
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of DNA-encoded libraries based on a benzodiazepine and a pyrazolopyrimidine scaffold†‡
M. Klika
Škopić§
a,
O.
Bugain§
a,
K.
Jung
a,
S.
Onstein
a,
S.
Brandherm
a,
T.
Kalliokoski
b and
A.
Brunschweiger
*a
aFaculty of Chemistry and Chemical Biology, Technical University of Dortmund, Otto-Hahn-Straße 6, D-44227 Dortmund, Germany. E-mail: andreas.brunschweiger@tu-dortmund.de; Fax: +49 231 7557080; Tel: +49 231 7557085
bLead Discovery Center GmbH, Otto-Hahn-Straße 15, D-44227 Dortmund, Germany
First published on 27th July 2016
Abstract
Selection-based screening of large DNA-encoded libraries of drug-like small molecules is a validated method to identify bioactive compounds. Among the chemical space of bioactive compounds certain scaffold structures are well represented. These are commonly called “privileged scaffolds”. We have synthesized DNA-encoded libraries based on two representatives of these scaffolds, a benzodiazepine and a pyrazolopyrimidine, and additionally a third library based on propargyl glycine. All three core structures possess a carboxylic acid to couple them to aminolinker-modified DNA. For subsequent library synthesis they contained an amino function to which a set of carboxylic acid building blocks were coupled, and a terminal alkyne that was reacted with a set of azides to furnish triazoles. The two sets of building blocks, 114 carboxylic acids and 104 azides, were selected with the help of chemoinformatic methods in order to control the physicochemical properties of the final libraries, remove unwanted substructures, and maximize diversity. The set of building blocks contained desthiobiotin allowing for validation of library synthesis. The DNA-encoded libraries were synthesized by split-and-pool combinatorial chemistry yielding three libraries that contain 28.254 compounds together. For DNA barcoding, 5′-phosphorylated double-stranded coding DNA sequences with four base overhangs were ligated with T4 ligase. The resulting DNA-encoded libraries were compared to bioactivity databases and, though being based on core structures well-established in medicinal chemistry, showed novelty with respect to the known bioactive chemical space.
Introduction
The selection of large, pooled DNA-encoded libraries (DELs) of drug-like small molecules is a validated method for the target-based identification of bioactive compounds.1 It is an operationally simple, economic screening technology that allows to interrogate large chemical space in a single experiment. The method holds promise to make a contribution to the validation of the plethora of putative drug targets emerging from the “-omics” technologies.2 Indeed, several valuable chemical biology probes that aid in gaining a deeper understanding of pathophysiology trace their ancestry to screening of DELs.3 Analysis of the chemical space of bioactive compounds has revealed that certain scaffold structures are highly represented.4 In the literature these are often referred to as “privileged scaffolds”. A scaffold is here defined as the core element of a molecule to which the substituents are connected. One of the archetypal privileged scaffolds is the benzodiazepine core 5 (Fig. 1)5 which can be found in hundreds of bioactive compounds, among them natural products such as asperlicin C, clinical candidates, and approved drugs. An exemplary structure of this compound class is the clinically evaluated farnesyltransferase inhibitor BMS214662 (2, Fig. 1A).6 | ||
| Fig. 1 (A) The approved drug ibrutinib 1 and the clinical candidate BMS214662 2; (B) reduction of these compounds to the core structures 3–5; (C) core structures functionalized for encoded library synthesis 6–8; (D) DNA-encoded libraries 9–11 based on the structures 6–8. | ||
While the benzodiazepine core targets proteins from diverse families thus meeting the initial definition of the term “privileged”, i.e. a structure capable of binding multiple targets,5 the pyrazolopyrimidine 4, exemplified by the approved kinase inhibitor ibrutinib 1,7 falls into a different class of privileged scaffolds. This structure resembles the nucleobase adenine and is thus biased towards adenine (–nucleotide) binding sites, e.g. in kinases.8,9
Several strategies have been developed for the synthesis of DNA-encoded libraries.1,3 These libraries can be furnished in a templated manner,10 exemplified by synthesis of encoded libraries of macrocycles and the yoctoreactor approach. However, templated synthesis requires coupling of all building blocks to oligonucleotides prior library synthesis, and building blocks used for library synthesis need to be bifunctional. Also, oligonucleotides can be used to encode and to template building blocks for fragment screening, e.g. in the ESAC (Encoded Self-Assembling Chemical libraries) format.11 The most common format for DNA-encoded library synthesis is the combinatorial split-and-pool approach with iteration of synthesis and encoding steps.12 The synthesis steps are recorded by chemical or enzymatic DNA-ligation techniques; Klenow fill-in is an efficient method to encode libraries composed of two building blocks.12,13 The present libraries were planned to consist of three building blocks. Each contains a central scaffold serving as vector for two sets of building blocks, thus enzymatic ligation of double-stranded, 5′-phosphorylated DNA sequences using T4 DNA ligase which efficiently connects was chosen for encoding. There is only a limited repertoire of organic synthesis reactions available that are amenable for DEL synthesis: mostly, carbonyl reactions such as amide bond or (thio)urea formation and reductive amination, C–C cross coupling reactions, e.g. the Suzuki reaction, nucleophilic (aromatic) substitution of reactive halides, and the Cu(I)-catalyzed azide–alkyne cycloaddition were used for library synthesis.1,3 These reactions allow for appendage of building blocks to properly functionalized structures. Synthesis strategies to substituted (hetero)cyclic structures from simple starting materials are less established for DNA-encoded library synthesis, and encompass for instance the Diels–Alder reaction, condensation reactions leading e.g. to benzimidazoles and imidazolidinones, and lately also a cascade reaction to spirocyclic structures.14–16
For the synthesis of the present libraries, compounds 1 and 2 were reduced to their core pyrazolopyrimidine structure 4 and benzodiazepine 5 (Fig. 1B),17 respectively. They, and the amino acid 6, which served initially to develop the library synthesis strategy, display functionalities for encoded library synthesis with DNA-compatible preparative organic synthesis methods (7, 8, Fig. 1C): a carboxylic acid to couple the scaffolds to 5′-aminolinker modified DNA, an Fmoc-protected amine for appendage of carboxylic acid building blocks by amide coupling, and a terminal alkyne for appendage of azide building blocks by Cu(I) catalyzed azide–alkyne cycloaddition (CuAAC), respectively. Two orthogonal reactions were chosen for library synthesis as this obviated the requirement of additional protective group chemistry. Amide synthesis is a workhorse reaction in the synthesis of DELs for which thousands of carboxylic acid building blocks are available.18–20 The CuAAC was less employed in reported DEL synthesis, although this reaction is compatible with DNA,21 has been extensively used in the synthesis of bioactive compounds,22–25 has broad functional group tolerance, does not demand protective group chemistry, and is a high-yielding reaction, which is important for library quality, i.e. an even distribution of the individual library members.26 Moreover, although only few azides are available,20 they are readily accessible from abundantly available precursors such as aryl amines and halides.27In situ synthesis of reactants was described in only a few reports on DNA encoded library synthesis.14,26,28 To the set of building blocks elected for library synthesis we added the streptavidin binder desthiobiotin to validate library synthesis.12b
Here, we show the synthesis of compounds 7 and 8, the chemoinformatics-supported selection of building blocks, the evaluation of these building blocks and the synthesis of three encoded libraries 9–11 (Fig. 1D) that each consist of a central scaffold structure serving as vector to project two sets of building blocks.
