temperatures. After 189 °C, metallic nanoparticles are formed and then converted to ‘bulk’ ruthenium dioxide. | ||
After this transition at 179 °C, the Ru–Al feature is insignificant in the magnitude of the k2 weighted Fourier transform. This shows that the ruthenium no-longer occupies framework sites, thus has formed an extra-framework species on being extruded from the framework. Both these factors account for the formation of small metal nanoparticles (Fig. S5†). The appearance of the metallic nanoparticles was a rapid process, as spectrum RuAlPO-5-O-179 was changing during collection and could not be isolated.
Increasing the temperature beyond 200 °C, results in a gradual reduction of the first shell Ru–Ru contribution, whereas oxidic features (both the Ru–O 1st coordination shell at 1.6 Å and the Ru–Ru 2nd coordination shell after 3 Å) become more prominent. This shows that the nanoparticles are oxidized to a bulk oxidic RuO2 phase.40 Monitoring the main-edge position (defined here as the energy at which the normalized absorption is equal to 0.5) as a function of temperature, provides an insight into the average oxidation state of ruthenium. This reveals that the average oxidation-state of the RuAlPO-5-AS species lies between Ru(III) and Ru(IV).35,36 There is a negligible change in main-edge position before 179 °C, suggesting higher temperatures are required for activation. However, where the phase-transition occurs between 189 and 202 °C the average oxidation-state decreases, as some of the ruthenium ions are reduced to ruthenium metal (Fig. S6†). Increasing the temperature beyond 200 °C led to an increase in the main-edge position (converging on the RuO2 value), and the oxidic character of the ruthenium, as the newly formed ruthenium metal species is oxidized (Fig. S7†).
These findings are corroborated by linear combination analysis using three components, RuAlPO-5-AS (initial stage – isolated oxidic Ru species incorporated into the framework), RuO2 (final stage – extra-framework Ru oxide) and ruthenium metal (intermediate stage). The RuAlPO-5-AS component decreases rapidly after 150 °C, as ruthenium extrudes from the framework. This is accompanied by a sharp increase in the metallic character, showing that the ruthenium has formed metallic nanoparticles. As the temperature increases beyond 200 °C the RuO2 component begins to dominate, as the nanoparticles are oxidized to extra-framework RuO2, showing that the XANES and EXAFS findings are in good agreement. Transmission electron microscopy (TEM) was used to directly observe the formation of extra-framework ruthenium species (Fig. 5 and S9–S12†). Over the studied temperature range (200, 300 and 400 °C) only small nanoparticle species were observed (between 1.5 and 2.5 nm). This is in good agreement with the XAFS findings, which suggest that the ruthenium nanoparticles form bulk ruthenium oxide, instead of aggregating as ruthenium metal, as shown in the EDS images (Fig. S10–S12†).
 | ||
| Fig. 5 TEM images showing the stability of ruthenium nanoparticles with increased temperatures. | ||
Whilst different catalytic species can be formed by controlling the annealing temperature, diverse species can be achieved by changing the annealing environment. Utilizing an inert atmosphere promotes different behaviour (Fig. 6), preventing the formation of bulk ruthenium oxide (Fig. 4). Under inert conditions, the isolated oxidic Ru framework-incorporated species is converted to bulk metal (Fig. S8†). This behaviour differs from the oxidic annealing, whereby this transition occurs more rapidly.