Results and discussion
Synthesis of trifunctionalized tetrahydrobenzodiazepine and pyrazolopyrimidine scaffolds
The functionalized pyrazolopyrimidine 7 was synthesized in straightforward manner from 6-aminopyrazolopyrimidine 12 (Fig. 2A). A Mitsunobu reaction with protected serine introduced the required carboxylic acid and amine functionalities to the core heterocycle, and iodination of the heterocycle and Sonogashira coupling with TMS-protected acetylene introduced the terminal alkyne moiety, yielding compound 16 in a few steps. Protective group chemistry then gave the properly substituted and protected scaffold 7.9 However, in the basic deprotection step to compound 16a (Fig. 2A, and ESI‡ part) that removed both the TMS-group and the tert-butyl ester, we noticed racemization of the amino substituent. The tetrahydrobenzodiazepine was accessed through a somewhat lengthier synthesis route that started with the bromination of isatoic anhydride 17 (Fig. 2B), the condensation of this product (18) with glycine methylester to the core heterocycle 19, and the reduction of the carbonyl groups of 19 yielding the tetrahydrobenzodiazepine structure 20.6,17 This was N-protected with a boc-group and reacted with acrylic acid tert-butylester by Heck reaction to introduce the carboxylic acid function. Protection of the amino function was needed to prevent N-alkylation by the acrylic acid ester. The Heck reaction was followed by hydrogenation of the double bond to remove the α,β-unsaturated Michael acceptor function (23). Alkylation of the aryl amine with propargyl bromide introduced the terminal alkyne. Finally, protective group chemistry furnished the target compound 8.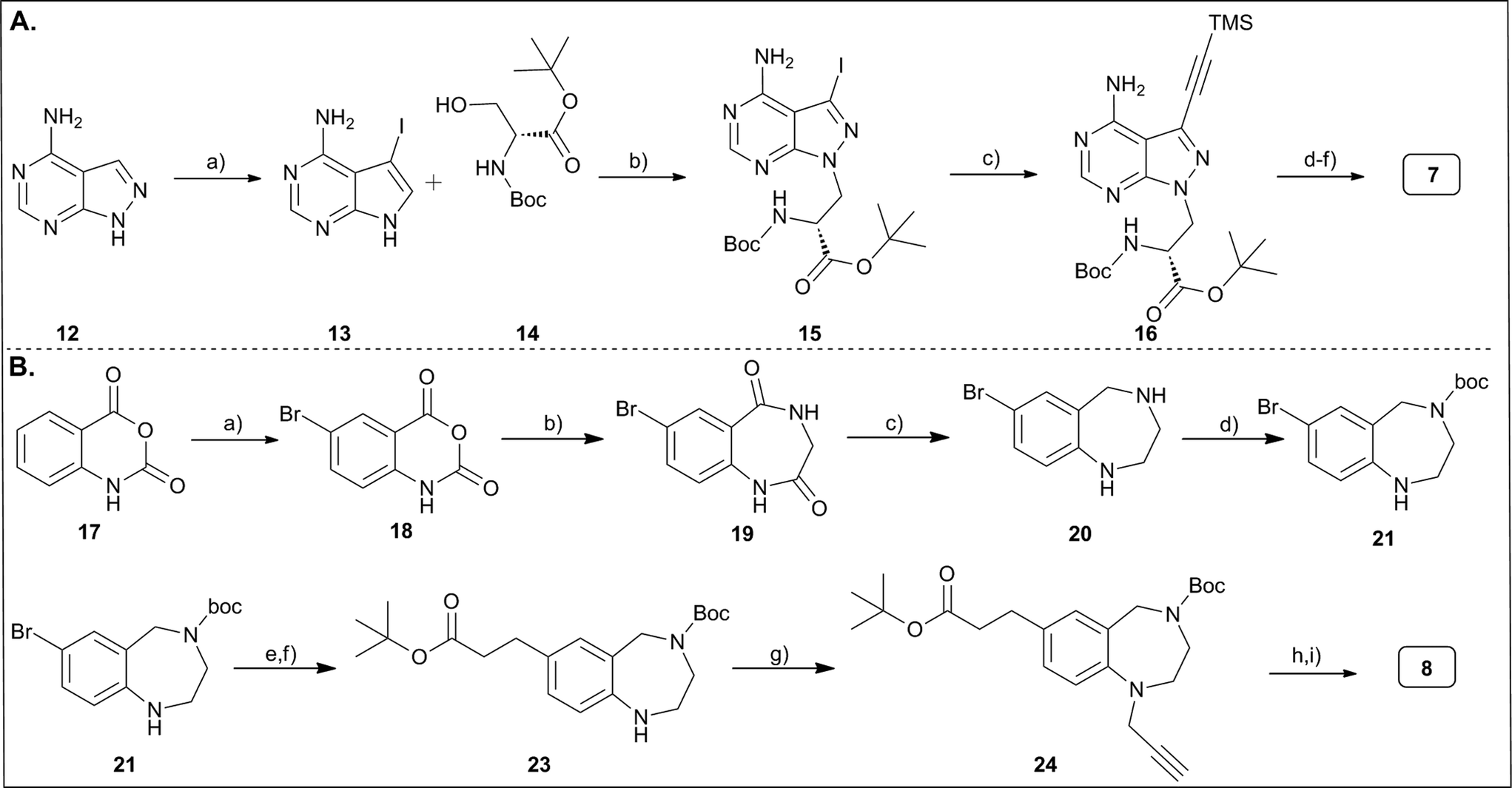 | ||
| Fig. 2 Synthesis of privileged scaffolds functionalized for combinatorial DEL-synthesis. (A) Synthesis of aminopyrazolopyrimidine 7. Reagents and conditions: a) N-iodosuccinimide, DMF, 80 °C, 14 h; b) PPh3, DIAD, THF, rt, 16 h; c) Pd(PPh3)4, CuI, Et3N, TMS-acetylene, DMF, rt, 18 h; d) K2CO3, MeOH, rt, 16 h; e) TFA, DCM, rt, 16 h; f) NaHCO3,Fmoc-OSu, H2O, 1,4-dioxane, rt, 16 h. (B) Synthesis of tetrahydrobenzodiazepine 8. Reagents and conditions: a) bromine in H2O at 50 °C for 1 h; b) glycine in H2O, Et3N, rt, 4 h; c) BH3 in dry THF, reflux, 18 h; d) “diboc” in dry MeOH, rt, 18 h; e) Pd(OAc)2, P(O-tol)3, tert-butyl acrylate, Et3N, dry CH3CN, 100 °C, 18 h; f) Pd/C, dry MeOH, rt, 18 h; g) Cs2CO3, propargyl bromide, dry DMF, 60 °C, 18 h; h) TFA in dry CH2Cl2, rt, 19 h; i) Fmoc-Osu, NaHCO3, H2O, dry 1,4-dioxane, rt, 18 h. | ||
Chemoinformatics-supported selection of carboxylic acid and azide building blocks for library synthesis
The scaffolds 6–8 allow for combinatorial substitution with building blocks by amide synthesis and by Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC). Both reactions are high yielding and have a broad scope, two properties that are important for sampling chemical space, and also for library quality (Fig. 3).1,3,18,19,26 | ||
| Fig. 3 Chemoinformatic filtering cascade to select from a large pool of commercially available building blocks a diverse set of carboxylic acids and halides endowing the projected DELs with defined properties. | ||
However, as only a limited number of azides are commercially available,20 we chose to use aliphatic and benzylic halides, and to convert these in situ into azides.27 There are thousands of aliphatic/benzylic halides and carboxylic acid building blocks available for library synthesis,20 so in order to facilitate the building block selection, we applied a chemoinformatic filtering cascade to a database of commercially available chemicals. For the calculation of the physicochemical properties the free carboxylic acid of compounds 6–8 was substituted with an ethyl amide. In the first step, we removed those building blocks that would yield library members with physicochemical properties outside pre-defined values: the calculated log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of all library members was to fall into the range of −2–5, and the molecular weight was not to exceed 650 Da including the linker structure connecting the core structure to the DNA barcode. In the second step, we removed building blocks that may show unwanted reactivity, for instance redox reactions or covalent reactions with the DNA barcode or the target (PAINS).29 Then, from the remaining two sets of building blocks, we made a selection of each 150 chemicals with RDKit diversity picker using Morgan fingerprints in order to maximize the diversity of the screening library.30,31 Finally, the collections of carboxylic acids and halides were manually curated by removing overly expensive compounds, and those that would likely not react or give rise to side products, for instance acids with an unprotected aliphatic amino group, dicarboxylic acids, or dihalides. We finally arrived at a set of 114 carboxylic acids (compounds A-DF, Table S1, ESI‡) and 104 halides (compounds 1–104, Table S1, ESI‡) for the synthesis of the DNA-encoded libraries 9–11 (Fig. 1). Within both sets of building blocks, aliphatic, cyclic aliphatic, aromatic and heteroaromatic structures, most of them substituted with heteroatoms, some of them displaying functional groups, are presented. Inspection of these fragment-sized building blocks (Tables S1 and S2‡) revealed a number of structures that can be found in bioactive compounds: for instance, the benzopyrazole AI (Table S1‡) is a fragment binding to the kinase CDK2;32 dihydrouracil BK was identified as binding motif for members of the family of PARP enzymes from a screen of a DNA-encoded library,33 and the uracil BN is a feature of a compound binding to the activated complement factor C3d.34 Other fragments as for instance the indole R, the benzofuran AC and the benzimidazole 75 (Table S2‡) are found in numerous bioactive compounds. Although the projected DNA-encoded libraries 9–11 are based on scaffolds that constitute core