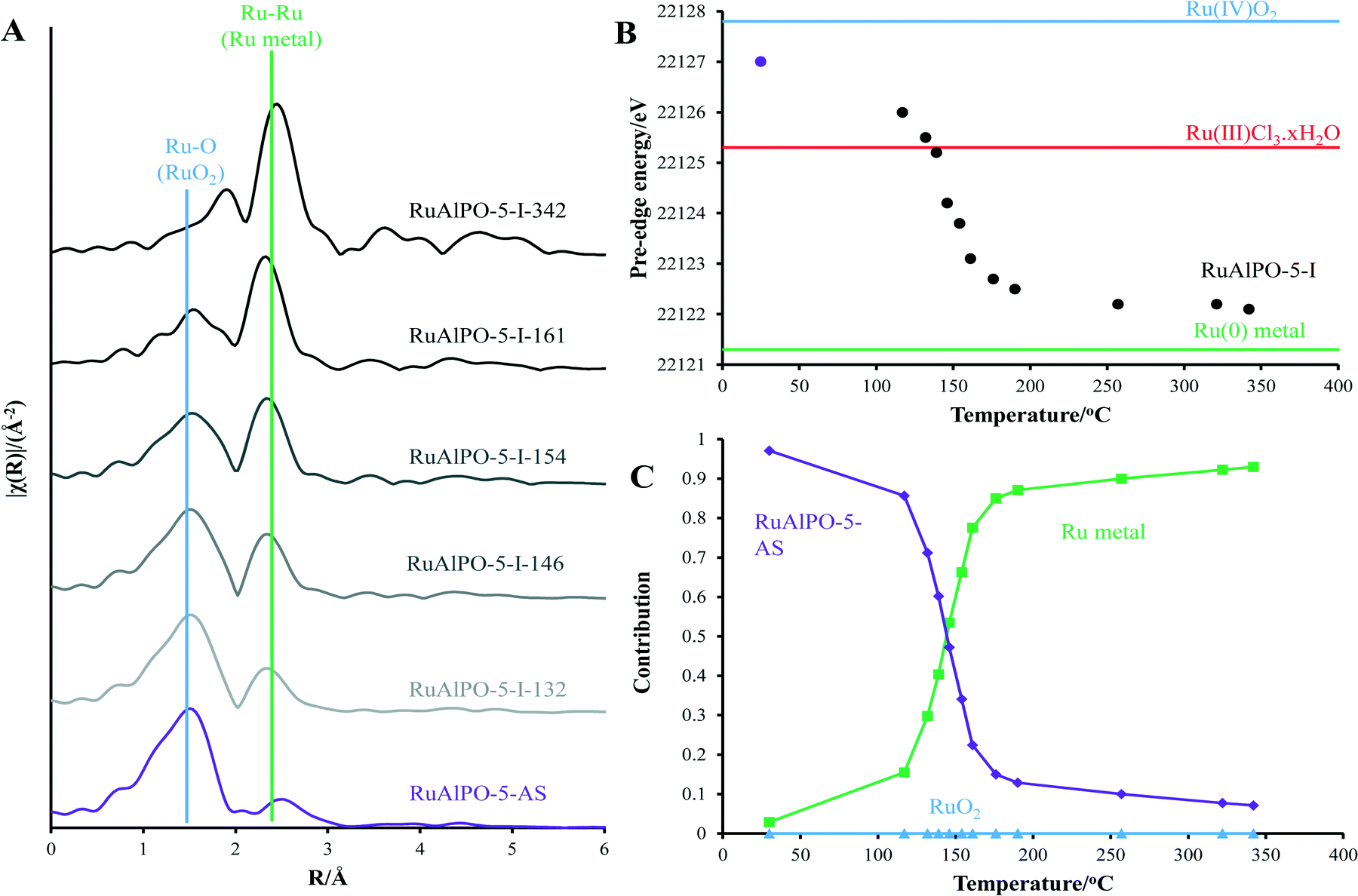 | ||
| Fig. 6 Variations in A) magnitude of the k2 weighted Fourier transform, B) pre-edge position (assigned as normalized absorption equal to 0.5) and C) linear combination coefficients for RuAlPO-5 annealed under inert conditions at different temperatures. In this case a gradual transition from isolated oxidic Ru framework-incorporated species to bulk metal ruthenium is observed between 120 and 180 °C. | ||
The oxidative removal of the SDA under oxidic conditions gives rise to an instantaneous response as the hydrogen generated is consumed by the ruthenium ions. Hydrogen is not produced under inert annealing conditions, so the transition is more gradual than the oxidative environment. This shows that the rapid extrusion of ruthenium under the oxidative environment was caused by the combustion (and not just the removal) of the SDA to liberate H2 gas. As the inert conditions cannot cause combustion, the resulting transition is more gradual. Features attributable to long range order only become visible at higher-temperatures (RuAlPO-I-321). However this cannot necessarily be attributed to a change in particle size, as the metallic component is considerably weaker at lower temperatures. Comparing the final temperature (RuAlPO-I-321) with the metallic ruthenium foil standard shows that longer-range features corresponding to further coordination shells are present (Fig. S8†). These features are not as defined as the metallic ruthenium foil, which can be seen in the 1st shell EXAFS fitting parameters (Table 2). RuAlPO-I-321 has both a lower coordination number than the metallic Ru foil, indicative of smaller sized particles (Table 2).41 TEM images (Fig. 7 and S13†) confirm that under inert annealing conditions metal nanoparticles (<5 nm in size) are present. At higher temperatures these species begin to aggregate and form larger extra-framework species, but a significant amount of small nanoparticles still remain at 400 °C. These images confirm that ruthenium initially forms nanoparticle species on extrusion from the framework under inert annealing conditions, which then aggregate, though a significant proportion of the smaller nanoparticles are still intact, as ruthenium is gradually extruded from the framework. These findings agree with the XAFS results (Fig. 6), which showed the gradual appearance of ruthenium metal at higher temperatures, however the particle size was significantly smaller than ruthenium foil.