structures of several bioactive compounds, and include several fragments that are well presented in bioactive molecules, they sample chemical space that is hitherto not covered. The novelty of the compounds was assessed by first enumerating all of the compounds produced by the three scaffolds and the selected building blocks and then calculating the nearest-neighbour (NN) similarity of these structures using Morgan- and Feat Morgan-fingerprints to the bioactivity database ChEMBL 21 (∼2 million compounds), the patent database SureChEMBL (April 2016 edition, ∼16 million compounds) and the bioactivity database PubChem (March 2016 edition, ∼89 million compounds). The NN-Tanimoto similarities from these large databases to the generated library are shown in Table 1. For instance, the nearest neighbour to library 10, which is based on the aminopyrazolopyrimidine, an adenine-mimicking scaffold often found in kinase inhibitors, is in the ChEMBL database compound CHEMBL2023774. This compound displays the same 1,3-disubstituted aminopyrazolopyrimidine core scaffold, and inhibited the Src kinase.35 However, the maximum Morgan/Feat Morgan–Tanimoto-similarity of the chemical space described in the bioactivity databases to the libraries 9–11 is generally rather low. Surprisingly, it is much higher to library 9 than to the pyrazolopyrimidine- and benzodiazepine-based libraries 10 and 11. This can be judged either beneficial, as these libraries cover novel chemical space which is desirable for a screening collection, or detrimental, as the DNA-compatible chemistry used to substitute the scaffolds 4 and 5 might bias the libraries towards chemical space that was recently described as “dark chemical matter”, compound classes in screening library collections that rarely show biological activity.36
P of all library members was to fall into the range of −2–5, and the molecular weight was not to exceed 650 Da including the linker structure connecting the core structure to the DNA barcode. In the second step, we removed building blocks that may show unwanted reactivity, for instance redox reactions or covalent reactions with the DNA barcode or the target (PAINS).29 Then, from the remaining two sets of building blocks, we made a selection of each 150 chemicals with RDKit diversity picker using Morgan fingerprints in order to maximize the diversity of the screening library.30,31 Finally, the collections of carboxylic acids and halides were manually curated by removing overly expensive compounds, and those that would likely not react or give rise to side products, for instance acids with an unprotected aliphatic amino group, dicarboxylic acids, or dihalides. We finally arrived at a set of 114 carboxylic acids (compounds A-DF, Table S1, ESI‡) and 104 halides (compounds 1–104, Table S1, ESI‡) for the synthesis of the DNA-encoded libraries 9–11 (Fig. 1). Within both sets of building blocks, aliphatic, cyclic aliphatic, aromatic and heteroaromatic structures, most of them substituted with heteroatoms, some of them displaying functional groups, are presented. Inspection of these fragment-sized building blocks (Tables S1 and S2‡) revealed a number of structures that can be found in bioactive compounds: for instance, the benzopyrazole AI (Table S1‡) is a fragment binding to the kinase CDK2;32 dihydrouracil BK was identified as binding motif for members of the family of PARP enzymes from a screen of a DNA-encoded library,33 and the uracil BN is a feature of a compound binding to the activated complement factor C3d.34 Other fragments as for instance the indole R, the benzofuran AC and the benzimidazole 75 (Table S2‡) are found in numerous bioactive compounds. Although the projected DNA-encoded libraries 9–11 are based on scaffolds that constitute core structures of several bioactive compounds, and include several fragments that are well presented in bioactive molecules, they sample chemical space that is hitherto not covered. The novelty of the compounds was assessed by first enumerating all of the compounds produced by the three scaffolds and the selected building blocks and then calculating the nearest-neighbour (NN) similarity of these structures using Morgan- and Feat Morgan-fingerprints to the bioactivity database ChEMBL 21 (∼2 million compounds), the patent database SureChEMBL (April 2016 edition, ∼16 million compounds) and the bioactivity database PubChem (March 2016 edition, ∼89 million compounds). The NN-Tanimoto similarities from these large databases to the generated library are shown in Table 1. For instance, the nearest neighbour to library 10, which is based on the aminopyrazolopyrimidine, an adenine-mimicking scaffold often found in kinase inhibitors, is in the ChEMBL database compound CHEMBL2023774. This compound displays the same 1,3-disubstituted aminopyrazolopyrimidine core scaffold, and inhibited the Src kinase.35 However, the maximum Morgan/Feat Morgan–Tanimoto-similarity of the chemical space described in the bioactivity databases to the libraries 9–11 is generally rather low. Surprisingly, it is much higher to library 9 than to the pyrazolopyrimidine- and benzodiazepine-based libraries 10 and 11. This can be judged either beneficial, as these libraries cover novel chemical space which is desirable for a screening collection, or detrimental, as the DNA-compatible chemistry used to substitute the scaffolds 4 and 5 might bias the libraries towards chemical space that was recently described as “dark chemical matter”, compound classes in screening library collections that rarely show biological activity.36
| Descriptor | Library 9 | Library 10 | Library 11 |
|---|---|---|---|
|
a Mean molecular weight (MW), calculated logP (clogP), fraction of the sp3-carbons (Fsp3), topical polar surface area (TPSA), number of rotatable bonds (NROT), number of hydrogen bond acceptors (HBA) and number of hydrogen bond donors (HBD). All of these values were calculated using the combination of RDKit and ChemFP.
b Maximum Morgan/Feat Morgan–Tanimoto-similarity in parenthesis.
|
|||
| NN-ChEMBL | CHEMBL2023-774 (0.688) | CHEMBL1683-305 (0.597) | CHEMBL1922-546 (0.569) |
| NN-Sure-ChEMBL | SCHEMBL1262 7717 (0.750) | SCHEMBL1316 0649 (0.605) | SCHEMBL1328 8590 (0.600) |
| NN-Pub-Chem | 98041443 (0.780) |
44235677 (0.685) |
42499651 (0.645) |
| Mean MW | 433 Da | 566 Da | 578 Da |
| Mean clogP |
1.3 | 1.2 | 3.6 |
| Mean Fsp3 | 0.483 | 0.387 | 0.454 |
| Mean TPSA | 120b Å | 190b Å | 115b Å |
| Mean NROT | 10 | 11 | 11 |
| Mean HBA | 7 | 12 | 8 |
| Mean HBD | 3 | 4 | 2 |
The properties of the three libraries were analyzed by typical descriptors that were found statistically associated with peroral bioavailability (Table 1):37,38 molecular weight, calculated logP to assess the mean lipophilicity of the libraries, the topological surface area, the number of rotatable bonds, the number of hydrogen bond donors and acceptors (mean values given). The fraction of sp3-hybridized carbon atoms has been suggested as a metric associated with clinical success.39 While the mean values of all parameters of library 9 fall into ranges statistically associated with peroral bioavailability, libraries 10 and 11 that are based on larger scaffold structures show a higher mean molecular weight and the aminopyrazolopyrimidine library 10 has a higher mean topical polar surface area.
Large diversity in the shapes of the molecules is also a desired feature in a screening library.40,41 Here, the shape diversity of the libraries was investigated by first generating single low-energy conformation for each of the library compounds using Schrödinger Suite 2016.1 and then calculating two 3D-diversity metrics for the molecules: the normalized principal moments of inertia ratios (NPRs) and plane of best fit score (PBF score). Briefly, NPRs are numeric values that describe the overall three-dimensional shapes of the library molecules. When these values are plotted against each other, they form a triangle where the corners represent rods, spheres and disks (for further information, see ref. 40). PBF score describes how different conformation of a molecule is from its 2D representation. It is a value of usually between zero and two for drug-like molecules, the higher value indicating higher 3D character (additional information is available in ref. 41) The NPR-plots show clearly that all libraries 9–11 cover a wide range of shape diversity as libraries 9–11 all focus on different regions of NPR-space (Fig. S17‡). In addition, most of the compounds have PBF-score above 1, which indicates a high 3D-character for the DEL library (Fig. S18‡).
Synthesis of the DNA-encoded libraries 9–11
The synthesis of the DNA-encoded libraries 9–11 was performed as outlined in Fig. 4: scaffold structures 6–8 (Fig. 1) were coupled to a fully protected, solid phase-bound 5′-aminolinker modified single-strand DNA sequence, and the Fmoc-group was removed (27–29).19,33 Then, 114 carboxylic acid building blocks A-DJ (Table S1‡) were reacted with the three DNA-conjugates 27–29. All conjugates 27A-DJ–29A-DJ were cleaved from the solid phase and each DNA-conjugate was purified to a single peak by ion pair reversed phase HPLC in order to synthesize the DNA-encoded library from a uniform set of DNA-conjugates which we deemed beneficial for library quality.26 These purified conjugates were then encoded with double-stranded DNA sequences by two successive DNA ligation reactions with T4 ligase. The library synthesis was concluded with appendage of a second set of azide building blocks 1–104 (Table S2‡) by copper(I)-catalyzed alkyne–azide cycloaddition on DEAE sepharose.27,42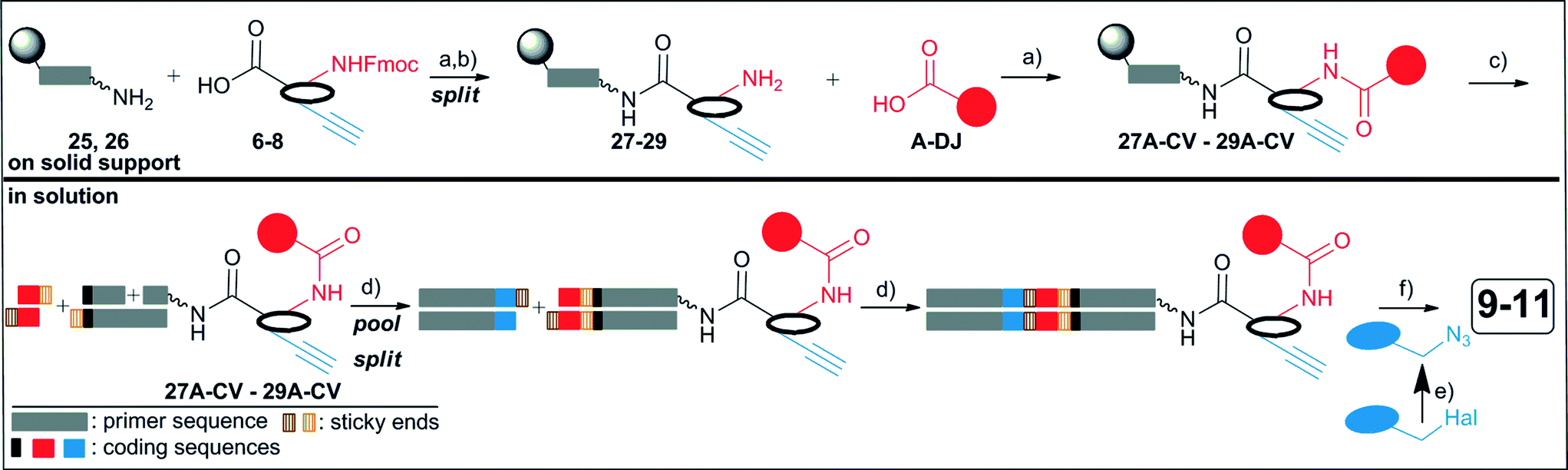 | ||
| Fig. 4 Synthesis of the DEL by a two stage synthesis strategy. a) HATU; b) piperidine in DMF; c) aq. NH3/aq. MeNH2; d) encoding by T4 DNA ligation; e) NaN3 in DMF; f) DEAE sepharose, Cu(I), TBTA, Na-ascorbate. | ||
Library synthesis was initiated with coupling of a protected amino-PEG-carboxylic acid using HATU as coupling reagent to a solid phase-coupled 5′-C6-aminolinker modified 23mer DNA containing the primer and scaffold code by amide synthesis on the 1 μmol-scale (see ESI‡). Prior removal of the protective group, unreacted amines were capped with acetic acid anhydride. We found both MMt-amino-PEG(8)-linker and Fmoc-protected amino-PEG(4)-linker suitable (Table S1‡). However, prolonged deprotection of the Fmoc-group with piperidine/DMF led to formation of a lipophilic side product. This side product was likely due to a transamidation reaction and has also been noticed by others.43 Reducing the deprotection time to 5 minutes suppressed formation of this side product effectively. In the next step, compounds 6–8 were coupled to the DNA-PEG-linker conjugates 25 (from the MMt-protected PEG-linker) and 26 (from the Fmoc-protected PEG-linker) on the 1 μmol-scale, unreacted amines were again capped, and the Fmoc-group of the scaffolds