| System | Abs Sc | N | R/Å | 2σ2/Å2 | E f/eV | R factor |
|---|---|---|---|---|---|---|
| Fitting parameters: S02 = 0.85, as deduced by Ru foil standard; fit range: range 3 < k < 10, 1.15 < R < 3.85; number of independent points = 11.8. | ||||||
| Ru foil | Ru–Ru | 11.6(5) | 2.67(1) | 0.008(3) | 3.6(6) | 0.002 |
| RuAlPO-I-321 | Ru–Ru | 6.8(2) | 2.66(1) | 0.016(1) | 2.7(7) | 0.012 |
 | ||
| Fig. 7 TEM images showing that aggregation of extra-framework ruthenium metal species with increasing temperature under inert conditions. | ||
Contrasting catalytic species annealed at the same temperature, but under different environments, highlights how the precise nature of the active site can be controlled. A metallic phase was formed at around 190 °C regardless of the environment, suggesting that the extrusion process is initiated by increased temperatures, though this transition is more rapid in the oxidative case as the species was short-lived, emphasising the need for in situ studies (Fig. 8A). Once extruded, these particles are susceptible to oxidation, as seen by the appearance of ruthenium oxide at 320 °C (Fig. 8B). The different ruthenium species formed were tested for their catalytic efficacy in the oxidation of cyclohexane to KA-oil. The annealing study (above) highlights the availability of a wide-range of ruthenium species, allowing the catalytic sites to be tailored for this demanding reaction, involving C–H activation. Considering the product yields, it is clear that the annealing protocols strongly influence the catalytic efficiency of RuAlPO-5 (Fig. 9).
 | ||
| Fig. 8 Magnitude of the k2 weighted Fourier transform for the EXAFS data, of RuAlPO-5 under inert (red) and oxidative (blue) annealing conditions A) 190 and B) 320 °C. | ||
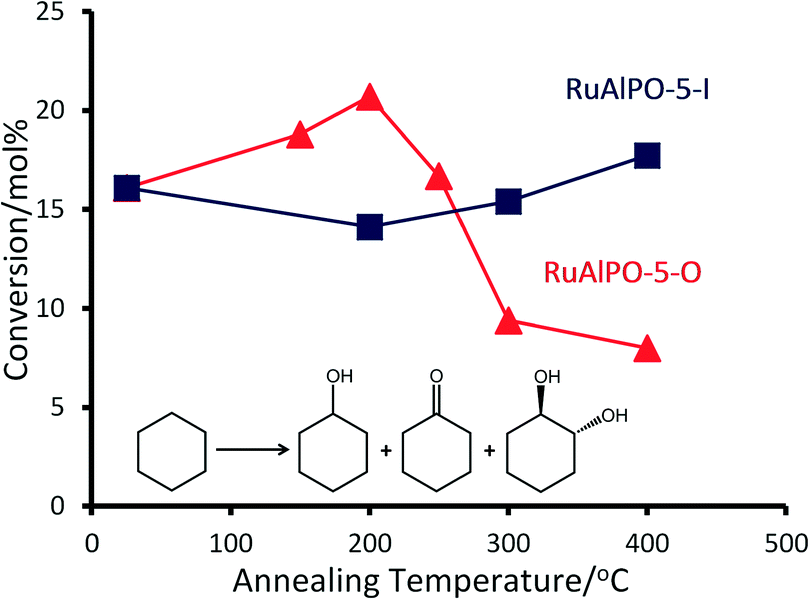 | ||
| Fig. 9 Comparing the effect of annealing temperature under oxidative and inert conditions on the conversion of RuAlPO-5 for the oxidation of cyclohexane. Conditions: 13 mmol cyclohexane, 13 mmol TBHP (70 wt% in H2O), 0.05 g of RuAlPO-5 and 5 ml of acetone, 70 °C, 6 hours. | ||
For the RuAlPO-5-O series, maximum values for product yield and peroxide efficiency (Fig. 10 and S14†) are achieved at 200 °C. Both yields and efficiency decline upon increased annealing temperatures. The selectivity to cyclohexanol is fairly consistent across this series, with the exception of the 200 °C system, which shows a notable drop in selectivity. However, subtle trends in cyclohexanone and trans-1,2-diol (diol) selectivity are observed (Fig. 10). GC-MS data found no traces of other products in the reaction mixture. Initially the as-synthesized material heavily favours the diol (selectivity 52.4 mol%), with considerably lower selectivity to cyclohexanone (19.8 mol%). As the annealing temperature increases, there is a concomitant increase in cyclohexanone formation, at the expense of the diol. Thus dehydrogenation of the activated cyclohexanol is favoured over repeated C–H activation. This is direct evidence that the ruthenium sites can be controlled to tailor their activity, which also influences the mechanistic pathway and particle sizes.
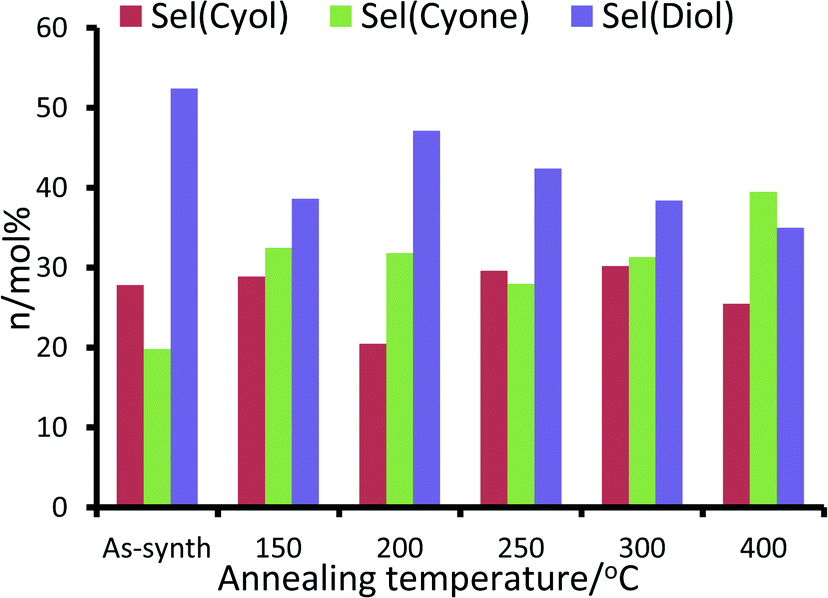 | ||
| Fig. 10 Comparing the effect of annealing temperature under oxidative and conditions on the reactivity of RuAlPO-5 for the oxidation of cyclohexane. Conditions: 13 mmol cyclohexane, 13 mmol TBHP (70 wt% in H2O), 0.05 g of RuAlPO-5 and 5 ml of acetone, 70 °C, 6 hours. | ||
The isolated framework-substituted ruthenium atoms show reasonable conversion (RuAlPO-5-AS conversion 16.1 mol%) and the trends in catalytic performance can be directly linked to the XAFS findings. The XAFS and TEM images confirm that, as the annealing temperature reaches 200 °C, the framework ruthenium species are extruded to the surface, forming small (<5 nm) metallic nanoparticles. This maxima of conversion coincides with the highest fraction of metallic ruthenium (Fig. 4C), which further vindicates the superior performance of the smaller, extruded ruthenium nanoparticles. Increasing the annealing temperature beyond 200 °C leads to the formation of extra-framework RuO2 species, which can be directly attributable to the inferior catalytic performance associated with bulk RuO2.