was removed with piperidine/DMF. Library synthesis continued with the parallel coupling of the carboxylic acid building blocks A-DJ (Table S1‡). It was most convenient to couple sets of 20 carboxylic acids to the three DNA conjugates 27–29, i.e. performing in total 60 reactions in parallel. Each solid phase (400 nmol, ca. 16 mg) containing the conjugates 27–29 was split in 20 nmol aliquots into a 96 well plate by suspending in the bulk solid phase in DMF and splitting the suspension (see ESI‡). Then, 20 carboxylic acid building blocks (Table S1‡) were coupled in parallel to each DNA scaffold conjugate using HATU as coupling reagent. The solid support-bound DNA conjugates were transferred to a 96 well filter plate, thoroughly washed, and deprotected and cleaved from the CPG with aq. ammonia/methylamine on the filter plate which was connected to a receiver plate and sealed for this purpose. The conjugates were then purified by ion pair reverse phase HPLC. Evaluating this process after coupling two sets of carboxylic acid building blocks, i.e. 40 carboxylic acids, we noted that ten (H, W, AC, AD, AE, CW, CX, CY, CZ, DA, Table S1‡) of these building blocks did not yield the target amide products. As efficient Fmoc-peptide chemistry has been reported for the synthesis of peptide conjugates of shorter solid support-bound DNA oligonucleotides,43 we changed the initial sequence from a 23mer DNA to a shorter 14mer. This shorter DNA-sequence allowed us to obtain amide coupling products from five of the ten carboxylic acid building blocks (H, W, AC, AD, AE). However, lack of detection of an amide coupling product could either be due to low reactivity of the carboxylic acid or to susceptibility of the amide bond to hydrolytic cleavage in the deprotection step.
Having coupled the whole set of 114 carboxylic acid building blocks A-DJ (Table S1‡) to the three DNA-conjugates 27–29 we obtained 99 products for the DNA-amino acid conjugate 27, 84 products for the DNA-pyrazolopyrimidine conjugate 28, and 94 products for DNA-benzodiazepine conjugate 29. For a statistic see Fig. S1.‡ The coupling efficiencies were very variable, while the amino acid 27 and the secondary amine of the benzodiazepine 29 yielded more than 80 of the target amides with good conversion rates, the pyrazolopyrimidine gave only 52 amides in acceptable to good yields. In hindsight, the varying coupling efficiency of the carboxylic acids justified the effort to purify and isolate every conjugate. HPLC analysis indicated a purity of more than 95% of the ion pair chromatography-purified conjugates 27A-CV–29A-CV and MALDI MS analysis confirmed the identity of the products (Table S1,‡ exemplary HPLC traces of amide coupling products: Fig. S19–S28‡). Interestingly, while most building blocks gave a shift to longer retention times, a number of building blocks caused a shift to shorter retention times. These were polar structures such as the primary amide H and the dihydrouracil BL. Thus, through HPLC-purification and isolation of the DNA-conjugates 27A-CV–29A-CV we obtained a uniform set of 277 DNA-small molecule conjugates for encoding and combinatorial library synthesis.
The second set of building blocks was introduced by Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC). As the analysis of complex mixtures of hundreds of DNA conjugates is not a trivial task, we evaluated the reactivity of the in situ synthesized azides with a DNA–alkyne conjugate 30 (Table S2‡) prior library synthesis.12a,33 Each 400 pmol of DNA–alkyne conjugate 30 was immobilized on DEAE sepharose in 96 well plates,42 while the azides were prepared by substitution of the halides with NaN3/tetrabutylammonium iodide.27 The cycloaddition reaction was then performed according to a previously established procedure with a 2500 fold excess of the azide at 45 °C for 16 h.27 Prior to elution of the triazole products, the resin was extensively washed to remove the excess of reactants and reagents. A 1 N buffered aqueous solution of EDTA was used to remove Cu-ion contaminants as these might compromise DNA stability during storage. The conjugates were analyzed by MALDI MS (Table S2‡). From the 104 azides tested in this experiment, 102 yielded the triazole products, for a statistical analysis, see Fig. S1.‡ For 82 azides complete conversion was detected by MALDI MS, while in case of azide building blocks 10, 16, 38, 44, 51, 53, 62, 68, 75, 76, 77, 101, 102 and 104 we noticed incomplete conversion, yet the yield for these building blocks exceeded 50% as estimated by MALDI MS analysis which we judged sufficient for library synthesis.12a These building blocks are mostly heterocyclic structures. However, only azides 6 and 87 did not yield the target triazoles at all. We then tested also a set of azide building blocks with a DNA conjugate of the amino acid 6 (31, see ESI,‡ Fig. S29) and detected quantitative conversion to the target triazoles by CuAAC.
The target DNA-encoded libraries 9–11 were encoded by T4 ligation of 5′-phosphorylated dsDNA containing overhangs.12a,c The conditions for enzymatic ligation were optimized with dsDNA sequences (Fig. S2 and S7, Tables S5 and S6‡) that contained tetramer overhangs. Several parameters were tested in combinatorial manner: ligation time, temperature, different buffer systems, ratio of 5′-phosphorylated to non-phosphorylated counter strand, and concentration of the T4 ligase (Table S4, Fig. S3–S6 and S8‡). Suitable conditions for ligation of dsDNA sequences were found for encoding of the DNA-encoded libraries: equal amounts of 5′-phosphorylated to non-phosphorylated counter strand, use of a 10 fold concentrated buffer that allowed for higher concentrations of the enzyme and the dsDNA, and a ligation temperature of 25 °C. The dsDNAs were ligated either for 4 hours or overnight with equal efficiency.
A chemically synthesized 69mer dsDNA VI/VI′ (Table S8‡) that served as surrogate of the DNA-encoded library was used to evaluate the primer efficiency. Even a high concentration of 100 pM template DNA VI/VI′ required more than 10 cycles of amplification with its primer pair VIII/IX (Table S8‡) to detect initiation of amplification with SYBR green, and 1 pM template required 17 cycles to detect initiation of amplification, while with the primer pair VIII/IX (Table S8‡) alone we detected initiation of amplification already at 21 cycles (Fig. S9a‡) due to formation of primer dimers. We therefore tested a number of primer pairs, arriving at the sequences X/XI which amplified its template DNA VII/VII′ much more efficiently (Fig. S9b‡) and showed no formation of primer dimers over 30 cycles of amplification. Thus, an optimized ligation protocol and efficient primer pairs were established. These were confirmed by comparison of the amplification plots (Fig. S12‡) of equal amounts of the chemically synthesized reference template DNA VII/VII′ (Table S8‡) and the DNA duplex 29CP-XVI/XVII (sequence: see Table S10‡) containing the same primer and coding sequence that was accessed from the desthiobiotin-substituted benzodiazepine DNA-conjugate 29CP (Table S1‡) through the optimized ligation protocol (Fig. S9, and S10, sequences see Table S10, for the ligation scheme see Fig. S13 and S14‡).
With the optimized conditions for T4 DNA ligation, a validated, efficient pair of primers, a purified set of DNA-small molecule conjugates 27A-CV–29A-CV, and a validated set of azide building blocks in hand we synthesized the three DNA-encoded libraries 9–11 (Fig. 4). In the first encoding step, an amount of 40 pmol of each purified and characterized DNA conjugate 27A-CV–29A-CV was ligated in one pot with a 5′-phosphorylated dsDNA that contained the optimized primer sequence and the code of the scaffold, and 5′-phosphorylated short 12mer dsDNA sequences encoding the building blocks A-CV with T4 DNA ligase (scheme for encoding: Fig. S13 and S14‡). The ligation reactions from each scaffold 27–29 were pooled and the DNA was precipitated with 70% aqueous ethanol for overnight. The pellets were re-dissolved, an aliquot of each pooled library was taken and analyzed to confirm successful ligation (Fig. S15‡) and again precipitated for two hours with 70% aqueous ethanol. The pellet was dissolved in water, and split into 102 wells of two 96 well plates, and, after 5′-phosphorylation with polynucleotide kinase, ligated with a set of 102 dsDNA sequences containing the code for the azide building blocks 1–102 and the reverse PCR primer. These 102 dsDNA sequences were added in 2 fold excess to drive the ligation to completion. The encoded DNA-conjugates were directly transferred to DEAE sepharose and reacted with the azides 1–102 as described above. After the reaction, the library was eluted from the resin, pooled, and twice precipitated. The pellet was dissolved, and an aliquot was analyzed by gel analysis (Fig. S16‡) and by qPCR (Table S9‡). The qPCR analysis indicated a loss of one ct-value, as compared the chemically synthesized reference DNA VII/VII′, i.e. a loss of 50% of amplifiable DNA. This loss might be due to some degradation of the DNA because of Cu(I)-mediated oxidation and fragmentation.
Finally, the encoded desthiobiotin conjugate 29CP-XVI/XVII and the chemically synthesized non-modified DNA VII/VII′ were used to establish the selection assay with streptavidin beads as a model system.12b Both the protein binder 29CP-XVI/XVII and the negative control dsDNA VII/VII′ were incubated at a 100 pM concentration with the beads. The beads were washed eight times with washing buffer, and then heat-denatured to release the oligonucleotides. These were incubated with a fresh batch of streptavidin beads and again washed eight times with washing buffer. The desthiobiotin conjugate was recovered with only a minor loss of DNA (−0.3 ct values) while the amount of the non-modified DNA was reduced by 5 ct values, i.e. by ca. 97%. In other words, the binder was enriched by a factor of 23 (Fig. 5).