Notable trends are observed, as the catalytic performance varies with temperature for inert annealing, which broadly shows an increase in activity with increased annealing temperature (Fig. 9 and S15†). These trends extend beyond experimental error and are reproducible between batches (Table S2†). XAFS data confirms that under inert conditions, higher temperatures result in a greater fraction of metallic ruthenium, which has been shown to improve catalytic efficacy. Increasing annealing temperatures extrudes ruthenium more effectively from the framework, with increased aggregation of the metallic ruthenium. Despite the aggregation, a significant quantity of nanoparticles are still present, which account for the increased conversion. The selectivity profiles for the as-synthesized, oxidative and inert series are disparate (Fig. 10 and S16†). As the annealing temperature increases the inert series progresses from predominantly forming the diol, to favouring KA oil formation. As the temperature increases the selectivity profile also shows a strong resemblance to the RuAlPO-5-O-200 system, confirming that analogous catalytic species are present. The observed low selectivity to cyclohexanol shows that metallic ruthenium species are versatile, and can activate a range of different functional groups, leading to the formation of cyclohexanone and the diol. In contrast, bulk oxidic ruthenium hinders the formation of cyclohexanone. This was confirmed (Fig. 11 and S17†) by contrasting the catalytic efficiency of the optimized system (RuAlPO-5-O-200) with the synthesis precursor (RuCl3·xH2O) and a range of bulk oxides (RuO2 and RuO2·xH2O).
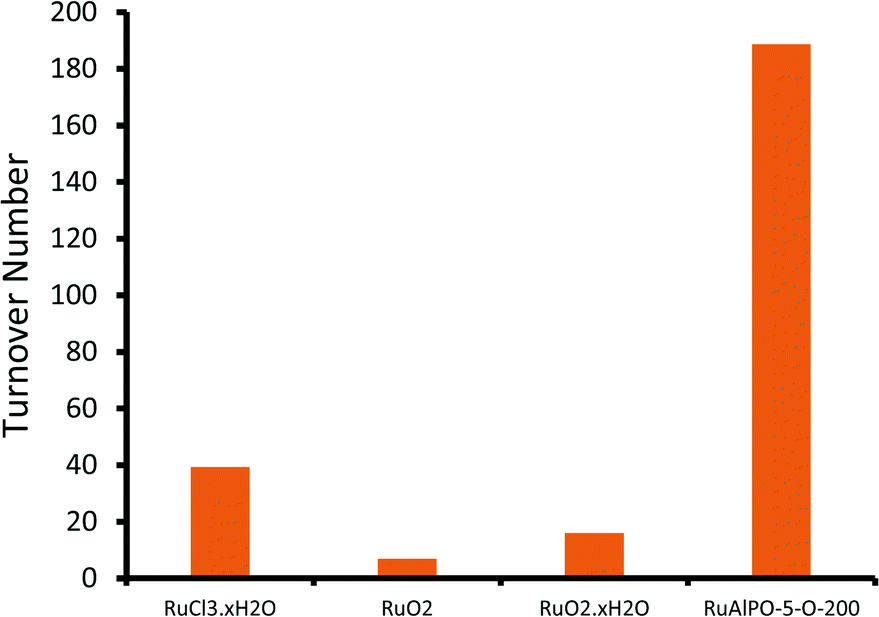 | ||
| Fig. 11 Comparing the catalytic behaviour of standard ruthenium materials on the conversion of RuAlPO-5 for the oxidation of cyclohexane. Conditions: 13 mmol cyclohexane, 13 mmol TBHP (70 wt% in H2O), 0.05 g of RuAlPO-5 and 5 ml of acetone, 70 °C, 6 hours. | ||
The superior activity of the RuAlPO-5-O-200 system vindicates our synthetic methodology. Thus isolated ruthenium species, introduced to the confines of a nanoporous aluminophosphate through one-pot hydrothermal synthesis, serve as a precursor for generating catalytically-active ruthenium species. XAFS and TEM findings reveal that these species can be selectively formed through dextrous control of post-synthesis thermal treatments, leading to enhanced catalytic activity in demanding C–H activations.