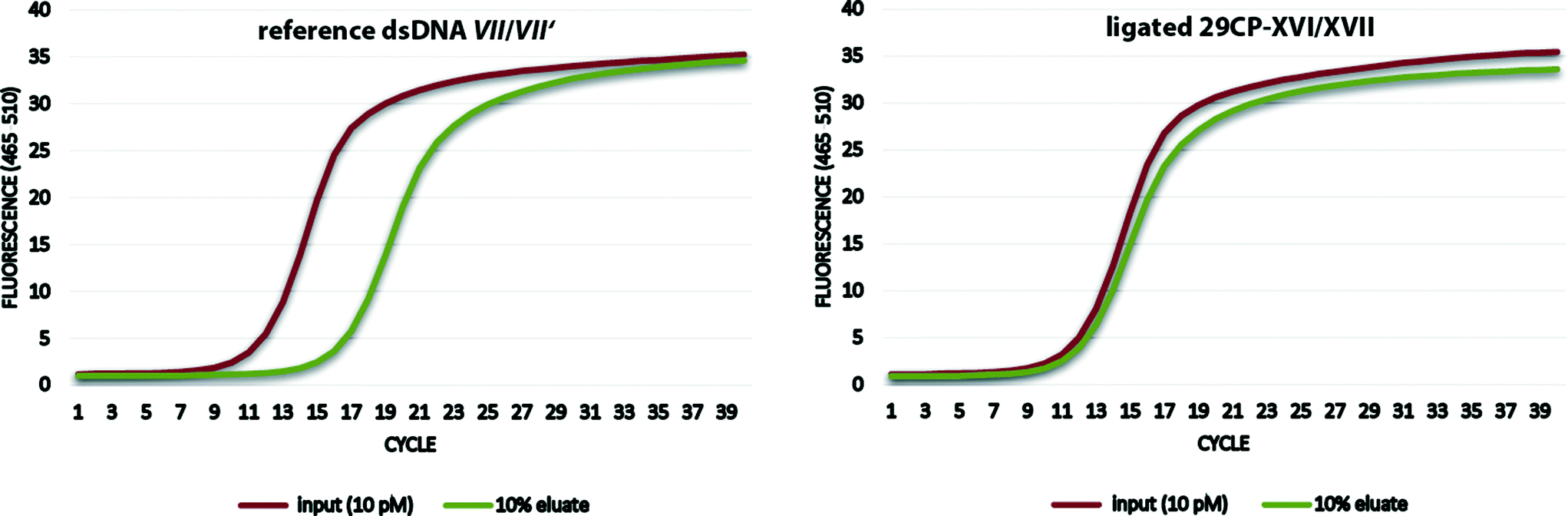 | ||
| Fig. 5 qPCR-analysis of the selection of the synthetic reference dsDNA VII/VII′ versus the encoded compound 29CP-XVI/XVII on streptavidin beads. | ||
Conclusions
We have designed and synthesized three DNA-encoded libraries 9–11 containing 28.254 molecules. The libraries are based on three core structures, two of them heterocycles that are found in many bioactive molecules: the kinase-targeted pyrazolopyrimidine 4, and the tetrahydrobenzodiazepine 5 (Fig. 1). These scaffolds were substituted with functional groups (6–8) allowing for combinatorial DNA-encoded library synthesis using high-yielding DNA-compatible reactions with broad reactant scope: amide coupling and Cu(I)-catalyzed azide–alkyne cycloaddition. The substituents for library synthesis were selected with the aid of chemoinformatic tools to filter out unwanted structural motifs such as PAINS and to control the physicochemical properties of the library members. The structural diversity of the substituents to be introduced into the library was maximized subsequent to the filtering steps. The chemical space of DNA-encoded libraries 9–11 showed low similarity to three databases of bioactive compounds indicating that these libraries covered novel chemical space.The synthesis of the DEL was initiated with the coupling of the scaffolds 6–8 to 5′-aminolinker-modified DNA on solid support. The first set of 114 carboxylic acid building blocks was appended to the DNA-scaffold conjugates by amide synthesis on the solid phase. These DNA conjugates were purified by ion-pair chromatography, and characterized. Roughly 80% of the carboxylic acids yielded the target products, though with variable yields justifying the effort to purify this first set of DNA conjugates. Library synthesis commenced with combinatorial ligation of coding dsDNA sequences with four-nucleotide overhangs by an optimized T4 DNA ligation protocol. The synthesis of the libraries was concluded with the introduction of a set of 102 validated azide building blocks. Finally, one library member containing desthiobiotin was used to validate the synthesis and encoding strategy through a successful selection on its target protein streptavidin.12b Currently, we are using the synthesized DNA-encoded libraries to identify novel binders for target proteins.
Acknowledgements
This work was supported by the German Federal Ministry of Education and Research (BMBF) Grant 131605. ChemAxon is acknowledged for the academic license for some of the software used in the chemoinformatics filtering cascade (http://www.chemaxon.com). The authors declare no competing interest.References
- (a) R. M. Franzini, D. Neri and J. Scheuermann, Acc. Chem. Res., 2014, 47, 1247–1255 CrossRef CAS PubMed; (b) M. A. Clark, Curr. Opin. Chem. Biol., 2010, 14, 396–403 CrossRef CAS PubMed; (c) R. E. Kleiner, C. E. Dumelin and D. R. Liu, Chem. Soc. Rev., 2011, 40, 5707–5717 RSC.
- (a) A. Brunschweiger and J. Hall, ChemMedChem, 2012, 7, 194–203 CrossRef CAS PubMed; (b) A. M. Edwards, R. Isserlin, G. D. Bader, S. V. Frye, T. M. Willson and F. H. Yu, Nature, 2011, 47, 163–165 CrossRef PubMed.
- H. Salamon, M. Klika Škopić, K. Jung, O. Bugain and A. Brunschweiger, ACS Chem. Biol., 2016, 11, 296–307 CrossRef CAS PubMed.
- (a) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed; (b) Y. Hu, D. Stumpfe and J. Bajorath, J. Med. Chem., 2016, 59, 4062–4076 CrossRef CAS PubMed; (c) Y. Hu and J. Bajorath, ChemMedChem, 2010, 5, 187–190 CrossRef CAS PubMed; (d) L. Costantino and D. Barlocco, Curr. Med. Chem., 2006, 13, 65–85 CrossRef CAS PubMed; (e) G. Müller, Drug Discovery Today, 2003, 8, 681–691 CrossRef.
- B. E. Evans, K. E. Rittle, M. G. Bock, R. M. DiPardo, R. M. Freidinger, W. L. Whitter, G. F. Lundell, D. F. Veber and P. S. Anderson, J. Med. Chem., 1988, 31, 2235–2246 CrossRef CAS PubMed.
- J. T. Hunt, C. Z. Ding, R. Batorsky, M. Bednarz, R. Bhide, Y. Cho, S. Chong, S. Chao, J. Gullo-Brown, P. Guo, S. H. Kim, F. Y. F. Lee, K. Leftheris, A. Miller, T. Mitt, M. Patel, B. A. Penhallow, C. Ricca, W. C. Rose, R. Schmidt, W. A. Slusarchyk, G. Vite and V. Manne, J. Med. Chem., 2000, 43, 3587–3595 CrossRef CAS PubMed.
- F. Cameron and M. Sanford, Drugs, 2014, 74, 263–271 CrossRef CAS PubMed.
- O. Prien, ChemBioChem, 2005, 6, 500–505 CrossRef CAS PubMed.
- M. Klein, P. Dinér, D. Dorin-Semblat, C. Doerig and M. Grøtli, Org. Biomol. Chem., 2009, 7, 3421–3429 CAS.
- (a) M. H. Hansen, P. Blakskjaer, L. K. Petersen, T. H. Hansen, J. W. Højfeldt, K. V. Gothelf and N. J. Hansen, J. Am. Chem. Soc., 2009, 131, 1322–1327 CrossRef CAS PubMed; (b) Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CrossRef CAS PubMed.
- (a) S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Nat. Biotechnol., 2004, 22, 568–574 CrossRef CAS PubMed; (b) J. Scheuermann and D. Neri, Curr. Opin. Chem. Biol., 2015, 26, 99–103 CrossRef CAS PubMed.
- (a) M. A. Clark, R. A. Acharya, C. C. Arico-Muendel, A. L. Belyanskaya, D. R. Benjamin, N. R. Carlson, P. A. Centrella, C. H. Chiu, S. P. Creaser, J. W. Cuozzo, C. P. Davie, Y. Ding, G. J. Franklin, K. D. Franzen, M. L. Gefter, S. P. Hale, N. J. Hansen, D. I. Israel, J. Jiang, M. J. Kavarana, M. S. Kelley, C. S. Kollmann, F. Li, K. Lind, S. Mataruse, P. F. Medeiros, J. A. Messer, P. Myers, H. O'Keefe, M. C. Oliff, C. E. Rise, A. L. Satz, S. R. Skinner, J. L. Svendsen, L. Tang, K. van Vloten, R. W. Wagner, G. Yao, B. Zhao and B. A. Morgan, Nat. Chem. Biol., 2009, 5, 647–654 CrossRef CAS PubMed; (b) L. Mannocci, Y. Zhang, J. Scheuermanna, M. Leimbacher, G. De Bellis, E. Rizzi, C. Dumelin, S. Melkko and D. Neri, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17670–17675 CrossRef CAS PubMed; (c) A. B. MacConnell, P. J. McEnaney, V. J. Cavett and B. M. Paegel, ACS Comb. Sci., 2015, 17, 518–534 CrossRef CAS PubMed.
- A. Litovchick, C. E. Dumelin, S. Habeshian, D. Gikunju, M. A. Guie, P. Centrella, Y. Zhang, E. A. Sigel, J. W. Cuozzo, A. D. Keefe and M. A. Clark, Sci. Rep., 2015, 5, 10916 CrossRef CAS PubMed.
- A. L. Satz, J. Cai, Y. Chen, R. Goodnow, F. Gruber, A. Kowalczyk, A. Petersen, G. Naderi-Oboodi, L. Orzechowski and Q. Strebel, Bioconjugate Chem., 2015, 26, 1623–1632 CrossRef CAS PubMed.
- F. Buller, Y. Zhang, J. Scheuermann, J. Schäfer, P. Bühlmann and D. Neri, Chem. Biol., 2009, 16, 1075–1086 CrossRef CAS PubMed.
- X. Tian, G. S. Basarab, N. Selmi, T. Kogej, Y. Zhang, M. Clark and R. A. Goodnow Jr., Med. Chem. Commun., 2016, 7, 1316–1322 RSC.
- E. Anouk Stigter, Z. Guo, R. S. Bon, Y. W. Wu, A. Choidas, A. Wolf, S. Menninger, H. Waldmann, W. Blankenfeld and R. S. Goody, J. Med. Chem., 2012, 55, 8330–8340 CrossRef PubMed.
- R. M. Franzini and C. Randolph, J. Med. Chem., 2016, 59, 6629–6644 CrossRef CAS PubMed.
- (a) I. Schwope, C. F. Bleczinski and C. Richert, J. Org. Chem., 1999, 64, 4749–4761 CrossRef CAS PubMed; (b) R. M. Franzini, F. Samain, M. Abd Elrahman, G. Mikutis, A. Nauer, M. Zimmermann, J. Scheuermann, J. Hall and D. Neri, Bioconjugate Chem., 2014, 25, 1453–1461 CrossRef CAS PubMed.
- T. Kalliokoski, ACS Comb. Sci., 2015, 17, 600–607 CrossRef CAS PubMed.
- A. Litovchick, C. E. Dumelin, S. Habeshian, D. Gikunju, M. A. Guié, P. Centrella, Y. Zhang, E. A. Sigel, J. W. Cuozzo, A. D. Keefe and M. A. Clark, Sci. Rep., 2015, 5, 10916 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- J. Hou, X. Liu, J. Shen, G. Zhao and P. G. Wang, Expert Opin. Drug Discovery, 2012, 7, 489–501 CrossRef CAS PubMed.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278–308 CrossRef CAS PubMed.
- A. L. Satz, ACS Chem. Biol., 2015, 10, 2237–2245 CrossRef CAS PubMed.
- A. Brunschweiger, L. F. Gebert, M. Lucic, U. Pradère, H. Jahns, C. Berk, J. Hunziker and J. Hall, Chem. Commun., 2016, 52, 156–159 RSC.
- F. Buller, L. Mannocci, Y. Zhang, C. E. Dumelin, J. Scheuermann and D. Neri, Bioorg. Med. Chem. Lett., 2008, 18, 5926–5931 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- RDKit: Open-source cheminformatics, http://www.rdkit.org Search PubMed.
- (a) D. Rogers, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed; (b) M. Ashton, J. Barnard, F. Casset, M. Charlton, G. Downs, D. Gorse, J. Holliday, R. Lahana and P. Willett, Quant. Struct.-Act. Relat., 2002, 21, 598–604 CrossRef CAS.
- P. G. Wyatt, A. J. Woodhead, V. Berdini, J. A. Boulstridge, M. G. Carr, D. M. Cross, D. J. Davis, L. A. Devine, T. R. Early, R. E. Feltell, E. J. Lewis, R. L. McMenamin, E. F. Navarro, M. A. O'Brien, M. O'Reilly, M. Reule, G. Saxty, L. C. Seavers, D. M. Smith, M. S. Squires, G. Trewartha, M. T. Walker and A. J. Woolford, J. Med. Chem., 2008, 51, 4986–4999 CrossRef CAS PubMed.
- R. M. Franzini, T. Ekblad, N. Zhong, M. Wichert, W. Decurtins, A. Nauer, M. Zimmermann, F. Samain, J. Scheuermann, P. J. Brown, J. Hall, S. Gräslund, H. Schüler and D. Neri, Angew. Chem., Int. Ed., 2015, 54, 3927–3931 CrossRef CAS PubMed.
- R. D. Jr Gorham, V. Nuñez, J. H. Lin, S. H. Rooijakkers, V. I. Vullev and D. Morikis, J. Med. Chem., 2015, 58, 9535–9545 CrossRef PubMed.
- A. Kumar, I. Ahmad, B. S. Chhikara, R. Tiwari, D. Mandal and K. Parang, Bioorg. Med. Chem. Lett., 2011, 21, 1342–1346 CrossRef CAS PubMed.
- (a) A. M. Wassermann, E. Lounkine, D. Hoepfner, G. Le Goff, F. J. King, C. Studer, J. M. Peltier, M. L. Grippo, V. Prindle, J. Tao, A. Schuffenhauer, I. M. Wallace, S. Chen, P. Krastel, A. Cobos-Correa, C. N. Parker, J. W. Davies and M. Glick, Nat. Chem. Biol., 2015, 11, 958–966 CrossRef CAS PubMed; (b) O. Eidam and A. L. Satz, Med. Chem. Commun., 2016, 7, 1323–1331 RSC.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6725–6756 CrossRef PubMed.
- W. H. Sauer and M. K. Schwarz, J. Chem. Inf. Model., 2003, 43, 987–1003 CrossRef CAS PubMed.
- N. C. Firth, N. Brown and J. Blagg, J. Chem. Inf. Model., 2012, 52, 2516–2525 CrossRef CAS PubMed.
- D. R. Halpin and P. B. Harbury, PLoS Biol., 2004, 2, 1015–1021 CAS.
- C. N. Tetzlaff, I. Schwope, C. F. Bleczinski, J. A. Steinberg and C. Richert, Tetrahedron Lett., 1998, 39, 4215–4218 CrossRef CAS.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00243a |
| § Equal contribution. |
| This journal is © The Royal Society of Chemistry 2016 |