Conclusions
In order to understand the nature and behaviour of a catalytic active site at the molecular level, in situ spectroscopy is fast becoming a necessity, in observing rapid structural changes, or identifying catalytically active intermediates. To this end we have employed a combined catalysis and XAFS study to demonstrate how the precise environment of a metallic ruthenium species can be controlled and suitably tailored. It was shown that in the initial as-synthesized form, ruthenium is isolated within the confines of an AlPO framework, existing as an oxidic ruthenium site. The distortion created by the isolated ruthenium species allows it to extrude from the framework (as seen by XAFS and TEM) within a temperature range of 150–200 °C, associated with the removal of the stabilising SDA. The combustion of the SDA serves as a source of hydrogen, rapidly reducing (and extruding) the ruthenium to form small (<3 nm) nanoparticles with enhanced catalytic potential for the oxidation of inert hydrocarbons. Under oxidative conditions, such sites agglomerate and form catalytically inactive RuO2 species. Inert annealing conditions preserved the metallic phase, resulting in increased activity. Therefore it has been shown that both the nature and the morphology of active sites can be controlled through meticulous selection of synthetic protocols, leading to a better understanding of structure–property relationships that will aid the rational design of catalytic systems in the future.Acknowledgements
The authors wish to acknowledge the Diamond Light Source for provision of beamtime (SP8071). The RCaH are also acknowledged for use of facilities and support of their staff. PW and RR wish to thank EPSRC for funding (EP I019693 and EP/K014714/1). MEP wishes to thank Honeywell LLC for studentship funding. We are also grateful to Cardiff University Microscopy facility for the additional TEM analysis.Notes and references
- L. Kesavan, R. Tiruvalam, M. H. Ab Rahim, M. I. bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195 CrossRef CAS PubMed.
- L. Balducci, D. Bianchi, R. Bortolo, R. D'Aloiso, M. Ricci, R. Tassinari and R. Ungarelli, Angew. Chem., 2003, 115, 5087 CrossRef.
- R. Schlogl, Top. Catal., 2011, 54, 627 CrossRef.
- N. V. Beznis, A. N. C. van Laak, B. M. Weckhuysen and J. H. Bitter, Microporous Mesoporous Mater., 2011, 138, 176 CrossRef CAS.
- T. Naota, H. Takaya and S. I. Murahashi, Chem. Rev., 1998, 98, 2599 CrossRef CAS PubMed.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2003, 42, 1480 CrossRef CAS PubMed.
- I. E. Marko, P. R. Giles, M. Tsukazaki, I. Chelle-Regnaut, C. J. Urch and S. M. Brown, J. Am. Chem. Soc., 1997, 119, 12661 CrossRef CAS.
- V. I. Parvulescu, S. Coman, P. Palade, D. Macovei, C. M. Teodorescu, G. Filoti, R. Molina, G. Poncelet and F. E. Wagner, Appl. Surf. Sci., 1999, 141, 164 CrossRef CAS.
- R. Raja, M. E. Potter and S. H. Newland, Chem. Commun., 2014, 50, 5940 RSC.
- J. Gaudet, K. K. Bando, Z. Song, T. Fujitani, W. Zhang, D. S. Su and S. T. Oyama, J. Catal., 2011, 1, 40 CrossRef.
- M. Neurock, J. Catal., 2003, 216, 73 CrossRef CAS.
- J. Perez-Ramirez, J. C. Groen, A. Bruckner, M. S. Kumar, U. Bentrup, M. N. Debbagh and L. A. Villaescusa, J. Catal., 2005, 232, 318 CrossRef CAS.
- M. Muller, G. Harvey and R. Prins, Microporous Mesoporous Mater., 2000, 34, 135 CrossRef CAS.
- R. D. Adams, M. Chen, G. Elpitiya, M. E. Potter and R. Raja, ACS Catal., 2013, 3, 3106 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, P. Miedziak, E. Ntainjua, J. K. Edwards, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2008, 10, 1921 RSC.
- T. Maschmeyer, F. Rey, G. Sankar and J. M. Thomas, Nature, 1995, 378, 159 CrossRef CAS.
- C. S. Hinde, G. Collins, S. Van Aswegen, J. D. Holmes, T. S. A. Hor and R. Raja, Dalton Trans., 2013, 42, 12600 RSC.
- T. Gutel, C. C. Santini, K. Philippot, A. Padau, K. Pelzer, B. Chaudret, Y. Chauvin and J. M. Basset, J. Mater. Chem., 2009, 19, 3624 RSC.
- Y. Zhu, Z. N. Kong, L. P. Stubbs, H. Lin, S. Shen, E. V. Anslyn and J. A. Maguire, ChemSusChem, 2010, 3, 67 CrossRef CAS PubMed.
- P. S. Campbell, C. S. Santini, D. Bochu, B. Fenet, K. Philippot, B. Chaudret, A. A. H. Padau and Y. A. Chauvin, Phys. Chem. Chem. Phys., 2010, 12, 4217 RSC.
- S. H. Joo, J. Y. Park, J. R. Renzas, D. R. Butcher, W. Huang and G. A. Somorjai, Nano Lett., 2010, 10, 2709 CrossRef CAS PubMed.
- S. Fiechter, I. Dorbandt, P. Bogdanoff, G. Zehl, H. Schulenburg and H. Tributsch, J. Phys. Chem. C, 2007, 111, 477 CAS.
- L. O. Paz-Borbon, A. Hellman, J. M. Thomas and H. Gronbeck, Phys. Chem. Chem. Phys., 2013, 15, 9694 RSC.
- A. B. Hungria, R. Raja, R. D. Adams, B. Captain, J. M. Thomas, P. Midgley, V. Golovko and B. F. G. Johnson, Angew. Chem., Int. Ed., 2006, 45, 4782 CrossRef CAS PubMed.
- R. M. Leithall, V. N. Shetti, S. Maurelli, M. Chiesa, E. Gianotti and R. Raja, J. Am. Chem. Soc., 2013, 135, 2915 CrossRef CAS PubMed.
- J. Paterson, M. E. Potter, E. Gianotti and R. Raja, Chem. Commun., 2011, 47, 517 RSC.
- N. R. Shiju, S. Fiddy, O. Sonntag, M. Stockenhuber and G. Sankar, Chem. Commun., 2006, 4955 RSC.
- P. A. Barrett, G. Sankar, C. R. A. Catlow and J. M. Thomas, J. Phys. Chem., 1996, 100, 8977 CrossRef CAS.
- L. Li, G. D. Li, C. Yan, X. Y. Mu, X. L. Pan, X. X. Zou, K. X. Wang and J. S. Chen, Angew. Chem., 2011, 123, 8449 CrossRef.
- M. E. Potter, A. J. Paterson and R. Raja, ACS Catal., 2012, 2, 2446 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- X. Y. Quek, R. Pestman, R. A. van Santen and E. J. M. Hensen, Catal. Sci. Technol., 2014, 4, 3510 CAS.
- D. A. McKeown, P. L. Hagans, L. P. L. Carette, A. E. Russell, K. E. Swider and D. R. Rolison, J. Phys. Chem. B, 1999, 103, 4825–4832 CrossRef CAS.
- C. I. Hiley, M. R. Lees, J. M. Fisher, D. Thompsett, S. Agrestini, R. I. Smith and R. I. Walton, Angew. Chem., Int. Ed., 2014, 53, 4423 CrossRef CAS PubMed.
- J. Zhang, B. Sun, Z. Guan, H. Wang, H. Bao, Y. Haung, J. Qiao and G. Zhou, Environ. Sci. Technol., 2013, 47, 13011 CrossRef CAS PubMed.
- L. Gomez-Hortiguela, F. Cora, G. Sankar, C. M. Zicovich-Wilson and C. R. A. Catlow, Chem. – Eur. J., 2010, 16, 13638 CrossRef CAS PubMed.
- J. M. Thomas, R. Raja, G. Sankar and R. G. Bell, Nature, 1998, 398, 227 CrossRef.
- M. E. Potter, A. J. Paterson, B. Mishra, S. D. Kelly, S. R. Bare, F. Cora, A. B. Levy and R. Raja, J. Am. Chem. Soc., 2015, 137, 8534 CrossRef CAS PubMed.
- S. Altwasser, R. Glaser, A. Sulaiman Lo, P. H. Liu, K. J. Chao and J. Weitkamp, Microporous Mesoporous Mater., 2006, 89, 109 CrossRef CAS.
- A. I. Frenkel, C. W. Hills and R. G. Nuzzo, J. Phys. Chem. B, 2001, 105, 12689 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6me00061d |
| This journal is © The Royal Society of Chemistry 2016 |